

Abstract
Members of the monoamine oxidase family of flavoproteins catalyze the oxidation of primary and secondary amines, polyamines, amino acids, and methylated lysine side chains in proteins. The enzymes have similar overall structures, with conserved FAD-binding domains and varied substrate-binding sites. Multiple mechanisms have been proposed for the catalytic reactions of these enzymes. The present review compares the structures of different members of the family and the various mechanistic proposals.
Keywords: monoamine oxidase, flavoproteins, polyamine oxidase, spermine oxidase, L-amino acid oxidase, lysine-specific demethylase, protein structure, enzyme mechanism
Introduction
Monoamine oxidase (MAO) family members oxidize a variety of amine substrates, including small-molecule monoamines and polyamines and modified amino acids within proteins. Enzymes belonging to the MAO family share similar overall structures (Figure 1), with nearly identical FAD binding domains resembling the folding pattern of p-hydroxybenzoate hydroxylase (PHBH) (1, 2), but contain varied substrate-binding sites (Figure 2). As flavoprotein oxidases, they catalyze substrate oxidation via two half-reactions; in the reductive half-reaction, the flavin cofactor is reduced when it accepts a hydride equivalent from the substrate, while in the oxidative step, the reduced flavin is reoxidized by molecular oxygen (Scheme 1).
Figure 1.
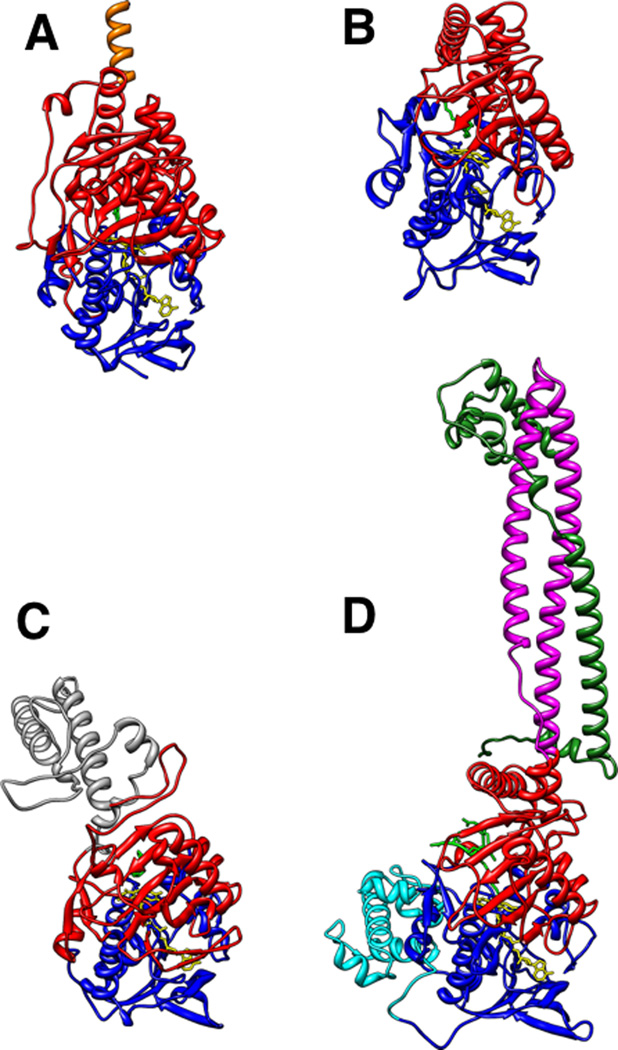
Ribbon representations of human MAO B (A), maize PAO (B), bacterial LAAO (C), and human LSD1 (D) were generated using the program Chimera (115) and the following Protein Data Bank (PDB) files: 1OJA, 1H83, 2JB2, and 2UXN. The FAD-binding domains are colored blue and substrate-binding domains are red; substrate analogs or inhibitors are colored green and the flavin cofactors are yellow. The C-terminal membrane-binding α-helix of MAO B is colored orange; the helical domain of LAAO is grey. The tower domain of LSD1 (magenta) is shown interacting with a portion of CoREST, colored dark green, and the SANT domain of LSD1 is cyan.
Figure 2.

The active sites of human MAO B (A, PDB file 1OJA), maize PAO (B, PDB file 1B5Q), bacterial LAAO (C, PDB file 2JB2), and human LSD1 (D, PDB file 2UXN) with bound substrates or inhibitors (green).
Scheme 1.

Due to the ability of the flavin cofactor to accept one or two electrons, several mechanisms have been proposed for the transfer of electrons from the substrate to the cofactor (Scheme 2). The single electron transfer mechanism (Scheme 2A) involves formation of semiquinone flavin and aminium cation radical intermediates, with subsequent transfer of a hydrogen atom equivalent (3). Direct hydrogen atom transfer from the substrate α-carbon to the flavin either directly or a via non-flavin radical (Scheme 2B) is another possible mechanism for substrate oxidation (4). Variations of a nucleophilic mechanism (Scheme 2C), in which the amino group of the substrate attacks the C4a of the flavin, forming a covalent intermediate, followed by proton abstraction by an active site base, have also been proposed (5). Finally, the reaction could occur by direct hydride transfer from the substrate to the flavin (Scheme 2D) (6).
Scheme 2.

While the flavin increases the number of possible enzymatic mechanisms, its presence simplifies the study of flavin-dependent reactions due to its characteristic spectrum, which changes as the isoalloxazine ring system gains and loses electrons. The spectrum of oxidized flavin has maxima around 380 and 460 nm, which diminish when the cofactor is reduced. Monitoring the change in absorbance during a reaction allows for measurement of individual kinetic rate constants and facilitates detection of reaction intermediates. Furthermore, even though the monoamine oxidase family members interact with two substrates, an amine and oxygen, the oxidation of the amine substrate is effectively irreversible (7), and oxygen only reacts with the reduced enzyme, making the analysis of steady-state kinetic parameters less complicated.
Monoamine Oxidases A and B
Monoamine oxidases A and B (MAO A and MAO B) are outer mitochondrial membrane proteins that catalyze the oxidation of primary, secondary, and tertiary amines, including several neurotransmitters, to the corresponding imines; the oxidized products are hydrolyzed nonenzymatically to the respective aldehydes or ketones (8) (Scheme 3). The two enzymes share 70% amino acid identity, and both contain a covalently-bound FAD cofactor attached to an enzyme cysteine via the 8α-methylene of the isoalloxazine ring (9). MAO A metabolizes serotonin (5-hydroxytryptamine), norepinephrine, and dopamine, while MAO B preferentially oxidizes benzylamine, dopamine and phenylethylamine and only metabolizes norepinephrine and serotonin slowly (10–13). MAO B also forms the neurotoxin 1-methyl-4-phenyl-pyridinium, a causative agent of Parkinson’s disease, from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (14). Inhibitors of the monoamine oxidases have been used clinically for the treatment of depression, as well as Parkinson’s, Alzheimer’s and other neurodegenerative diseases (15, 16).
Scheme 3.

Both MAOs are composed of an FAD-binding domain, conserved among a number of other flavoprotein oxidases, a substrate-binding domain, and a membrane-binding domain (Figure 1A) (17, 18). While NMR studies have demonstrated that both forms of rat and human MAOs exist as dimers in solution (19), human MAO A crystallizes as a monomer (20). Both MAOs bind the outer mitochondrial membrane through a C-terminal α-helical region, with additional membrane interactions occurring with other hydrophobic residues (17, 18, 21). The FAD isoalloxazine ring exists in a strained, puckered state, in contrast to its planar conformation in solution; this difference has been suggested to make covalent addition at N5 or C4a more favorable by making these flavin atoms more “sp3-like” (18).
The substrate-binding sites of MAO A and MAO B (Figure 2A) are both mainly hydrophobic, encased by predominantly aromatic and aliphatic residues (17, 18). A notable exception is a conserved lysine (Lys296 in MAO B and Lys305 in MAO A) that interacts with a water molecule that also binds the N5 atom of the flavin cofactor (22). Tyrosines 398 and 435 in MAO B (Tyr407 and Tyr444 in MAO A) are located on opposite sides of covalently bound inhibitors and substrates in all of the MAOs, forming an aromatic “sandwich”, and mutation of these residues alters activity (23–25). These tyrosines have been proposed to orient the substrate for oxidation or to activate the amine by enhancing its nucleophilicity (26). The substrate-binding site of human MAO A consists of a cavity approximately 400 Å3 in size (27, 28), while in MAO B a smaller hydrophobic cavity, termed the “entrance cavity”, is positioned between the main substrate-binding site and the protein surface; rotation of an isoleucine residue (Ile199) allows the two cavities to be fused into one larger 700 Å3 site (22). The cavities remain separated or become joined depending on the nature of the substrate or inhibitor present (29). In MAO A, a phenylalanine (Phe208) replaces Ile199 of MAO B (Figure 3). Another difference in the substrate-binding sites of the two enzymes is Ile335 in MAO A vs. Tyr326 in MAO B; these residues contribute to the substrate and inhibitor selectivity of the two enzymes) (25, 30). While structural studies initially suggested that substrate specificity differences were due to the conformation of a six-residue loop (residues 210–216 in human MAO A and 201–206 of human MAO B) (20, 28), subsequent studies showed that the loop is in the same conformation in both enzymes (Figure 3) (30).
Figure 3.

An overlay of the substrate-binding sites of human MAO B (PDB file 1OJA) and human MAO A (PDB file 2Z5X). The figure highlights residue differences in the active sites of these two enzymes that affect substrate specificity, and identifies the substrate-cavity forming loop that appears to be conserved in the two enzymes. MAO B is colored blue, and Ile199, residues 201–206, and Tyr326, are colored red; the inhibitor isatin is green. MAO A is pictured in dark grey, with Phe208, residues 210–216, and Ile335 colored orange; the inhibitor harmine is cyan. The flavin cofactor is shown in yellow.
MAOs preferentially bind the substrates with the amino group in the neutral from (31), consistent with deprotonation of the amine being required for oxidation (32). Cleavage of the amine substrate CH bond is rate-limiting with benzylamines (31) and phenethylamines (33) as substrates for MAO A, and with a series benzylamine (7) and MPTP analogs (34) for MAO B.
Much of the initial mechanistic work with MAOs A and B was interpreted as favoring mechanisms in which a substrate radical was formed, either by single electron (Scheme 2A) or hydrogen atom transfer (Scheme 2B). The inactivation of the MAOs by cyclopropyl inhibitors is consistent with formation of a substrate radical (3, 35–37), leading Silverman and coworkers to propose a single electron transfer mechanism for oxidation of substrates, similar to the electrochemical oxidation of amines (3). Further support for the single electron transfer mechanism came from Yue et al. (38), who reported resonance Raman spectra consistent with an anionic flavin semiquinone in MAO B; however, they were not able to observe a flavin radical by EPR spectroscopy, a more definitive approach. Evidence of a radical was later found in the EPR spectrum of MAO B; however, this spectrum was not altered by either the inhibitor pargyline or by substrates and was not equivalent to that of the semiquinone formed by photoreduction, suggesting that the signal arose from another source (39). Miller and colleagues similarly failed to find any evidence of a radical in the EPR spectra of MAO A or MAO B (31), further suggesting that the previous results were the result of an impurity. Finally, stopped-flow absorbance spectroscopy of flavin reduction failed to detect an intermediate radical (7, 31). It is possible that the flavin semiquinone and aminium cation radical do not accumulate during the reaction, making them difficult to detect; thus, lack of evidence for an intermediate does not ensure that it does not form during the reaction. Nevertheless, lack of a consistently observable flavin semiquinone, either by EPR spectroscopy or during stopped-flow analyses, provides a strong argument against any single electron mechanism. Furthermore, the thermodynamic barrier to single electron transfer is quite high, with the high redox potential of amines (Em = + 1.5 V) making them unlikely to be oxidized by the flavin cofactor (Em = −0.2 to 0 V), and there is no alternate oxidant strong enough to abstract an electron from the amine substrate (31). Although substrate binding might increase the redox potential of the flavin cofactor in MAO A and B (40), the difference in potentials still remains unfavorable.
An alternative version of the hydrogen atom transfer mechanism (Scheme 2B) utilizes an active site radical rather than the flavin for abstraction of the hydrogen atom; spectroscopic evidence for tyrosyl radical formation was described by Rigby et al. for MAO A (41). While three tyrosines are located in the vicinity of the active site (Tyr69, Tyr407, and Tyr444 in MAO A and Tyr60, Tyr398, and Tyr435 in MAO B), the data were most consistent with radical formation at Tyr407 (Tyr398 in MAO B). Mutagenesis studies, however, showed that the Y444F mutation affects activity more significantly than the Y407F mutation (25), and mechanistic studies with the Y444F mutant established that mutating that residue did not alter the mechanism of C-H cleavage (23). Subsequent studies with mouse polyamine oxidase (PAO), another MAO family member, showed that mutation of PAO Tyr430, homologous to MAO A Tyr407, to phenylalanine only resulted in a 6-fold decrease in activity, again suggesting that tyrosyl radical formation was unlikely during catalysis for that enzyme (42). Thus, the functional evidence does not support the involvement of a tyrosine radical in amine oxidation by the MAO family.
Quantitative structure-activity relation studies (QSARs) of the MAOs have yielded conflicting results. Substituent σ values are a quantitative measure of the electron-donating or withdrawing properties of the substituent. Comparing rates with the σ values for a series of substrates can indicate whether development of charge occurs in the transition state for the reaction. A positive correlation, or ρ value, corresponds to a development of negative charge, whereas a negative ρ value is associated with a buildup of positive charge; lack of a correlation, or a ρ value around zero, indicates little charge development. Studies with a series of para- and meta-substituted benzylamine analogs with MAO B (7), as well as with a series of phenethylamine analogs and MAO A (33), showed no correlation of activity with the substrate σ values, suggesting that no charge builds up during the reaction. Development of negative charge would be expected with mechanisms involving proton abstraction, namely single electron transfer and nucleophilic mechanisms; thus, these studies instead support direct hydrogen atom abstraction from the substrate or a hydride transfer mechanism. In contrast, in QSAR studies with MAO A and a series of 16 para-substituted benzylamine analogs, the activity correlated positively with the substituent σ values, with a ρ value of 2.0 for 12 of the analogs studied, but a ρ value of 0.5 for the remaining four analogs; the reason for the discrepancies was not clear. These results suggest a buildup of negative charge on the substrate, consistent with one version of the nucleophilic mechanism (Scheme 2C) (31); neither a flavin-substrate adduct nor a radical intermediate was observed during the reaction. Miller and Edmondson (31) attributed the differences between the results of QSAR studies with benzylamines for MAO A and B to different timing of proton abstraction in the transition state. The transition state for MAO B would be early, so that the small amount of negative charge on the benzyl carbon could be offset by positive charge created on the amine nitrogen. In MAO A, abstraction would occur later, so the charge development would be more sensitive to the substituent. Furthermore, these authors argued that substrate binding differences in the two enzymes could change the orientation of the π-orbitals of the benzene ring with respect to the π orbital of the breaking C-H bond, decreasing or preventing transmission of electronic effects from the substituents to the amino group, leading to small ρ values in MAO B. The differences in the results for MAO A with benzylamines and phenethylamines were attributed to reduction of the substituent electronic effects in the phenylethylamine analogs from the methylene group between the aromatic ring and the reaction site causing electronic effects to be undetectable (33); However, this effect should only be 2–3 fold (43). An alternative explanation to the different ρ values in the reactions of MAO A and B with benzylamines is that different steps in the overall reaction are determining the rate of the reaction. As noted above, the neutral amine is the substrate for MAO, and the pKa values of benzylamines are affected by substituents in the aromatic ring (44). The reactions of benzylamines with MAO A and B were studied at a pH below the pKa of benzylamine even when bound in the enzyme active site (32), so that the ρ value would reflect the fraction of the substrate that is deprotonated in addition to the rate constant for amine oxidation. Different contributions from these two effects could account for the different results with MAO A and B.
Support for the nucleophilic mechanism initially came from model studies with methyllumiflavins, which oxidize amines via a mechanism that involves a covalent intermediate between the flavin and amine, with subsequent proton transfer from the substrate (5). Proton transfer requires the presence of an active site base; a base strong enough to accept a proton from the benzyl substrate, however, is missing in either MAO. To circumvent the lack of the required base, Miller and Edmondson (31) proposed an altered nucleophilic mechanism in which amine attack of the flavin C4a leads to formation of a strong base at N5 of the flavin, which then accepts a proton from the substrate (Scheme 2C).
At present, the nucleophilic and radical mechanisms both have strong advocates, but there is a lack of definitive evidence to support either. In addition, neither fully accounts for all the mechanistic data on MAOs. A more complete mechanistic picture for this enzyme family comes from consideration of the other family members described below.
The mechanism of oxidation of reduced MAO by O2 has not been studied in detail. When monitored by stopped-flow absorbance, the reactions of reduced flavoprotein oxidases with O2 are typically found to be single exponential processes with rate constants directly proportional to the O2 concentration and no discernible intermediates. Based on model studies (45), the oxidation reaction is generally assumed to involve two single-electron transfers, forming a short-lived superoxide-flavin semiquinone radical pair (46). More detailed studies of the oxidation mechanism have been carried out with flavoprotein oxidases from other structural families. Protonation of an active site histidine in glucose oxidase is required for rapid reaction of the reduced enzyme with O2 (47), presumably by stabilizing the negative charge on oxygen as it is converted to superoxide anion. A lysine in monomeric sarcosine oxidase similarly must be protonated for rapid oxidation of the reduced form of that enzyme (48, 49). This lysine is also part of a water channel that extends to the surface and may provide the proton needed in the oxidation reaction. Whether the active site lysine in MAO (Figure 2A) has a similar role has not been established.
Polyamine Oxidases
Polyamine oxidases (PAOs) participate in the catabolism of spermine and spermidine and their acetyl derivatives. Increased levels of the polyamines spermine and spermidine are found in rapidly proliferating cells, and polyamines are essential for cell growth (50), yet excess accumulation of polyamines in cells causes cytotoxicity (51). The polyamine metabolic pathways are deregulated in several cancers, and polyamine analogs can act as antineoplastic agents (52–54) by altering the regulation of polyamine metabolic and catabolic pathways, ultimately reducing the availability of the natural polyamines. The term PAO is often reserved for enzymes preferring acetylated spermine or spermidine, while enzymes that preferentially oxidize spermine itself are referred to as spermine oxidases (SMOs). This terminology is not consistently used, resulting in some confusion as to the preferred reaction of individual enzymes. Mammalian PAOs convert N1-acetylspermine and N1-acetylspermidine to spermidine and putrescine, respectively, plus N-acetyl-3-aminopropanaldehyde and H2O2 (Scheme 4) (55, 56). In contrast, plant (57) and protozoan (58) PAOs oxidize their substrates on the endo side of the secondary N(4)-amino group, oxidizing spermine and spermidine to 3-aminopropyl-4-aminobutyraldehyde and 4-aminobutyraldehyde, respectively, plus 1,3-diaminopropane and H2O2 (Scheme 4). While mammalian polyamine oxidase is constitutively expressed, the expression of mammalian SMO is induced by polyamines, including antitumor polyamine analogs (59–61). Induction of SMO also has cytotoxic effects, possibly due to the increase in production of H2O2 (62, 63). Conversely, treatment of SMO with the competitive inhibitor MDL72527 inhibits production of H2O2, leading to decreased oxidative DNA damage and thereby decreasing the mutagenic changes associated with cancer progression (64).
Scheme 4.

Structures are only available for PAOs from maize and yeast. Unlike the MAOs, maize PAO is monomeric and contains a non-covalently bound FAD (Figure 1B) (65–67). A structure of maize PAO in complex with MDL72527 (Figure 2B) reveals several similarities with the structure of the MAOs, including homologous FAD-binding sites, a bent orientation for the flavin cofactor, the presence of the water molecule that interacts with a conserved lysine residue (Lys 300 in maize PAO) and the N5 of the flavin cofactor, and the “aromatic sandwich” for the reactive nitrogen of the substrate (66). Unlike the MAOs, the substrate-binding site consists of a U-shaped cavity that is approximately 30 Å long (66). The cavity has several acidic residues at one entrance that likely guide the positively charged substrate to the active site, while the other entrance is slightly more narrow and is lined by backbone carbonyl groups (66). The inhibitor forms a series of hydrogen bonds and van der Waals interactions with the protein.
The yeast PAO Fms1 oxidizes spermine and N1-acetylspermine, but not spermidine, forming spermidine and 3-aminopropanal or N-acetyl-3-aminopropanaldehyde, respectively, and functions in pantothenic acid production (68). Fms1 and maize PAO share only 20% amino acid sequence identity, but have very similar overall structures (69). Unlike maize PAO, Fms1 crystallizes as a dimer and also forms a dimer in solution, but like maize PAO the enzyme contains a tunnel with two entrances that forms the substrate-binding site (69). The substrate-binding site is hydrophilic on each of the two ends and hydrophobic in the middle, with the substrate bound through hydrogen bonds and hydrophobic interactions. Lys296 of Fms1 is conserved with Lys300 of maize PAO, and the FAD-binding domain is similar to that of the MAOs. The FAD cofactor, however, is planar.
Structural modeling of mouse SMO based on the structure of maize PAO suggests that the overall structural features, including the FAD and substrate-binding sites, are generally the same, although the substrate and inhibitor specificities differ (60, 70). Like the MAOs, the reaction kinetic mechanism is ping-pong for both Fms1 and human SMO, but the rate-limiting step is product release for these two enzymes (71, 72).
Mouse PAO, human SMO, and the yeast PAO Fms1 differ from one another in the protonation states of their substrate nitrogens required for optimal activity (Scheme 5). For all three enzymes, results of pH studies are consistent with a requirement that the substrate nitrogen at the site of C-H bond cleavage be uncharged for oxidation (71–73), consistent with observations with the MAOs. Whether the remaining nitrogens must be neutral or charged differs among the three enzymes, as shown in Scheme 5 for spermine. For human SMO, the effects of pH on steady-state and rapid-reaction kinetic parameters are consistent with the reactive form of spermine having all three non-reacting nitrogen atoms protonated (72). Similar analyses of Fms1 support a preference for the substrate form with only two protonated nitrogens (71). Finally, mouse PAO preferentially binds the singly charged forms of substrates (73). These differences likely play a role in determining the substrate specificity of the enzymes. For example N1-acetylspermine is a 50-fold worse substrate for SMO than spermine; this makes sense because the N1 atom of N1-acetylspermine cannot be protonated (72). Fms1 prefers N1-acetylspermine over spermine by less than 10-fold (71); this is consistent with the requirement that N1 be neutral. Finally, mouse PAO prefers N1-acetylspermine over spermine by over 100-fold (73); this can be attributed to the extra protonatable nitrogen in the latter.
Scheme 5.

QSAR studies with mouse PAO have been used to differentiate among the various proposed mechanisms for amine oxidation. With N,N’-dibenzyl-1,4-diaminobutanes as the substrate, substrate oxidation is the rate-limiting step for enzyme. The effect of substituents of seven para-substituted N,N’-dibenzyl-1,4-diaminobutanes on activity yielded small negative ρ values (−0.59 at pH 8.6 and −0.09 at pH 6.6), consistent with little or no accumulation of charge in the transition state and with either a hydride transfer mechanism or a hydrogen atom abstraction mechanism (74). This is similar to the results with MAO B with benzylamines (7) and with MAO A with phenethylamines (33). The more positive ρ value at pH 6.6 in the case of PAO can be attributed to the effect of the substrate protonation state when the reactions are carried out below the pKa of 8 for the substrate bound to the enzyme (74). As is the case with MAO A and B, rapid reaction studies of amine oxidation by PAO, SMO, and Fms1 failed to detect a semiquinone or flavin adduct intermediate (71–74), contrary to the expectations of radical and nucleophilic mechanisms. Thus, the data for the PAOs are fully consistent with a hydride transfer mechanism (Scheme 2D).
The role of the conserved lysine-water-flavin N5 atom interaction has been studied by site-directed mutagenesis in mouse and maize PAO with conflicting results. In maize polyamine oxidase, mutagenesis of this lysine to methionine results in a 1400-fold decrease in the rate constant for flavin reduction (75), suggesting that this residue plays an essential role in substrate oxidation in members of this family of enzymes. In mouse PAO, however, the similar mutagenesis of the conserved lysine to methionine has no effect on the kinetics of reduction (73). Instead, the mutation decreases the rate of flavin reoxidation; the effects of pH and D2O as solvent have been interpreted in favor of a mechanism in which the neutral form of the lysine coordinates a water molecule that accepts a proton during flavin oxidation (76). The need for a neutral lysine is in contrast to the need for a positively charged residue for rapid reaction of sarcosine and glucose oxidase with O2, so that this conserved lysine is not playing such a role in mouse PAO. The reason for the divergent effects of the identical mutation in the maize and mouse enzymes is not clear, but may reflect differences in the specificities of the plant and animal enzymes.
L-Amino Acid Oxidases
L-Amino acid oxidases (LAAOs) catalyze the oxidative deamination of an L-amino acid substrate to an α-keto acid, ammonia, and hydrogen peroxide (Scheme 6). The structures of the enzymes from the snake Calloselasma rhodostoma and the bacterium Rhodococcus opacus (Figure 1D) have been solved in the presence of ligands and substrates and establish that the LAAOs are in the MAO structural family (77–79). The structures show that the enzymes are both dimers, but dimerize differently. LAAO from R. opacus contains a helical domain responsible for dimerization, while the enzyme from C. rhodostoma dimerizes via interactions between residues in several different domains (Figure 4). Both enzymes contain a FAD-binding site similar to those in other members of the MAO family, as well as the conserved lysine residue near the N5 atom of the FAD (Figure 2C). The deep substrate-binding site in LAAO resembles the substrate-binding tunnel in PAO, but in LAAO the tunnel is only open on one end. LAAOs prefer hydrophobic amino acids, including phenylalanine, tryptophan, and tyrosine, as substrates (80); this is reminiscent of the preference of MAOs for amines with aromatic groups such as serotonin and catecholamines. Consistent with this substrate preference, the active site contains a hydrophobic pocket for the substrate side chain. LAAOs differ from MAOs and PAOs in that the substrates contain a carboxylate in addition to an amino moiety. The active sites of LAAOs contain a conserved arginine that forms a salt bridge with the carboxylate and a conserved tyrosine that forms a hydrogen bond with it (77, 78). The substrate-binding site is a mirror image of that in D-amino acid oxidase (DAAO), a member of a different structural family of flavin amine oxidases that catalyzes the same chemistry; a significant number of mechanistic studies have led to the conclusion that hydride transfer is the mechanism for substrate oxidation by DAAO (81, 82).
Scheme 6.

Figure 4.

An overlay of the structures of monomers (A) and dimers (B) of L-amino acid oxidases (LAAOs) from the snake C. rhodostoma (red, PDB file 2IID) and the bacterium R. opacus (blue, PDB file 2JB2).
Tryptophan monooxygenase (TMO) from Pseudomonas savastonai is representative of a subfamily of LAAOs that catalyzes the oxidative decarboxylation of amino acids; the physiological reaction catalyzed by the enzyme is the oxidation of tryptophan to indoleacetamide, carbon dioxide, and water (Scheme 7) in the two-step pathway for production of indoleacetic acid (83). Homologous genes have been identified in other bacteria (84).While the structure of (TMO) has not been solved, modeling studies demonstrated that LAAO and TMO are homologous proteins, and results of mutagenesis support this conclusion (85–87). Like other LAAOs, the enzyme most efficiently oxidizes amino acids with large hydrophobic side chains, such as tryptophan and phenylalanine, although methionine and alanine are also substrates (88).
Scheme 7.

The mechanism of amine oxidation by TMO has been studied in much more detail than that of the other LAAOs, making this a model enzyme for LAAOs in general. Similar to the PAOs, product release is rate-limiting for turnover of the physiological substrate tryptophan (89). The pH dependence of kinetic parameters established that the neutral form of the amine is required for catalysis, similar to the situation with MAO and PAOs (86, 90). No intermediates have been detected by stopped-flow absorbance spectroscopy during the oxidation of amino acids by TMO (88, 90). A series of deuterium and nitrogen kinetic isotope effects established that rehybridization of the substrate nitrogen atom occurs during C-H bond cleavage, in contrast to the expectation for the nucleophilic mechanism of Scheme 2C (90). Ab initio calculations of the transition states and energetics of the different mechanisms in Scheme 2 showed that the magnitudes of the isotope effects measured for TMO are only consistent with a hydride transfer mechanism (91).
Lysine-Specific Demethylase
Lysine-specific demethylase 1 (LSD1) removes methyl groups from lysyl residues in the N-terminal tail of histone H3 (92, 93) and the tumor suppressor protein p53 (94) by catalyzing oxidation of the carbon-nitrogen bond between the methyl group and the epsilon amine of the lysine, forming an imine intermediate that is nonenzymatically hydrolyzed to produce formaldehyde and the demethylated lysine (Scheme 8). The enzyme plays a role in epigenetic regulation of gene expression in cells, modulating cellular activities including growth and differentiation (95–98). Altered expression of LSD1 has been correlated with proliferation of neuroblastoma (99) and prostate cancer (100). Nonselective MAO inhibitors, such as tranylcypromine and propargylamine, as well as more specific mechanism-based inhibitors, inactivate LSD1 (101–106).
Scheme 8.

The structure of LSD1 (Figure 1) has been solved with and without peptide substrates and in the presence of inhibitors (102, 103, 107, 108). The amine oxidase domain of LSD1, which is homologous to the other MAO family members (66, 107), is divided by an insert, called the tower domain, that protrudes out of the core of LSD1 and interacts with CoREST, a regulatory protein (Figure 1D) (107, 108). LSD1 also contains an N-terminal SWIRM domain with affinity for DNA (109). LSD1 has a large substrate-binding pocket that accommodates the N-terminal tail of the histone, which appears to fold inside the substrate-binding cavity, forming several interactions with active-site residues (103, 107, 110). LSD1 is very specific in its histone demethylase activity, exclusively demethylating lysine 4 of histone H3 in vitro, with activity towards lysine 9 of histone H3 reported only in the presence of the androgen receptor (111). The interactions between substrate and enzyme position the fourth lysine of the peptide substrate directly in front of the flavin cofactor (107). LSD1 also contains the conserved active site lysine (Lys661) (Figure 2D) (112), and mutation of this residue to alanine eliminates LSD1 activity (110). LSD1 lacks the “aromatic cage” of MAO. In LSD1, Tyr761 corresponds to one of the aromatic residues in MAO; however, a threonine replaces the other (113).
Mechanistic studies with LSD1 are more difficult, due to the complexity of the substrate; thus, kinetic studies have been carried out with peptide substrates corresponding to the N-terminal tail of histone H3. The turnover rate for LSD1 is about 200 to 1000 times slower than that for other amine oxidases (113). Due to its regulatory activities, LSD1 may have evolved for increased specificity, not optimum activity. C-H bond cleavage is the rate-limiting step in the oxidation of a 21-mer peptide by LSD1 (114). Just as with the other members of the family, stopped-flow studies failed to show evidence of intermediates between oxidized and reduced enzyme during oxidation of the peptide substrate by LSD1. Based on these results and analyses of kinetic isotope effects on LSD1 kinetics, it was concluded that the mechanism of amine oxidation by LSD1 is hydride transfer (114).
Expert Opinion
Because of its physiological importance, the mechanism and structure of monoamine oxidase has been subject to intense study for decades. In recent years it has become apparent that enzymes with similar structures catalyze the oxidation of other amine substrates, presumably using a common mechanism. The monoamine oxidase family members share structural features, including a conserved FAD-binding domain and a lysine-water-flavin triad. The substrate-binding sites, however, reflect the different substrates. In each case, there is evidence that the deprotonated amine is the functional substrate. While, nucleophilic and radical mechanisms have been proposed for oxidation of amines by MAO, the accumulation of structural and mechanistic evidence supports a common hydride transfer mechanism for all members of the MAO family.
Outlook
The last decade has seen major advances in our understanding of the MAO family of flavoproteins. Future studies are likely to focus on obtaining additional structural information and development of specific inhibitors of the different family members. These efforts will complement continuing studies of the mechanism of these enzymes and provide insight into the structural bases for the differences in specificity of the family members. The growing interest in the mechanism by which flavoprotein oxidases catalyze the reaction of the reduced flavin with oxygen will likely lead to studies of the oxidative half-reaction of the MAO family.
Highlights.
The monoamine oxidase family of flavoenzymes catalyze the oxidation of primary and secondary amines, polyamines and amino acids.
Despite low levels of sequence identity, structures of the MAO family are homologous, with a conserved FAD-binding site and a novel lysine-water-flavin interaction.
The substrate binding sites of family members are quite diverse, corresponding to the varied structures of the substrates.
Mechanistic studies support a common hydride transfer mechanism for all members of the MAO family.
Acknowledgements
The research described here from the authors’ laboratory was supported in part by NIH grants R01 GM058698 and F32 GM097762.
Abbreviations
- MAO
monoamine oxidase
- PHBH
p-hydroxybenzoate hydroxylase
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- QSAR
quantitative structure-activity relation
- PAO
polyamine oxidase
- SMO
spermine oxidase
- LAAO
L-amino acid oxidase
- DAAO
D-amino acid oxidase
- TMO
tryptophan monooxygenase
- LSD
lysine-specific demethylase
References
- 1.Wierenga RK, de Jong RJ, Kalk KH, Hol WGJ, Drenth J. Crystal structure of p-hydroxybenzoate hydroxylase. J. Mol. Biol. 1979;131:55–73. doi: 10.1016/0022-2836(79)90301-2. [DOI] [PubMed] [Google Scholar]
- 2.Schreuder HA, van der Laan JM, Hol WGJ, Drenth J. Crystal structure of p-hydroxybenzoate hydroxylase complexed with its reaction product 3,4-dihydroxybenzoate. J.Mol.Biol. 1988;199:637–648. doi: 10.1016/0022-2836(88)90307-5. [DOI] [PubMed] [Google Scholar]
- 3.Silverman RB, Hoffman SJ, Catus WB. A mechanism for mitochondrial monoamine oxidase catalyzed amine oxidation. J. Am. Chem. Soc. 1980;102:7126–7128. [Google Scholar]
- 4.Edmondson DE. Aminium cation radical mechanism proposed for monoamine oxidase B catalysis: are there alternatives? Xenobiotica. 1995;25:735–753. doi: 10.3109/00498259509061889. [DOI] [PubMed] [Google Scholar]
- 5.Kim J-M, Bogdan MA, Mariano PS. Mechanistic analysis of the 3-methyllumiflavin-promoted oxidative deamination of benzylamine. A potential model for monoamine oxidase catalysis. J.Am.Chem.Soc. 1993;115:10591–10595. [Google Scholar]
- 6.Fitzpatrick PF. Oxidation of amines by flavoproteins. Arch. Biochem. Biophys. 2010;493:13–25. doi: 10.1016/j.abb.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker MC, Edmondson DE. Structure-activity relationships in the oxidation of benzylamine analogues by bovine liver mitochondrial monoamine oxidase B. Biochemistry. 1994;33:7088–7098. doi: 10.1021/bi00189a011. [DOI] [PubMed] [Google Scholar]
- 8.Edmondson DE, Bhattacharyya AK, Walker MC. Spectral and kinetic studies of imine product formation in the oxidation of p-(N,N-dimethylamino) benzylamine analogues by monoamine oxidase B. Biochemistry. 1993;32:5196–5202. doi: 10.1021/bi00070a031. [DOI] [PubMed] [Google Scholar]
- 9.Bach RD, Andres JL, Su M-D, McDouall JJW. Theoretical model for electrophilic oxygen atom insertion into hydrocarbons. J. Am. Chem. Soc. 1993;115:5768–5775. [Google Scholar]
- 10.Fowler CJ, Benedetti MS. The Metabolism of Dopamine by Both Forms of Monoamine Oxidase in the Rat Brain and Its Inhibition by Cimoxatone. J. Neurochem. 1983;40:1534–1541. doi: 10.1111/j.1471-4159.1983.tb08123.x. [DOI] [PubMed] [Google Scholar]
- 11.McCauley R, Racker E. Separation of two monoamine oxidases from bovine bran. Mol. Cell. Biochem. 1973;1:73–81. doi: 10.1007/BF01659940. [DOI] [PubMed] [Google Scholar]
- 12.Yang H-TT, Neff NH. β-Phenethylamine: a specific substrate for type B monoamine oxidase of brain. J. Pharmacol. Exp. Ther. 1973;187:365–371. [PubMed] [Google Scholar]
- 13.Hall DWR, Logan BW, Parsons GH. Further studies on the inhibition of monoamine oxidase by M & B 9302 (clorgyline)--I: Substrate specificity in various mammalian species. Biochem. Pharmacol. 1969;18:1447–1454. doi: 10.1016/0006-2952(69)90258-5. [DOI] [PubMed] [Google Scholar]
- 14.Chiba K, Trevor A, Castagnoli N. Metabolism of the neurotoxic tertiary amine, MPTP, by brain monoamine oxidase. Biochem. Biophys. Res. Commun. 1984;120:574–578. doi: 10.1016/0006-291x(84)91293-2. [DOI] [PubMed] [Google Scholar]
- 15.Cesura AM, Pletscher A. The new generation of monoamine oxidase inhibitors. Progress in Drug Research. 1992;38:171–297. doi: 10.1007/978-3-0348-7141-9_3. [DOI] [PubMed] [Google Scholar]
- 16.Youdim MBH, Edmondson D, Tipton KF. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006;7:295–309. doi: 10.1038/nrn1883. [DOI] [PubMed] [Google Scholar]
- 17.Ma J, Yoshimura M, Yamashita E, Nakagawa A, Ito A, Tsukihara T. Structure of rat monoamine oxidase A and its specific recognitions for substrates and inhibitors. J. Mol. Biol. 2004;338:103–114. doi: 10.1016/j.jmb.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 18.Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002;9:22–26. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- 19.Upadhyay AK, Borbat PP, Wang J, Freed JH, Edmondson DE. Determination of the Oligomeric States of Human and Rat Monoamine Oxidases in the Outer Mitochondrial Membrane and Octyl β-d-Glucopyranoside Micelles Using Pulsed Dipolar Electron Spin Resonance Spectroscopy. Biochemistry. 2008;47:1554–1566. doi: 10.1021/bi7021377. [DOI] [PubMed] [Google Scholar]
- 20.De Colibus L, Li M, Binda C, Lustig A, Edmondson DE, Mattevi A. Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. Proc. Nat. Acad. Sci. USA. 2005;102:12684–12689. doi: 10.1073/pnas.0505975102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rebrin I, Geha RM, Chen K, Shih JC. Effects of Carboxyl-terminal Truncations on the Activity and Solubility of Human Monoamine Oxidase B. J. Biol. Chem. 2001;276:29499–29506. doi: 10.1074/jbc.M100431200. [DOI] [PubMed] [Google Scholar]
- 22.Binda C, Li M, Hubalek F, Restelli N, Edmondson D, Mattevi A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. USA. 2003;100:9750–9755. doi: 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nandigama RK, Miller JR, Edmondson D. Loss of serotonin oxidation as a component of the altered substrate specificity in the Y444F mutant of recombinant human liver MAO A. Biochemistry. 2001;40:14839–14846. doi: 10.1021/bi011113d. [DOI] [PubMed] [Google Scholar]
- 24.Li M, Binda C, Mattevi A, Edmondson DE. Functional role of the "aromatic cage" in human monoamine oxidase B: structures and catalytic properties of Tyr435 mutant proteins. Biochemistry. 2006;45:4775–4784. doi: 10.1021/bi051847g. [DOI] [PubMed] [Google Scholar]
- 25.Geha RM, Chen K, Wouters J, Ooms F, Shih JC. Analysis of conserved active site residues in monoamine oxidase A and B and their three-dimensional molecular modeling. J. Biol. Chem. 2002;277:17209–17216. doi: 10.1074/jbc.M110920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alieva I, Mustafayeva N, Gojayev N. Conformation analysis of the N-terminal sequence Met1-Val60 of the tyrosine hydroxylase. J. Mol. Struct. 2006;785:76–84. [Google Scholar]
- 27.Binda C, Wang J, Li M, Hubalek F, Mattevi A, Edmondson DE. Structural and Mechanistic Studies of Arylalkylhydrazine Inhibition of Human Monoamine Oxidases A and B. Biochemistry. 2008;47:5616–5625. doi: 10.1021/bi8002814. [DOI] [PubMed] [Google Scholar]
- 28.Edmondson DE, Binda C, Mattevi A. Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Arch. Biochem. Biophys. 2007;464:269–276. doi: 10.1016/j.abb.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hubálek F, Binda C, Khalil A, Li M, Mattevi A, Castagnoli N, Edmondson DE. Demonstration of Isoleucine 199 as a Structural Determinant for the Selective Inhibition of Human Monoamine Oxidase B by Specific Reversible Inhibitors. J. Biol. Chem. 2005;280:15761–15766. doi: 10.1074/jbc.M500949200. [DOI] [PubMed] [Google Scholar]
- 30.Son S-Y, Ma J, Kondou Y, Yoshimura M, Yamashita E, Tsukihara T. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Nat. Acad. Sci. USA. 2008;105:5739–5744. doi: 10.1073/pnas.0710626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller JR, Edmondson DE. Structure-activity relationships in the oxidation of para-substituted benzylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry. 1999;38:13670–13683. doi: 10.1021/bi990920y. [DOI] [PubMed] [Google Scholar]
- 32.Dunn RV, Marshall KR, Munro AW, Scrutton NS. The pH dependence of kinetic isotope effects in monoamine oxidase A indicates stabilization of the neutral amine in the enzyme-substrate complex. FEBS Journal. 2008;275:3850–3858. doi: 10.1111/j.1742-4658.2008.06532.x. [DOI] [PubMed] [Google Scholar]
- 33.Nandigama RK, Edmondson DE. Structure-activity relations in the oxidation of phenethylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry. 2000;39:15258–15265. doi: 10.1021/bi001957h. [DOI] [PubMed] [Google Scholar]
- 34.Pretorius A, Ogunrombi MO, Terre'Blanche G, Castagnoli N, Jr, Bergh JJ, Petzer JP. Deuterium isotope effects for the oxidation of 1-methyl-3-phenyl-3-pyrrolinyl analogues by monoamine oxidase B. Biorg. Med. Chem. 2008;16:8813–8817. doi: 10.1016/j.bmc.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 35.Silverman RB. Effect of α-methylation on inactivation of monoamine oxidase by N-cyclopropylbenzylamine. Biochemistry. 1984;23:5206–5213. doi: 10.1021/bi00317a019. [DOI] [PubMed] [Google Scholar]
- 36.Silverman RB, Zieske PA. Mechanism of inactivation of monoamine oxidase by 1-phenylcyclopropylamine. Biochemistry. 1985;24:2128–2138. doi: 10.1021/bi00330a005. [DOI] [PubMed] [Google Scholar]
- 37.Silverman RB, Hoffman SJ. Mechanism of inactivation of mitochondrial monoamine oxidase by N-cyclopropyl-N-arylalkyl amines. J. Am. Chem. Soc. 1980;102:884–886. [Google Scholar]
- 38.Yue KT, Bhattacharyya AK, Zhelyaskov VR, Edmondson DE. Resonance Raman Spectroscopic Evidence for an Anionic Flavin Semiquinone in Bovine Liver Monoamine Oxidase. Arch. Biochem. Biophys. 1993;300:178–185. doi: 10.1006/abbi.1993.1025. [DOI] [PubMed] [Google Scholar]
- 39.DeRose VJ, Woo JCG, Hawe WP, Hoffman BM, Silverman RB, Yelekci K. Observation of a flavin semiquinone in the resting state of monoamine oxidase B by electron paramagnetic resonance and electron nuclear double resonance spectroscopy. Biochemistry. 1996;35:11085–11091. doi: 10.1021/bi960749f. [DOI] [PubMed] [Google Scholar]
- 40.Sablin SO, Ramsay RR. Substrates but not inhibitors alter the redox potentials of monoamine oxidases. Antioxid. Redox Signal. 2001;3:723–729. doi: 10.1089/15230860152664920. [DOI] [PubMed] [Google Scholar]
- 41.Rigby SE, Hynson RM, Ramsay RR, Munro AW, Scrutton NS. A stable tyrosyl radical in monoamine oxidase A. J. Biol. Chem. 2005;280:4627–4631. doi: 10.1074/jbc.M410596200. [DOI] [PubMed] [Google Scholar]
- 42.Royo M, Fitzpatrick PF. Mechanistic studies of mouse polyamine oxidase with N1,N12-bisethylspermine as a substrate. Biochemistry. 2005;44:7079–7084. doi: 10.1021/bi050347k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaffe HH. Theoretical Considerations Concerning Hammett's Equation. IV. Calculation of ρ-Values. J. Chem. Phys. 1953;21:415–419. [Google Scholar]
- 44.Peters FB, Johnson HW., Jr Ionization constants of substituted 2-aminoacetanilides and benzylamines. Transmission of electronic effects through amide links. J. Org. Chem. 1975;40:1517–1519. [Google Scholar]
- 45.Eberlein G, Bruice TC. The chemistry of a 1,5-diblocked flavin. 2. Proton and electron transfer steps in the reaction of dihydroflavins with oxygen. J. Am. Chem. Soc. 1983;105:6685–6697. [Google Scholar]
- 46.Massey V. Activation of molecular oxygen by flavins and flavoproteins. J.Biol.Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- 47.Roth JP, Klinman JP. Catalysis of electron transfer during the activation of O2 by the flavoprotein glucose oxidase. Proc. Natl. Acad. Sci. USA. 2003;100:62–67. doi: 10.1073/pnas.252644599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jorns MS, Chen Z-w, Mathews FS. Structural Characterization of Mutations at the Oxygen Activation Site in Monomeric Sarcosine Oxidase. Biochemistry. 2010 doi: 10.1021/bi100160j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao G, Bruckner RC, Jorns MS. Identification of the Oxygen Activation Site in Monomeric Sarcosine Oxidase: Role of Lys265 in Catalysis. Biochemistry. 2008;47:9124–9135. doi: 10.1021/bi8008642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabor CW, Tabor H. Polyamines. Ann. Rev. Biochem. 1984;53:749–790. doi: 10.1146/annurev.bi.53.070184.003533. [DOI] [PubMed] [Google Scholar]
- 51.Hu RH, Pegg AE. Rapid induction of apoptosis by deregulated uptake of polyamine analogues. Biochem. J. 1997;328:307–316. doi: 10.1042/bj3280307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Casero RA, Woster PM. Recent Advances in the Development of Polyamine Analogues as Antitumor Agents. J. Med. Chem. 2009;52:4551–4573. doi: 10.1021/jm900187v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Porter CW. Effect of polyamine analogues and inhibition of polyamine oxidase on spermidine/spermine N1-acetyltransferase activity and cell proliferation. Anticancer Res. 1986;6:525–542. [Google Scholar]
- 54.Marton LJ, Pegg AE. Polyamines as targets for therapeutic intervention. Ann.Rev.Pharmacol.Toxicol. 1995;35:55–91. doi: 10.1146/annurev.pa.35.040195.000415. [DOI] [PubMed] [Google Scholar]
- 55.Holtta E. Oxidation of spermidine and spermine in rat liver: purification and properties of polyamine oxidase. Biochemistry. 1977;16:91–100. doi: 10.1021/bi00620a015. [DOI] [PubMed] [Google Scholar]
- 56.Wu T, Yankovskaya V, McIntire WS. Cloning, sequencing, and heterologous expression of the murine peroxisomal flavoprotein, N1-acetylated polyamine oxidase. J. Biol. Chem. 2003;278:20514–20525. doi: 10.1074/jbc.M302149200. [DOI] [PubMed] [Google Scholar]
- 57.Federico R, Ercolini L, Laurenzi M, Angelini R. Oxidation of acetylpolyamines by maize polyamine oxidase. Phytochemistry. 1996;43:339–341. [Google Scholar]
- 58.Kim BG. Polyamine metabolism in Acanthamoeba: polyamine content and synthesis of ornithine, putrescine, and diaminopropane. J. Protozoology. 1987;34:728–784. doi: 10.1111/j.1550-7408.1987.tb03175.x. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, Murray-Stewart T, Devereux W, Hacker A, Frydman B, Woster PM, Casero RA., Jr Properties of purified recombinant human polyamine oxidase, PAOh1/SMO. Biochem. Biophys. Res. Commun. 2003;304:605–611. doi: 10.1016/s0006-291x(03)00636-3. [DOI] [PubMed] [Google Scholar]
- 60.Cervelli M, Polticelli F, Federico R, Mariottini P. Heterologous expression and characterization of mouse spermine oxidase. J. Biol. Chem. 2003;278:5271–5276. doi: 10.1074/jbc.M207888200. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y, Devereux W, Woster PM, Stewart TM, Hacker A, Casero RA., Jr Cloning and characterization of a human polyamine oxidase that is inducible by polyamine analogue exposure. Cancer Res. 2001;61:5370–5373. [PubMed] [Google Scholar]
- 62.Pledgie A, Huang Y, Hacker A, Zhang Z, Woster PM, Davidson NE, Casero RA., Jr Spermine oxidase SMO(PAOh1), not N1-acetylpolyamine oxidase PAO, is the primary source of cytotoxic H2O2 in polyamine analogue-treated human breast cancer cell lines. J. Biol. Chem. 2005;280:39843–39851. doi: 10.1074/jbc.M508177200. [DOI] [PubMed] [Google Scholar]
- 63.Ha HC, Woster PM, Yager JD, Casero RA., Jr The role of polyamine catabolism in polyamine analogue-induced programmed cell death. Proc. Natl. Acad. Sci. U S A. 1997;94:11557–11562. doi: 10.1073/pnas.94.21.11557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Babbar N, Casero RA., Jr Tumor Necrosis Factor-α Increases Reactive Oxygen Species by Inducing Spermine Oxidase in Human Lung Epithelial Cells: A Potential Mechanism for Inflammation-Induced Carcinogenesis. Cancer Res. 2006;66:11125–11130. doi: 10.1158/0008-5472.CAN-06-3174. [DOI] [PubMed] [Google Scholar]
- 65.Tavladoraki P, Schininá ME, Cecconi F, Di Agostino S, Manera F, Rea G, Mariottini P, Federico R, Angelini R. Maize polyamine oxidase: primary structure from protein and cDNA sequencing. FEBS Lett. 1998;426:62–66. doi: 10.1016/s0014-5793(98)00311-1. [DOI] [PubMed] [Google Scholar]
- 66.Binda C, Coda A, Angelini R, Federico R, Ascenzi P, Mattevi A. A 30 Å long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Structure. 1999;7:265–276. doi: 10.1016/s0969-2126(99)80037-9. [DOI] [PubMed] [Google Scholar]
- 67.Binda C, Angelini R, Federico R, Ascenzi P, Mattevi A. Structural bases for inhibitor binding and catalysis in polyamine oxidase. Biochemistry. 2001;40:2766–2776. doi: 10.1021/bi002751j. [DOI] [PubMed] [Google Scholar]
- 68.Landry J, Sternglanz R. Yeast Fms1 is a FAD-utilizing polyamine oxidase. Biochem. Biophys. Res. Commun. 2003;303:771–776. doi: 10.1016/s0006-291x(03)00416-9. [DOI] [PubMed] [Google Scholar]
- 69.Huang Q, Liu Q, Hao Q. Crystal structures of Fms1 and its complex with spermine reveal substrate specificity. J. Mol. Biol. 2005;348:951–959. doi: 10.1016/j.jmb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 70.Bianchi M, Polticelli F, Ascenzi P, Botta M, Federico R, Mariottini P, Cona A. Inhibition of polyamine and spermine oxidases by polyamine analogues. FEBS. J. 2006;273:1115–1123. doi: 10.1111/j.1742-4658.2006.05137.x. [DOI] [PubMed] [Google Scholar]
- 71.Adachi MS, Torres JM, Fitzpatrick PF. Mechanistic Studies of the Yeast Polyamine Oxidase Fms1: Kinetic Mechanism, Substrate Specificity, and pH Dependence. Biochemistry. 2010;49:10440–10448. doi: 10.1021/bi1016099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adachi MS, Juarez PR, Fitzpatrick PF. Mechanistic Studies of Human Spermine Oxidase: Kinetic Mechanism and pH Effects. Biochemistry. 2010;49:386–392. doi: 10.1021/bi9017945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Henderson Pozzi M, Gawandi V, Fitzpatrick PF. pH Dependence of a Mammalian Polyamine Oxidase: Insights into Substrate Specificity and the Role of Lysine 315. Biochemistry. 2009;48:1508–1516. doi: 10.1021/bi802227m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Henderson Pozzi M, Gawandi V, Fitzpatrick PF. Mechanistic Studies of para-Substituted N,N,-Dibenzyl-1,4-diaminobutanes as Substrates for a Mammalian Polyamine Oxidase. Biochemistry. 2009;48:12305–12313. doi: 10.1021/bi901694s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Polticelli F, Basran J, Faso C, Cona A, Minervini G, Angelini R, Federico R, Scrutton NS, Tavladoraki P. Lys300 plays a major role in the catalytic mechanism of maize polyamine oxidase. Biochemistry. 2005;44:16108–16120. doi: 10.1021/bi050983i. [DOI] [PubMed] [Google Scholar]
- 76.Henderson Pozzi M, Fitzpatrick PF. A lysine conserved in the monoamine oxidase family is involved in oxidation of the reduced flavin in mouse polyamine oxidase. Arch. Biochem. Biophys. 2010;498:83–88. doi: 10.1016/j.abb.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pawelek PD, Cheah J, Coulombe R, Macheroux P, Ghisla S, Vrielink A. The structure of L-amino acid oxidase reveals the substrate trajectory into an enantiomerically conserved active site. EMBO J. 2000;19:4204–4215. doi: 10.1093/emboj/19.16.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moustafa IM, Foster S, Lyubimov AY, Vrielink A. Crystal structure of LAAO from Calloselasma rhodostoma with an L-phenylalanine substrate: insights into structure and mechanism. J. Mol. Biol. 2006;364:991–1002. doi: 10.1016/j.jmb.2006.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Faust A, Niefind K, Hummel W, Schomburg D. The structure of a bacterial L-amino acid oxidase from Rhodococcus opacus gives new evidence for the hydride mechanism for dehydrogenation. J. Mol. Biol. 2007;367:234–248. doi: 10.1016/j.jmb.2006.11.071. [DOI] [PubMed] [Google Scholar]
- 80.Curti B, Ronchi S, Simonetta MP. D- and L-amino acid oxidases. In: Müller F, editor. Chemistry and Biochemistry of Flavoenzymes. Vol III. Boca Raton: CRC Press; 1991. pp. 69–94. [Google Scholar]
- 81.Fitzpatrick PF. Substrate dehydrogenation by flavoproteins. Acc. Chem. Res. 2001;34:299–307. doi: 10.1021/ar0000511. [DOI] [PubMed] [Google Scholar]
- 82.Fitzpatrick PF. Carbanion versus hydride transfer mechanisms in flavoprotein-catalyzed dehydrogenations. Bioorg. Chem. 2004;32:125–139. doi: 10.1016/j.bioorg.2003.02.001. [DOI] [PubMed] [Google Scholar]
- 83.Magie AR, Wilson EE, Kosuge T. Indoleacetamide as an intermediate in the synthesis of indoleacetic acid in Pseudomonas savastanoi. Science. 1963;141:1281–1282. doi: 10.1126/science.141.3587.1281. [DOI] [PubMed] [Google Scholar]
- 84.Thomashow MF, Hugly S, Buchholz WG, Thomashow LS. Molecular basis for the auxin-independent phenotype of crown gall tumor tissues. Science. 1986;231:616–618. doi: 10.1126/science.3511528. [DOI] [PubMed] [Google Scholar]
- 85.Sobrado P, Fitzpatrick PF. Analysis of the roles of amino acid residues in the flavoprotein tryptophan 2-monooxygenase modified by 2-oxo-3-pentynoate: characterization of His338, Cys339, and Cys511 mutant enzymes. Arch.Biochem.Biophys. 2002;402:24–30. doi: 10.1016/S0003-9861(02)00063-2. [DOI] [PubMed] [Google Scholar]
- 86.Sobrado P, Fitzpatrick P. Analysis of the role of the active site residue Arg98 in the flavoprotein tryptophan 2-monooxygenase, a member of the L-amino oxidase family. Biochemistry. 2003;42:13826–13832. doi: 10.1021/bi035299n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sobrado P, Fitzpatrick PF. Identification of Tyr413 as an active site residue in the flavoprotein tryptophan 2-monooxygenase and analysis of its contribution to catalysis. Biochemistry. 2003;42:13833–13838. doi: 10.1021/bi035300i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Emanuele JJ, Jr, Fitzpatrick PF. Mechanistic studies of the flavoprotein tryptophan 2-monooxygenase. 1. Kinetic mechanism. Biochemistry. 1995;34:3710–3715. doi: 10.1021/bi00011a028. [DOI] [PubMed] [Google Scholar]
- 89.Emanuele JJ, Jr, Fitzpatrick PF. Mechanistic studies of the flavoprotein tryptophan 2-monooxygenase. 2. pH and kinetic isotope effects. Biochemistry. 1995;34:3716–3723. doi: 10.1021/bi00011a029. [DOI] [PubMed] [Google Scholar]
- 90.Ralph EC, Anderson MA, Cleland WW, Fitzpatrick PF. Mechanistic studies of the flavoenzyme tryptophan 2-monooxygenase: Deuterium and 15N kinetic isotope effects on alanine oxidation by an L-amino acid oxidase. Biochemistry. 2006;45:15844–15852. doi: 10.1021/bi061894o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ralph EC, Hirschi JS, Anderson MA, Cleland WW, Singleton DA, Fitzpatrick PF. Insights into the mechanism of flavoprotein-catalyzed amine oxidation from nitrogen isotope effects on the reaction of N-methyltryptophan oxidase. Biochemistry. 2007;46:7655–7664. doi: 10.1021/bi700482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 93.Forneris F, Binda C, Vanoni MA, Mattevi A, Battaglioli E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005;579:2203–2207. doi: 10.1016/j.febslet.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 94.Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, Berger SL. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- 95.Zhu X, Wang J, Ju BG, Rosenfeld MG. Signaling and epigenetic regulation of pituitary development. Curr. Opin. Cell Biol. 2007;19:605–611. doi: 10.1016/j.ceb.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Su ST, Ying HY, Chiu YK, Lin FR, Chen MY, Lin KI. Involvement of histone demethylase LSD1 in Blimp-1-mediated gene repression during plasma cell differentiation. Mol. Cell Biol. 2009;29:1421–1431. doi: 10.1128/MCB.01158-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic Regulation of Hematopoietic Differentiation by Gfi-1 and Gfi-1b Is Mediated by the Cofactors CoREST and LSD1. Mol. Cell. 2007;27:562–572. doi: 10.1016/j.molcel.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 98.Hakimi M-A, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R. A core–BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc. Natl. Acad. Sci. U S A. 2002;99:7420–7425. doi: 10.1073/pnas.112008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, Metzger E, Schule R, Eggert A, Buettner R, Kirfel J. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69:2065–2071. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 100.Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J, Metzger E, Schule R, Buettner R. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006;66:11341–11347. doi: 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- 101.Culhane JC, Szewczuk LM, Liu X, Da G, Marmorstein R, Cole PA. A mechanism-based inactivator for histone demethylase LSD1. J. Am. Chem. Soc. 2006;128:4536–4537. doi: 10.1021/ja0602748. [DOI] [PubMed] [Google Scholar]
- 102.Szewczuk LM, Culhane JC, Yang M, Majumdar A, Yu H, Cole PA. Mechanistic analysis of a suicide inactivator of histone demethylase LSD1. Biochemistry. 2007;46:6892–6902. doi: 10.1021/bi700414b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, Cole PA, Yu H. Structural Basis for the Inhibition of the LSD1 Histone Demethylase by the Antidepressant trans-2-Phenylcyclopropylamine. Biochemistry. 2007;46:8058–8065. doi: 10.1021/bi700664y. [DOI] [PubMed] [Google Scholar]
- 104.Gooden DM, Schmidt DMZ, Pollock JA, Kabadi AM, McCafferty DG. Facile synthesis of substituted trans-2-arylcyclopropylamine inhibitors of the human histone demethylase LSD1 and monoamine oxidases A and B. Bioorg. Med. Chem. Lett. 2008;18:3047–3051. doi: 10.1016/j.bmcl.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schmidt DMZ, McCafferty DG. trans-2-Phenylcyclopropylamine Is a Mechanism-Based Inactivator of the Histone Demethylase LSD1. Biochemistry. 2007;46:4408–4416. doi: 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- 106.Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 Lysine 4 Demethylation Is a Target of Nonselective Antidepressive Medications. Chem. Biol. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 107.Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. Structural Basis of LSD1-CoREST Selectivity in Histone H3 Recognition. J. Biol. Chem. 2007;282:20070–20074. doi: 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
- 108.Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1) Proc. Natl. Acad. Sci. USA. 2006;103:13956–13961. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Da G, Lenkart J, Zhao K, Shiekhattar R, Cairns BR, Marmorstein R. Structure and function of the SWIRM domain, a conserved protein module found in chromatin regulatory complexes. Proc. Natl. Acad. Sci. U S A. 2006;103 doi: 10.1073/pnas.0510949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Stavropoulos P, Blobel G, Hoelz A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat. Struct. Mol. Biol. 2006;13:626–632. doi: 10.1038/nsmb1113. [DOI] [PubMed] [Google Scholar]
- 111.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AHFM, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 112.Mimasu S, Sengoku T, Fukuzawa S, Umehara T, Yokoyama S. Crystal structure of histone demethylase LSD1 and tranylcypromine at 2.25 Å. Biochem. Biophys. Res. Commun. 2008;366:15–22. doi: 10.1016/j.bbrc.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 113.Forneris F, Battaglioli E, Mattevi A, Binda C. New roles of flavoproteins in molecular cell biology: Histone demethylase LSD1 and chromatin. FEBS Journal. 2009;276:4304–4312. doi: 10.1111/j.1742-4658.2009.07142.x. [DOI] [PubMed] [Google Scholar]
- 114.Gaweska H, Henderson Pozzi M, Schmidt DMZ, McCafferty DG, Fitzpatrick PF. Use of pH and Kinetic Isotope Effects to Establish Chemistry As Rate-Limiting in Oxidation of a Peptide Substrate by LSD1. Biochemistry. 2009;48:5440–5445. doi: 10.1021/bi900499w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]