

Abstract
Systemic lupus erythematosus (SLE) is an autoimmune disease of unknown origin affecting virtually all organ systems. Beyond genetic and environmental factors, cytokine imbalances contribute to immune dysfunction, trigger inflammation, and induce organ damage. The key cytokine that is involved in SLE pathogenesis is interferon alpha. Interferon secretion is induced by immune complexes and leads to upregulation of several inflammatory proteins, which account for the so-called IFN signature that can be found in the majority of SLE PBMCs. Additionally IL-6 and IFN-y as well as T-cell-derived cytokines like IL-17, IL-21, and IL-2 are dysregulated in SLE. The latter induce a T-cell phenotype that is characterized by enhanced B-cell help and enhanced secretion of proinflammatory cytokines but reduced induction of suppressive T cells and activation-induced cell death. This paper will focus on these cytokines and highlights pathophysiological approaches and therapeutic potential.
1. Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune disease of unknown origin affecting virtually every organ in the human body. SLE is primarily caused by autoantibodies and immune complex deposition. Enhanced apoptosis in conjunction with defective clearance of apoptotic cells results in occurrence of high levels of autoantibodies [1]. Deregulated cytokine production contributes to immune dysfunction and mediates tissue inflammation and organ damage. Inflammatory cytokines, like type I and type II interferons and interleukin-6 (IL-6), IL-1, and tumor necrosis factor-alpha (TNF-α) as well as immunomodulatory cytokines like IL-10 and TGF-β, have been identified as important players in SLE. Apart from those IL-21 and IL-17 have been lately identified to play a relevant role in autoimmunity, while recent findings regarding IL-2 brought this cytokine back in focus of SLE research. Beside interferons this paper will highlight some recent advances of IL-6, IL-21, IL-17, and IL-2 research with regard to SLE.
2. Type I Interferons
Type I interferons (IFNs) are important cytokines, whose most prominent function is to mediate the early immune response to viral infections. Viral RNA and DNA are recognized by Toll-like receptors (TLRs) and trigger IFN release of leukocytes. Although all leukocytes produce IFN, plasmacytoid dendritic cells (pDCs) are the main producer [2]. PDCs are a rare cell population. Only 0,2–0,8% of peripheral mononuclear cells (PBMCs) are pDCs, but their capacity to produce IFN is unique and 100–200 times enhanced compared to any other cell type [3, 4]. The ability to release such high amounts of IFN might be caused by the fact that pDCs constitutively express Toll-like receptor 7 (TLR7) and Toll-like receptor 9 (TLR9) [5]. After secretion IFN binds its heteromeric type I IFN receptor on target cells, transduces signals mainly via JAK/STAT pathways, and initiates gene transcription of so-called interferon-stimulated genes [6]. Microarray analysis detected >300 genes induced by interferon [7]. By activation of genes which are responsible for antimicrobial responses, antigen processing, and inflammation, IFNs exert several immunomodulatory effects and are therefore supposed to be key cytokines not only in the innate immune system but also in adaptive immune responses [8]. The central role of IFN in SLE has been confirmed by several observations.
Many of the symptoms that SLE patients develop are congruent with symptoms of patients suffering from influenza or as a side effect of interferon-alpha (IFN-α) therapy. Fever, fatigue, and leukopenia are some examples. SLE patients often show enhanced IFN-α serum levels [9], and the IFN levels correlate with anti-dsDNA production and disease activity [10]. Furthermore, IFN-α therapy may lead to autoantibody production and an SLE-like syndrome [11, 12]. Genetic association studies of patients with SLE identified several genes, amongst which components of the upstream and downstream pathways of type I interferon are the most frequently found [13] including Signal Transducer and Activator of Transcription 4 (STAT4) and interferon regulatory factor 5 (IRF5) [14–16]. STAT4 interacts with type I interferon receptors and is directly involved in IFN signaling. IRF5 is a transcription factor which induces IFN transcription in response to TLR signaling. In fact the IRF5 risk haplotype in SLE patients is associated with high serum IFN-α activity [17]. These genetic association studies are in accordance with the fundamental observations identified by gene expression profiling of SLE PBMCs in the group of Virginia Pascual. These experiments demonstrate a significant upregulation of interferon-regulated gene transcripts in adult and paediatric SLE PBMCs [18, 19]. This characteristic is referred to as the “interferon signature” and assessed as a new biomarker for disease activity [13].
These observations raised the questions of how the IFN signature in SLE patients develops and how IFNs are involved in pathogenesis of SLE. A hallmark of SLE is the formation of immune complexes (ICs). One cause of immune complex formation is an increased apoptosis and defective clearance of apoptotic material on the one hand and high occurrence of autoantibodies on the other hand [1]. In 1998 Cederblad et al. observed the production of IFN-α by PBMCs when serum samples from SLE patients were used as culture supplement [20]. Further studies showed that immune complexes induce IFN-α production by pDCs [21–24]. Immune complexes are internalized after binding Fc gamma RIIa on the surface of pDCs and activate TLR9 and TLR7 in the endosomal compartment, which induces secretion of IFN-α [25]. Indeed pDC are reduced in SLE blood [20], but this reduction might be related to enhanced recruitment to tissues [26, 27].
The overproduction of IFNs in SLE exerts wide effects, which result in the above-mentioned IFN signature. We would like to accent a few of these effects which were intensively observed and papered by Obermoser and Pascual [13].
First IFN-α promotes feedback loops by induction of TLR7 in pDCs, mDCs, and monocytes which enhance synthesis of IFN [28]. Secondly IFNs contribute to disruption of peripheral tolerance by promoting DC maturation (mDC) and thereby reducing numbers of immature DCs. Immature DCs are important to keep up immune tolerance by induction and maintenance of regulatory T cells. In addition immature DCs promote anergy and deletion of self-reactive T cells by presenting self-peptide MHC complexes in the absence of costimulatory signals to self-reactive T cells [29]. Activated and self-reactive T cells provide help for B cells. Thirdly mDCs can also directly enhance selection and survival of autoreative B cells by producing B-cell activating factor (BAFF) [30]. This cytokine belongs to the family of B-lymphocyte stimulators (BLySs) and contributes to survival of B cells [31]. Finally IFN-α drives disease activity by enhancing cytotoxicity of CD8 T cells [32] and also directly increases numbers of autoreactive CD4 T cells by upregulation of the costimulatory molecules CD80 and CD86 on antigen-presenting cells (APCs) [13]. Therefore, activation of the IFN system by ICs as endogenous IFN inducers in SLE patients generates a self-reinforcing trial which Rönnblom and Alm describe as a vicious circle (Figure 1) [8].
Figure 1.
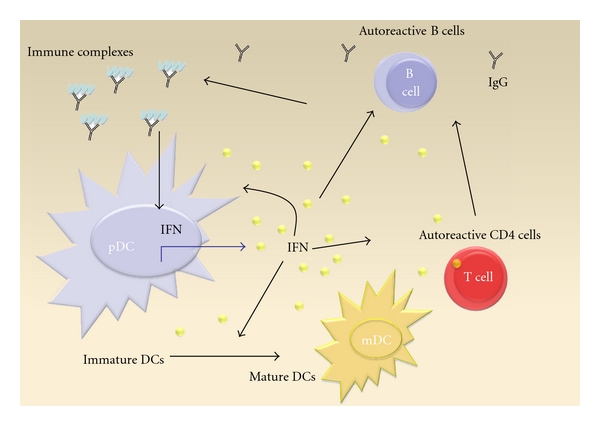
The vicious circle of IFN signaling in SLE: ICs bind to Fc gamma RII receptors on pDCs and reach the endosomes where they are recognized by TLRs. TLRs transduce signals to the nucleus which induce transcription of IFN. IFN secretion enhances expression of its own receptor on pDCs, mDCs, and monocytes. Furthermore, IFN promotes maturation of DCs which leads to disruption of peripheral tolerance and activation of autoreactive CD4 T helper cells. The appearance of autoreactive CD4 T cells is further amplified by upregulation of CD80 and CD86 on APCs. This results in enhanced B cell help by autoreactive CD4 cells, which is again sustained by an upregulation of BAFF. The increased formation of autoreactive B cells triggers appearance of ICs and further IFN release.
Therapeutical targets which disrupt this circle are subjects of intensive research. The widely used and old drug resochin changes the pH of the endosomes and therefore the affinity of TLR7 and TLR9 towards ICs. Specific inhibitors of TLR7 and TLR9 have already been tested in animal models [33]. Antibodies to block IFN-alpha (Sifalimumab, Rontalizumab) are currently being tested in clinical trials [34]. In a phase I trial treatment of SLE patients with an anti-IFN-α monoclonal antibody influenced interferon signature and skin lesions of these patients [35].
3. Interleukin-6
IL-6 is produced in many cell types, like monocytes, fibroblasts, endothelial cells, and also T and B lymphocytes [36] and has a range of biological activities on various target cells. IL-6 serves as a differentiation factor for several haematopoetic cells. Differentiation of B cells in plasma cells and induction of IgG production is induced by IL-6 [37] as well as differentiation and proliferation of T cells [38] and macrophages [39]. Further effects of IL-6 are bone marrow stem cell maturation, activation of neutrophils, and stimulation of the production of platelets from megacaryocytes and osteoclast differentiation [40]. IL-6 is the major hepatocyte stimulation factor and induces acute-phase proteins [41].
IL-6 signaling occurs via its heteromeric receptor complex, which consists of two glycoproteins, an IL-6-specific binding chain (IL-6R) and a signal transducing chain (gp130) [42]. Binding of IL-6 on IL-6R triggers dimerisation of gp130, which results in activation of JAK1 and tyrosine phosphorylation of gp130. This activates the ERK/MAPK signaling pathway and p-STAT3-mediated pathways [43]. IL-6R expression is limited to several cells, but a so-called gp130 transsignaling occurs when IL-6 binds a soluble IL-6R form and then interacts with the more unique expressed gp130 (Figure 2) [44].
Figure 2.
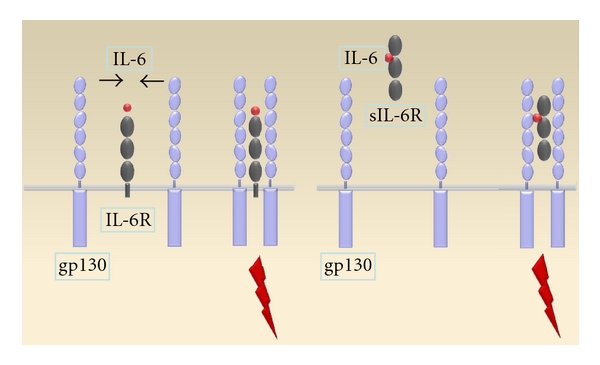
Model of gp130 transsignaling. IL-6 signaling occurs by binding its membrane bound receptor (IL-6R) in target cells and subsequent dimerization of gp130 (Figure on the left). Cells which do not express IL-6R can also be susceptible to IL-6 via soluble IL-6 receptors that dimerize with membrane bound gp130 (Figure on the right).
Murine lupus models indicate the involvement of IL-6 in B-cell hyperactivation and onset of autoimmune disease. In Mrl/lpr mice IL-6 and soluble IL-6R serum levels are increased related to age [45, 46]. IL-6-deficient Mrl/lpr mice show a delayed onset of lupus nephritis and prolonged survival [47]. IL-6 receptor blockade suppressed IgG antibody production in NZB/W F1 mice and development of autoimmune disease [48, 49], whereas exogenous administration of IL-6 accelerates glomerulonephritis in NZB/W F1 mice. Recent investigations suggest that IL-6 blockade not only targets autoreactive B cells but also inhibits autoreactive T cells in NZB/W F1 mice [50]. Next to its effects on B cells IL-6 is a key cytokine that determines T-cell differentiation of naïve T cells into so-called regulatory T cells with a suppressive phenotype or into T cells with a proinflammatory Th17 phenotype. Since IL-6R blockade in mouse model of arthritis inhibited the differentiation of Th17 cells [51], effects of IL-6 blockade on T-cell responses and therefore benefits for autoimmune diseases should also be taken into consideration [52].
Patients with active SLE have increased IL-6 serum levels [53, 54] which in some studies correlated with disease activity [53] or anti-DNA levels [40, 54]. Elevated IL-6 levels are associated with B-cell hyperactivity and autoantibody production [40] and secretion of IgG anti-DNA antibodies were reduced by neutralizing IL-6 and restored by adding exogenous IL-6 in vitro [40, 53]. In addition to its systemic effects IL-6 also has a role in local inflammation, for example, in lupus nephritis and is supposed to be involved in mesangial cell proliferation, one of the hallmarks of proliferative lupus nephritis [40]. Patients with active lupus nephritis show elevated urinary IL-6 secretion [55, 56], and the expression of IL-6 is increased along glomerular and tubulus tissue in lupus nephritis kidneys in situ [57]. IL-6 is increased during cardiopulmonary complications of SLE [58], and SLE patients with neuropsychiatric syndromes show elevated IL-6 levels in the cerebrospinal fluid [59]. Current investigations also indicated the involvement of IL-6 in joint damage in SLE patients [60].
As IL-6 exerts systemic effects and also mediates local inflammation, IL-6 targeting therapy, which has been shown to be efficacious in inflammatory autoimmune diseases [61], might also be promising in the treatment of SLE patients. Tocilizumab is a humanized monoclonal antibody, which inhibits IL-6 signaling by binding IL-6R and soluble IL-6 receptors. It was recently tested in an open label-phase 1 dosage escalation study in SLE patients. The results are promising regarding decreased levels of anti-dsDNA antibodies and of acute-phase reactants in Tocilizumab treated patients [62].
4. Interferon-Gamma
Interferon-gamma (IFN-γ) activates macrophages at the site of inflammation, contributes to cytotoxic T-cell activity, has antiviral capacities, and is strongly associated with Th1 responses. It induces differentiation of naïve T cells into Th1 cells and triggers Th1 differentiation in an autocrine manner. IFN-γ signaling induces phosphorylation of STAT1 which leads to expression of the Th1-lineage-specific transcription factor T-bet and subsequent expression of IFN-γ [63].
Due to the fact that Th1-mediated effects can explain many features of autoimmune diseases, IFN-γ became an archetypical inducer of organ-specific autoimmunity [64]. IFN-γ might contribute to autoimmune disease by inducing production of IgG2a and IgG3 isotype antibodies that activate complement and furthermore by activating macrophages and promoting tissue inflammation. However, in autoimmune models like experimental autoimmune encephalomyelitis [65] and collagen-induced Arthritis [64, 66], IFN-γ-deficient mice are more susceptible; therefore, the role of IFN-γ is not proinflammatory per se. Recent studies detected diverse mechanisms via which IFN-γ might counteract inflammatory pathways (review in [67]). One important mechanism might be that IFN-γ inhibits the development of autoimmune-related Th17 cells [67, 68].
The role of IFN-γ in SLE was analyzed in several mouse models. T-helper cells expressing IFN-γ correlate with age and development of disease in NZB/W F1 mice [69]. Additionally treatment of NZB/W F1 mice with recombinant IFN-γ accelerated development of disease, while administration of monoclonal antibodies against IFN-γ resulted in remission of disease [70]. Furthermore, IFN-γR-deficient NZB/W F1 mice show reduced glomerulonephritis and reduced serum concentration of anti-dsDNA antibodies [71].
IFN-γ-deficient Mrl/lpr mice are prevented from early death and have reduced lymphadenopathy and reduced glomerulonephritis [72]. Treatment with a cDNA encoding IFN-γR/Fc reduces disease manifestations [73]. However, treatment of Mrl/lpr mice with recombinant IFN-γ leads to dichotomic effects. While treatment at an early age proves to be protective, treatment later in life accelerates disease manifestations [74]. In a pristine-induced lupus model IFN-γ deficient BALB/c mice are protected from renal disease [75]. Several studies on lupus models suggest that an imbalance towards Th1 dominance plays a role in acceleration of disease [76–78]. In human patients with SLE a disbalance in mechanisms that regulate Th1 and Th17 cells with an enhanced expression of Th17 cells was observed [79], which was partially aggravated by the use of glucocorticoids [80]. Recent studies detected unusual IFN-γ and IL-17 double-positive T cells [81] which indicates a quite complex and not yet understood plasticity of Th1 and Th17 cells [82]. The complex role of IFN-γ in SLE is underscored by contradictory clinical studies that find a correlation between serum IFN-γ level and disease activity and a correlation between IFN-γ expression and severity of lupus nephritis while others show decreased IFN-γ levels in lupus nephritis [83, 84]. Nevertheless, AMG-811, a human monoclonal antibody to IFN-γ, is under investigation in a phase Ib study in SLE patients [34].
5. Interleukin-2
T cells are the main producer and responder cells of interleukin-2 (IL-2). IL-2 production is induced after T-cell receptor (TCR) activation, induces itself in paracrine and autocrine loops, and also upregulates surface expression of its receptor. IL-2 was initially discovered as a cytokine which drives clonal expansion of T cells, but the phenotypes of IL-2-deficient or IL-2-receptor- (IL-2R-) deficient mice expand the tasks and impact of IL-2 [85].
Mice with IL-2 or IL-2R deficiency show an enlargement of peripheral lymphoid organs (lymphadenopathy and splenomegaly) and impaired activation-induced cell death (AICD) and develop autoimmune disorders [86, 87]. In addition to this, a defective production of IL-2 is observed in several murine models of autoimmune diseases [88] including three well-established lupus models. In all of these models the production of IL-2 is reduced once disease starts to appear [89–91].
These observations are somewhat inconsistent with the view of IL-2 as growth factor for T cells and raise the question of how loss of IL-2 is connected with loss of immunotolerance. Interestingly, IL-2 deficiency in mouse models is paralleled by reduced levels of regulatory T cells (Tregs). Therefore, the uncontrolled activation of B and T cells in the absence of IL-2 might be caused by deficiencies of regulatory T cells in these mice. Direct evidence that regulatory T cells depend on IL-2 comes from experiments which show that IL-2 is required for homeostatic maintenance of regulatory T cells [92] as well as for their thymic development and IL-2 also directly affects suppressive function of regulatory T cells [93]. In addition to its effect on regulatory T cells, it was very recently discovered that IL-2 also affects Th17 cells. This highly proinflammatory T-cell subset is linked to many autoimmune diseases. IL-2 limits production of IL-17 in vivo and in vitro, and low levels of IL-2 favour occurrence of Th17 cells [94]. IL-2-deficient mice show enhanced serum levels of IL-17 and a higher number of IL-17 producing T cells in peripheral lymph nodes. Laurence et al. showed by adoptive transfer experiments that the IL-17 overproduction is not caused by a secondary manifestation of disease, but directly due to deficiency of IL-2 [95].
It is therefore currently accepted that IL-2, beyond its role as a growth factor, is important to maintain functionality and homeostasis of regulatory T cells on the one hand and to inhibit production of IL-17 on the other hand. As a consequence IL-2 appears to be a crucial cytokine to prevent formation of autoimmunity.
In accordance with this SLE T cells show reduced IL-2 production [96–98] and IL-2 deficiency is also paralleled by low numbers of regulatory T cells in SLE patients [99]. The molecular mechanism of the IL-2 defect in SLE is caused amongst others by overexpression of cAMP response element modulator alpha (CREMα), a transcription factor which binds to the IL-2 promoter and inhibits IL-2 transcription. Anti TCR/CD3 antibodies present in SLE sera induce expression of CREMα, which leads to an increased CREMα binding to the IL-2 promoter and decreased IL-2 production [100]. We recently showed that increased CREMα expression is the result of enhanced CREMα promoter activity in SLE T cells and CREMα promoter activity correlates with disease activity [101]. Interestingly the defective IL-2 production of SLE T cells can be restored by introducing a plasmid encoding antisense CREMα into these cells [102]. The IL-2 activating transcription factor CRE-binding protein (CREB) shares the same binding site on the IL-2 promotor and is displaced by CREM in SLE cells possibly because of high levels of CREM [103]. Furthermore, diminished activity of CREB, caused by increased levels of the serine/threonine phosphatase PP2a, the phosphatase that is responsible for dephosphorylation of CREB, contributes to reduced production of IL-2 [104]. It is not clear whether lower IL-2 levels in SLE also contribute to enhanced IL-17 levels, but the ratio of Treg to Th17 cells in SLE patients with active disease is significantly lower than that in healthy controls and inversely correlates with the severity of active SLE [105]. IL-2 is also involved in activation-induced cell death (AICD). AICD is a controlled apoptotic mechanism by which excess effector cells are eliminated and it is regulated by CD95 and TNFR1 [106–108]. This process is affected in SLE patients, in whom T cells are more resistant to AICD [109, 110] resulting in persistence of autoreactive T cells. Furthermore, IL-2 is also important for the development of CD8 T-cell cytoxicity. Cytotoxic T cells (CTL) destroy virus-infected T cells and are important to defend infections. Some SLE patients develop cytotoxic defects, while a lot of SLE patients suffer from increased mortality and morbidity during infections [109].
Altogether defective IL-2 production in SLE T cells seems to contribute to several immune alterations including reduced numbers and function of regulatory T cells, decreased AICD, decreased CTL responses and to upregulation of IL-17 production [109]. This raised the question whether compensation of low IL-2 levels by adding exogenous IL-2 would result in lower disease activity [111].
Humrich et al. treated lupus prone mice with IL-2. In the IL-2 treated mice the homeostatic balance of Treg and T effector cells was re-established and impeded disease progression [112]. However, the half live of exogenous cytokines in vivo is quite short, while IL-2 in complexes with an antibody is more functional [111]. These complexes can prevent type 1 diabetes [113] and suppress experimental myasthenia [114]. Furthermore in vivo expansion of regulatory T cells with IL-2/IL-2mAB complexes induces resistance to experimental autoimmune encephalomyelitis [115].
Therefore IL-2 seems to have a therapeutic potential to treat autoimmune diseases, but the activity of IL-2 as growth factor bears a risk. IL-2 has been used as adjuvant for treatment of patients with renal cancer albeit with considerable side effects. The effect of IL-2 seems to depend on the administered dose, it is possible that low doses favour Tregs, while high doses favor memory/effector cell function [111].
Recently published data from Liao et al. further expand the impact of IL-2 to a cytokine that in addition to its influence on regulatory T cell and Th17 cells broadly regulates T helper cell differentiation [116]. Further investigations is needed to understand the several and sometimes ambivalent roles of IL-2. It should be taken into consideration to therapeutically influence mechanisms upstream from IL-2, which are responsible for reduced IL-2 expression in SLE.
6. Interleukin-21
IL-21 is produced by a range of differentiated CD4+ T cell subsets and natural killer (NK)T cells [117]. IL-21 signals through a heterodimeric receptor, which is formed by common gamma chain (shared with IL-2, IL-4, IL-7, IL-9, IL-13 and IL-15 receptors) and an IL-21 specific receptor (IL-21R) [118, 119]. Since IL-21R is expressed on CD4+, CD8+ T cells, B cells, NK cells, dendritic cells, macrophages and keratinocytes [118], IL-21 acts on a range of lymphoid lineages and exerts pleiotropic effects. We will give a short numeration of its effects on immune cells. IL-21 is a stimulator of CD8+ T cell proliferation. In synergy with IL-15 and IL-7 it promotes CD8+ T cell expansion [117, 120, 121]. IL-21 drives differentiation of naïve T cells into Th17 cells [122]. IL-21 is induced by IL-6 and RORγt and stabilizes and maintains Th17 cells by upregulating its own expression and the expression of IL-23R [117, 121]. Induced regulatory T cells are negatively regulated by IL-21, as IL-21 downregulates FoxP3 induction in TGF-β stimulated cells [122]. Furthermore IL-21 counteracts suppressive effects of Tregs, however it is not known if IL-21 acts on Tregs or CD4+ T cells in this circumstances [123]. Furthermore IL-21 plays a role in follicular T helper cell (Tfh) development and is necessary for germinal center (GC) formation [124, 125]. GCs can be the origin of autoantibodies and abnormalities in GCs can lead to aberrant selection of autoreactive B cells and might contribute to autoimmunity [126]. IL-21 effects on B cells are context-depended. IL-21 has a role in B cell activation and differentiation of plasma cells that produce IgG [127], but also induces apoptosis of resting and activated B cells [128]. IL-21 without antigen or in the presence of a non-specific polyclonal signal induces deletion of autoreactive B cells. IL-21 in context of a specific antigen and T cell interaction leads to expansion of responding cells [118]. IL-21 can also act anti-inflammatory, it inhibits dendritic cell maturation and stimulates IL-10 production [129, 130].
SLE patients have higher serum levels of IL-21, while IL-21 and IL-21R polymorphisms are associated with susceptibility to SLE [131, 132]. A subset of patients with SLE shows increased numbers of circulating CD4+ CXCR5+ cells (Tfh cells) [133]. The sanroque mouse bears a mutation in a gene that negatively regulates Tfh cell development. These mice develop lupus-like symptoms, paralleled by an overproduction of IL-21 and increased levels of Tfh cells [134]. Mrl/lpr mice show increasing numbers of Tfh cells and extrafollicular T helper cells with age and disease development [135]. Mrl/lpr mice treated with IL-21R/Fc to block IL-21 signaling displayed reduced level of autoantibodies and SLE-like symptoms [136]. The lupus mouse BXSB.B6-Yaa+ shows increased IL-21 mRNA levels compared to wildtype mice [125] and disease was prevented by genetic deletion of IL-21R in these mice [137]. Notably treatment of BXSB.B6-Yaa+ mice with an IL-21R/Fc fragment negatively influenced survival early on and positively influenced survival at later stages of disease [138]. Because of these pleiotropic effects, it remains debatable if IL-21 blockade might be useful to treat SLE.
7. Interleukin-17
IL-17 is produced by several T-cell subsets including T helper cells (CD4+ T cells), cytotoxic T cells (CD8+ T cells), double-negative (CD4−CD8−CD3+) T cells, gamma-delta T cells but also by natural killer (NK) cells and neutrophils [139]. A new CD4+ T-cell subset, which preferentially produces IL-17 but not IL-4 or IFN-γ, is termed Th17 cells. Beyond IL-17a and IL-17f these cells produce IL-22 and IL-21. Important factors for the differentiation of murine as well as human Th17 cells include IL-6, IL-21, and IL-1β together with TGF-β [122, 140–146]. In addition to these cytokines, IL-23 is crucial for expansion and maintenance of Th17 cells [147]. Th17 cells are involved in the immune response against bacteria, like Citrobacter, Klebsiella pneumoniae, and Borrelia burgdoerferi and against fungi like Candida albicans [64]. Some of these infections cannot be cleared by Th1 or Th2 cells. Beyond these protective roles, IL-17 and Th17 cells contribute to tissue inflammation and organ damage in autoimmune diseases by triggering chronic inflammation [148].
IL-17 exerts several effects and affects several cell types (Table 1). IL-17 receptors are broadly expressed not only on immune cells but also on epithelial and endothelial cells [139, 149–151]. IL-17 signaling through these receptors increases production of chemokines (interleukin-8 (IL-8), monocyte chemoattractant protein-1, growth-related oncogene protein-alpha), which leads to recruitment of monocytes and neutrophils into the inflamed tissue [152–154]. Moreover, IL-17 also induces T-cell infiltration by upregulating the expression of intercellular adhesion molecule 1 (ICAM-1) [155]. IL-17 induces secretion of many proinflammatory proteins, among them prostaglandin E2, granulocyte-macrophage colony-stimulating factor (GM-CSF), and granulocyte colony stimulating factor [155–157], and also cytokines which induce a positive feedback loop and lead to further IL-17 production like interleukin-6 (IL-6), IL-1β (interleukin-1 beta) and IL-21 (interleukin-21) [148]. Recent experiments provide evidence that IL-17 alone or in synergy with BAFF also promotes B-cell differentiation and autoantibody production [158, 159].
Table 1.
IL-17 exerts effects on several cell types and tissues.
| T cells | Induces production of proinflammatory IL-6, IL-1beta, and IL-21, providing a feedback loop [148] |
| Enhances recruitment of T cells to inflamed tissue [155] | |
| B cells | Drives B-cell differentiation into plasma cells and production of autoantibodies [158, 159] |
| Monocytes | Enhances migration to inflamed tissue [173] |
| Epithelial/ endothelial cells |
Induces increased production of chemokines and upregulation of adhesion molecules [152] |
| Neutrophils | Enhances migration to inflamed tissue [154] |
SLE patients have raised serum levels of IL-17. Enhanced percentages of IL-17 producing cells [160–164] and plasma IL-17 levels correlate with disease activity [162]. One source of IL-17 in SLE patients is double-negative T cells (DNTs) [164]. SLE patients have expanded numbers of double-negative T cells (DNTs) compared to healthy individuals [165]. IL-17 producing cells infiltrate skin, lung, and kidneys of SLE and lupus nephritis (LN) patients [160, 165–167] and most likely contribute to organ damage by exerting-above-mentioned effects. Evidence that IL-17 also contributes to B cell activation in LN comes from in vitro experiments with PBMCs [168]. These experiments document that IL-17 induces induction of IgG and anti-dsDNA production.
In the last years the Mrl/lpr mice model provided some evidence for the functional contribution of IL-17 to disease progression and organ damage. Mrl/lpr mice have increased numbers of double-negative T cells (DNTs), which produce high amounts of IL-17 and expression of IL-17, and IL-23 receptor (IL-23R) increases with disease progression [169]. Lymphoid cells from Mrl/lpr mice can induce nephritis in nonautoimmune species after IL-23 in vitro treatment [169]. After ischemic reperfusion of the gut, enhanced IL-17-mediated tissue injury was observed in Mrl/lpr mice [170]. Splenocytes from SNF1 (New Zealand Black x SWR F1) mice secrete higher levels of IL-17 than nonautoimmune B6 mice [171]. In congruence with observation from the Mrl/lpr model IL-17-producing T cells are detected in kidneys affected by nephritis [171]. BXD2 mice express high levels of IL-17 in serum and increased numbers of IL-17+ cells in the spleen [172], which form spontaneous autoreactive germinal centers in concert with IL-17R expressing B cells. These features could be blocked by inhibition or deletion of the IL-17 receptor [172].
Although these data indicate that IL-17 plays a role in pathogenesis of autoimmune diseases, it is not clear whether targeting IL-17 is suited to treat SLE. Next to Th17 cell other T-cell subsets like Th1 cells crossregulate each other [158]. In a graft-versus-host-disease model the absence of donor Th17 cells leads to an exacerbated disease by augmented Th1 differentiation [174]. More importantly there is a reciprocal relationship between regulatory T cells and Th17 cells. Recent studies showed that increases in Th17 cells are directly correlated with the depletion of Treg cells during SLE flares [160]. It is therefore suggested to consider possibilities to recover the balance between Th17 and regulatory T cells to treat SLE and other autoimmune diseases [148, 175]. In fact Tregs and Th17 cells can be generated from the same cell. TGF-beta induces the differentiation of Treg cells from naïve T cells; however, the addition of IL-6 or IL-21 results in Th17 differentiation [140, 176, 177]. The lineage transcription factors of Th17 and Treg cells, RoRγT/RORα and FoxP3, respectively, bind each other and inhibit each other's function [178, 179]. IL-2 is an indispensable growth factor for Tregs but inhibits Th17 differentiation [94, 95], and IL-21 promotes Th17 differentiation and inhibits the induction of regulatory T cells [122]. Finally Tregs treated with IL-6 can produce IL-17 [180–182] and can convert into IL-17 producing autoimmune effector cells [183]. The balance of Th17 and Treg cells is regulated by several transcription factors which are activated in a context-dependent manner depending on external cytokines. The cytokine environment in SLE is ideal for the generation of Th17 cells [184]. Low levels of IL-2, enhanced production of IL-21 and IL-6 [53, 185] might lead to enhanced IL-17 levels. We do not know if Tregs lose expression of FoxP3 and become IL-17-producing cells during SLE flares. But the cytokine milieu apparent in SLE patients could theoretically facilitate this phenomenon.
Future investigations might shed light on the question whether IL-17 blockade or blockade of cytokines or transcription factors that regulate Th17-Treg homeostasis will be useful to treat SLE.
8. Concluding Remarks
Cytokines are important mediators of intercellular communication and orchestrate the interaction of immune cells during immune responses. In SLE several cytokines are involved in general immune dysregulation and also in local inflammation which leads to tissue injury and organ damage. Here we summarized recent advances in the studies of some cytokines, which contribute to SLE pathogenesis. It is widely accepted that interferons have a crucial role in the pathogenesis of SLE. The therapeutic blockade of the IFN driven vicious circle might be one of the most promising anti-cytokine therapies in the future. Furthermore, an aberrant SLE T-cell phenotype which is characterized by a dysregulated production of IL-17 and IL-21 and low production of IL-2 also aggravates disease pathology (Figure 3). These cytokines exert pleiotropic pathogenic effects, which make them potential targets in SLE.
Figure 3.
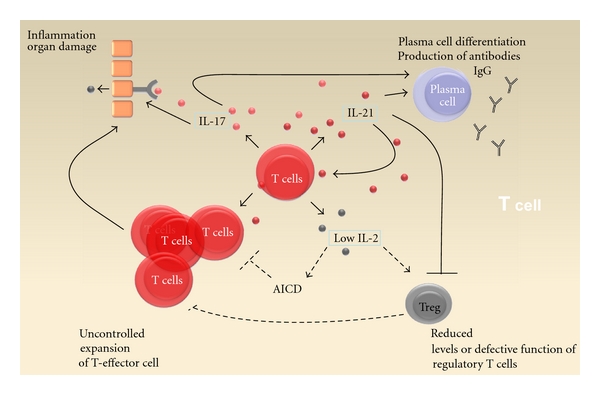
Dysregulated cytokine expression by T cells contributes to pathogenesis of SLE. SLE T cells secrete enhanced levels of IL-17 and IL-21 compared to healthy persons. IL-17 induces secretion of chemokines and other proinflammatory cytokines and therefore participates in tissue inflammation and organ damage. IL-21 and IL-17 both promote differentiation of B cells into plasma cells and production of IgG antibodies. IL-21 further maintains and expands occurrence of Th17 cells. In contrast SLE T cells have a defective production of IL-2, which leads to reduced level of regulatory T cells and defective function of T cells, which might also be caused by IL-21. Since IL-2 is crucial for AICD, low levels of IL-2 might be responsible for reduced AICD leading to expansion of autoreactive T cells, which further trigger B-cell activation and tissue inflammation.
Abbreviations
- AICD:
Activation-induced cell death
- APCs:
Antigen-presenting cells
- BAFF:
B-cell activating factor
- BLyS:
B-lymphocyte stimulator
- CREB:
cAMP response element binding protein
- CREM:
cAMP response element modulator
- CTL:
Cytotoxic T cells
- DNTs:
Double-negative T cells
- GC:
Germinal center
- IC:
Immune complex
- IFN:
Here referred as interferone type I
- IFN-α:
Interferon-alpha
- IFN-γ:
Interferon-gamma
- IL:
Interleukin
- IL-…R:
IL-…receptor
- IL-21R:
IL-21 receptor
- IRF5:
Interferon regulatory factor 5
- JAK:
Janus kinase
- LN:
Lupus nephritis
- NK:
Natural killer cell
- NKT:
Natural killer T cell
- PBMC:
Peripheral mononuclear cells
- pDCs:
Plasmacytoid dendritic cells
- SLE:
Systemic lupus erythematosus
- STAT:
Signal transducer and activator of transcription
- TCR:
T-cell receptor
- Tfh:
Follicular helper cell
- TLR:
Toll-like receptor
- TNF-α:
Tumor necrosis factor-alpha
- Tregs:
Regulatory T cells.
References
- 1.Herrmann M, Voll RE, Kalden JR. Etiopathogenesis of systemic lupus erythematosus. Immunology Today. 2000;21(9):424–426. doi: 10.1016/s0167-5699(00)01675-3. [DOI] [PubMed] [Google Scholar]
- 2.Fitzgerald-Bocarsly P, Dai J, Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine and Growth Factor Reviews. 2008;19(1):3–19. doi: 10.1016/j.cytogfr.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284(5421):1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 4.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annual Review of Immunology. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 5.Kadowaki N, Ho S, Antonenko S, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. Journal of Experimental Medicine. 2001;194(6):863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haque SJ, Williams BRG. Signal transduction in the interferon system. Seminars in Oncology. 1998;25(supplement 1):14–22. [PubMed] [Google Scholar]
- 7.de Veer MJ, Holko M, Frevel M, et al. Functional classification of interferon-stimulated genes identified using microarrays. Journal of Leukocyte Biology. 2001;69(6):912–920. [PubMed] [Google Scholar]
- 8.Rönnblom L, Alm GV. Systemic lupus erythematosus and the type I interferon system. Arthritis Research and Therapy. 2003;5(2):68–75. doi: 10.1186/ar625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ytterberg SR, Schnitzer TJ. Serum interferon levels in patients with systemic lupus erythematosus. Arthritis & Rheumatism. 1982;25(4):401–406. doi: 10.1002/art.1780250407. [DOI] [PubMed] [Google Scholar]
- 10.Bengtsson AA, Sturfelt G, Truedsson L, et al. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus. 2000;9(9):664–671. doi: 10.1191/096120300674499064. [DOI] [PubMed] [Google Scholar]
- 11.Ioannou Y, Isenberg DA. Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis & Rheumatism. 2000;43(7):1431–1442. doi: 10.1002/1529-0131(200007)43:7<1431::AID-ANR3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 12.Ronnblom LE, Oberg KE, Alm GV. Possible induction of systemic lupus erythematosus by interferon α-treatment in a patient with a malignant carcinoid tumour. Journal of Internal Medicine. 1990;227(3):207–210. doi: 10.1111/j.1365-2796.1990.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 13.Obermoser G, Pascual V. The interferon-α signature of systemic lupus erythematosus. Lupus. 2010;19(9):1012–1019. doi: 10.1177/0961203310371161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gateva V, Sandling JK, Hom G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nature Genetics. 2009;41(11):1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sigurdsson S, Nordmark G, Göring HHH, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. American Journal of Human Genetics. 2005;76(3):528–537. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nature Genetics. 2008;40(2):204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-α activity in systemic lupus erythematosus patients. Arthritis & Rheumatism. 2008;58(8):2481–2487. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. Journal of Experimental Medicine. 2003;197(6):711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cederblad B, Blomberg S, Vallin H, Perers A, Alm GV, Rönnblom L. Patients with systemic lupus erythematosus have reduced numbers of circulating natural interferon-α-producing cells. Journal of Autoimmunity. 1998;11(5):465–470. doi: 10.1006/jaut.1998.0215. [DOI] [PubMed] [Google Scholar]
- 21.Vallin H, Blomberg S, Alm GV, Cederblad B, Rönnblom L. Patients with systemic lupus erythematosus (SLE) have a circulating inducer of interferon-alpha (IFN-α) production acting on leucocytes resembling immature dendritic cells. Clinical and Experimental Immunology. 1999;115(1):196–202. doi: 10.1046/j.1365-2249.1999.00772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vallin H, Perers A, Alm GV, Rönnblom L. Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-α inducer in systemic lupus erythematosus. Journal of Immunology. 1999;163(11):6306–6313. [PubMed] [Google Scholar]
- 23.Rönnblom L, Alm GV. A pivotal role for the natural interferon α-producing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus. Journal of Experimental Medicine. 2001;194(12):F59–F63. doi: 10.1084/jem.194.12.f59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lövgren T, Eloranta M-L, Båve U, Alm GV, Rönnblom L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis & Rheumatism. 2004;50(6):1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 25.Båve U, Magnusson M, Eloranta M-L, Perers A, Alm GV, Rönnblom L. FcγRIIa is expressed on natural IFN-α-producing cells (plasmacytoid dendritic cells) and is required for the IFN-α production induced by apoptotic cells combined with Lupus IgG. Journal of Immunology. 2003;171(6):3296–3302. doi: 10.4049/jimmunol.171.6.3296. [DOI] [PubMed] [Google Scholar]
- 26.Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid dendritic cells (natural interferon-α/β-producing cells) accumulate in cutaneous lupus erythematosus lesions. American Journal of Pathology. 2001;159(1):237–243. doi: 10.1016/s0002-9440(10)61689-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blomberg S, Eloranta ML, Cederblad B, Nordlind K, Alm GV, Rönnblom L. Presence of cutaneous interferon-α producing cells in patients with systemic lupus erythematosus. Lupus. 2001;10(7):484–490. doi: 10.1191/096120301678416042. [DOI] [PubMed] [Google Scholar]
- 28.Ganguly D, Chamilos G, Lande R, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. Journal of Experimental Medicine. 2009;206(9):1983–1994. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annual Review of Immunology. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 30.Boulé MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. Journal of Experimental Medicine. 2004;199(12):1631–1640. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacKay F, Figgett WA, Saulep D, Lepage M, Hibbs ML. B-cell stage and context-dependent requirements for survival signals from BAFF and the B-cell receptor. Immunological Reviews. 2010;237(1):205–225. doi: 10.1111/j.1600-065X.2010.00944.x. [DOI] [PubMed] [Google Scholar]
- 32.Blanco P, Pitard V, Viallard JF, Taupin JL, Pellegrin JL, Moreau JF. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis & Rheumatism. 2005;52(1):201–211. doi: 10.1002/art.20745. [DOI] [PubMed] [Google Scholar]
- 33.Barrat FJ, Meeker T, Gregorio J, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. Journal of Experimental Medicine. 2005;202(8):1131–1139. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.La Cava A. Anticytokine therapies in systemic lupus erythematosus. Immunotherapy. 2010;2(4):575–582. doi: 10.2217/imt.10.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao Y, Richman L, Higgs BW, et al. Neutralization of interferon-α/β-inducible genes and downstream effect in a phase I trial of an anti-interferon-α monoclonal antibody in systemic lupus erythematosus. Arthritis & Rheumatism. 2009;60(6):1785–1796. doi: 10.1002/art.24557. [DOI] [PubMed] [Google Scholar]
- 36.Hirano T. Interleukin 6 and its receptor: ten years later. International Reviews of Immunology. 1998;16(3-4):249–284. doi: 10.3109/08830189809042997. [DOI] [PubMed] [Google Scholar]
- 37.Muraguchi A, Hirano T, Tang B, et al. The essential role of B cell stimulatory factor 2 (BSF-2/IL-6) for the terminal differentiation of B cells. Journal of Experimental Medicine. 1988;167(2):332–344. doi: 10.1084/jem.167.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lotz M, Jirik F, Kabouridis P, et al. B cell stimulating factor 2/interleukin 6 is a costimulant for human thymocytes and T lymphocytes. Journal of Experimental Medicine. 1988;167(3):1253–1258. doi: 10.1084/jem.167.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sachs L, Lotem J, Shabo Y. The molecular regulators of macrophage and granulocyte development: role of MGI-2/IL-6. Annals of the New York Academy of Sciences. 1989;557:417–437. doi: 10.1111/j.1749-6632.1989.tb24035.x. [DOI] [PubMed] [Google Scholar]
- 40.Tackey E, Lipsky PE, Illei GG. Rationale for interleukin-6 blockade in systemic lupus erythematosus. Lupus. 2004;13(5):339–343. doi: 10.1191/0961203304lu1023oa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochemical Journal. 1990;265(3):621–636. doi: 10.1042/bj2650621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kishimoto T. Interleukin-6: from basic science to medicine: 40 years in immunology. Annual Review of Immunology. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- 43.Hirano T, Nakajima K, Hibi M. Signaling mechanisms through gp130: a model of the cytokine system. Cytokine and Growth Factor Reviews. 1997;8(4):241–252. doi: 10.1016/s1359-6101(98)80005-1. [DOI] [PubMed] [Google Scholar]
- 44.Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 1990;63(6):1149–1157. doi: 10.1016/0092-8674(90)90411-7. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki H, Yasukawa K, Saito T, et al. Serum soluble interleukin-6 receptor in MRL/lpr mice is elevated with age and mediates the interleukin-6 signal. European Journal of Immunology. 1993;23(5):1078–1082. doi: 10.1002/eji.1830230515. [DOI] [PubMed] [Google Scholar]
- 46.Tang B, Matsuda T, Akira S, et al. Age-associated increase in interleukin 6 in MRL/lpr mice. International Immunology. 1991;3(3):273–278. doi: 10.1093/intimm/3.3.273. [DOI] [PubMed] [Google Scholar]
- 47.Cash H, Relle M, Menke J, et al. Interleukin 6 (IL-6) deficiency delays lupus nephritis in MRL-Fas lpr mice: the IL-6 pathway as a new therapeutic target in treatment of autoimmune kidney disease in systemic lupus erythematosus. Journal of Rheumatology. 2010;37(1):60–70. doi: 10.3899/jrheum.090194. [DOI] [PubMed] [Google Scholar]
- 48.Mihara M, Takagi N, Takeda Y, Ohsugi Y. IL-6 receptor blockage inhibits the onset of autoimmune kidney disease in NZB/WF1 mice. Clinical and Experimental Immunology. 1998;112(3):397–402. doi: 10.1046/j.1365-2249.1998.00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finck BK, Chan B, Wofsy D. Interleukin 6 promotes murine lupus in NZB/NZW F1 mice. Journal of Clinical Investigation. 1994;94(2):585–591. doi: 10.1172/JCI117373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang B, Gardner DB, Griswold DE, Bugelski PJ, Song XYR. Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology. 2006;119(3):296–305. doi: 10.1111/j.1365-2567.2006.02433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujimoto M, Serada S, Mihara M, et al. Interleukin-6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis & Rheumatism. 2008;58(12):3710–3719. doi: 10.1002/art.24126. [DOI] [PubMed] [Google Scholar]
- 52.Mihara M, Ohsugi Y, Kishimoto T. Evidence for the role of Th17 cell inhibition in the prevention of autoimmune diseases by antiinterluekin-6 receptor antibody. BioFactors. 2009;35(1):47–51. doi: 10.1002/biof.9. [DOI] [PubMed] [Google Scholar]
- 53.Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR. Elevated levels of endogenous IL-6 in systemic lupus erythematosus: a putative role in pathogenesis. Journal of Immunology. 1991;147(1):117–123. [PubMed] [Google Scholar]
- 54.Grondal G, Gunnarsson I, Ronnelid J, Rogberg S, Klareskog L, Lundberg I. Cytokine production, serum levels and disease activity in systemic lupus erythematosus. Clinical and Experimental Rheumatology. 2000;18(5):565–570. [PubMed] [Google Scholar]
- 55.Iwano M, Dohi K, Hirata E, et al. Urinary levels of IL-6 in patients with active lupus nephritis. Clinical Nephrology. 1993;40(1):16–21. [PubMed] [Google Scholar]
- 56.Tsai CY, Wu TH, Yu CL, Lu JY, Tsai YY. Increased excretions of β2-microglobulin, IL-6, and IL-8 and decreased excretion of Tamm-Horsfall glycoprotein in urine of patients with active lupus nephritis. Nephron. 2000;85(3):207–214. doi: 10.1159/000045663. [DOI] [PubMed] [Google Scholar]
- 57.Fukatsu A, Matsuo S, Tamai H, Sakamoto N, Matsuda T, Hirano T. Distribution of interleukin-6 in normal and diseased human kidney. Laboratory Investigation. 1991;65(1):61–66. [PubMed] [Google Scholar]
- 58.Yoshio T, Masuyama JI, Kohda N, et al. Association of interleukin 6 release from endothelial cells and pulmonary hypertension in SLE. Journal of Rheumatology. 1997;24(3):489–495. [PubMed] [Google Scholar]
- 59.Alcocer-Varela J, Aleman-Hoey D, Alarcon-Segovia D. Interleukin-1 and interleukin-6 activities are increased in the cerebrospinal fluid of patients with CNS lupus erythematosus and correlate with local late T-cell activation markers. Lupus. 1992;1(2):111–117. doi: 10.1177/096120339200100209. [DOI] [PubMed] [Google Scholar]
- 60.Eilertsen GØ, Nikolaisen C, Becker-Merok A, Nossent JC. Interleukin-6 promotes arthritis and joint deformation in patients with systemic lupus erythematosus. Lupus. 2011;20(6):607–613. doi: 10.1177/0961203310392432. [DOI] [PubMed] [Google Scholar]
- 61.Mihara M, Nishimoto N, Ohsugi Y. The therapy of autoimmune diseases by anti-interleukin-6 receptor antibody. Expert Opinion on Biological Therapy. 2005;5(5):683–690. doi: 10.1517/14712598.5.5.683. [DOI] [PubMed] [Google Scholar]
- 62.Illei GG, Shirota Y, Yarboro CH, et al. Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis & Rheumatism. 2010;62(2):542–552. doi: 10.1002/art.27221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang J, Murphy TL, Ouyang W, Murphy KM. Induction of interferon-γ production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. European Journal of Immunology. 1999;29(2):548–555. doi: 10.1002/(SICI)1521-4141(199902)29:02<548::AID-IMMU548>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 64.Jäger A, Kuchroo VK. Effector and regulatory T-cell subsets in autoimmunity and tissue inflammation. Scandinavian Journal of Immunology. 2010;72(3):173–184. doi: 10.1111/j.1365-3083.2010.02432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferber IA, Brocke S, Taylor-Edwards C, et al. Mice with a disrupted IFN-γ gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) Journal of Immunology. 1996;156(1):5–7. [PubMed] [Google Scholar]
- 66.Matthys P, Vermeire K, Mitera T, Heremans H, Huang S, Billiau A. Anti-IL-12 antibody prevents the development and progression of collagen-induced arthritis in IFN-γ receptor-deficient mice. European Journal of Immunology. 1998;28(7):2143–2151. doi: 10.1002/(SICI)1521-4141(199807)28:07<2143::AID-IMMU2143>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 67.Kelchtermans H, Billiau A, Matthys P. How interferon-γ keeps autoimmune diseases in check. Trends in Immunology. 2008;29(10):479–486. doi: 10.1016/j.it.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 68.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nature Immunology. 2005;6(11):1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Enghard P, Langnickel D, Riemekasten G. T cell cytokine imbalance towards production of IFN-γ and IL-10 in NZB/W F1 lupus-prone mice is associated with autoantibody levels and nephritis. Scandinavian Journal of Rheumatology. 2006;35(3):209–216. doi: 10.1080/03009740500417791. [DOI] [PubMed] [Google Scholar]
- 70.Jacob CO, van der Meide PH, McDevitt HO. In vivo treatment of (NZB x NZW)F1 lupus-like nephritis with monoclonal antibody to γ interferon. Journal of Experimental Medicine. 1987;166(3):798–803. doi: 10.1084/jem.166.3.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haas C, Ryffel B, Le Hir M. IFN-γ receptor deletion prevents autoantibody production and glomerulonephritis in lupus-prone (NZB x NZW)F1 mice. Journal of Immunology. 1998;160(8):3713–3718. [PubMed] [Google Scholar]
- 72.Balomenos D, Rumold R, Theofilopoulos AN. Interferon-γ is required for lupus-like disease and lymphoaccumulation in MRL-lpr mice. Journal of Clinical Investigation. 1998;101(2):364–371. doi: 10.1172/JCI750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lawson BR, Prud’homme GJ, Chang Y, et al. Treatment of murine lupus with cDNA encoding IFN-γR/Fc. Journal of Clinical Investigation. 2000;106(2):207–215. doi: 10.1172/JCI10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nicoletti F, Di Marco R, Zaccone P, et al. Dichotomic effects of IFN-γ on the development of systemic lupus erythematosus-like syndrome in MRL-lpr/lpr mice. European Journal of Immunology. 2000;30(2):438–447. doi: 10.1002/1521-4141(200002)30:2<438::AID-IMMU438>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 75.Richards HB, Satoh M, Jennette JC, Croker BP, Yoshida H, Reeves WH. Interferon-γ is required for lupus nephritis in mice treated with the hydrocarbon oil pristane. Kidney International. 2001;60(6):2173–2180. doi: 10.1046/j.1523-1755.2001.00045.x. [DOI] [PubMed] [Google Scholar]
- 76.Takahashi S, Fossati L, Iwamoto M, et al. Imbalance towards Th1 predominance is associated with acceleration of lupus-like autoimmune syndrome in MRL mice. Journal of Clinical Investigation. 1996;97(7):1597–1604. doi: 10.1172/JCI118584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tokano Y, Morimoto S, Kaneko H, et al. Levels of IL-12 in the sera of patients with systemic lupus erythematosus (SLE)—relation to Th1- and Th2-derived cytokines. Clinical and Experimental Immunology. 1999;116(1):169–173. doi: 10.1046/j.1365-2249.1999.00862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tucci M, Lombardi L, Richards HB, Dammacco F, Silvestris F. Overexpression of interleukin-12 and T helper 1 predominance in lupus nephritis. Clinical and Experimental Immunology. 2008;154(2):247–254. doi: 10.1111/j.1365-2249.2008.03758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shah K, Lee WW, Lee SH, et al. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Research & Therapy. 2010;12(2):p. R53. doi: 10.1186/ar2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Prado C, de Paz B, Gomez J, Lopez P, Rodriguez-Carrio J, Suarez A. Glucocorticoids enhance Th17/Th1 imbalance and signal transducer and activator of transcription 3 expression in systemic lupus erythematosus patients. Rheumatology. 2011;50(10):1794–1801. doi: 10.1093/rheumatology/ker227. [DOI] [PubMed] [Google Scholar]
- 81.Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human Th17 cells. Journal of Experimental Medicine. 2007;204(8):1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-γ: implications for immune responses and autoimmune diseases. Immunity. 2009;31(4):539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Uhm WS, Na K, Song GW, et al. Cytokine balance in kidney tissue from lupus nephritis patients. Rheumatology. 2003;42(8):935–938. doi: 10.1093/rheumatology/keg255. [DOI] [PubMed] [Google Scholar]
- 84.Min DJ, Cho ML, Cho CS, et al. Decreased production of interleukin-12 and interferon-γ is associated with renal involvement in systemic lupus erythematosus. Scandinavian Journal of Rheumatology. 2001;30(3):159–163. doi: 10.1080/030097401300162932. [DOI] [PubMed] [Google Scholar]
- 85.Malek TR. The biology of interleukin-2. Annual Review of Immunology. 2008;26:453–479. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 86.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75(2):253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 87.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3(4):521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- 88.Dauphinee MJ, Kipper SB, Wofsy D, Talal N. Interleukin 2 deficiency is a common feature of autoimmune mice. Journal of Immunology. 1981;127(6):2483–2487. [PubMed] [Google Scholar]
- 89.Altman A, Theofilopoulos AN, Weiner R. Analysis of T cell function in autoimmune murine strains. Defects in production of and responsiveness to interleukin 2. Journal of Experimental Medicine. 1981;154(3):791–808. doi: 10.1084/jem.154.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wofsy D, Murphy ED, Roths JB. Deficient interleukin 2 activity in MRL/Mp and C57BL/6J mice bearing the lpr gene. Journal of Experimental Medicine. 1981;154(5):1671–1680. doi: 10.1084/jem.154.5.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Davignon JL, Cohen PL, Eisenberg RA. Rapid T cell receptor modulation accompanies lack of in vitro mitogenic responsiveness of double negative T cells to anti-CD3 monoclonal antibody in MRL/Mp-lpr/lpr mice. Journal of Immunology. 1988;141(6):1848–1854. [PubMed] [Google Scholar]
- 92.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3+ CD25+ CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. Journal of Experimental Medicine. 2005;201(5):723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brandenburg S, Takahashi T, de la Rosa M, et al. IL-2 induces in vivo suppression by CD4+CD25+Foxp3+ regulatory T cells. European Journal of Immunology. 2008;38(6):1643–1653. doi: 10.1002/eji.200737791. [DOI] [PubMed] [Google Scholar]
- 94.Yang X-P, Ghoreschi K, Steward-Tharp SM, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nature Immunology. 2011;12(3):247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Laurence A, Tato CM, Davidson TS, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26(3):371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 96.Alcocer-Varela J, Alarcon-Segovia D. Decreased production of and response to interleukin-2 by cultured lymphocytes from patients with systemic lupus erythematosus. Journal of Clinical Investigation. 1982;69(6):1388–1392. doi: 10.1172/JCI110579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.de Faucal P, Godard A, Peyrat MA. Impaired IL2 production by lymphocytes of patients with systemic lupus erythematosus. Annales d'Immunologie. 1984;135(2):161–172. doi: 10.1016/s0769-2625(84)81108-3. [DOI] [PubMed] [Google Scholar]
- 98.Linker Israeli M, Bakke AC, Kitridou RC. Defective production of interleukin 1 and interleukin 2 in patients with systemic lupus erythematosus (SLE) Journal of Immunology. 1983;130(6):2651–2655. [PubMed] [Google Scholar]
- 99.Miyara M, Amoura Z, Parizot C, et al. Global natural regulatory T cell depletion in active systemic lupus erythematosus. Journal of Immunology. 2005;175(12):8392–8400. doi: 10.4049/jimmunol.175.12.8392. [DOI] [PubMed] [Google Scholar]
- 100.Juang YT, Wang Y, Solomou EE, et al. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. Journal of Clinical Investigation. 2005;115(4):996–1005. doi: 10.1172/JCI200522854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Juang Y-T, Rauen T, Wang Y, et al. Transcriptional activation of the cAMP-responsive modulator promoter in human T cells is regulated by protein phosphatase 2A-mediated dephosphorylation of SP-1 and reflects disease activity in patients with systemic lupus erythematosus. Journal of Biological Chemistry. 2011;286(3):1795–1801. doi: 10.1074/jbc.M110.166785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tenbrock K, Juang YT, Gourley MF, Nambiar MP, Tsokos GC. Antisense cyclic adenosine 5′-monophosphate response element modulator up-regulates IL-2 in T cells from patients with systemic lupus erythematosus. Journal of Immunology. 2002;169(8):4147–4152. doi: 10.4049/jimmunol.169.8.4147. [DOI] [PubMed] [Google Scholar]
- 103.Solomou EE, Juang YT, Gourley M, Kammer GM, Tsokos GC. Molecular basis of deficient IL-2 production in T cells from patients with SLE: cAMP responsive element modulator (CREM) binds to the -180 site of the IL-2 promoter instead of cAMP responsive element binding protein (CREB) Arthritis & Rheumatism. 2000;43(9):S238–S238. [Google Scholar]
- 104.Katsiari CG, Kyttaris VC, Juang YT, Tsokos GC. Protein phosphatase 2A (PP2A): a novel negative regulator of IL-2 production in patients with systemic lupus erythematosus (SLE) Arthritis & Rheumatism. 2005;52(9):S490–S491. doi: 10.1172/JCI24895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ma J, Yu J, Tao X, Cai L, Wang J, Zheng SG. The imbalance between regulatory and IL-17-secreting CD4+ T cells in lupus patients. Clinical Rheumatology. 2010;29(11):1251–1258. doi: 10.1007/s10067-010-1510-7. [DOI] [PubMed] [Google Scholar]
- 106.Dai Z, Arakelov A, Wagener M, Konieczny BT. The role of the common cytokine receptor γ-chain in regulating IL-2-dependent, activation-induced CD8+ T cell death. Journal of Immunology. 1999;163(6):3131–3137. [PubMed] [Google Scholar]
- 107.Sytwu HK, Liblau RS, McDevitt HO. The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity. 1996;5(1):17–30. doi: 10.1016/s1074-7613(00)80306-4. [DOI] [PubMed] [Google Scholar]
- 108.Dhein J, Walczak H, Baumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373(6513):438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 109.Lieberman LA, Tsokos GC. The IL-2 defect in systemic lupus erythematosus disease has an expansive effect on host immunity. Journal of Biomedicine and Biotechnology. 2010;2010:6 pages. doi: 10.1155/2010/740619. Article ID 740619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kovacs B, Vassilopoulos D, Vogelgesang SA, Tsokos GC. Defective CD3-mediated cell death in activated T cells from patients with systemic lupus erythematosus: role of decreased intracellular TNF-α. Clinical Immunology and Immunopathology. 1996;81(3):293–302. doi: 10.1006/clin.1996.0192. [DOI] [PubMed] [Google Scholar]
- 111.Dooms H, Abbas AK. Revisiting the role of IL-2 in autoimmunity. European Journal of Immunology. 2010;40(6):1538–1540. doi: 10.1002/eji.201040617. [DOI] [PubMed] [Google Scholar]
- 112.Humrich JY, Morbach H, Undeutsch R, et al. Homeostatic imbalance of regulatory and effector T cells due to IL-2 deprivation amplifies murine lupus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(1):204–209. doi: 10.1073/pnas.0903158107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tang Q, Adams JY, Penaranda C, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28(5):687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu R, Zhou Q, La Cava A, Campagnolo DI, van Kaer L, Shi FD. Expansion of regulatory T cells via IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia. European Journal of Immunology. 2010;40(6):1577–1589. doi: 10.1002/eji.200939792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Webster KE, Walters S, Kohler RE, et al. In vivo expansion of t reg cells with il-2-mab complexes: induction of resistance to eae and long-term acceptance of islet allografts without immunosuppression. Journal of Experimental Medicine. 2009;206(4):751–760. doi: 10.1084/jem.20082824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liao W, Lin J-X, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nature Immunology. 2011;12(6):551–559. doi: 10.1038/ni.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leonard WJ, Zeng R, Spolski R. Interleukin 21: a cytokine/cytokine receptor system that has come of age. Journal of Leukocyte Biology. 2008;84(2):348–356. doi: 10.1189/jlb.0308149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Spolski R, Leonard WJ. The Yin and Yang of interleukin-21 in allergy, autoimmunity and cancer. Current Opinion in Immunology. 2008;20(3):295–301. doi: 10.1016/j.coi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Monteleone G, Pallone F, MacDonald TT. Interleukin-21: a critical regulator of the balance between effector and regulatory T-cell responses. Trends in Immunology. 2008;29(6):290–294. doi: 10.1016/j.it.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 120.Liu S, Lizée G, Lou Y, et al. IL-21 synergizes with IL-7 to augment expansion and anti-tumor function of cytotoxic T cells. International Immunology. 2007;19(10):1213–1221. doi: 10.1093/intimm/dxm093. [DOI] [PubMed] [Google Scholar]
- 121.Wu Z, Kim HP, Xue HH, Liu H, Zhao K, Leonard WJ. Interleukin-21 receptor gene induction in human T cells is mediated by T-cell receptor-induced Sp1 activity. Molecular and Cellular Biology. 2005;25(22):9741–9752. doi: 10.1128/MCB.25.22.9741-9752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Korn T, Bettelli E, Gao W, et al. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature. 2007;448(7152):484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Peluso I, Fantini MC, Fina D, et al. IL-21 counteracts the regulatory T cell-mediated suppression of human CD4+ T lymphocytes. Journal of Immunology. 2007;178(2):732–739. doi: 10.4049/jimmunol.178.2.732. [DOI] [PubMed] [Google Scholar]
- 124.Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity. 2008;29(1):127–137. doi: 10.1016/j.immuni.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 125.Ozaki K, Spolski R, Ettinger R, et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. Journal of Immunology. 2004;173(9):5361–5371. doi: 10.4049/jimmunol.173.9.5361. [DOI] [PubMed] [Google Scholar]
- 126.Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nature Reviews Immunology. 2009;9(12):845–857. doi: 10.1038/nri2637. [DOI] [PubMed] [Google Scholar]
- 127.Kuchen S, Robbins R, Sims GP, et al. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. Journal of Immunology. 2007;179(9):5886–5896. doi: 10.4049/jimmunol.179.9.5886. [DOI] [PubMed] [Google Scholar]
- 128.Mehta DS, Wurster AL, Grusby MJ. Biology of IL-21 and the IL-21 receptor. Immunological Reviews. 2004;202:84–95. doi: 10.1111/j.0105-2896.2004.00201.x. [DOI] [PubMed] [Google Scholar]
- 129.Brandt K, Bulfone-Paus S, Foster DC, Rückert R. Interleukin-21 inhibits dendritic cell activation and maturation. Blood. 2003;102(12):4090–4098. doi: 10.1182/blood-2003-03-0669. [DOI] [PubMed] [Google Scholar]
- 130.Spolski R, Kim HP, Zhu W, Levy DE, Leonard WJ. IL-21 mediates suppressive effects via its induction of IL-10. Journal of Immunology. 2009;182(5):2859–2867. doi: 10.4049/jimmunol.0802978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sawalha AH, Kaufman KM, Kelly JA, et al. Genetic association of interleukin-21 polymorphisms with systemic lupus erythematosus. Annals of the Rheumatic Diseases. 2008;67(4):458–461. doi: 10.1136/ard.2007.075424. [DOI] [PubMed] [Google Scholar]
- 132.Webb R, Merrill JT, Kelly JA, et al. A polymorphism within IL21R confers risk for systemic lupus erythematosus. Arthritis & Rheumatism. 2009;60(8):2402–2407. doi: 10.1002/art.24658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Simpson N, Gatenby PA, Wilson A, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis & Rheumatism. 2010;62(1):234–244. doi: 10.1002/art.25032. [DOI] [PubMed] [Google Scholar]
- 134.Vinuesa CG, Cook MC, Angelucci C, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435(7041):452–458. doi: 10.1038/nature03555. [DOI] [PubMed] [Google Scholar]
- 135.Odegard JM, Marks BR, Diplacido LD, et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. Journal of Experimental Medicine. 2008;205(12):2873–2886. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Herber D, Brown TP, Liang S, Young DA, Collins M, Dunussi-Joannopoulos K. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. Journal of Immunology. 2007;178(6):3822–3830. doi: 10.4049/jimmunol.178.6.3822. [DOI] [PubMed] [Google Scholar]
- 137.Bubier JA, Sproule TJ, Foreman O, et al. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(5):1518–1523. doi: 10.1073/pnas.0807309106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bubier JA, Bennett SM, Sproule TJ, et al. Treatment of BXSB-Yaa mice with IL-21R-Fc fusion protein minimally attenuates systemic lupus erythematosus. Annals of the New York Academy of Sciences. 2007;1110:590–601. doi: 10.1196/annals.1423.063. [DOI] [PubMed] [Google Scholar]
- 139.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annual Review of Immunology. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 140.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 141.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 142.Mangan PR, Harrington LE, O’Quinn DB, et al. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441(7090):231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 143.Nurieva R, Yang XO, Martinez G, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448(7152):480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 144.Zhou L, Ivanov II, Spolski R, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nature Immunology. 2007;8(9):967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 145.Manel N, Unutmaz D, Littman DR. The differentiation of human TH-17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγt. Nature Immunology. 2008;9(6):641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yang L, Anderson DE, Baecher-Allan C, et al. IL-21 and TGF-β are required for differentiation of human TH17 cells. Nature. 2008;454(7202):350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. Journal of Experimental Medicine. 2005;201(2):233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. The New England Journal of Medicine. 2009;361(9):848–898. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 149.Toy D, Kugler D, Wolfson M, et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. Journal of Immunology. 2006;177(1):36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 150.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annual Review of Immunology. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 151.Aggarwal S, Gurney AL. IL-17: prototype member of an emerging cytokine family. Journal of Leukocyte Biology. 2002;71(1):1–8. [PubMed] [Google Scholar]
- 152.Laan M, Lötvall J, Chung KF, Lindén A. IL-17-induced cytokine release in human bronchial epithelial cells in vitro: role of mitogen-activated protein (MAP) kinases. British Journal of Pharmacology. 2001;133(1):200–206. doi: 10.1038/sj.bjp.0704063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Woltman AM, de Haij S, Boonstra JG, Gobin SJP, Daha MR, van Kooten C. Interleukin-17 and CD40-Ligand synergistically enhance cytokine and chemokine production by renal epithelial cells. Journal of the American Society of Nephrology. 2000;11(11):2044–2055. doi: 10.1681/ASN.V11112044. [DOI] [PubMed] [Google Scholar]
- 154.Witowski J, Pawlaczyk K, Breborowicz A, et al. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GROα chemokine from mesothelial cells. Journal of Immunology. 2000;165(10):5814–5821. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]
- 155.Albanesi C, Cavani A, Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonist effects with IFN-γ and TNF-α. Journal of Immunology. 1999;162(1):494–502. [PubMed] [Google Scholar]
- 156.Schwarzenberger P, Huang W, Peng Y, et al. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. Journal of Immunology. 2000;164(9):4783–4789. doi: 10.4049/jimmunol.164.9.4783. [DOI] [PubMed] [Google Scholar]
- 157.Cai XY, Gommoll CP, Justice L, Narula SK, Fine JS. Regulation of granulocyte colony-stimulating factor gene expression by interleukin-17. Immunology Letters. 1998;62(1):51–58. doi: 10.1016/s0165-2478(98)00027-3. [DOI] [PubMed] [Google Scholar]
- 158.Mitsdoerffer M, Lee Y, Jäger A, et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(32):14292–14297. doi: 10.1073/pnas.1009234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Doreau A, Belot A, Bastid J, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nature Immunology. 2009;10(7):778–785. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- 160.Yang J, Chu Y, Yang X, et al. Th17 and natural treg cell population dynamics in systemic lupus erythematosus. Arthritis & Rheumatism. 2009;60(5):1472–1483. doi: 10.1002/art.24499. [DOI] [PubMed] [Google Scholar]
- 161.Wong CK, Ho CY, Li EK, Lam CWK. Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus. 2000;9(8):589–593. doi: 10.1191/096120300678828703. [DOI] [PubMed] [Google Scholar]
- 162.Wong CK, Lit LCW, Tam LS, Li EKM, Wong PTY, Lam CWK. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clinical Immunology. 2008;127(3):385–393. doi: 10.1016/j.clim.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 163.Nalbandian A, Crispín JC, Tsokos GC. Interleukin-17 and systemic lupus erythematosus: current concepts. Clinical and Experimental Immunology. 2009;157(2):209–215. doi: 10.1111/j.1365-2249.2009.03944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Crispín JC, Oukka M, Bayliss G, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. Journal of Immunology. 2008;181(12):8761–8766. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Crispín JC, Tsokos GC. Human TCR-αβ+CD4−CD8− T cells can derive from CD8+ T cells and display an inflammatory effector phenotype. Journal of Immunology. 2009;183(7):4675–4681. doi: 10.4049/jimmunol.0901533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang Y, Ito S, Chino Y, et al. Laser microdissection-based analysis of cytokine balance in the kidneys of patients with lupus nephritis. Clinical and Experimental Immunology. 2010;159(1):1–10. doi: 10.1111/j.1365-2249.2009.04031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kwan BCH, Tam LS, Lai KB, et al. The gene expression of type 17 T-helper cell-related cytokines in the urinary sediment of patients with systemic lupus erythematosus. Rheumatology. 2009;48(12):1491–1497. doi: 10.1093/rheumatology/kep255. [DOI] [PubMed] [Google Scholar]
- 168.Dong G, Ye R, Shi W, et al. IL-17 induces autoantibody overproduction and peripheral blood mononuclear cell overexpression of IL-6 in lupus nephritis patients. Chinese Medical Journal. 2003;116(4):543–548. [PubMed] [Google Scholar]
- 169.Zhang Z, Kyttaris VC, Tsokos GC. The role of IL-23/IL-17 axis in lupus nephritis. Journal of Immunology. 2009;183(5):3160–3169. doi: 10.4049/jimmunol.0900385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Edgerton C, Crispín JC, Moratz CM, et al. IL-17 producing CD4+ T cells mediate accelerated ischemia/reperfusion-induced injury in autoimmunity-prone mice. Clinical Immunology. 2009;130(3):313–321. doi: 10.1016/j.clim.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kang HK, Liu M, Datta SK. Low-dose peptide tolerance therapy of lupus generates plasmacytoid dendritic cells that cause expansion of autoantigen-specific regulatory T cells and contraction of inflammatory Th17 cells. Journal of Immunology. 2007;178(12):7849–7858. doi: 10.4049/jimmunol.178.12.7849. [DOI] [PubMed] [Google Scholar]
- 172.Hsu HC, Yang PA, Wang J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nature Immunology. 2008;9(2):166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 173.Shahrara S, Pickens SR, Dorfleutner A, Pope RM. IL-17 induces monocyte migration in rheumatoid arthritis. Journal of Immunology. 2009;182(6):3884–3891. doi: 10.4049/jimmunol.0802246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yi T, Zhao D, Lin CL, et al. Absence of donor Thl7 leads to augmented Thl differentiation and exacerbated acute graft-versus-host disease. Blood. 2008;112(5):2101–2110. doi: 10.1182/blood-2007-12-126987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yang J, Yang X, Zou H, Chu Y, Li M. Recovery of the immune balance between Th17 and regulatory T cells as a treatment for systemic lupus erythematosus. Rheumatology. 2011;50(8):1366–1372. doi: 10.1093/rheumatology/ker116. [DOI] [PubMed] [Google Scholar]
- 176.Bettelli E, Oukka M, Kuchroo VK. TH-17 cells in the circle of immunity and autoimmunity. Nature Immunology. 2007;8(4):345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 177.McGeachy MJ, Bak-Jensen KS, Chen Y, et al. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nature Immunology. 2007;8(12):1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 178.Zhou L, Lopes JE, Chong MMW, et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature. 2008;453(7192):236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Du J, Huang C, Zhou B, Ziegler SF. Isoform-specific inhibition of RORα-mediated transcriptional activation by human FOXP3. Journal of Immunology. 2008;180(7):4785–4792. doi: 10.4049/jimmunol.180.7.4785. [DOI] [PubMed] [Google Scholar]
- 180.Yang XO, Nurieva R, Martinez GJ, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29(1):44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lochner M, Peduto L, Cherrier M, et al. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORγt+ T cells. Journal of Experimental Medicine. 2008;205(6):1381–1393. doi: 10.1084/jem.20080034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Voo KS, Wang YH, Santori FR, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(12):4793–4798. doi: 10.1073/pnas.0900408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Radhakrishnan S, Cabrera R, Schenk EL, et al. Reprogrammed FoxP3+ T regulatory cells become IL-17+ antigen-specific autoimmune effectors in vitro and in vivo. Journal of Immunology. 2008;181(5):3137–3147. doi: 10.4049/jimmunol.181.5.3137. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 184.Crispín JC, Kyttaris VC, Juang Y-T, Tsokos GC. Systemic lupus erythematosus: new molecular targets. Annals of the Rheumatic Diseases. 2007;66(supplement 3):iii65–iii69. doi: 10.1136/ard.2007.078493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Li J, Pan HF, Cen H, et al. Interleukin-21 as a potential therapeutic target for systemic lupus erythematosus. Molecular Biology Reports. 2010;38(6):4077–4081. doi: 10.1007/s11033-010-0527-y. [DOI] [PubMed] [Google Scholar]