

Abstract
CP-31398 (N′-[2-[2-(4-methoxyphenyl)ethenyl]-4-quinazolinyl]-N,N-dimethyl-1,3-propanediamine dihydrochloride) is a styrylquinazoline that stabilizes the DNA binding conformation of p53, thereby maintaining the activity of p53 as a transcription factor and tumor suppressor. In consideration of the potential use of p53 stabilizers for cancer prevention and therapy, 28-day studies (with recovery) were performed to characterize the toxicity of CP-31398 in rats and dogs. In the rat study, groups of 15 CD rats/sex received daily gavage exposure to CP-31398 at 0, 40, 80, or 160 mg/kg/day (0, 240, 480, or 960 mg/m2/day). In the dog study, groups of five beagle dogs received daily gavage exposure to CP-31398 at 0, 10, 20, or 40 mg/kg/day (0, 200, 400, or 800 mg/m2/day). The high dose of CP-31398 induced mortality in both species: seven male rats and four female rats died as a result of hepatic infarcts, and two female dogs died as a result of hepatic necrosis without evidence of thrombosis. No deaths were seen in the mid- or low dose groups in either species. In dogs, sporadic emesis was seen in the high dose and mid dose groups, and reductions in body weight gain were observed in all drug-exposed groups. CP-31398 induced mild anemia in both species; clinical pathology data also demonstrated hepatic toxicity, renal toxicity, inflammatory reactions, and coagulopathies in rats in the high dose and mid dose groups. Treatment-related microscopic changes in high dose and mid dose rats were identified in the liver, kidney, heart, bone marrow, lung, adrenals, spleen, thymus, skeletal muscle, and ovary; microscopic changes in the liver, heart, lung, and adrenals persisted through the recovery period. In dogs, microscopic changes were identified in the central nervous system, lung, and liver; changes in all tissues remained at the end of the recovery period. The liver is the primary site of limiting toxicity for CP-31398 in rats, and is also a key site of toxicity in dogs. The Maximum Tolerated Dose (MTD) for subchronic oral administration of CP-31398 is 80 mg/kg/day (480 mg/m2/day) in rats and 20 mg/kg/day (400 mg/m2/day) in dogs. Although only modest and apparently reversible toxicities (microscopic changes in rats; reductions in body weight gain and alterations in red cell parameters in dogs) were seen in the low dose groups, No Observed Adverse Effect Levels (NOAELs) for CP-31398 could not be established for either species. The toxicity of CP-31398 suggests that this agent may not be suitable for use in cancer prevention. However, should in vivo antitumor efficacy be achievable at doses that do not induce limiting toxicity, CP-31398 may have utility as a cancer therapeutic. Modification of the primary sites of CP-31398 metabolism (N-demethylation of the alkyl side chain; hydroxylation and O-demethylation of the styryl benzene group) may result in the development of CP-31398 analogs with comparable pharmacologic activity and reduced toxicity.
Keywords: CP-31398; N′-[2-[2-(4-methoxyphenyl)ethenyl]-4-quinazolinyl]-N,N-dimethyl-1,3-propanediamine dihydrochloride; p53; chemopreventive agent; chemotherapeutic agent
1. Introduction
p53 regulates a broad range of signal transduction pathways that are linked to differentiation, development, and carcinogenesis (Harris and Levine, 2005). Normal (wild-type) p53 protein is a tumor suppressor; this activity may involve maintenance of genetic stability, suppression of cell proliferation, induction of apoptosis, inhibition of angiogenesis, and/or modulation of other cancer-associated cellular processes (Wang et al., 2003, Molchadsky et al., 2010). As a result of these diverse regulatory functions, wild-type p53 has been characterized as a “gate keeper” to cell proliferation and neoplastic development (Levine, 1997).
The activity of wild-type p53 requires its binding to DNA in a sequence-specific manner (Kern et al., 1991). p53 binding to DNA is critically dependent on the structural integrity of its DNA binding site; mutations inducing conformational changes in the DNA binding domain of the p53 protein result in loss of both DNA binding and tumor suppressor activity (Kern et al., 1991; Foster et al., 1999; Wang et al., 2003).
Loss of p53 function is an important step in carcinogenesis in many sites (Hollstein et al., 1991; Greenblatt et al., 1994). Inactivating mutations in the p53 gene are present in approximately 50% of human cancers (Joerger and Fersht, 2007), and p53 function is often partially lost in tumors that do not contain a p53 mutation (Cheok et al., 2011). As such, maintenance or restoration of wild-type p53 activity presents an attractive target for the design of drugs for cancer prevention and therapy (Wang et al., 2003; Joerger and Fersht, 2007; Brown et al., 2009).
The hypothesis that restoration of normal p53 function is a viable approach to cancer prevention and therapy is supported by data from studies using either gene therapy or pharmacologic interventions to maintain p53 activity. Reintroduction of the wild-type p53 gene into p53-deficient cells or animals via gene therapy can restore p53 function and eradicate tumors in several experimental models (Harris et al., 1996; Horowitz, 1999). In consideration of drug delivery and other challenges associated with gene therapy, however, recent focus has shifted to small molecule therapeutics that may stabilize or restore normal p53 function (Brown et al., 2011; Kim and Dass, 2011). Several small molecules that stabilize or restore wild-type p53 activity demonstrate significant anticancer activity in preclinical models (Foster et al., 1999; Cheok et al., 2011)
CP-31398 [N′-[2-[2-(4-methoxyphenyl)ethenyl]-4-quinazolinyl]-N,N-dimethyl-1,3-propanediamine dihydrochloride; Fig. 1] is a synthetic styrylquinazoline that can restore a wild-type-associated epitope to the DNA-binding site of the mutant p53 protein (Foster et al., 1999). In vitro, CP-31398 restores the DNA-binding activity of mutant p53, and can induce growth arrest or apoptosis in human cancer cells expressing mutant p53 protein (Foster et al., 1999; Demma et al., 2004; Roh et al., 2011). CP-31398 can also increase both the levels and activity of wild-type p53 in cells that do not carry p53 mutations (Luu et al., 2002; Takimoto et al., 2002). Furthermore, CP-31398 stabilizes exogenous p53 in a broad range of human cell lines, including cells that express normal (wild-type) p53 protein, cells that express mutant p53 protein, and cells that are p53 null (Wang et al., 2003).
Fig. 1.
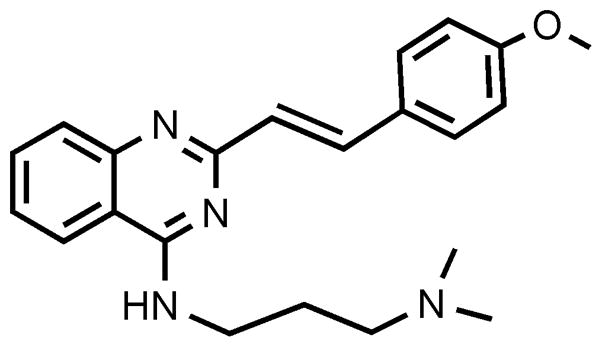
Chemical structure of CP-31398.
In vivo, CP-31398 demonstrates chemopreventive and chemotherapeutic activity in experimental models for cancer of the colon and other tissues. CP-31398 confers significant protection against the development of colon cancer in APC (min+/−) mice (Rao et al., 2008); inhibits the induction of colon cancer in rats by the chemical carcinogen, azoxymethane (Rao et al., 2009); and inhibits the induction of skin cancer in mice by UVB radiation (Tang et al., 2007). When administered in a therapeutic mode, CP-31398 inhibits the growth of human tumor xenografts in mice (Xu et al., 2010).
In consideration of the demonstrated chemopreventive and chemotherapeutic activities of CP-31398 in preclinical models, the compound is in preclinical development as a prototype p53 stabilizing agent. The present study was performed to characterize the toxicity of CP-31398 following its subchronic oral administration to rats and dogs.
2. Materials and methods
2.1. Animal welfare considerations
Prior to the initiation of in vivo experimentation, study protocols were reviewed and approved by the IIT Research Institute Institutional Animal Care and Use Committee. All aspects of the program involving animal care, use, and welfare were performed in compliance with United States Department of Agriculture regulations and the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996). Both studies were conducted in full compliance with the Good Laboratory Practice regulations of the United States Food and Drug Administration (21 CFR Part 58).
2.2. Test article
CP-31398 (> 99% purity; Indofine Chemical Co., Hillsborough, NJ) was obtained through the Chemopreventive Agent Repository maintained by the Division of Cancer Prevention, National Cancer Institute. Bulk CP-31398 was protected from light and stored desiccated at 2–8°C under nitrogen. Dosing formulations were prepared by dissolving CP-31398 in ASTM Type 1 water; after preparation, dosing formulations were stored under nitrogen at 2–8°C until used. CP-31398 was protected from light during all dose preparation, storage, and dose administration operations.
2.3. Twenty-eight day oral toxicity study (with recovery) in rats
Male and female CD rats (Crl:CD® [SD]IGS) were received at approximately five to six weeks of age from virus-free colonies maintained at Charles River Laboratories (Portage, MI). Rats were housed individually in suspended stainless steel cages in a temperature-controlled room maintained on a 12-hour light/dark cycle, and were held in quarantine for approximately two weeks prior to the initiation of dosing. With the exception of overnight fasts prior to scheduled necropsies, rats were allowed free access to Certified Rodent Diet 5002 (PMI Nutrition International, Brentwood, MO) throughout the study. City of Chicago drinking water was supplied to rats ad libitum using an automatic watering system.
After release from quarantine, rats were assigned to groups of 15/sex using a computer-based procedure that blocks for body weights. Within each dose group, 10 rats/sex were assigned to the Main Study cohort (scheduled for necropsy on Study Day 29), and five rats/sex were assigned to the Recovery cohort (scheduled for necropsy on Study Day 43). After group assignment, rats received daily oral (gavage) administration of CP-31398 (in ASTM water) at doses of 0 (control), 40, 80, or 160 mg/kg/day (0, 240, 480, or 960 mg/m2/day) for 28 days; a constant dosing volume of 10 ml/kg/day was used.
Doses were selected for the 28-day definitive toxicity study in rats on the basis of the results of a preliminary 14-day range-finding study in which groups of 5 rats/sex received oral exposure to CP-31398 at doses of up to 500 mg/kg/day. In this study, administration of CP-31398 at 500 mg/kg/day induced 100% mortality, and drug administration at 250 mg/kg/day induced mortality in 0/5 male rats and 1/5 female rats. At both dose levels, mortality was associated with severe coagulation necrosis in the liver (hepatic infarcts); hepatic infarcts were also a common microscopic finding in surviving rats receiving drug at 250 mg/kg/day. By contrast, toxicity in rats receiving CP-31398 at 100 mg/kg/day was limited to minimal to mild microscopic alterations in the liver (hypertrophy and vacuolation of Kupffer cells) and heart (cytoplasmic alterations); no clear evidence of drug-related toxicity was seen in rats receiving CP-31398 at 50 mg/kg/day. On this basis, doses for the definitive study were selected with the expectation that significant toxicity would be seen in rats exposed to the high dose (160 mg/kg/day) of CP-31398, and that little or no toxicity would be seen at the low dose (40 mg/kg/day).
Throughout the study, rats were observed a minimum of twice daily to monitor their general health status; detailed clinical examinations and measurements of body weight and food consumption were performed weekly. Indirect funduscopic ophthalmic examinations were performed on all animals during the quarantine period (pre-test) and during the final week of the treatment period. A Functional Observational Battery (FOB) was performed on five rats/sex/group at pretest, during Weeks 1 and 4 of the treatment period, and during the final week of the recovery period; during Weeks 1 and 4, FOBs were performed at two to four hours post-dosing. Evaluations in the FOB included home cage observation, hand-held observation, open field observation (mobility/gait), body weight, body temperature, eye blink, pupil response, tail pinch, hind limb extension, hearing (click response), vision, catalepsy, righting reflex, grip strength (forelimb and hind limb), and foot splay.
Blood samples for clinical chemistry, hematology, and coagulation evaluations were collected from fasted rats prior to their scheduled necropsy. Clinical pathology assays were performed using automated instruments (Synchron CX5 Clinical Chemistry Analyzer [Beckman Instruments, Brea, CA]; Advia System 120 Hematology Analyzer [Bayer Corp., Tarrytown, NY]; STA Compact CT automatic coagulation analyzer [Diagnostica Stago, Parsippany, NJ]).
On Study Day 29, surviving Main Study rats were humanely euthanized with sodium pentobarbital followed by exsanguinations, and were subjected to a complete necropsy with tissue collection. Surviving Recovery rats in each dose group were euthanized and submitted for necropsy on Study Day 43 (following a 14-day recovery period). At both scheduled necropsies, weights of the adrenals, brain, heart, kidneys, liver, ovaries/testes, spleen, thyroid, and uterus were collected from each animal. All gross lesions plus approximately 45 tissues per rat were collected and fixed in 10% neutral buffered formalin. All tissues from all rats in the high dose and vehicle control groups were processed by routine histologic methods, cut at 5 μm, stained with hematoxylin and eosin, and evaluated microscopically. Microscopic evaluation of tissues from rats in the mid and low dose groups in the Main Study and from all Recovery animals was limited to gross lesions and identified target tissues.
2.4. Metabolite profiling of CP-31398 in rats
Blood samples for plasma drug level analysis and metabolite profiling were collected from cohorts of four rats/sex/group at 1, 4, 8, 12, and 24 hours post-dosing on Study Day 1 and during Week 4 of CP-31398 administration; samples analyzed for metabolite profiles reflect a subset of samples that were collected to characterize CP-31398 pharmacokinetics (data reported elsewhere).
Levels of CP-31398 and metabolites in blood samples were quantitated by high performance liquid chromatography/quantitative time-of-flight mass spectrometry (LC/QTOF), using analytical methods developed in our laboratory (Muzzio et al., 2011). Separations were performed using an Agilent 1200 LC (Agilent Technologies, Wilmington, DE) and a Luna 3 μ PFP(2) 100 Å, 100 × 2.0 mm column (Phenomenex, Torrance, CA). The mobile phases consisted of Solvent A (50 mM ammonium acetate in water/methanol/acetonnitrile [v:v:v::90:6:4]) and Solvent B (50 mM ammonium acetate in water/methanol/acetonnitrile [v:v:v::10:54:36)). The separation program was A:B::80:20 for 16 minutes and A:B::5:95 for 6 minutes, followed by 6 minutes of post-run time at A:B::80:20. Parent compound and metabolites were identified and quantitated using an Agilent 6250 QTOF mass spectrometer with electrospray ionization; data were analyzed using Agilent Mass Hunter software (revision B.01.03).
CP-31398 has a formula weight of 362.46804 daltons and a mono-isotopic mass of 362.21066 daltons. Under the analytical conditions used, CP-31398 was detected in the LC/QTOF system as the protonated molecular ion, with an exact mass of 363.21794 and a retention time of approximately 18 minutes. Metabolites would be expected to produce ions with molecular weight shifts characteristic of the biotransformation of the parent molecule; ions resulting from common biotransformation reactions (e.g., hydroxylation, demethylation, demethylation + hydroxylation, etc.) were detected by inspecting the total ion chromatogram file for the suspected metabolite protonated molecular ions. When such ions were found, the extracted ion chromatogram was integrated and the concentration of the metabolite was estimated using the calibration curve established for CP-31398.
2.5. Twenty-eight day oral toxicity study (with recovery) in dogs
Male and female purebred beagle dogs were received at approximately six months of age from Ridglan Farms, Inc. (Mount Horeb, WI), and were held in quarantine for three weeks prior to assignment to experimental groups. Throughout the study, dogs were housed individually in floor-level pens in a temperature-controlled room maintained on a 12-hour light/dark cycle. Dogs were provided with 400 g of Certified Canine Diet 5007 (PMI Nutrition International, Inc.) for a minimum of two hours each day, and were permitted free access to City of Chicago drinking water supplied via an automatic watering system.
After release from quarantine, dogs were assigned to groups of five/sex (three Main Study dogs/sex [scheduled for necropsy on Day 29] + two Recovery dogs/sex [scheduled for necropsy on Day 43]) using a computerized procedure that blocks for body weight. Dogs received daily oral (gavage) exposure to CP-31398 at doses of 0 (control), 10, 20, or 40 mg/kg/day (0, 200, 400, or 800 mg/m2/day) for 28 consecutive days, using a dosing volume of 2 ml/kg.
Doses of CP-31398 for the 28-day definitive toxicity study in dogs were selected on the basis of the results of a preliminary 14-day range-finding study in which groups of 1 dog/sex received oral administration of CP-31398 at doses of up to 50 mg/kg/day. In this range-finding study, administration of CP-31398 at 50 mg/kg/day induced mortality in one male dog; the probable cause of death was hepatic necrosis. Drug-related toxicity in dogs exposed to CP-31398 at 25 mg/kg/day was limited to minimal cytoplasmic alterations in the liver (basophilic clumping, cytoplasmic clearing); no evidence of toxicity was seen in dogs receiving CP-31398 at 5 or 10 mg/kg/day for 14 days. On the basis of these results, doses for the definitive toxicity study were selected with the expectation that clear evidence of CP-31398 toxicity would be seen in dogs receiving the high dose (40 mg/kg/day) of CP-31398, and that little or no toxicity would be seen in dogs receiving the low dose (10 mg/kg/day).
Dogs were observed a minimum of twice daily to monitor their general health status. Detailed clinical examinations and body weight measurements were performed weekly, and food consumption was measured daily. Blood samples for clinical chemistry, hematology, and coagulation evaluations were collected from all surviving dogs during pre-test and on Study Days 4, 8, 15, 22, 29, and 43. Clinical chemistry, hematology, and coagulation assays were performed as described for the 28-day toxicity study in rats. Urine samples were collected on the same schedule and analyzed by dipstick and microscopy.
Surviving Main Study dogs were humanely euthanized by barbiturate overdose on Study Day 29, and were subjected to a complete necropsy with tissue collection; this process was repeated on Study Day 43 for dogs in the Recovery groups. Weights of the adrenals, brain, heart, kidneys, liver, spleen, testes, thymus, and thyroids were collected at necropsy, and all gross lesions and approximately 45 tissues were collected from each animal and fixed in 10% neutral buffered formalin. All tissues collected from dogs in the high dose and control groups in the Main Study were processed by routine histologic methods, cut at 5 μm, stained with hematoxylin and eosin, and evaluated microscopically. Microscopic evaluation of tissues from Main Study dogs in the mid and low dose groups and from Recovery dogs in all dose groups were limited to gross lesions and identified target tissues.
2.6. Metabolite profiling of CP-31398 in dogs
Blood samples for plasma drug level analysis and metabolite profiling were collected from all CP-31398-treated dogs at 0.5, 4, and 12 hours post-dosing on Study Days 1 and 25. Levels of CP-31398 and metabolites in blood samples were quantitated by LC/QTOF, as described for rats in Section 2.4.
2.7. Statistical analyses
Statistical evaluations of continuous data were performed by analysis of variance (ANOVA), with post-hoc analyses performed using Dunnett’s test. Incidence data were compared by χ2 analysis or Fischer’s Exact Test. A minimum significance level of p < 0.05 was used in all comparisons.
3. Results
3.1. Subchronic oral toxicity of CP-31398 in rats
3.1.1. Mortality
Drug-related mortality was seen in both sexes of rats receiving the high dose (160 mg/kg body weight/day) of CP-31398. In the high dose group, 7/15 males and 4/15 females died or were euthanized in extremis during the dosing period; the earliest death was seen on Day 12, while the last death occurred on Day 29, prior to the scheduled Main Study necropsy. No mortality was seen in rats receiving CP-31398 at either the mid or low doses, or in vehicle controls.
3.1.2. In-life evaluations
Clinical signs indicative of generalized stress were observed in nearly all animals in the high dose group. Clinical signs included chromodacryorrhea, rough coat, hypoactivity, hunched posture, and coldness to touch; male rats appeared to be affected more severely than were females. No clinical evidence of toxicity was observed in rats in either the mid or low dose groups, or in vehicle controls.
Daily oral administration of CP-31398 induced dose-related reductions in group mean body weight in both sexes. In comparison to vehicle controls, statistically significant reductions in mean body weight were seen in both sexes in the high dose group during Weeks 3 and 4 of dosing (Figs. 2 and 3). Significant reductions in mean body weight in high dose male rats persisted through the recovery period (Fig. 2), while body weight effects in high dose females were largely reversed during recovery (Fig. 3).
Fig. 2.

Group mean body weights in male rats exposed to CP-31398 for 28 days.
Fig. 3.
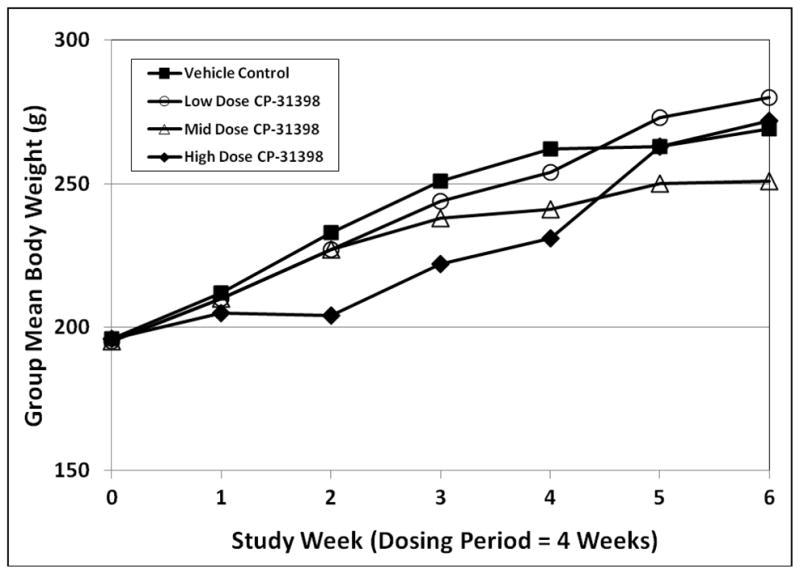
Group mean body weights in female rats exposed to CP-31398 for 28 days.
At the mid dose of CP-31398, statistically significant reductions in group mean body weight were seen only in female rats at Week 4 of dosing (Fig. 3). No statistically significant differences in body weight were seen in mid dose male rats at any time during the study. The low dose of CP-31398 had no effect on mean body weight or body weight gain in either sex.
The effects of CP-31398 on food consumption generally mirrored its effects on body weight. In the high dose group, male rats demonstrated statistically significant decreases in food consumption during each week of the dosing period and during the first week of recovery. Female rats in the high dose group demonstrated significant decreases in food consumption during Weeks 2, 3, and 4 of dosing. In the mid dose group, statistically significant decreases in food consumption were seen in male rats only during the first week of dosing and in female rats only during the final week of dosing. The low dose of CP-31398 had no effect on food consumption in either sex.
No evidence of ocular or central nervous system (CNS) toxicity was identified in any treated animal through ophthalmologic examinations and behavioral assessments performed using the FOB.
3.1.3. Clinical pathology
The results of clinical chemistry evaluations identified the liver as a primary site of CP-31398 toxicity, and the kidney as a secondary side of agent toxicity. At the end of the exposure period, dose-related increases in alkaline phosphatase (ALP), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) were seen in the mid and high dose groups in both sexes (Table 1). High dose male rats also demonstrated statistically significant increases in blood urea nitrogen (BUN) and BUN/creatinine ratios. Alterations in clinical chemistry parameters indicative of hepatic toxicity were reversed during the 14-day recovery period in the mid dose group, but not in the high dose group (data not shown). The low dose of CP-31398 did not induce any pattern of alterations in clinical chemistry parameters that suggested organ-specific toxic effects.
Table 1.
Selected Clinical Chemistry Findings in Rats Exposed to CP-31398 for 28 Days
| Group | Sex | Dose (mg/kg/day) | ALP (IU/L) | ALT (IU/L) | AST (IU/L) | BUN (mg/dL) | BUN/CRE Ratio |
|---|---|---|---|---|---|---|---|
| 1 | Male | 0 | 165 ± 34 | 34 ± 7 | 90 ± 15 | 11 ± 2 | 21.7 ± 3.7 |
| 2 | Male | 40 | 170 ± 52 | 33 ± 5 | 89 ± 8 | 10 ± 2 | 19.0 ± 4.2 |
| 3 | Male | 80 | 181 ± 48 | 68 ± 53* | 156 ± 64* | 12 ± 2 | 22.0 ± 3.9 |
| 4 | Male | 160 | 464 ± 141* | 230 ± 336* | 1227 ± 1109* | 17 ± 3* | 39.4 ± 7.4* |
| 1 | Female | 0 | 111 ± 21 | 30 ± 8 | 89 ± 12 | 14 ± 4 | 22.0 ± 5.0 |
| 2 | Female | 40 | 125 ± 55 | 29 ± 8 | 94 ± 7 | 13 ± 2 | 20.7 ± 3.4 |
| 3 | Female | 80 | 129 ± 41 | 127 ± 224* | 405 ± 566* | 15 ± 2 | 23.6 ± 3.6 |
| 4 | Female | 160 | 406 ± 207* | 167 ± 133* | 1020 ± 700* | 11 ± 2 | 23.8 ± 4.3 |
p < 0.05 versus sex-matched vehicle controls
Hematology evaluations identified statistically significant, dose-related increases in white blood cells (WBC) counts at Day 29 (Table 2) and at the end of the recovery period (data not shown) in both sexes exposed to CP-31398. These results are interpreted as inflammatory responses associated with hepatic necrosis induced by the drug. The high dose of CP-31398 induced mild anemia in both sexes, as indicated by modest but statistically significant decreases in several red blood cell (RBC) parameters (Table 2); the effects on RBC parameters remained at the end of the recovery period. The low dose of CP-31398 had no significant effects on hematologic parameters in either sex.
Table 2.
Selected Hematology Findings in Rats Exposed to CP-31398 for 28 Days
| Group | Sex | Dose (mg/kg/day) | WBC Count (103/μL) | RBC Count (106/μL) | Hemoglobin (g/dL) | Hematocrit (%) |
|---|---|---|---|---|---|---|
| 1 | Male | 0 | 12.7 ± 1.7 | 8.0 ± 0.4 | 14.8 ± 0.6 | 46.1 ± 2.2 |
| 2 | Male | 40 | 15.3 ± 3.5 | 8.2 ± 0.5 | 15.2 ± 0.6 | 47.3 ± 2.6 |
| 3 | Male | 80 | 17.9 ± 4.7* | 8.1 ± 0.5 | 15.2 ± 0.7 | 47.4 ± 2.6 |
| 4 | Male | 160 | 60.3 ± 21.0* | 7.1 ± 0.9* | 12.4 ± 1.7* | 40.3 ± 5.3* |
| 1 | Female | 0 | 11.8 ± 2.2 | 7.9 ± 0.3 | 14.7 ± 0.4 | 44.8 ± 1.2 |
| 2 | Female | 40 | 12.3 ± 3.0 | 7.9 ± 0.4 | 14.7 ± 0.7 | 44.8 ± 2.2 |
| 3 | Female | 80 | 18.9 ± 4.5* | 8.2 ± 0.5 | 14.7 ± 1.0 | 44.6 ± 2.5 |
| 4 | Female | 160 | 37.0 ± 12.0* | 6.3 ± 0.7* | 10.4 ± 1.1* | 34.0 ± 3.8* |
p < 0.05 versus sex-matched vehicle controls
Subchronic oral administration of CP-31398 induced dose-related increases in prothrombin time, activated partial thromboplastin time, and fibrinogen in both sexes (Table 3); alterations in coagulation parameters were interpreted as being secondary to hepatic toxicity. Although changes in coagulation parameters were somewhat greater in high dose males than in females, significant differences in the mid dose group were seen in females only. These effects persisted in the high dose group at the end of the recovery period.
Table 3.
Coagulation Parameters in Rats Exposed to CP-31398 for 28 Days
| Group | Sex | Dose (mg/kg/day) | Prothrombin Time (seconds) | Activated Partial Thromboplastin Time (seconds) | Fibrinogen (mg/dL) |
|---|---|---|---|---|---|
| 1 | Male | 0 | 17.8 ± 1.2 | 18.4 ± 1.6 | 302 ± 27 |
| 2 | Male | 40 | 17.0 ± 0.8 | 17.3 ± 0.9 | 315 ± 22 |
| 3 | Male | 80 | 16.8 ± 0.7 | 19.1 ± 3.0 | 306 ± 28 |
| 4 | Male | 160 | 21.1 ± 2.0* | 40.5 ± 10.4* | 457 ± 56* |
| 1 | Female | 0 | 16.9 ± 0.5 | 15.1 ± 1.1 | 273 ± 35 |
| 2 | Female | 40 | 16.8 ± 0.8 | 15.1 ± 1.8 | 248 ± 22 |
| 3 | Female | 80 | 18.8 ± 1.4* | 23.9 ± 6.1* | 254 ± 84 |
| 4 | Female | 160 | 19.3 ± 1.2* | 34.2 ± 8.1* | 394 ± 33* |
p < 0.05 versus sex-matched vehicle controls
3.1.4. Gross and microscopic pathology
At the Main Study (Day 29) necropsy, dose-related alterations in absolute and relative organ weights were seen in several tissues in rats exposed to CP-31398. The most dramatic effect of CP-31398 on organ weights was a statistically significant, dose-related hepatomegaly in high dose males and in mid and high dose females (Table 4). Other dose-related changes in organ weights included statistically significant increases in adrenal weights in the high dose group in both sexes and in mid dose females; statistically significant increases in lung weights in the mid and high dose groups (both sexes); dose-related splenomegaly in both sexes; a significant decrease in mean testicular weight in the high dose group; and a significant decrease in mean thymus weight in both sexes in the high dose group. With the exception of the effect on thymus weights, changes in organ weights induced by the high dose of CP-31398 persisted through the recovery period.
Table 4.
Selected Organ Weights in Rats Exposed to CP-31398 for 28 Days
| Group | Sex | Dose (mg/kg/day) | Liver Weights | Lung Weights | Adrenal Weights | |||
|---|---|---|---|---|---|---|---|---|
| Absolute (g) | Relative (% of Body Weight) | Absolute (g) | Relative (%of Body Weight) | Absolute (g) | Relative (%of Body Weight) | |||
| 1 | Male | 0 | 11.7 ± 1.7 | 3.05 ± 0.38 | 1.58 ± 0.22 | 0.41 ± 0.05 | 0.074 ± 0.033 | 0.019 ± 0.009 |
| 2 | Male | 40 | 10.9 ± 1.3 | 3.02 ± 0.24 | 1.69 ± 0.29 | 0.47 ± 0.06 | 0.095 ± 0.034 | 0.026 ± 0.009 |
| 3 | Male | 80 | 12.7 ± 2.0 | 3.48 ± 0.36 | 2.06 ± 0.21 | 0.58 ± 0.07 | 0.100 ± 0.023 | 0.028 ± 0.007 |
| 4 | Male | 160 | 28.6 ± 4.4* | 10.97 ± 1.12* | 2.43 ± 0.19* | 0.94 ± 0.13* | 0.199 ± 0.052* | 0.076 ± 0.018* |
| 1 | Female | 0 | 7.4 ± 0.6 | 3.00 ± 0.19 | 1.26 ± 0.15 | 0.51 ± 0.06 | 0.076 ± 0.015 | 0.031 ± 0.006 |
| 2 | Female | 40 | 7.5 ± 0.8 | 3.22 ± 0.26 | 1.26 ± 0.12 | 0.54 ± 0.05 | 0.078 ± 0.018 | 0.034 ± 0.007 |
| 3 | Female | 80 | 9.6 ± 1.1* | 4.37 ± 0.76* | 1.52 ± 0.14 | 0.69 ± 0.08 | 0.093 ± 0.012 | 0.042 ± 0.007* |
| 4 | Female | 160 | 23.5 ± 5.3* | 11.23 ± 2.77* | 2.00 ± 0.25 | 0.95 ± 0.12* | 0.172 ± 0.034* | 0.082 ± 0.017* |
p < 0.05 versus sex-matched vehicle controls
Numerous target tissues were identified on the basis of gross and microscopic changes in early death animals (found dead or euthanized in extremis) and in rats surviving until the Main Study necropsy. Target tissues that were identified during histopathologic evaluation of tissues included the liver, heart, lung, skeletal muscle, thymus, spleen, adrenal, kidney, ovary, lymph nodes, and bone marrow.
Dose-limiting pathology was identified in the liver in the high dose group, with less severe hepatic alterations seen at the mid dose. At the Main Study necropsy, hepatomegaly and altered hepatic pigmentation were common findings in both sexes at the mid and high doses; these gross findings were correlated with increased absolute and relative liver weights in both sexes. This gross pathology was associated with microscopic diagnoses of submassive coagulative necrosis (infarcts; Fig. 4) in both sexes; in some animals, areas of necrosis were separated by broad bands of fibrosis. In the high dose group, hepatic infarcts with fibrosis were present in all early deaths, and in all rats of both sexes necropsied on Day 29; infarcts and/or evidence of hepatic fibrosis were also identified in the high dose group at the end of the 14-day recovery period. Hepatic infarcts of mild severity were identified at the Main Study necropsy in 2/10 female rats (but 0/10 male rats) in the mid dose group; although no hepatic infarcts were identified in either sex in the mid dose group after the recovery period, 1/5 female rats in the mid dose Recovery group demonstrated mild hepatic fibrosis. Other hepatic changes identified in the mid- and high dose groups included bile duct hyperplasia (both sexes in high dose group; females only in mid dose group); Kupffer cell hypertrophy (both sexes); and deposition of basophilic material in Kupffer cells (both sexes). Changes in Kupffer cells were present in only one female rat in the low dose group.
Fig. 4.
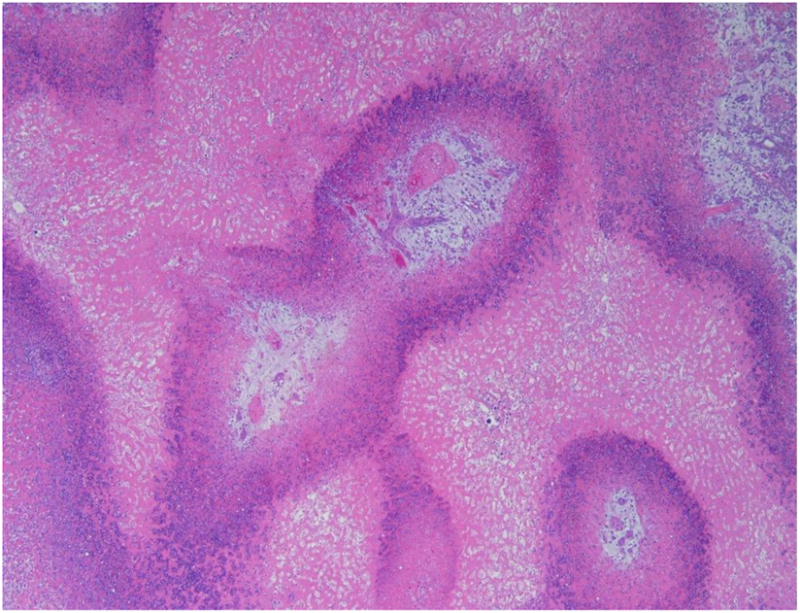
Representative photomicrograph demonstrating submassive coagulative necrosis in the liver of a female rat receiving the high dose (160 mg/kg/day) of CP-31398 for 28 days. Note infiltrates of viable and necrotic neutrophils in peripheral regions (hematoxylin and eosin).
Increased incidences of cardiomyopathy were observed in male rats exposed to all dose levels of CP-31398, and in female rats exposed to CP-31398 at the mid and high dose levels. At the high dose, valvular endocardosis was also present in both sexes, but was more prominent in female rats. Both cardiac effects were also identified in high dose animals at the end of the recovery period.
In the lung, increased numbers of alveolar macrophages and proteinaceous material in the alveoli were observed in male rats in all dose groups and in female rats in the mid and high dose groups. These changes may reflect alveolar edema and a response by normal histiocytes to clear the fluid, diagnoses that are consistent with the increases in absolute and relative lung weights observed in both sexes. Alveolar histiocytosis remained in mid and high dose animals evaluated at the end of the recovery period.
Animals of both sexes in the high dose group demonstrated thymic atrophy, which could reflect either a direct toxic effect of CP-31398 or a reaction to stress. In addition, many animals in the high dose group demonstrated degeneration of the myofibers in skeletal muscle with associated infiltrates of mononuclear cells. Degeneration of the myofibers was more common in males than in females; neither this change, nor the thymic atrophy discussed above, was seen in rats in either the mid- or low dose groups.
3.2. Subchronic oral toxicity of CP-31398 in dogs
3.2.1. Mortality
Drug-related mortality was seen in the high dose (40 mg/kg/day) group in dogs, as 2/5 female dogs died on Study Days 28 and 29. One of the dogs that died was hypoactive prior to death. No mortality was seen in male dogs in the high dose group, or in either sex in groups exposed to the mid- or low doses of CP-31398.
3.2.2. In-life evaluations
All dogs in the high- (40 mg/kg/day) and mid dose (20 mg/kg/day) groups demonstrated emesis on various days throughout the dosing period. Emesis was also observed sporadically in 3/5 male and 4/5 female dogs in the low dose (10 mg/kg/day) group. Other clinical observations included discolored (yellow) gums in all dogs in the high dose group and in 2/5 males in the mid dose group; this change was still present in high dose dogs at the end of the recovery period.
Female dogs in the mid- and high dose groups and male dogs in all dose groups demonstrated statistically significant reductions in total body weight gain throughout the 28-day dosing period (Figs. 5 and 6). In male dogs, mean body weight in the low dose group returned to control levels during the recovery period; however, mean body weights in the mid- and high dose groups remained below that of the vehicle control group. In female dogs, body weight gains in all drug-treated groups during the recovery period exceeded that of the vehicle control group; at the end of the recovery period, mean body weights in female dogs remained suppressed only in the high dose group.
Fig. 5.
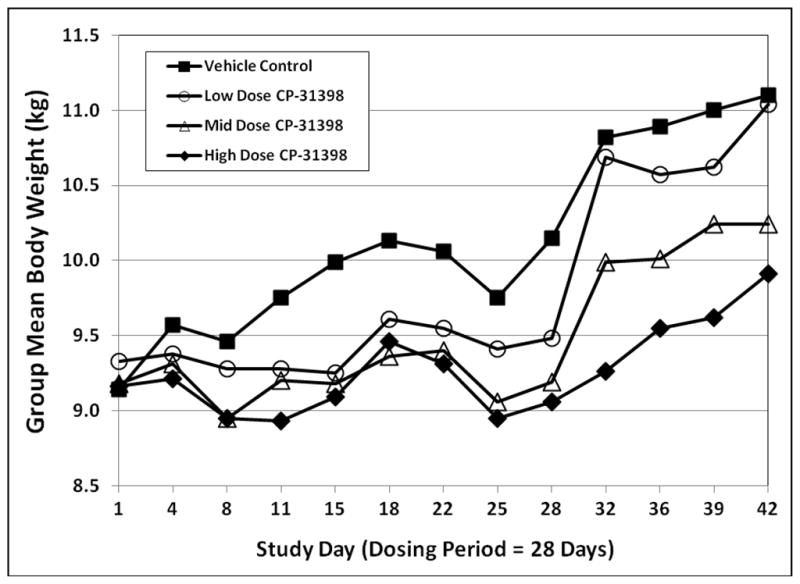
Group mean body weights in male dogs exposed to CP-31398 for 28 days.
Fig. 6.

Group mean body weights in female dogs exposed to CP-31398 for 28 days.
As was seen with rats, the effects of CP-31398 on food consumption in dogs generally mirrored its effects on body weight. Male and female dogs in the mid- and high dose groups demonstrated treatment-related decreases in food consumption at various times during the dosing period. These decreases were correlated with overall reductions in body weight gain.
3.2.3. Clinical pathology
Statistically significant changes in various clinical chemistry parameters were seen in all dose groups at one or more time points (Days 4, 8, 15, 22, 29, and 43) at which clinical pathology tests were performed (Table 5). However, these data provided little consistent evidence to identify changes in organ function in dogs exposed to CP-31398. Most changes in clinical chemistry parameters were interpreted as sporadic or incidental findings; the few parameters (albumin levels, globulin levels, and albumin:globulin ratio) in which changes were considered to be treatment-related were interpreted as being of minimal toxicologic significance.
Table 5.
Statistically Significant Clinical Chemistry Findings in Dogs Receiving CP-31398
| Sex | Day | Parameter(s) increased in Groups Exposed to CP- 31398 | Group(s) | Parameter(s) Decreased in Groups Exposed to CP-31398 | Group(s) |
|---|---|---|---|---|---|
| Male | 4 | None | -- | Pi | Mid, High |
| Male | 8 | ALT | High | Pi ALP |
Low, Mid Mid, High |
| Male | 15 | None | -- | Pi Glucose |
Low, Mid, High Mid, High |
| Male | 22 | None | -- | None | -- |
| Male | 29 | BUN/creatinine ratio | High | None | -- |
| Male | 43 | BUN | High | Pi A/G ratio |
Mid High |
| Female | 4 | ALT | High | None | -- |
| Female | 8 | K+ ALT |
High High |
ALP Glucose |
Low, Mid, High Mid, High |
| Female | 15 | K+ | Mid, High | Pi | Mid, High |
| Female | 22 | Cl- Globulin |
High Low, Mid |
Albumin A/G ratio Glucose |
High Low, Mid Low, Mid, High |
| Female | 29 | Globulin Triglycerides |
Low, High High |
A/G ratio | Low, High |
| Female | 43 | None | -- | None | -- |
Evidence of modest anemia was present in all dose groups throughout the dosing period, but was statistically significant only in female dogs in the low and mid dose groups (Day 29 data are presented in Table 6). The lack of significant changes in female dogs in the high dose group, and in male dogs in all dose groups may be related to dose-related emesis seen in dogs treated with CP-31398. No clear pattern of effects on coagulation parameters was seen in any group (data not shown).
Table 6.
Selected Hematology Findings in Dogs Receiving CP-31398 for 28 Days
| Group | Sex | CP-31398 (mg/kg/day) | WBC Count (103/μL) | RBC Count (106/μL) | Hemoglobin (g/dL) | Hematocrit (%) |
|---|---|---|---|---|---|---|
| 1 | Male | 0 | 14.0 ± 5.9 | 6.7 ± 0.6 | 14.4 ± 1.3 | 45.5 ± 4.2 |
| 2 | Male | 10 | 13.3 ± 1.7 | 6.2 ± 0.5 | 13.5 ± 0.8 | 42.0 ± 2.7 |
| 3 | Male | 20 | 12.1 ± 2.8 | 6.1 ± 0.2 | 13.4 ± 0.5 | 41.4 ± 1.9 |
| 4 | Male | 40 | 12.9 ± 3.5 | 6.0 ± 0.3 | 13.1 ± 0.8 | 40.9 ± 2.5 |
| 1 | Female | 0 | 11.3 ± 1.1 | 6.7 ± 0.4 | 15.1 ± 0.7 | 46.0 ± 2.4 |
| 2 | Female | 10 | 11.7 ± 1.3 | 6.3 ± 0.2 | 13.7 ± 0.6* | 42.4 ± 1.9* |
| 3 | Female | 20 | 10.8 ± 1.7 | 6.2 ± 0.2 | 13.5 ± 0.3* | 41.5 ± 0.8* |
| 4 | Female | 40 | 12.8 ± 1.6 | 6.4 ± 0.2 | 14.1 ± 0.6 | 43.0 ± 1.4 |
p < 0.05 versus sex-matched vehicle controls
3.2.4. Gross and microscopic pathology
At the Main Study (Day 29) necropsy, a trend towards decreased absolute kidney and heart weights was observed in female, but not male, dogs exposed to CP-31398 (Table 7). Differences in absolute organ weights appear to reflect changes in body weight, since relative organ weights (as % of body weight) did not differ in these groups. No statistically significant evidence of hepatomegaly was seen in dogs of either sex.
Table 7.
Selected Day 29 Organ Weights in Dogs Receiving CP-31398 for 28 Days
| Group | Sex | CP-31398 Dose (mg/kg/day) | Left Kidney Weights | Heart Weights | Liver Weights | |||
|---|---|---|---|---|---|---|---|---|
| Absolute (g) | Relative (% of Body Wt) | Absolute (g) | Relative (% of Body Wt) | Absolute (g) | Relative (% of Body Wt) | |||
| 1 | M | 0 | 22.3 ± 4.3 | 0.24 ± 0.03 | 70.2 ± 4.1 | 0.76 ± 0.04 | 270.4 ± 32.6 | 2.92 ± 0.28 |
| 2 | M | 10 | 23.1 ± 3.6 | 0.26 ± 0.05 | 63.9 ± 5.7 | 0.72 ± 0.05 | 269.9 ± 43.1 | 3.04 ± 0.49 |
| 3 | M | 20 | 22.4 ± 6.8 | 0.25 ± 0.05 | 68.1 ± 13.3 | 0.77 ± 0.05 | 278.9 ± 58.0 | 3.15 ± 0.23 |
| 4 | M | 40 | 23.4 ± 3.2 | 0.27 ± 0.01 | 67.1 ± 5.3 | 0.76 ± 0.04 | 298.9 ± 55.9 | 3.37 ± 0.26 |
| 1 | F | 0 | 23.9 ± 1.3 | 0.27 ± 0.03 | 74.2 ± 2.1 | 0.84 ± 0.07 | 261.6 ± 21.2 | 2.96 ± 0.32 |
| 2 | F | 10 | 19.8 ± 1.5* | 0.26 ± 0.02 | 60.0 ± 5.3* | 0.77 ± 0.09 | 223.7 ± 25.5 | 2.88 ± 0.21 |
| 3 | F | 20 | 21.4 ± 0.7 | 0.28 ± 0.02 | 60.3 ± 2.0* | 0.78 ± 0.10 | 248.6 ± 11.3 | 3.21 ± 0.01 |
| 4 | F | 40 | 18.7 (n = 1) | 0.25 | 52.4 (n=1) | 0.71 | 209.3 (n=1) | 2.85 |
p < 0.05 versus sex-matched vehicle controls
Gross pathology was unremarkable in all dogs exposed to CP-31398, and no gross lesions attributed to exposure to the test article were identified in any treated animal. Both early death animals (female dogs in the high dose group that died on Study Days 28 and 29) demonstrated mild multifocal necrosis of the liver; hepatic necrosis did not demonstrate any consistent pattern of zonal orientation. In both animals, hepatic necrosis was identified as contributing to the death.
At the Day 29 (Main Study) necropsy, drug-related lesions were identified in the brain, spinal cord, lung, liver, and spleen. Drug-related lesions identified in the CNS, lung, and liver at the Day 29 necropsy were also seen in dogs undergoing necropsy after the 14-day recovery period.
In the brain, minimal to mild gliosis was identified as a treatment-related lesion in both sexes. In male dogs, minimal gliosis was present in 1/3 dogs in the mid dose group, and mild gliosis was present in 1/3 dogs in the high dose group. In female dogs, minimal gliosis was seen in 1/3 dogs in the low dose group and in 2/3 dogs in the mid dose group. This change was identified in 1/2 dogs in each sex at the recovery necropsy. In the brain and spinal cord, minimal to mild perivascular lymphocyte infiltration was present in all dose groups. This was identified as a treatment-related lesion, and remained in the mid- and high dose groups at the end of the recovery period.
In the lung, minimal to mild alveolar histiocytosis was present in all dose groups at the Day 29 necropsy; this change was not identified in the low- or mid dose groups at the recovery necropsy, but remained in one male and two female dogs in the high dose group. In the liver, minimal to mild deposition of basophilic material in the hepatocyte was identified in all dose groups, and persisted through the recovery period. In the spleen, minimal deposition of basophilic material in macrophages was identified in 1/3 male dogs and 2/3 female dogs in the high dose group only; this change was also seen at the end of the recovery period.
3.3. Plasma drug levels and metabolic profiles of CP-31398 in rats
In rats receiving daily oral exposure to CP-31398, peak plasma drug levels were identified at 2 or 4 hours after dosing. Plasma drug levels increased with increasing dose; within a dose group, plasma drug levels did not differ significantly between male and female rats. At 4 hours post-dosing on Day 1 (the Tmax in most groups), mean plasma CP-31398 levels were 65 ± 17 and 76 ± 11 ng/ml in male and female rats, respectively. Mean plasma drug levels at 4 hours were 96 ± 16 ng/ml in male rats and 127 ± 45 ng/ml in female rats in the mid dose group, and 155 ± 15 ng/ml and 148 ± 5 ng/ml in male and female rats in the high dose group.
Plasma drug levels measured during Week 4 were approximately two- to three-fold higher than those measured on Day 1. At four hours post-dosing during Week 4, mean plasma CP-31398 levels were 119 ± 37 and 145 ± 23 ng/ml in male and female rats, respectively. In the mid dose group, mean plasma drug levels at 4 hours were 251 ± 35 ng/ml in male rats and 224 ± 43 ng/ml in female rats. In the high dose group, mean plasma drug levels at four hours post-dosing during Week 4 were 512 ± 149 ng/ml and 427 ± 145 ng/ml in male and female rats, respectively.
In rats, the major metabolites of CP-31398 represented the products of biotransformation reactions that included hydroxylation, N- and O-demethylation, and O-demethylation + hydroxylation; several positional isomers are possible for some metabolites. Male and female rats demonstrated comparable patterns of CP-31398 metabolism, and metabolite profiles were quantitatively similar in both sexes in samples collected on Day 1 and during the fourth week of CP-31398 exposure.
A summary structure chart for CP-31398 and major metabolites identified is provided in Fig. 7. The primary metabolite identified in rats was the product of N-demethylation; this metabolite was approximately 1.5 to 3 times as abundant as were the O-demethyl metabolite and the total levels of two hydroxy isomers. The O-demethylhydroxy metabolite was less abundant in rats, as it was present at approximately 3 to 6 times lower levels than was the O-demethyl metabolite.
Fig. 7.

Major metabolites of CP-31398 identified in rats in rats and dogs.
I = CP-31398
II = hydroxy CP-31398 (structural isomers may exist)
III = O-demethyl CP-31398
IV = N-demethyl CP-31398
V = hydroxy O-demethy CP-31398 (rats only; structural isomers may exist)
VI = didemethyl CP-31398 (dogs only)
3.4. Plasma drug levels and metabolic profiles of CP-31398 in dogs
On an mg/m2 basis, doses of CP-31398 administered to dogs were 83.3% of the doses administered to rats. In spite of this relatively modest difference in administered doses of CP-31398, plasma drug levels in dogs were 5- to more than 10-fold lower than in rats. As was seen in rats, plasma drug levels in dogs did increase with increasing dose, and were generally comparable in males and females.
At 4 hours post-dosing on Day 1, mean plasma CP-31398 levels were 5.0 ± 1.7 and 1.7 ± 0.4 ng/ml in male and female dogs, respectively. Mean plasma drug levels at 4 hours were 14.3 ± 4.0 ng/ml in male dogs and 20.7 ± 5.0 ng/ml in female dogs in the mid dose group, and 26.8 ± 17 ng/ml and 32.2 ± 17.5 ng/ml in male and female dogs in the high dose group.
Plasma drug levels measured in dogs during Week 4 were higher than those measured on Day 1, but the differences in comparison to Day 1 were smaller than seen in rats. At four hours post-dosing during Week 4, mean plasma CP-31398 levels in male and female dogs were 6.7 ± 2.1 and 8.0 ± 0.6 ng/ml, respectively. In the mid dose group, mean plasma drug levels at four hours were 10.8 ± 2.7 ng/ml in males and 27.2 ± 19.7 ng/ml in females. In the high dose group, mean plasma drug levels at four hours post-dosing during Week 4 were 36.6 ± 5.79 ng/ml and 42.8 ± 5.1 ng/ml in male and female dogs, respectively.
In dogs, hydroxylated and N- and O-demethylated metabolites of CP-31398 were identified, as was a didemethylated metabolite that was not present in rats; the O-demethylhydroxy metabolite that was identified in rats was not present in measureable quantities in dogs. In samples collected from dogs on Day 1 of dosing, hydroxylated metabolites of CP-31398 were most abundant. Levels of hydroxylated metabolites were approximately three-fold greater than the levels of N-demethylated and O-demethylated metabolites; the levels of N-demethyl and O-demethyl metabolites were approximately equal. The didemethylated metabolite was less abundant, and represented approximately one-third to one-half of the levels of the demethylated metabolites. On Day 25, the hydroxylated and N-demethylated metabolites were found in approximately equal quantities; the O-demethylated metabolite was approximately one-half as abundant. As was the case on Day 1, levels of the didemethyl metabolite were approximately one-half of the levels of the product(s) of O-demethylation.
4. Discussion
The high doses of CP-31398 used in the subchronic oral toxicity studies in rats (160 mg/kg/day [960 mg/m2/day]) and in dogs (40 mg/kg/day [800 mg/m2/day]) both exceeded the maximum tolerated dose (MTD) for this agent. Daily gavage administration of the high dose of CP-31398 induced mortality in 2/5 female dogs; both deaths were associated with hepatic necrosis. In rats, submassive coagulation necrosis (hepatic infarct) was identified in all eleven early deaths (7/15 male rats, 4/15 female rats) in the study. Although microscopic lesions were identified in several other tissues in rats receiving the high dose of CP-31398, the observed hepatic infarcts clearly identify the liver as the most sensitive target tissue for CP-31398 toxicity in rats, and identify hepatic toxicity as the proximal cause of death in these animals.
At the mid doses (80 mg/kg/day in rats; 20 mg/kg/day in dogs), no mortality or other evidence of dose-limiting toxicity was identified in either species. On this basis, the mid doses used in each study were identified as the MTD for oral administration of CP-31398 for 28 days. On a mg/m2 basis, the MTDs for subchronic oral administration of CP-31398 are comparable in the two species (rats: 480 mg/m2/day; dogs: 400 mg/m2/day).
The mid dose of CP-31398 reduced mean body weight gain in female rats; this effect persisted through the recovery period. In addition, the mid dose of CP-31398 increased mean liver weights in female rats and increased plasma transaminase levels in both sexes, suggesting that the liver is a site of CP-31398 toxicity in the mid dose group. These data were confirmed and extended by microscopic evaluation of tissues collected at the Day 29 necropsy; microscopic evidence of hepatic toxicity (infarcts [mild severity], fibrosis [mild to moderate severity], and bile duct hyperplasia [minimal to mild severity]) was identified in 2/10 female rats in the mid dose group. Hepatic fibrosis was also seen at the recovery necropsy in 1/5 female rats in the mid dose group.
Dogs receiving subchronic oral exposure to CP-31398 at the mid dose (20 mg/kg/day [400 mg/m 2/day]) demonstrated mild anemia and statistically significant reductions in body weight gain; neither effect was completely reversed during the recovery period. The liver was not a site of critical microscopic changes in dogs in the mid dose group; notable microscopic lesions identified in dogs in this group included minimal gliosis in the brain and minimal to mild perivascular lymphocytes in the brain and spinal cord; perivascular lymphocytes in the brain and spinal cord were also identified in recovery dogs in this dose group.
Although no clinical evidence of toxicity was identified in rats receiving subchronic oral exposure to CP-31398 at the low dose (40 mg/kg/day [240 mg/m2/day], low incidences of several apparently reversible microscopic changes (alveolar histiocytosis, adrenocortical vacuolation, and endocardiosis in the heart valve) were seen. Dogs in the low dose group (10 mg/kg/day [200 mg/m 2/day]) demonstrated reduced body weight gain, mild anemia, and reversible microscopic changes of minimal severity in the brain and spinal cord (perivascular lymphocytes) and lung (alveolar histiocytosis). Although the toxicologic significance of the changes seen in the low dose groups in both rats and dogs is considered to be modest, the presence of these alterations demonstrates that both of the low doses are above the No Observed Adverse Effect Level (NOAEL) for subchronic oral administration of CP-31398 in these species.
The challenges posed by agent toxicology are substantially different for the development of novel cancer chemopreventive agents than they are for cancer therapeutics. In most cases, chemopreventive agents are designed to be administered to individuals who have an elevated cancer risk, but are otherwise healthy. In such a population, a successful outcome is the statistical demonstration of a reduced incidence of cancer in the treated population. In general, it is not possible to identify specific individuals for whom a chemopreventive agent has actually protected against the induction of malignancy (individuals who would have otherwise developed cancer but did not as a result of the chemopreventive intervention) as opposed to members of the population who would have not developed cancer, regardless of the intervention. Because chemopreventive agents are designed for administration to otherwise healthy individuals, and because a successful outcome is based on demonstrated protection in a population rather than in a specific individual, chemopreventive agents must be designed and developed with the goal of identifying pharmacologically active doses that induce zero or minimal toxicity. Should a chemopreventive agent induce any toxicity in an otherwise healthy individual, that individual becomes less likely to maintain compliance and chemopreventive activity may be lost.
By contrast, cancer therapeutics are designed to be administered to patients with existing malignant disease. Because the consequences of intervention failure or lack of compliance by cancer patients may be both immediate and severe, a far greater level of agent toxicity is generally tolerated for cancer therapeutic agents than for cancer chemopreventive drugs. Toxicity in cancer chemotherapy patients is also actively managed by clinical oncologists, and the potential use of combination regimens may reduce the toxicity of a chemotherapeutic agent while maintaining much or all of its therapeutic activity. As a result, although a key goal in the development of cancer chemotherapeutics is to maximize therapeutic ratios, the induction of toxicity by a pharmacologically active dose of a chemotherapeutic drug is seen as a less significant limitation to its clinical use than is the induction of toxicity by a chemopreventive agent.
The results of the present toxicology studies suggest that human chemoprevention trials with CP-31398 would likely be initiated using a dose ≤ 400 mg/m2/day. In a chemoprevention study performed using a rat model, Rao et al. (2009) reported that dietary administration of CP-31398 (150 or 300 mg/kg diet) conferred statistically significant protection against colon carcinogenesis; the 300/mg diet level of CP-31398 used in that study produces plasma drug levels that are comparable to those seen in the low dose group (40 mg/kg/day; 240 mg/m2/day) in the present toxicology study in rats. On this basis, CP-31398 demonstrates chemopreventive efficacy in rats at plasma drug levels at which only limited toxicity is seen. However, although the toxicity induced in rats exposed to this dose of CP-31398 was modest, this dose was considered to be above the NOAEL. Should comparable toxicity be induced by similar plasma drug levels of CP-31398 in otherwise healthy individuals, oral administration of this agent may be considered unsuitable for use in chemoprevention clinical trials.
In chemotherapy studies performed using human tumor xenograft models in mice, administration of CP-31398 at 100 or 200 mg/kg/day (300 or 600 mg/m2/day) inhibited the growth and/or induced the regression of DLD-1 colon carcinomas and A375.S2 melanomas (Foster et al., 1999). Although no published data are available to link plasma drug levels to chemotherapeutic efficacy in xenograft or other tumor models, interspecies extrapolations demonstrate that chemotherapeutic activity in mice is seen at a CP-31398 dose (300 mg/m2/day) that is below the MTD for CP-31398 in both rats (480 mg/m2/day) and dogs (400 mg/m2/day). These results demonstrate that tumor growth inhibition and/or tumor regression can be achieved by CP-31398 doses that are below the MTD for this agent. Further reductions in agent toxicity might be achievable through the use of combination drug regimens and/or the active management of CP-31398 toxicity.
Although its substantial toxicity in the liver and other tissues in rats and dogs make CP-31398 an unlikely agent for further development for cancer prevention, cancer chemotherapeutic activity can be achieved at CP-31398 doses that are well below the MTD. Further, the results of the present toxicology studies suggest that the most significant hepatic toxicities of CP-31398 can be titrated out at low doses. On this basis, this agent may have utility as either a cancer therapeutic or as adjunctive therapy for cancer.
CP-31398 was originally identified in a high throughput screening system for its ability to restore the wild-type conformation to the DNA binding domain of p53 (Foster et al., 1999). Toxicity was not considered in this screening system, and although CP-31398 demonstrates useful activity in preclinical models for cancer chemoprevention and cancer therapy, the pharmacologic activity of this agent is clearly limited by hepatic and other toxicities. However, the possibility remains that structural modifications of CP-31398 may generate congeners with equivalent or better pharmacologic activity and reduced toxicity. In the present studies, we identified two major sites of CP-31398 metabolism: N-demethylation of the alkyl side chain generates the most abundant metabolite of CP-31398 in both rats and dogs, while the styryl benzene group undergoes both hydroxylation and O-demethylation to form a series of more polar metabolites. Although the relationships between the metabolism and biological activities of CP-31398 have not been elucidated, it is possible that the toxicity of this agent may be reduced by modification of either or both sites of metabolism.
Acknowledgments
This work was supported by contract N01-CN-43304 (HHSN261200433004C) from the National Cancer Institute, Division of Health and Human Services.
The authors thank Ms. Nicole Kozub, Ms. Sheila Huang, and Ms. Gail Dianis for excellent technical assistance and Ms. Leigh Ann Senoussi for assistance in manuscript preparation.
Abbreviations
- FOB
functional observational battery
- LC/QTOF
liquid chromatography/quantitative time-of-flight mass spectrometry
- ANOVA
analysis of variance
- CNS
central nervous system
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BUN
blood urea nitrogen
- WBC
white blood cells
- RBC
red blood cells
- MTD
maximum tolerated dose
- NOAEL
no observed adverse effect level
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
William D. Johnson, Email: wjohnson@iitri.org.
Miguel Muzzio, Email: mmuzzio@iitri.org.
Carol J. Detrisac, Email: Carol.Detrisac@crl.com.
Izet M. Kapetanovic, Email: kapetani@mail.nih.gov.
Levy Kopelovich, Email: kopelovl@mail.nih.gov.
David L. McCormick, Email: dmccormick@iitri.org.
References
- Brown CJ, Cheok CF, Verma CS, Lane DP. Reactivation of p53: from peptides to small molecules. Trends Pharmacol Sci. 2011;32:53–62. doi: 10.1016/j.tips.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nat Rev Clin Oncol. 2011;8:25–37. doi: 10.1038/nrclinonc.2010.174. [DOI] [PubMed] [Google Scholar]
- Demma JJ, Wong S, Maxwell E, Dasmahapatra B. CP-31398 restores DNA-binding activity to mutant p53 in vitro but does not affect p53 homologs p63 and p73. J Biol Chem. 2004;279:45887–45896. doi: 10.1074/jbc.M401854200. [DOI] [PubMed] [Google Scholar]
- Foster BS, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science. 1999;286:2507–2510. doi: 10.1126/science.286.5449.2507. [DOI] [PubMed] [Google Scholar]
- Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54:4855–4878. [PubMed] [Google Scholar]
- Harris MP, Sutjipto S, Wills KN, Hancock W, Cornell D, Johnson DE, Gregory RJ, Shepard HM, Maneval DC. Adenovirus-mediated p53 gene transfer inhibits growth of human tumor cells expressing mutant p53 protein. Cancer Gene Ther. 1996;3:121–130. [PubMed] [Google Scholar]
- Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- Horowitz J. Adenovirus-mediated p53 gene therapy: overview of preclinical studies and potential clinical applications. Curr Opin Mol Ther. 1999;1:500–509. [PubMed] [Google Scholar]
- Joerger AC, Fersht AR. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene. 2007;26:2226–2242. doi: 10.1038/sj.onc.1210291. [DOI] [PubMed] [Google Scholar]
- Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friendman P, Prives C, Vogelstein B. Identification of p53 as a sequence-specific DNA-binding protein. Science. 1991;252:1708–1711. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- Kim SH, Dass CR. p53-targeted cancer pharmacotherapy: move towards small molecule compounds. J Pharm Pharmacol. 2011;63:605–610. doi: 10.1111/j.2042-7158.2010.01248.x. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Luu Y, Bush J, Cheung KJ, Jr, Li G. The p53 stabilizing compound CP-31398 induces apoptosis by activating the intrinsic Bax/mitochondrial/caspase-9 pathway. Exp Cell Res. 2002;276:214–222. doi: 10.1006/excr.2002.5526. [DOI] [PubMed] [Google Scholar]
- Molchadsky A, Rivlin N, Brosh R, Rotter V, Sarig R. p53 is balancing development, differentiation and de-differentiation to assure cancer prevention. Carcinogenesis. 2010;31:1501–1508. doi: 10.1093/carcin/bgq101. [DOI] [PubMed] [Google Scholar]
- Muzzio M, Huang Z, Johnson WD, McCormick DL, Kapetanovic IM. Determination and stability of CP-31398 in plasma from experimental animals by LC-MS/MS. J Pharm Biomed Anal. 2011 doi: 10.1016/j.jpba.2011.07.013. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao CV, Steele VE, Swamy MV, Patlolla JM, Guruswamy S, Kopelovich L. Inhibition of azoxymethane-induced colorectal cancer by CP-31398, a TP53 modulator, alone or in combination with low doses of Celecoxib in male F34 rats. Cancer Res. 2009;69:8175–8182. doi: 10.1158/0008-5472.CAN-09-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao CV, Swamy MV, Patlolla JM, Kopelovich L. Suppression of familial adenomatous polyposis by CP-31398, a TP53 modulator in APCmin/+ mice. Cancer Res. 2008;68:7670–7675. doi: 10.1158/0008-5472.CAN-08-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JL, Kang SK, Minn I, Califano JA, Sidransky D, Koch WM. p53-Reactivating small molecules induce apoptosis and enhance chemotherapic cytotoxicity in head and neck squamous cell carcinoma. Oral Oncol. 2011;47:8–15. doi: 10.1016/j.oraloncology.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto R, Wang W, Dicker DT, Rastinejad F, Lyssikatos J, El-Deiry WS. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis in human cancer cells and can stabilize wild-type p53 protein. Cancer Biol Ther. 2002;1:47–55. doi: 10.4161/cbt.1.1.41. [DOI] [PubMed] [Google Scholar]
- Tang X, Zhu Y, Han L, Kim AL, Kopelovich L, Bickers DR, Athar M. CP-31398 restores mutant p53 tumor suppressor function and inhibits UVB-induced skin carcinogenesis in mice. J Clin Invest. 2007;117:3753–3764. doi: 10.1172/JCI32481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Rastinejad F, El-Deiry WS. Restoring p53-dependent tumor suppression. Cancer Biol Ther. 2003;2:S55–63. [PubMed] [Google Scholar]
- Xu J, Timares L, Heilpern C, Weng Z, Li C, Xu H, Pressey JG, Elmets CA, Kopelvich L, Athar M. Targeting wild-type and mutant p53 with small molecule CP-31398 blocks the growth of rhabdomyosarcoma by inducing reactive oxygen species-dependent apoptosis. Cancer Res. 2010;70:6566–6576. doi: 10.1158/0008-5472.CAN-10-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]