

Background: The stability of Gli proteins is important for the outcome of Hedgehog signaling.
Results: Dzip1 is involved in a novel Gli turnover mechanism. This function of Dzip1 is regulated by CK2 and PP2A-dependent reversible phosphorylation.
Conclusion: The antagonistic action of PP2A and CK2 controls the phosphorylation and Gli turnover function of Dzip1.
Significance: Our studies have identified a novel Gli regulatory mechanism.
Keywords: Development, Hedgehog, Phosphorylation, PP2A, Protein Degradation, B56, CK2, Dzip1, Gli
Abstract
The Hedgehog (Hh) pathway is evolutionarily conserved and plays critical roles during embryonic development and adult tissue homeostasis. Defective Hh signaling has been linked to a wide range of birth defects and cancers. Hh family proteins regulate the expression of their downstream target genes through the control of proteolytic processing and the transcriptional activation function of Gli transcription factors. Although Hh-dependent regulation of Gli has been studied extensively, other Gli regulatory mechanisms remain relatively unappreciated. Here we report our identification of a novel signaling cascade that controls the stability of Gli proteins. This cascade consists of Daz interacting protein 1 (Dzip1), casein kinase 2 (CK2), and B56 containing protein phosphatase 2As (PP2As). We provide evidence that Dzip1 is involved in a novel Gli turnover pathway. We show that CK2 directly phosphorylates Dzip1 at four serine residues, Ser-664/665/706/714. B56-containing PP2As, through binding to a domain located between amino acid residue 474 and 550 of Dzip1, dephosphorylate Dzip1 on these CK2 sites. Our mutagenesis analysis further demonstrates that the unphosphorylatable form of Dzip1 is more potent in promoting Gli turnover. Consistently, we found that the stability of Gli proteins was decreased upon CK2 inhibition and increased by inhibition of B56-containing PP2As. Thus, reversible phosphorylation of Dzip1, which is controlled by the antagonistic action of CK2 and B56-containing PP2As, has an important impact on the stability of Gli transcription factors and Hh signaling.
Introduction
The Hh2 pathway is essential for embryogenesis and adult tissue homeostasis. Dysregulation of Hh signaling causes severe consequences ranging from birth defects to the development of basal cell carcinoma, medulloblastoma, and many other tumors (1–3). At the molecular level, the Cubitus interruptus (Ci)/Gli family of zinc finger transcription factors act at the downstream end of the Hh signaling cascade to control the expression of Hh target genes. In the absence of Hh signaling, Ci/Gli proteins undergo proteolytic cleavage, leading to production of the repressor forms of Ci/Gli (4–6). Hh family proteins operate the pathway by preventing proteolytic processing of Ci/Gli and converting the full-length Ci/Gli into transcriptional activators (7).
Emerging evidence suggests that the stability of Ci/Gli can be regulated through Hh-independent Gli regulatory mechanisms. In vertebrates, Gli processing requires intact primary cilium, a microtubule-based organelle on the cell surface that is also essential for cells to respond to Hh ligands. Loss of cilia compromises proteolytic processing of Gli3 and prevents the formation of Gli3 repressor (8–11). Another important Gli regulator in vertebrates is Suppressor of Fused (Sufu), which antagonizes the Spop/Cul3 ubiquitin ligase and protects Gli2 and Gli3 from proteasome degradation (12, 13). Interestingly, although Drosophila homologue of Spop plays important roles in controlling Ci degradation (14–16), Sufu is dispensable for Drosophila Hh signaling. Gli stability can be regulated by other ubiquitin ligases. It has been reported that Itch, a critical regulator of Notch, acts together with Numb to regulate Gli1 turnover (17, 18). Several motifs in Gli protein function as degradation signals to mediate proteasome degradation of Gli. Some of these motifs regulate the stability of Gli through as yet unclear mechanisms (19), arguing for the existence of more Ci/Gli regulatory pathways.
PP2A is one of the major Ser/Thr protein phosphatases. PP2A holoenzymes consist of a catalytic subunit (C), scaffold subunit (A), and variable regulatory subunit (B). The catalytic activity, subcellular localization, and substrate specificity of PP2A are regulated by multiple families of regulatory subunits, including B55/B, B56/B′, and PR72/B″ (20). PP2A is an important Ci/Gli regulator. In Drosophila, B56-containing PP2A is required for Hh signaling (21). WDB, a Drosophila B56 family member (22), dephosphorylates Ci and prevents Ci processing (23). In vertebrates, PP2A regulates Hh signaling at the level of Gli as well. It has been observed that PP2As regulate the nuclear localization of Gli3 (24, 25). We previously reported that B56ϵ positively regulates transcriptional activation function of Gli during Xenopus embryonic development (26). To understand the mechanisms by which PP2A regulates Gli, we performed a yeast 2-hybrid screen and identified Dzip1 as a substrate of B56-containing PP2As. Previous studies in zebrafish have revealed that Dzip1 plays a dual role in the Hh pathway. Dzip1 regulates ciliogenesis (27–29) and, hence, is required for Hh signaling (30–32). Interestingly, Dzip1 also acts upstream of Gli to inhibit the Hh pathway through an as yet unclear mechanism (30–32). In this paper, we report that Dzip1 is involved in a Gli turnover pathway. CK2 and B56-containing PP2As influence the Gli turnover function of Dzip1 by controlling reversible phosphorylation of Dzip1.
EXPERIMENTAL PROCEDURES
Plasmids and Antibodies
All Dzip1 constructs were derived from mouse Dzip1 (IMAGE: 6825425) and Xenopus Dzip1 (IMAGE: 5511284). Yeast expression constructs (Dzip1, xB56ϵ, hB56α, hB56β, hB56γ1, and xB56δ (33)) and Dzip1 mammalian expression plasmids were constructed by PCR amplification and standard cloning methods. Dzip1SA and single Ser→Ala mutations were generated by site-directed mutagenesis. Cloning details will be provided upon request.
Antibodies used in this study are: anti-FLAG (Sigma F1804 or F7425, 1:1000), anti-Myc (Sigma M5546, 1:1000), anti-B56ϵ (1:500) (33), anti-PP2Ac (Upstate #05-421, 1:2000), anti-PP2Aa (Upstate #05-657, 1:500), anti-β-tubulin (Sigma T5293, 1:1000), and anti-acetylated tubulin (Sigma 6–11B1, 1:1000).
Custom anti-Dzip1 antibodies were generated by New England Peptide. Briefly, a keyhole limpet hemocyanin-coupled peptide (Ac-CAVK(pS)DTDWTEG(pS)EMD-amide) was synthesized and used to immunize rabbits for generating anti-Dzip1 antiserum. Crude sera recognized both unphosphorylated and phosphorylated Dzip1 (supplemental Fig. S1A). After phosphopeptide affinity purification, eluates contained antibodies highly specific to Dzip1 that is phosphorylated on Ser-706/714 (supplemental Fig. S1B).
Cell Culture, Transfection, and Treatments
PC6-3, HEK293T, and NIH3T3 cells were cultured as described (34, 35). Lipofectamine 2000 (Invitrogen) was used for transfection. HEK293T cells were used for protein interaction and phosphorylation analysis. NIH3T3 cells were used for all Hh signaling experiments. To replace endogenous Aα with DTP177AAA or wild type (wt) Aα, PC6-3 cells were treated with 1 μg/ml doxycycline (Dox) for 4 days. For okadaic acid (OA) and 4,5,6,7-tetrabromobenzotriazole (TBB) treatment, unless otherwise specified, cells were treated with OA or TBB in DMEM containing 0.5% FBS 1 day after transfection and harvested 18 h after addition of the drug. For dual luciferase assay, cells were transfected with 8×Gli-Luc (200 ng) and pRL-TK (25 ng). Luciferase activities were measured using the dual-luciferase reporter assay system (Promega).
Yeast Two-hybrid Assay
Mouse brain and embryonic day 7 whole embryo cDNA libraries were screened using the full-length Xenopus B56ϵ as bait. A total of five millions colonies was screened. To assay the interaction between Dzip1 and B56s, yeast AH109 strain was transformed in combination of pGBKT7 or pGBKT7-Dzip1 with pACT2, pACT2-B56α, pACT2-B56β, pACT2-B56γ, pACT2-B56δ, or pACT2-B56ϵ. Growth of transformants on selective medium was assayed.
λ-Protein Phosphatase Treatment, in Vitro Kinase Assay, Microcystin Pulldown, GST Pulldown, Co-immunoprecipitation, and Western Blots
For λ-protein phosphatase treatment, lysates were treated with 400 units of λ-protein phosphatase (New England Biolabs, #P0753) for 2 h at 30 °C. GST-Dzip1 or Dzip1SA proteins were used for in vitro CK2 (New England Biolabs, #P6010) kinase assay as described (36). Microcystin pulldown was performed as described (37). GST pulldown, co-immunoprecipitation, and Western blots were performed as described (35). To detect interaction between endogenous Dzip1 and B56ϵ, whole brain protein extract from postnatal day 15 mouse were used. For protein phosphorylation analysis, cells were extracted with lysis buffer containing phosphatase inhibitor cocktails (Sigma). For detection of phosphorylated Dzip1 protein, lysates were separated on 5% SDS-PAGE.
Xenopus Embryos and Manipulations
Xenopus embryos were obtained as described. Microinjection, animal cap assays, and section were performed as described (38). The amounts of RNAs and morpholinos used in microinjection are described in the legends of Figs. 6 and 7. Sequences of Dzip1 morpholinos are: DMO1, 5′-AGCAGCCTCTTCTCCTTCTGCGCTG-3′; DMO2, 5′-AAAGGCATTCTCTTTCCTCCCGCTC-3′ (Gene Tools, Philomath, OR). Xenopus cilia were stained by labeling acetylated tubulin as described (39). Embryo lysates were prepared as described (40).
FIGURE 6.

Knockdown of Dzip1 stabilizes Gli proteins. A, a schematic drawing shows the design of Dzip1 morpholinos. B, injection of DMOs blocked the translation of xDzip1-myc in Xenopus embryos. Fertilized eggs were injected with DMO1 (20 ng), DMO2 (20 ng), or DMO1 + DMO2 (10 ng each). Subsequently, control and injected embryos were injected with 200 pg of xDzip1-myc RNA. Embryos were harvested at the gastrula stage and subjected to Western blot analysis. β-Tubulin is a control for loading. C, depletion of Dzip1 stabilizes Gli2 protein. RNAs encoding Myc-Gli2 (1 ng), Chd (50 pg), and Myc-GFP (50 pg) were injected into the animal pole of wild type embryos or embryos previously injected with DMO1 (20 ng), DMO2 (20 ng), or DMO1+DMO2 (10 ng each). Caps were dissected at stage 8/9 and harvested at stage 18 for Western blot analysis. D, overexpression of myc-xDzip1 rescues the effect of DMOs on the stability of Gli2. Control and DMOs (20 ng)-injected embryos were injected with Myc-Gli2 or Myc-Gli2 with various doses of myc-xDzip1 (100 and 250 pg). Caps were dissected at stage8/9. Western blot analysis was performed when control embryos reached stage 18. E, Gli2 protein is rapidly degraded around stage 13. Gli2 RNA was injected into control embryos or embryos previously injected with 20 ng of DMO. Caps were dissected at stage8/9 and harvested at various developmental stages as indicated. Injection of 20 ng of DMOs delayed Gli2 turnover. F, Western blot results show that knockdown of Dzip1 resulted in stabilization of Gli1, Gli2, and Gli3 in animal caps. G, real-time RT-PCR results show that depletion of Dzip1 enhanced Gli1-induced expression of foxA2 and ptc-1 in neuralized animal caps. The expression levels of foxA2 and ptc-1 were normalized to that of ODC.
FIGURE 7.
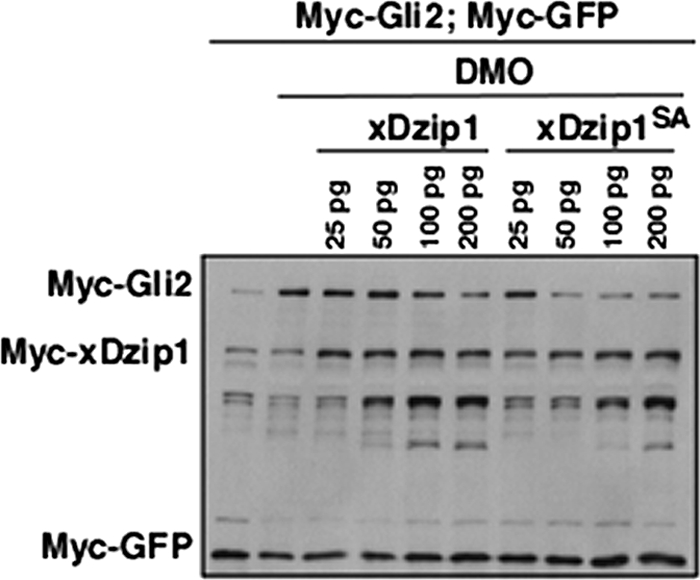
The unphosphorylatable form of Dzip1 is more potent in promoting Gli turnover. Western blot results show that xDzip1SA was more potent in rescuing DMOs-induced Gli2 stabilization. Embryo manipulation was similar to what was described in Fig. 6. Note that as little as 50 pg of xDzip1SA was sufficient for reversing the effect of DMO on Gli2. A similar rescue was achieved only when as much as 200 pg of xDzip1 was expressed.
Quantitative Real-time PCR
RNA purification and reverse transcription were performed as described (41). Real-time PCR reactions were performed in triplicate using SYBR Green master mix (Applied Biosystems) on the Applied Biosystems 7500 Real-time PCR System. Values were normalized to the control. Primers for Xenopus genes are: foxA2, 5′-gcatgaatcccatgaacacg-3′; 5′-gctcaggtgacttgcatgc-3′; ptc1, 5′-ggacaagaatcgcagagctg-3′; 5′-ggatgctcagggaacct-3′; odc, 5′-cagctagctgtggtgtgg-3′; 5′-caacatggaaactcacacc-3′. Primers for mouse genes are: gli1, 5′-tccctggtggctttcatcaact-3′; 5′-gcatcattgaaccccgagtaga-3′; ptc1, 5′-aggctctcctgcaacacctg-3′; 5′-gtaacctgtctccgtgataagttc-3′; β-actin, 5′-agagggaaatcgtgcgtgac-3′; 5′-caatagtgatgacctggccgt-3′.
RESULTS
Dephosphorylation of Dzip1 by B56-containing PP2As
In an effort to understand the mechanisms by which B56-containing PP2As regulate the Hh pathway, we performed a yeast two-hybrid screen using the full-length B56ϵ as bait. Several clones carrying various lengths of Dzip1 cDNA fragments were identified. Because Dzip1 is involved in Hh signaling (27–32), we chose to further characterize the potential interaction between B56ϵ and Dzip1.
The interaction between B56ϵ and Dzip1 was first analyzed by a GST pulldown assay. An myc-tagged Dzip1 was transfected into HEK293T cells. Cell lysate was incubated with bacterially expressed GST or GST-B56ϵ. We observed that Dzip1 was pulled down by GST-B56ϵ but not by GST (Fig. 1A, left panel). Consistently, myc-Dzip1 was immunoprecipitated by an anti-FLAG antibody when it was coexpressed with B56ϵ-FLAG in HEK293T cells (Fig. 1B), demonstrating that B56ϵ and Dzip1 interact with each other. We then generated Dzip1 deletion constructs (Fig. 1C) and mapped the B56ϵ binding domain of Dzip1. We found that the sequence between amino acid residues 474 and 550 of Dzip1 was sufficient and necessary for B56ϵ binding. GST-B56ϵ pulled down all Dzip1 deletions carrying this domain, including 1–550, 192–852, 285–550, and 474–550. In contrast, all deletions lacking this domain, including 550–852, 1–221, 192–362, and Δ474–550, were not pulled down by GST-B56ϵ (Fig. 1D). This indicates that 474–550 is the B56ϵ interacting domain (BID). Accordingly, Δ474–550 was designated as ΔBID. In a co-immunoprecipitation experiment, Dzip1, but not ΔBID, formed a complex with B56ϵ (Fig. 1E). We further determined whether the interaction between Dzip1 and B56ϵ occurs under physiological conditions. Indeed, endogenous Dzip1 was co-immunoprecipitated with endogenous B56ϵ from P15 whole mouse brain lysate (Fig. 1F). Thus, Dzip1 and B56ϵ physically interact with each other.
FIGURE 1.

Physical interaction between Dzip1 and B56ϵ. A, a GST pulldown (GST-PD) assay shows the interaction between B56ϵ and Dzip1. Lysate from myc-Dzip1-transfected cells was incubated with bacterially expressed GST and GST-B56ϵ. Proteins associated with GST and GST-B56ϵ were analyzed by Western blot and Coomassie Blue staining. Left, myc-Dzip1 in total cell lysate and after pulldown with GST and GST-B56ϵ. Right, a Coomassie Blue staining shows GST and GST-B56ϵ used in the GST pulldown. B, shown is co-immunoprecipitation (IP) of Dzip1 with B56ϵ. B56ϵ-FLAG and myc-Dzip1 were transfected either alone or in combination. Cells were harvested and subjected to co-immunoprecipitation. When co-expressed with B56ϵ-FLAG, myc-Dzip1 was immunoprecipitated by an anti-FLAG antibody. C, shown is a schematic representation of Dzip1 deletion constructs. Whether a Dzip1 construct interacts with B56ϵ in the GST pulldown assay is summarized by + (interaction detected) or – (interaction not detected). NLS, nuclear localization signal. D, shown is mapping the B56ϵ interaction domain of Dzip1. Dzip1 deletions were expressed in cells. A GST pulldown assay was performed to determine the interaction between B56ϵ and Dzip1 deletion constructs. Note that the full-length, 1–550, 192–852, 285–550, and 474–550 of Dzip1 were pulled down by GST-B56ϵ. In contrast, GST-B56ϵ failed to pull down 550–852, 1–221, 192–362, and Δ474–550 of Dzip1. E, BID is required for the binding between Dzip1 and B56ϵ. Myc-Dzip1 or ΔBID were transfected into HEK293 cells alone or together with B56ϵ-FLAG. After cells were harvested, a co-immunoprecipitation was performed using an anti-FLAG antibody. Note that the full-length Dzip1, but not ΔBID, formed a complex with B56ϵ. F, shown is the interaction between endogenous B56ϵ and Dzip1. Anti-B56ϵ antibody was used to immunoprecipitate endogenous B56ϵ from P15 mouse whole brain lysate. A Western blot was performed subsequently to monitor endogenous Dzip1 and B56ϵ. Heat-inactivated anti-B56ϵ antibody served as a negative control.
The interaction between Dzip1 and B56ϵ regulatory subunit of PP2A suggests that PP2A dephosphorylates Dzip1. To test this hypothesis, we first asked whether Dzip1 forms a complex with PP2A holoenzymes. A microcystin pulldown assay was performed to purify PP2Ac, the catalytic subunit of PP2A. We observed that Dzip1, but not EGFP, was co-purified with PP2Ac (Fig. 2A, top panel), demonstrating that Dzip1 forms a complex with PP2A holoenzymes. Moreover, we found that Dzip1 is a phosphoprotein. When separated on 5% SDS-PAGE, two Dzip1 bands were detected. The slower migrating band, which collapsed after a λ-protein phosphatase treatment, is the phosphorylated form of Dzip1 (Fig. 2B). When cells were treated with PP2A inhibitor OA, the amount of the phosphorylated Dzip1 was increased (Fig. 2C). We also noticed that ΔBID, which does not bind to B56ϵ, was hyperphosphorylated in transfected cells (data not shown, also see Fig. 3E). When expressed in Xenopus neural ectoderm, the majority of ΔBID existed in its phosphorylated form (Fig. 2D). Interestingly, the phosphorylation of Dzip1 is not regulated by Hh signaling. Overexpression of Shh in Xenopus embryos (data not shown) or stimulating cells with Smoothened agonist purmorphamine failed to alter the phosphorylation of Dzip1 (Fig. 2E). Purmorphamine treatment markedly induced the expression ptc-1 and gli1 (data not shown, also see Fig. 8D). It appears that PP2A maintains Dzip1 in its dephosphorylated form.
FIGURE 2.

Dzip1 is dephosphorylated by PP2A. A, interaction between Dzip1 and PP2Ac is shown. Myc-Dzip1 and myc-EGFP were transfected into HEK293 cells. Cell lysates were incubated with microcystin beads, which directly bind to PP2Ac. Myc-Dzip1, but not myc-EGFP, was pulled down (PD) by microcystin beads. B, Dzip1 is a phosphoprotein. Western blot results show the mobility change of Dzip1 on SDS-PAGE after a λ-protein phosphatase (λPPase) treatment. C, phosphorylation of Dzip1 is enhanced by inhibition of PP2A. Myc-Dzip1-transfected cells were either mock-treated (DMSO) or treated with OA (50 nm). Cell lysates were blotted with anti-myc antibody. D, ΔBID is hyperphosphorylated. Myc-Dzip1 and myc-ΔBID were expressed in Xenopus embryos. Embryo lysates were either treated with λ-protein phosphatase or left untreated. Western blot was performed subsequently to analyze the phosphorylation of Dzip1 and ΔBID. E, Dzip1 phosphorylation is independent of Hh signaling. Dzip1-transfected NIH3T3 cells were either mock-treated (DMSO) or treated with purmorphamine. Cell lysates were analyzed by a myc Western. Although purmorphamine treatment induced expression of ptc-1 and gli1 robustly (data not shown; also see Fig. 8D), phosphorylation of Dzip1 remained unchanged. Myc-EGFP in A, C, D, and E served as a control for transfection/microinjection and loading.
FIGURE 3.

Dephosphorylation of Dzip1 by B56-containing PP2As. A, shown is a summary of the interaction between B56s and Dzip1 in the yeast 2-hybrid system. The number of + symbols indicates the relative binding affinity, as judged by the growth rate of yeast transformants. B, Western blot results show that overexpression of B56ϵ resulted in dephosphorylation of Dzip1. C, Western blot results show dephosphorylation of Dzip1 by B56γ. D, Western blot results show the effect of B56γ overexpression on the phosphorylation status of ΔBID. E, depletion of B56s enhances the phosphorylation of Dzip1. PC6-3 cells were transfected with Dzip1 or ΔBID. Cells were then treated with Dox and subjected to Western blot analysis. Depletion of B56s by Dox-induced Aα → DTP177AAA exchange enhanced phosphorylation of Dzip1 (lanes 1 and 2) but not that of ΔBID (lane 3 and 4). Replacing endogenous Aα with a wt Aα failed to affect Dzip1 phosphorylation (lanes 5–8). Lacking an anti-pan-B56s antibody, we monitored the knockdown efficiency by measuring the level of B56ϵ. Myc-EGFP in B–E served as a control for transfection and loading.
FIGURE 8.

The antagonistic action of B56-containing PP2As and CK2 controls the stability of Gli proteins. A, knockdown of B56-containing PP2As in PC6-3 cells by Dox-induced Aα → DTP177AAA exchange enhanced the activity of Gli1 in an 8×Gli luciferase assay. Firefly luciferase activity was normalized to the activity of Renilla luciferase. B, Western blot results show that knockdown of B56-containing PP2As stabilized Gli1, Gli2, and Gli3. C, inhibition of CK2 in NIH3T3 cells by TBB treatment reduced the activity of Gli1 in an 8×Gli luciferase assay. D, inhibition of CK2 reduces purmorphamine-induced expression of gli1 and ptc-1. NIH3T3 cells were treated with purmorphamine alone (PM) or together with various doses of TBB. expression of ptc-1 and gli1 was assayed by real-time RT-PCR. E, Western blot results show that inhibition of CK2 by TBB in NIH3T3 cells decreased the stability of Gli1, Gli2, and Gli3, whereas inhibition of PP2A (OA treatment) increased the level of Gli proteins. When both CK2 and PP2A were inhibited, Gli proteins were degraded, similar to the effect seen when just CK2 was inhibited. F, shown is a working model for regulation of the Gli-destabilizing function of Dzip1 by reversible phosphorylation. CK2 phosphorylated Dzip1. The phosphorylated Dzip1 is less active. B56-containing PP2As, through binding to the BID, dephosphorylate Dzip1 and enhance the Gli-destabilizing function of Dzip1.
We then examined the effect of B56ϵ knockdown on Dzip1 phosphorylation. Surprisingly, we found that knockdown of B56ϵ in Xenopus embryos or HEK293T cells failed to cause any detectable change in the phosphorylation of Dzip1 (data not shown), raising the possibility that B56 family members function redundantly to dephosphorylate Dzip1. To test this hypothesis, we examined the interaction between Dzip1 and all five B56 family members using the yeast two-hybrid system. Indeed, co-transformation of Dzip1 with B56s supported the growth of yeast on selection medium (Fig. 3A). This indicates that all B56 family members are capable of binding to Dzip1, although the binding affinities vary as judged by the growth rate of yeast. When overexpressed in HEK293T cells, B56ϵ (Fig. 3B) or B56γ (Fig. 3C) dephosphorylated Dzip1 robustly. Overexpression of B56γ had no effect on the phosphorylation of ΔBID (Fig. 3D), demonstrating that dephosphorylation of Dzip1 by B56s requires physical interaction between Dzip1 and B56s. Overexpression of B56α, B56β, or B56δ had only marginal effects (data not shown). These results suggest that B56-containing PP2As can function redundantly to dephosphorylate Dzip1.
We next decided to knock down all five B56s and assayed Dzip1 phosphorylation. The experiment was performed in PC6-3 cells, a specially engineered system allowing Dox inducible knockdown of all five B56s (34). This system takes advantage of the fact that monomeric B56s are degraded rapidly through the proteasome pathway (34, 42, 43). When PC6-3 cells are treated with Dox, endogenous Aα is replaced with DTP177AAA, a mutant form of Aα deficient in binding of B56s but not other PP2A regulatory subunits. This leads to monomerization of B56s and ultimately knockdown of all B56s in PC6-3 cells (34, 44). We also performed our analysis in a control PC6-3 cell line, in which endogenous Aα is replaced with a wt exogenous Aα upon Dox treatment. As shown in Fig. 3E, knockdown of total B56s by Dox-induced Aα → DIP177AAA exchange increased the amount of phosphorylated Dzip1. In contrast, the phosphorylation status of ΔBID was not altered by Dox-induced knockdown of B56s. Furthermore, Dox-induced Aα → wt Aα exchange, which had no effect on B56s, failed to alter the phosphorylation of Dzip1. Thus, knockdown of total B56s enhances phosphorylation of Dzip1. Taken together, we conclude that B56-containing PP2As dephosphorylate Dzip1.
Phosphorylation of Dzip1 by CK2
Knowing that B56-containing PP2As dephosphorylate Dzip1, we set out to identify the sites of Dzip1 that are dephosphorylated by B56-containing PP2As. Several Dzip1 deletion constructs were generated (Fig. 4A, upper panel). The migration rate of protein on SDS-PAGE was used as a readout for phosphorylation of Dzip1 constructs. As shown in Fig. 4B, all Dzip1 constructs carrying amino acid residues 620–740 (550–852 and 620–740) were phosphorylated, whereas constructs lacking 620–740 (1–550, 550–641, and 739–852) migrated as a single band. The region of 620–740 contains 10 putative kinase recognition sites (Fig. 4A). We mutated these potential phosphorylation sites and assayed the phosphorylation of these Dzip1 mutants in HEK293T cells. Strikingly, mutation of Ser-664, -665, -706, and -714 significantly reduced the amount of phosphorylated Dzip1, whereas mutation of other sites had no effect. Dzip1SA, which carries Ser → Ala mutation on all four serine residues, was no longer band-shifted (Fig. 4C), indicating that Dzip1 is phosphorylated at Ser-664/665/706/714. We then determined whether B56-containing PP2As dephosphorylate Dzip1 at Ser-664/665/706/714. Knockdown of B56s in PC6-3 cells or treating cells with OA enhanced phosphorylation of wild type Dzip1. In contrast, PP2A inhibition failed to alter the phosphorylation status of Dzip1SA (Fig. 4, D and E). Based on these results, we conclude that B56-containing PP2As dephosphorylate Dzip1 at Ser-664/665/706/714.
FIGURE 4.

B56-containing PP2As dephosphorylated Dzip1 at Ser-664/665/706/714. A, shown is a schematic representation of Dzip1 deletion constructs used to identify phosphorylation sites of Dzip1. All potential phosphorylation sites within residue 620–740 are listed. The four CK2 sites are highlighted in boxes. aa, amino acids. NLS, nuclear localization signal. B, shown is phosphorylation of Dzip1 deletions. Dzip1 deletion constructs were transfected into HEK293T cells and subjected to Western blot analysis. All deletions carrying 620–740 are phosphorylated, as judged by mobility shift of proteins on SDS-PAGE. λPPase, λ-protein phosphatase. C, shown is phosphorylation of Dzip1 mutants. Dzip1 mutants were transfected into HEK293T cells and analyzed by Western blotting. Whether a protein is phosphorylated is judged by the mobility shift of protein on SDS-PAGE. Mutations of Ser-664, -665, -706, or -714, but not other potential kinase recognition sites, impaired phosphorylation of Dzip1. Dzip1 with Ser-664/665/706/714 mutated is denoted as Dzip1SA. D, knockdown of B56s had no effect on the phosphorylation status of Dzip1SA. Dzip1 and Dzip1SA were transfected into PC6-3 cells. Cells were treated with Dox to induce B56s knockdown. Phosphorylation of Dzip1 and Dzip1SA was assayed. Note that knockdown of B56s failed to alter the migration rate of Dzip1SA on SDS-PAGE. E, inhibition of PP2A had no effect on the phosphorylation status of Dzip1SA. Dzip1 and Dzip1SA were transfected into NIH3T3 cells. Cells were treated with OA to inhibit PP2A. Western blot was performed to assay the phosphorylation status of Dzip1 and Dzip1SA.
Intriguingly, Ser-664/665/706/714 are all putative CK2 recognition sites (Fig. 4A). These Dzip1 phosphorylation sites are conserved from zebrafish to mammals. To determine whether CK2 has the ability to phosphorylate Dzip1 at Ser-664/665/706/714, we performed an in vitro kinase assay using commercially purchased CK2 and bacterially expressed GST-Dzip1 (amino acid residues 650–730). We observed that GST-Dzip1, but not GST-Dzip1SA, was phosphorylated by CK2 in vitro (Fig. 5A, lanes 2 and 3). We further investigated CK2-dependent phosphorylation of Dzip1 in vivo by overexpression or pharmacological inhibition of CK2 in HEK293T cells. As shown in Fig. 5B, overexpression of CK2 (α + β) markedly enhanced phosphorylation of the wild type Dzip1 and increased the phosphorylation of S664/665A, S706A, and S714A to some degree but had no effect on the phosphorylation of Dzip1SA. Conversely, treating cells with TBB, a widely used CK2 inhibitor, decreased the amount of phosphorylated Dzip1 (Fig. 5C). Taken together, we conclude that CK2 phosphorylates Dzip1 at Ser-664/665/706/714.
FIGURE 5.

Ser-664/665/706/714 of Dzip1 are phosphorylated by CK2. A, CK2 phosphorylated GST-Dzip1 in an in vitro kinase assay. GST-Dzip1SA served as a negative control. B, shown is the effects of CK2 overexpression on the phosphorylation of Dzip1 and phospho mutants. Dzip1 and Dzip1 phospho mutants were either transfected alone or together with CK2. Western blot was performed to assay the phosphorylation of Dzip1 and Dzip1 mutants. Note that weak phosphorylation of S664A, S665A, S706A, and S714A was detected when CK2 was overexpressed. In contrast, overexpression of CK2 had no effect on Dzip1SA. C, shown is the effects of CK2 inhibition on the phosphorylation of Dzip1. Dzip1-transfected cells were treated with various doses of TBB. A Western blot was performed to evaluate the effects of CK2 inhibition on Dzip1 phosphorylation. D, the antagonistic action of CK2 and B56-containing PP2As determines the phosphorylation status of Dzip1. Dzip1 and ΔBID were transfected alone or with CK2 and B56ϵ/γ. A Western blot was performed to assay the phosphorylation of Dzip1. Phosphorylation of Dzip1 was increased upon overexpression of CK2 and decreased by B56ϵ or B56γ. In contrast, ΔBID can be phosphorylated by CK2 but cannot be dephosphorylated by B56-contaning PP2As. E, CK2 and PP2A regulate the phosphorylation of Dzip1 on Ser-706/714. Dzip1-transfected cells were left untreated or treated with various doses of OA. In parallel experiments, Dzip1 was transfected alone or together with CK2. The phosphorylation of Dzip1 was monitored using an antibody recognizing Dzip1 that is phosphorylated on Ser-706/714.
Ser-664/665/706/714 of Dzip1 are phosphorylated by CK2 and dephosphorylated by B56-containing PP2As. To better visualize this antagonistic action, we overexpressed CK2 and B56ϵ/γ individually or in combination and examined the effects on Dzip1 phosphorylation. As shown in Fig. 5D, the phosphorylation of Dzip1 was increased upon overexpression of CK2 and decreased when B56ϵ or B56γ were overexpressed. CK2-induced phosphorylation of Dzip1 was reversed by overexpression of B56ϵ or B56γ. In contrast, CK2-induced phosphorylation of ΔBID was not affected by co-expression of B56ϵ or B56γ, further reinforcing the importance of the physical interaction between Dzip1 and B56-containing PP2As.
To directly assay phosphorylation of Dzip1 on these CK2 sites, we generated an antibody that detects Dzip1 only when it was phosphorylated at Ser-706/714 (supplemental Fig. S1B). As shown is Fig. 5E, phosphorylation of Dzip1 on Ser-706/714 was significantly increased when cells were exposed to OA, the PP2A inhibitor. A similar result was observed when CK2 was overexpressed. Thus, the antagonistic action of PP2A and CK2 controls the phosphorylation status of Dzip1.
The Antagonistic Action of B56-containing PP2As and CK2 Controls the Gli-destabilizing Function of Dzip1
In Drosophila, inhibition of CK2 induces proteasome-dependent degradation of Ci (36). Recent studies indicate that in addition to its function in ciliogenesis (27–29), Dzip1 also plays a negative role in the Hh pathway. Loss of Dzip1 induces simultaneous Hh signaling in cells some distance away from Hh secreting cells. Importantly, ectopic Hh signaling in Dzip1 mutant is Gli-dependent (30–32). Because Dzip1 is phosphorylated by CK2, we asked whether Dzip1 is involved in a Gli-regulatory mechanism.
We designed two morpholinos (DMO1 and DMO2) (Fig. 6A, highlighted in boxes) and investigated functions of Dzip1 in Xenopus. These DMOs inhibited translation of a C-terminal myc-tagged xDzip1 efficiently (Fig. 6B). Strikingly, injection of DMOs markedly increased the protein level of overexpressed Gli2 (Fig. 6C). The effect was observed when DMOs were injected individually or in combination, indicating that stabilization of Gli2 by DMOs was not an off-target effect. In addition, we performed a rescue experiment. DMOs-induced Gli2 stabilization was reversed by an N-terminal Myc-tagged xDzip1, whose translation was not affected by DMOs (Fig. 6D), further proving the specificity of Dzip1 knockdown. Interestingly, we found that Gli2 was rapidly degraded from the late-gastrula stage in Xenopus embryos. Knockdown of Dzip1 had no effect on Gli2 during earlier stages but prevented the turnover of Gli2 at the late-gastrula stage (stage 13) (Fig. 6E). We assayed other Gli family members as well. As shown in Fig. 6F, Gli1 and Gli3 were also stabilized upon depletion of Dzip1. Consistent with these observations, we found that knockdown of Dzip1 enhanced Gli1-dependent transcription. Overexpression of a low dose of Gli1 (25 pg) in neuralized animal caps was not sufficient for the expression of foxA2 and ptc-1. In contrast, when the same amount of Gli1 was expressed in Dzip1-depleted caps, a robust induction of foxA2 and ptc-1 was detected (Fig. 6G). These results suggest that Dzip1 is involved in a Gli turnover pathway.
Dzip1 is required for ciliogenesis (27–29). Loss of cilia resulted in reduced proteolytic processing of Gli proteins in vertebrates (8–11). It is formally possible that stabilization of Gli in Dzip1-depleted animal caps is indirectly caused by defective ciliogenesis. Alternatively, Dzip1 may regulate Gli stability independent of its role in ciliogenesis. To distinguish between these possibilities, we carried out dose titration experiments to determine whether Dzip1-dependent ciliogenesis and Dzip1-dependent Gli turnover could be uncoupled. Thus, increasing amounts of DMOs were injected. Effects of DMOs injection on Gli2 stabilization and ciliogenesis were examined. As shown in supplemental Fig. S2A, injection of as little as 10 ng of DMOs stabilized Gli2 protein. Injection of higher doses of DMOs did not further increase the level of Gli2. To evaluate the effect of Dzip1 knockdown on ciliogenesis, we examined floor plate primary cilia and epidermis motile cilia in control and Dzip1-depleted samples. Intriguingly, little if any ciliogenesis defects were observed in embryos injected with 10 ng of DMOs (supplemental Fig. S2B), although cilia were affected in embryos injected with 30 ng of DMOs. Because a low dose of DMOs injection stabilizes Gli2 without causing detectable ciliogenesis defects, we conclude that Dzip1 regulates the stability of Gli proteins independent of its function in ciliogenesis.
We then investigated the significance of Ser-664/665/706/714 phosphorylation by titrating the doses of Dzip1 and Dzip1SA in rescue experiments. Consistent with the above results, knockdown of Dzip1 by DMOs injection stabilized Gli2 in neuralized animal caps. Although both Dzip1 and Dzip1SA could rescue the effect of DMOs on Gli2, the activity of Dzip1SA was more potent in this assay. A significant rescue was observed when as little as 50 pg of Dzip1SA RNA was injected. In contrast, a similar level of rescue was achieved only when a higher amount of Dzip1 RNA (200 pg) was injected (Fig. 7). Because the unphosphorylateable form of Dzip1 is more potent in restoring Dzip1-dependent Gli turnover, we conclude that dephosphorylation of Dzip1 at Ser-664/665/706/714 enhances the Gli-degrading function of Dzip1.
B56-containing PP2As and CK2 Antagonize Each Other to Control the Stability of Gli Proteins
The above observations demonstrate that B56-containing PP2As and CK2 antagonize each other to regulate the phosphorylation and Gli-destabilizing function of Dzip1. We thus directly investigated the functions of B56-containing PP2As and CK2 in controlling the level of Gli proteins and Hh signaling.
We first measured the effect of B56s knockdown on Hh signaling using 8×Gli-Luc, an Hh-responsive luciferase reporter carrying eight copies of Gli binding sites (45). Overexpression of Gli1 caused a dose-dependent activation of the 8×Gli-Luc in PC6-3 cells. Depletion of B56s by Dox-induced Aα → DIP177AAA exchange enhanced the activity of Gli1 in this assay (Fig. 8A). We then examined the stability of Gli proteins and found that the expression levels of Gli1, Gli2, and Gli3 were all increased upon B56s knockdown (Fig. 8B). Consistently, inhibition of total PP2A in NIH3T3 cells by an OA treatment increased the levels of Gli proteins (Fig. 8E). Thus, B56s-containing PP2As negatively regulate the stability of Gli proteins.
In parallel to the above studies, we investigated the function of CK2 in the regulation of Gli. TBB was used to inhibit the activity of CK2 in NIH3T3 cells. As shown in Fig. 8C, TBB treatment reduced Gli1-induced 8×Gli-Luc in a dose-dependent manner. In addition, TBB treatment reduced the expression of endogenous ptc-1 and gli1 in response to the Smoothened agonist purmorphamine (Fig. 8D). We found that inhibition of CK2 by TBB decreased the expression level of Gli proteins. Intriguingly, inhibition of CK2 blunted the effect of OA on the stability of Gli proteins (Fig. 8E). This demonstrates that CK2 antagonizes B56-containing PP2As to control the stability of Gli proteins.
DISCUSSION
B56-containing PP2As are important regulators of Ci/Gli. Drosophila B56 family has two members, WDB and PP2A-B′. WDB directly dephosphorylates Ci and inhibits Ci processing (23). Loss of WDB decreases the amount of full-length Ci (23) and impairs Hh signaling (21, 23). The vertebrate genome contains five B56 genes (20, 46). Our previous studies demonstrate that B56ϵ is required for the transcriptional activation function of Gli during Xenopus development (26).
In this paper we report a novel Gli inhibitory function of B56-containing PP2As. We show that B56-containing PP2As regulate the stability of Gli proteins. B56-containing PP2As function redundantly to dephosphorylate Dzip1, a protein required for Gli turnover. We were able to map B56s-sensitive phosphorylation sites to four serine residues, Ser-664/665/706/714. Strikingly, all these sites are phosphorylated by CK2, a protein kinase stabilizing Ci (36) and Gli proteins (Fig. 8). Our results argue that CK2-dependent phosphorylation decreases the Gli-destabilizing activity of Dzip1, as the unphosphorylatable form of Dzip1 is more potent in reversing Gli stabilization induced by Dzip1 knockdown (Fig. 7). It appears that B56-containing PP2As opposes CK2 to dephosphorylate Dzip1 and enhance the Gli-destabilizing function of Dzip1 (Fig. 8F). It is worth mentioning that overexpression of Dzip1, Dzip1SA, or ΔBID is not sufficient to reduce the levels of Gli proteins (data not shown), raising the possibility that B56-containing PP2As regulate the phosphorylation and function of other Gli regulators. Nevertheless, results presented here clearly demonstrate that B56-containing PP2As play an inhibitory role in the Hh pathway by negatively controlling the stability of Gli proteins.
Dzip1 plays a dual role in the Hh pathway. In zebrafish, loss of Dzip1 reduces the expression of Hh target genes in the floor plate and ventral neural tube. Intriguingly, loss of Dzip1 also causes up-regulation of Hh target genes in dorsal neural tube and somites, arguing for an inhibitory function of Dzip1 in Hh signaling (30–32). Subsequent investigation reveals that Dzip1 exhibits its positive role in Hh signaling through its function in ciliogenesis (27–29). However, the mechanism by which Dzip1 inhibits the Hh pathway remains a puzzle. Nevertheless, by analyzing Dzip1/Gli1 compound mutants, it has been found that the enhanced Hh signaling in Dzip1 mutant is Gli-dependent (30–32). Our results presented here provide mechanistic insights into the inhibitory role of Dzip1 in the Hh pathway. We show that Dzip1 is involved in a novel Gli turnover pathway. We found that the stability of Gli decreased dramatically from the late-gastrula stage (Fig. 6E). Although the mechanism by which Gli is degraded at the late-gastrula stage remains unclear, Dzip1 is clearly an important player of this Gli turnover pathway. Interestingly, a partial knockdown of Dzip1 induced Gli stabilization without causing any detectable cilia defect (supplemental Fig. S2). It seems that Dzip1 can control the stability of Gli independent of its function in ciliogenesis. In agreement with this view, earlier subcellular localization studies indicate that, in addition to cilia basal body, Dzip1 can be found in other cellular compartments. Dzip1 undergoes nuclear-cytoplasmic shuttling in response to changes in PKA activity in zebrafish paraxial mesodermal cells (31). In NIH3T3 cells, Dzip1 protein can be detected in many punctate vesicles, some of which are positive for the late endosome/lysosome marker Lamp1 (30). Further analysis will be needed to fully elucidate mechanism by which Dzip1 regulates Gli turnover.
Supplementary Material
Acknowledgments
We thank Drs. A. Ruiz I Altaba and I. Dominguez for providing Gli1, -2, and -3 and CK2 (α and β) plasmids, respectively. We are grateful to Drs. B. Mitchell and J. Stubbs for sharing cilia staining protocols. We thank Dr. J. P. Saint-Jeannet for reading the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM093217 (to J. Y.) and 1R01GM079684 (to J. J.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- Hh
- Hedgehog
- Dox
- doxycycline
- TBB
- 4,5,6,7-tetrabromobenzotriazole
- BID
- B56ϵ interacting domain
- PP2A
- protein phosphatase 2As
- OA
- okadaic acid
- Ci
- cubitus interruptus
- EGFP
- enhanced green fluorescent protein
- CK2
- casein kinase 2
- DOM
- Dzip1 Morpholino.
REFERENCES
- 1. Ingham P. W., McMahon A. P. (2001) Genes Dev. 15, 3059–3087 [DOI] [PubMed] [Google Scholar]
- 2. McMahon A. P., Ingham P. W., Tabin C. J. (2003) Curr. Top. Dev. Biol. 53, 1–114 [DOI] [PubMed] [Google Scholar]
- 3. Lum L., Beachy P. A. (2004) Science 304, 1755–1759 [DOI] [PubMed] [Google Scholar]
- 4. Aza-Blanc P., Ramírez-Weber F. A., Laget M. P., Schwartz C., Kornberg T. B. (1997) Cell 89, 1043–1053 [DOI] [PubMed] [Google Scholar]
- 5. Wang B., Fallon J. F., Beachy P. A. (2000) Cell 100, 423–434 [DOI] [PubMed] [Google Scholar]
- 6. Pan Y., Bai C. B., Joyner A. L., Wang B. (2006) Mol. Cell. Biol. 26, 3365–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiang J., Hui C. C. (2008) Dev. Cell 15, 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haycraft C. J., Banizs B., Aydin-Son Y., Zhang Q., Michaud E. J., Yoder B. K. (2005) PLoS Genet. 1, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huangfu D., Anderson K. V. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 11325–11330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu A., Wang B., Niswander L. A. (2005) Development 132, 3103–3111 [DOI] [PubMed] [Google Scholar]
- 11. May S. R., Ashique A. M., Karlen M., Wang B., Shen Y., Zarbalis K., Reiter J., Ericson J., Peterson A. S. (2005) Dev. Biol. 287, 378–389 [DOI] [PubMed] [Google Scholar]
- 12. Chen M. H., Wilson C. W., Li Y. J., Law K. K., Lu C. S., Gacayan R., Zhang X., Hui C. C., Chuang P. T. (2009) Genes Dev. 23, 1910–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang C., Pan Y., Wang B. (2010) Development 137, 2001–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Q., Zhang L., Wang B., Ou C. Y., Chien C. T., Jiang J. (2006) Dev. Cell 10, 719–729 [DOI] [PubMed] [Google Scholar]
- 15. Kent D., Bush E. W., Hooper J. E. (2006) Development 133, 2001–2010 [DOI] [PubMed] [Google Scholar]
- 16. Zhang Q., Shi Q., Chen Y., Yue T., Li S., Wang B., Jiang J. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 21191–21196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Di Marcotullio L., Greco A., Mazzà D., Canettieri G., Pietrosanti L., Infante P., Coni S., Moretti M., De Smaele E., Ferretti E., Screpanti I., Gulino A. (2011) Oncogene 30, 65–76 [DOI] [PubMed] [Google Scholar]
- 18. Di Marcotullio L., Ferretti E., Greco A., De Smaele E., Po A., Sico M. A., Alimandi M., Giannini G., Maroder M., Screpanti I., Gulino A. (2006) Nat. Cell Biol. 8, 1415–1423 [DOI] [PubMed] [Google Scholar]
- 19. Huntzicker E. G., Estay I. S., Zhen H., Lokteva L. A., Jackson P. K., Oro A. E. (2006) Genes Dev. 20, 276–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Virshup D. M., Shenolikar S. (2009) Mol. Cell 33, 537–545 [DOI] [PubMed] [Google Scholar]
- 21. Nybakken K., Vokes S. A., Lin T. Y., McMahon A. P., Perrimon N. (2005) Nat. Genet. 37, 1323–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hannus M., Feiguin F., Heisenberg C. P., Eaton S. (2002) Development 129, 3493–3503 [DOI] [PubMed] [Google Scholar]
- 23. Jia H., Liu Y., Yan W., Jia J. (2009) Development 136, 307–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krauss S., Foerster J., Schneider R., Schweiger S. (2008) Cancer Res. 68, 4658–4665 [DOI] [PubMed] [Google Scholar]
- 25. Krauss S., So J., Hambrock M., Köhler A., Kunath M., Scharff C., Wessling M., Grzeschik K. H., Schneider R., Schweiger S. (2009) PLoS ONE 4, e7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rorick A. M., Mei W., Liette N. L., Phiel C., El-Hodiri H. M., Yang J. (2007) Dev. Biol. 302, 477–493 [DOI] [PubMed] [Google Scholar]
- 27. Glazer A. M., Wilkinson A. W., Backer C. B., Lapan S. W., Gutzman J. H., Cheeseman I. M., Reddien P. W. (2010) Dev. Biol. 337, 148–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tay S. Y., Yu X., Wong K. N., Panse P., Ng C. P., Roy S. (2010) Dev. Dyn. 239, 527–534 [DOI] [PubMed] [Google Scholar]
- 29. Kim H. R., Richardson J., van Eeden F., Ingham P. W. (2010) BMC Biol. 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sekimizu K., Nishioka N., Sasaki H., Takeda H., Karlstrom R. O., Kawakami A. (2004) Development 131, 2521–2532 [DOI] [PubMed] [Google Scholar]
- 31. Wolff C., Roy S., Lewis K. E., Schauerte H., Joerg-Rauch G., Kirn A., Weiler C., Geisler R., Haffter P., Ingham P. W. (2004) Genes Dev. 18, 1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vokes S. A., McMahon A. P. (2004) Curr. Biol. 14, R668–R670 [DOI] [PubMed] [Google Scholar]
- 33. Jin Z., Shi J., Saraf A., Mei W., Zhu G. Z., Strack S., Yang J. (2009) J. Biol. Chem. 284, 7190–7200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Strack S., Cribbs J. T., Gomez L. (2004) J. Biol. Chem. 279, 47732–47739 [DOI] [PubMed] [Google Scholar]
- 35. Jin Z., Wallace L., Harper S. Q., Yang J. (2010) J. Biol. Chem. 285, 34493–34502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jia H., Liu Y., Xia R., Tong C., Yue T., Jiang J., Jia J. (2010) J. Biol. Chem. 285, 37218–37226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saraf A., Virshup D. M., Strack S. (2007) J. Biol. Chem. 282, 573–580 [DOI] [PubMed] [Google Scholar]
- 38. Sive H., Grainger R., Harland R. (2000) Early Development of Xenopus laevis; A Laboratory Manual, 1st Ed., pp. 91–184, Cold Spring Harbor Press, Cold Spring Harbor, New York [Google Scholar]
- 39. Stubbs J. L., Davidson L., Keller R., Kintner C. (2006) Development 133, 2507–2515 [DOI] [PubMed] [Google Scholar]
- 40. Yang J., Chan C. Y., Jiang B., Yu X., Zhu G. Z., Chen Y., Barnard J., Mei W. (2009) PLoS Genet. 5, e1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang J., Wu J., Tan C., Klein P. S. (2003) Development 130, 5569–5578 [DOI] [PubMed] [Google Scholar]
- 42. Li X., Scuderi A., Letsou A., Virshup D. M. (2002) Mol. Cell. Biol. 22, 3674–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Silverstein A. M., Barrow C. A., Davis A. J., Mumby M. C. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 4221–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Van Kanegan M. J., Adams D. G., Wadzinski B. E., Strack S. (2005) J. Biol. Chem. 280, 36029–36036 [DOI] [PubMed] [Google Scholar]
- 45. Sasaki H., Hui C., Nakafuku M., Kondoh H. (1997) Development 124, 1313–1322 [DOI] [PubMed] [Google Scholar]
- 46. Yang J., Phiel C. (2010) Life Sci. 87, 659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.