

Abstract
The syntheses of the natural product anibamine and its three olefin isomers have been achieved concisely and efficiently via highly regio- and stereo-selective reactions. The crucial steps included a regio-selective palladium-catalyzed alkynylation by Sonogashira coupling and a stereo-selective Suzuki coupling. Further conformation analyses and in vitro calcium mobilization studies were carried out to characterize the compounds' biological properties.
Introduction
Anibamine, a pyridine quarternary alkaloid isolated from Aniba panurensis,1,2 has been identified as a chemokine receptor CCR5 antagonist with an IC50 of 1 μM in inhibiting the binding of 125I-gp120 to the CCR5 receptor. Anibamine has a novel structural skeleton compared with other small molecule CCR5 antagonists identified through high-throughput screenings, such as Vicriviroc,3 Maraviroc,4 and TAK-7795 (Figure 1), all of which have been studied in anti-HIV therapeutics development.6 Recent studies7 also demonstrate that anibamine inhibits prostate cancer cell proliferation, adhesion, and invasion at micromolar to submicromolar levels. Preliminary in vivo studies further indicate that anibamine also inhibits prostate tumor growth in mice. Correspondingly, it has been found that the chemokine CCL5 and its receptor CCR5 may play important roles in cancer progression8 and TAK-779 blocked CCL5-induced invasion and proliferation of prostate cancer cells.9 Therefore, anibamine may serve as a new lead for the development of CCR5 antagonists with potential applications in both AIDS and cancer therapies.
Figure 1. Anibamine and some known CCR5 antagonists.

The first total synthesis of anibamine has been recently reported by our group.10 In this approach, the two ten-carbon side chains were introduced via the Wittig reagent of 1-bromononane and the 3,5-dialdehyde pyridine intermediate. Four isomers were obtained as a mixture from this reaction and were used in the next few steps without separation. At the last step, anibamine was isolated by preparative HPLC with 8% overall yield (10 steps). This synthetic route suffered from tedious separations in the last few steps and relatively low yield due to the limited stereo-selectivity of the Wittig reaction. At that time, the difficulties in obtaining the other isomers of anibamine (2, 3 and 4, Figure 2) prevented us from studying the effects of the double bond configuration and resulting conformation on the biological activities of the molecules. Here we report new, highly regio- and stereo-selective syntheses of anibamine and its three olefin isomers in a concise and efficient manner.
Figure 2. Anibamine and its three isomers.

The retrosynthetic analysis of anibamine is depicted in Scheme 1. In this route the two double bonds will be introduced stereo-selectively by adopting different Suzuki coupling conditions while a regio-selective Sonogashira coupling will be applied to introduce the side chain at the 2-position and form the five-member ring system.
Scheme 1. The Retrosynthetic Analysis of Anibamine.

Results and Discussion
The synthesis was initiated (Scheme 2) from commercially available 2-amino-4,6-dimethylpyridine that was brominated in glacial acetic acid to give the desired 3,5-dibromo-4,6-dimethyl-2-aminopyridine (5) in 75% yield.11 Diazotization of 5 under routine condition with Br2/NaNO2 in 48% HBr as solvent did not yield the desired product 6 due to the poor solubility of the starting material. Co-solvents such as DMSO12 or 1,4-dioxane13 were added with limited success. However, a modified Sandmeyer reaction14 by treating 5 with tert-butyl nitrite/CuBr2 in CHBr3 as the solvent, obtained the desired product 6 in a reasonable yield. The reaction was further improved to a yield of 88% by replacing CuBr2 with Br2.
Scheme 2. Preparation of Intermediate 7.

The regio-selective synthesis of the key intermediate 2-alkynyl-3,5-dibromo-4,6-dimethylpyridine (7) was achieved by reacting 6 with terminal acetylenes under palladium-catalyzed Sonogashira conditions15 with 91% yield (Scheme 2). In order to ascertain the regio-chemistry of the coupling product 7, it was hydrogenated over palladium/carbon to give 8. Simultaneously, 8 was prepared by another method starting with 2-amino-4,6-dimethylpyridine. The corresponding 2-bromopyridine (9) was obtained in a reasonable yield by diazotization in a bromine and 48% hydrobromic acid mixture.16 Further treatment of 9 with propargyl alcohol under palladium-catalyzed Sonogashira conditions gave the alkynylated pyridine 10 in 80% yield. Catalytic hydrogenation of the triple bond15a afforded the pyridylpropanol, whose NMR spectra were unequivocally consistent with the product 8. Thus, the regio-selective synthesis of 7 via a Sonogashira coupling condition was confirmed.
Next, the alkynyl bond of 7 was chemo-selectively reduced while minimizing the possible reductive debromination product.15a The hydrogenation of 7 over platinum oxide in ethanol was stopped after 2 equivalents of hydrogen were consumed yielding the key intermediate 3,5-dibromo-2-substituted-4,6-dimethylpyridine (11) in 73% yield (Scheme 3).
Scheme 3. The Synthesis of Anibamine.

To stereo-selectively introduce the two aliphatic side chains onto positions 3 and 5 of intermediate 11, excess organoborane reagent was used under Suzuki coupling conditions. Recent development17 on the utilization of alkyl- and alkenylboronic acids and esters suggested a convenient method for the preparation of diisopropyl (Z)-1-decenylboronate (12). Under standard Suzuki reaction conditions18 1.0 equiv of 11 and 4.0 equiv of 12 were coupled in the presence of Pd(OAc)2, PPh3 and Na2CO3 in a mixture of solvents (toluene : EtOH : water = 2 : 1 : 1) at refluxing temperature for 6h. Compound 13 was obtained stereo-selectively in an excellent yield. The crude 1H-NMR and 13C-NMR of the reaction showed no observable stereo-isomers.
Subsequently, the PMB group of 13 was removed under acidic conditions to give compound 14 in 80% yield. The ring-closure was achieved by treating 14 with methanesulfonylchloride and triethylamine at room temperature to give the natural product anibamine (1) in 71% yield (Scheme 3). The spectral properties of the synthesized target compound 1 were identical with those of the natural product.1
The (11E, 22E) isomer of anibamine (2) was prepared in a similar fashion to anibamine. (E)-dec-1-enylboronic acid (15) was readily obtained by the hydroboration of 1-decyne with BHBr2·SMe2, followed by hydrolysis.19 Under the same Suzuki cross-coupling reaction conditions, 1.0 equiv of 11 and 4.0 equiv of 15 were reacted to give the intermediate 16 stereo-selectively in an excellent yield. Then by following the same procedure described for 1, the (11E, 22E) isomer 2 was obtained in 64% yield via two steps from compound 16 (Scheme 4).
Scheme 4. The Synthesis of (11E, 22E)-Anibamine.

Both electronic and steric considerations suggest that the regio-selective introduction of the two side chains of isomers 3 and 4 would be challenging due to the similar reactivity of the two bromo-substitutions at the 3- and 5-positions of intermediate 11 toward coupling reactions. The electronic effects arising from substitutions on the pyridinyl ring system are similar and the steric hindrance effects of the bulky 2-position OPMB group would be diminished due to the relatively long aliphatic chain connection to the parent ring. Unsurprisingly, Suzuki coupling of 11 with an equivalent of 15 with varied temperatures and solvent systems gave products 18 and 19 in roughly similar amounts. The successful chromatographic separation of the two isomers, however, allowed the preparation of 3 and 4 in reasonably good yields (Scheme 5).
Scheme 5. The Synthesis of (11Z, 22E)-Anibamine and (11E, 22Z)-Anibamine.

In order to predict the influence of the double bond configuration of 1-4 with respect to interaction with the receptor CCR5, in silico conformation analyses were conducted. As shown in Figure 3, the conformations of the core portions of all four isomers were similar with varied, but somewhat kinked side chain orientations. Overall, however, their molecular shapes were quite similar and almost overlapped. To validate such results from the conformation analyses as if the binding affinity of these isomers to the receptor CCR5 would be influenced, further biological assay is deemed to be conducted.
Figure 3.
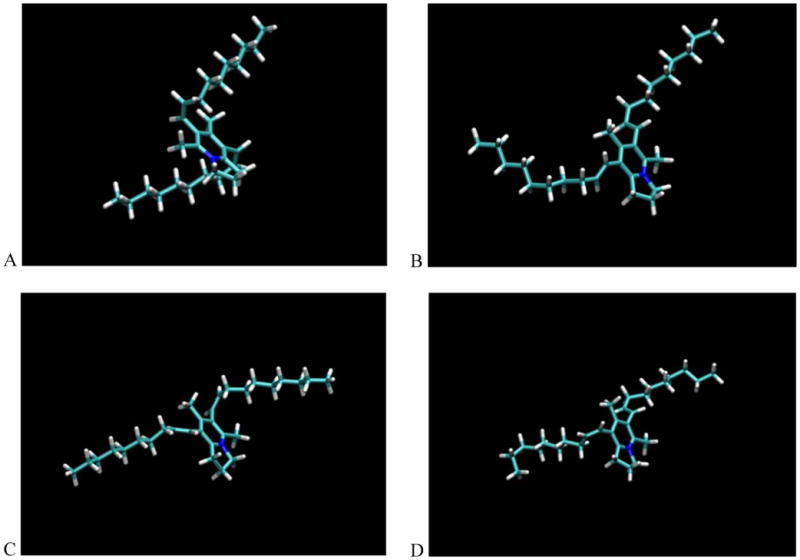
Conformation analyses of compound 1 (A), 2 (B), 3 (C), and 4 (D).
To experimentally characterize the influence of the double bond configuration of compound 1-4 on their affinity to the receptor, the calcium mobilization assay20 was adopted by using CCR5 MOLT-4 cell lines. The IC50 values of the calcium reflux inhibitory effect for all four isomers are summarized in Table 1. First, the comparable IC50 of anibamine (1) to its inhibitory binding affinity of RANTES at the receptor (1.0 μM) validated the calcium mobilization assay protocol. Second, not surprisingly, all four isomers showed similar inhibitory effect on calcium reflux, which indicates that their binding affinity, as well as possibly their binding mode to the receptor, is comparable. This supports the conformation analyses results: the double bond configuration does not significantly influence the overall conformation of the whole molecule because of the two long aliphatic chains.
Table 1.
The calcium mobilization inhibition assay results of all the four isomers.a
| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| IC50 | 5.43 | 6.53 | 9.23 | 10.09 |
| SEM | 0.91 | 1.79 | 0.59 | 3.87 |
Values shown were from at least three separate experiments performed in triplicate. The IC50 values were calculated using Prism.
Conclusion
In conclusion, the regio- and stereo-selective total synthesis of anibamine has been achieved in 7 steps with an overall yield of 23%, which is a three-fold improvement over previous results. Its three isomers were also obtained in similar yields. The methodology described above for the preparation of intermediate 7 and 11 will allow more convenient synthesis of a variety of anibamine derivatives for our further biological evaluations and structure-activity relationship studies. Conformational analyses of 1-4 are supported by the calcium mobilization screens, which together, suggest that the confirguration of the double bond may not critical for binding to the receptor. These results shed light on a possible conserved binding mode of these ligands to the receptor and will facilitate our next step molecular design in order to identify novel anibamine derivatives as the chemokine receptor CCR5 antagonists, and potential anti-HIV and anti-cancer agents.
Experimental Section
General information and methods
All reagents were used directly as obtained commercially unless otherwise noted. Melting points were determined on a melting point apparatus and are uncorrected. 1H NMR and 13C NMR spectra were taken on a 400 MHz spectrometer and tetramethylsilane was used as an internal standard. Infrared spectra were obtained on a FT-IR spectrometer. LC-MS was performed on a QTOF-2 instrument using ESI ion source. High-Resolution mass spectral analyses were performed on a QTOF-2 instrument using ESI ion source operated in positive ion mode. Column chromatography was performed on silica gel (grade 60 mesh). Preparative thin-layer chromatography (TLC) was performed on silica gel GF plates (1000 μm, 20 × 20 cm).
3,5-dibromo-4,6-dimethylpyridin-2-amine (5)
A solution of 2-amino-4,6-dimethylpyridine (2.44 g, 20 mmol) in 15 mL of glacial acetic acid under nitrogen was treated with a solution of 7.36 g (46 mmol) Br2 in 5 mL of glacial acetic acid over 15 minutes. The solution became a solid mass and was allowed to stand at r.t. for 1h. After cooling in an ice bath, the material was made alkaline with 20% cold NaOH solution. Then the mixture was extracted with CH2Cl2 (3×50 mL), the combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified on silica gel (Hexane-EtOAc, 2:1∼1:1) to afford the product (4.21 g, 75%) as a yellow solid. M.p. 136-137°C. (lit.11 M.p. 136-136.5°C). IR (KBr, cm-1) vmax: 3459, 3283, 3124, 1628, 1428, 659. 1H NMR (400 MHz, CDCl3) δ 4.86 (br, 2H), 2.53 (s, 3H), 2.49 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 153.9, 153.6, 147.0, 111.5, 103.9, 25.2, 24.3.
2,3,5-tribromo-4,6-dimethylpyridine (6)
To a solution of 5 (556 mg, 2 mmol) and Br2 (384 mg, 2.4 mmol) in bromoform (6 mL), was added dropwise BuNO2 (618 mg, 6 mmol) at r.t. The solution was stirred at r.t. for 2h. Then the solution was stopped by addition of saturated Na2CO3 solution, followed by extraction with EtOAc (3×40 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified on silica gel (Hexane-EtOAc, 40:1) to afford the product (600 mg, 88%) as a pale yellow solid. M.p. 124-125°C. (lit.16 M.p. 124-125°C). IR (KBr, cm-1) vmax: 1535, 1373, 1241, 1045, 847. 1H NMR (400 MHz, CDCl3) δ 2.68 (s, 3H), 2.63 (s, 3H). 13C NMR (100 MHz, CDCl3)δ 156.6, 149.6, 141.6, 123.3, 123.0, 26.1, 25.4.
2-(3-(4-methoxybenzyloxy)prop-1-ynyl)-3,5-dibromo-4,6-dimethylpyridine (7)
To a solution of p-methoxybenzylalcohol (3.32 g, 24 mmol) in anhydrous Et2O (24 mL) was added NaH (60%) (100 mg, 2.4 mmol) at r.t. under N2. After stirring for 30 minutes, the mixture was cooled to 0°C and trichloroacetonitrile (3.46 g, 24 mmol) was added. The resulting mixture was allowed to warm up slowly to room temperature and stirred for 4h. After concentrated to remove Et2O, the residue was dissolved in hexane (28 mL) and MeOH (0.12 mL). The suspension was filtered through celite. The filtrate was concentrated to give a yellow oil (6.68 g). The crude intermediate was dissolved in anhydrous dichloromethane (25 mL). Propargyl alcohol (896 mg, 16 mmol) was added to the reaction and the solution was cooled to 0°C and treated with a catalytic amount of 10-camphorsulfonic acid. The reaction was allowed to warm up slowly to r.t. and stirred overnight under N2 protection. The solution was filtered, washed with saturated aqueous NaHCO3, dried over anhydrous Na2SO4, filtered and concentrated. The crude product was purified on silica gel (Hexane-EtOAc, 8:1) to afford 1-methoxy-4-((prop-2-ynyloxy)methyl)benzene (2.42 g, 86%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.28 (m, 2H), 6.88 (m, 2H), 4.54 (s, 2H), 4.13 (d, J=2.4 Hz, 2H), 3.79 (s, 3H), 2.45 (t, J=2.4 Hz, 1H). (lit.10)
A solution of 6 (682 mg, 2.0 mmol), 1-methoxy-4-((prop-2-ynyloxy)methyl)benzene (422 mg, 2.4 mmol), CuI (19 mg, 0.1 mmol) and piperidine (510 mg, 6.0 mmol) in 15 mL THF was purged by the passage of nitrogen through the solution, and 140 mg (0.2 mmol) of PdCl2(PPh3)2 was added all at once. The reaction mixture was stirred at room temperature for 1h. Then the mixture was diluted with EtOAc, washed with water, saturated brine and dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified on silica gel (Hexane-EtOAc, 8:1) to afford the product (797 mg, 91%) as a pale yellow oil. IR (KBr, cm-1) vmax: 2921, 2850, 1736, 1512, 1359, 1245, 1043, 818. 1H NMR (400 MHz, CDCl3) δ 7.33 (m, 2H), 6.89 (m, 2H), 4.67 (s, 2H), 4.43 (s, 2H), 3.81 (s, 3H), 2.65 (s, 3H), 2.64 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 159.5, 156.6, 147.4, 140.8, 129.9, 129.9, 129.4, 123.8, 123.1, 113.9, 113.9, 90.3, 84.7, 71.3, 57.3, 55.3, 25.8, 24.8. HRMS m/z calcd for C18H18Br2NO2 [M+H]+ = 437.9704, found 437.9691.
3-(4,6-dimethylpyridin-2-yl)propan-1-ol (8)
Hydrogenation was conducted on a mixture of compound 7 (131 mg, 0.3 mmol), Pd/C (13 mg, 10%) in 16 mL MeOH/EtOAc (1:1) under 60 psi. H2 overnight. The reaction mixture was filtered and concentrated. The residue was purified on silica gel (DCM-MeOH, 10:1) to afford the product (45 mg, 91%) as a pale yellow oil. IR (KBr, cm-1) vmax: 3262, 2922, 2864, 1610, 1451, 1058, 843. 1H NMR (400 MHz, CDCl3) δ 6.83 (s, 2H), 4.55 (br s, 1H), 3.73 (t, J=5.6Hz, 2H), 2.92 (t, J=6.4Hz, 2H), 2.48 (s, 3H), 2.28 (s, 3H), 1.92∼1.98 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 160.2, 156.9, 148.5, 121.8, 121.2, 62.6, 35.6, 31.3, 23.7, 20.9. HRMS m/z calcd for C10H16NO [M+H]+ = 166.1232, found 166.1219.
2-bromo-4,6-dimethylpyridine (9)
To a solution of 2-amino-4,6-dimethylpyridine (610 mg, 5 mmol) in 2.5 mL HBr (48%), was added Br2 (2.21 g, 13.8 mmol) at -10°C, keeping the temperature below 0°C. NaNO2 (0.86 g, 12.5 mmol) was added with the reaction temperature being kept below 0°C. After a further 30 min, NaOH (2 g in 2 mL water) was added, keeping the temperature below 10°C and then more NaOH was added to make the solution strongly alkaline. Extracted with EtOAc (3×50 mL) and the combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated, the residue was purified on silica gel (Hexane-EtOAc, 6:1) to afford the product (626 mg, 68%) as a pale yellow oil. IR (KBr, cm-1) vmax: 2922, 1597, 1540, 1274, 1191, 1127, 825. 1H NMR (400 MHz, CDCl3) δ 7.13 (s, 1H), 6.92 (s, 1H), 2.48 (s, 3H), 2.28 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 159.4, 150.2, 141.4, 125.6, 123.3, 24.0, 20.6. (lit.16)
3-(4,6-dimethylpyridin-2-yl)prop-2-yn-1-ol (10)
A solution of 9 (370 mg, 2 mmol), prop-2-yn-1-ol (168 mg, 3 mmol), CuI (19 mg, 0.1 mmol) and piperidine (510 mg, 6 mmol) in 10 mL THF was purged by the passage of nitrogen through the solution, and 140 mg (0.2 mmol) of PdCl2(PPh3)2 was added all at once. The reaction mixture was stirred at room temperature for 3h. Then the mixture was diluted with EtOAc, washed with water, saturated brine and dried with Na2SO4, filtered and concentrated. The residue was purified on silica gel (EtOAc) to afford the product (257 mg, 80%) as a white solid. M.p. 93-94°C. IR (KBr, cm-1) vmax: 3195, 2916, 1604, 1025, 850. 1H NMR (400 MHz, CDCl3) δ 7.10 (s, 1H), 6.94 (s, 1H), 4.52 (br s, 2H), 2.79 (br s, 1H), 2.50 (s, 3H), 2.29 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 158.6, 147.8, 141.7, 125.2, 124.0, 87.1, 84.9, 51.3, 24.2, 20.7. HRMS m/z calcd for C10H12NO [M+H]+ = 162.0919, found 162.0909.
3-(4,6-dimethylpyridin-2-yl)propan-1-ol (8)
A solution of 10 (200 mg, 1.24 mmol) in 10 mL of ethanol and 0.2 mL of triethylamine was hydrogenated over 20 mg (0.09 mmol) of PtO2 overnight. The reaction mixture was filtered and concentrated. The residue was purified on silica gel (DCM-MeOH, 10:1) to afford the product (187 mg, 91%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 6.81 (s, 2H), 5.05 (br s, 1H), 3.71 (t, J=5.7Hz, 2H), 2.90 (t, J=6.5Hz, 2H), 2.47 (s, 3H), 2.27 (s, 3H), 1.91∼1.98 (m, 2H). The NMR spectra were unequivocally consistent with the product 8.
2-(3-(4-methoxybenzyloxy)propyl)-3,5-dibromo-4,6-dimethylpyridine (11)
A solution of 7 (746 mg, 1.71 mmol) in 10 mL of ethanol and 0.2 mL of triethylamine was hydrogenated over 16 mg (0.07 mmol) of PtO2 for 1h. The reaction mixture was filtered and concentrated. The residue was purified on silica gel (Hexane-EtOAc, 8:1) to afford the product (547 mg, 73%) as pale yellow oil. IR (KBr, cm-1) vmax: 2930, 2851, 1612, 1511, 1360, 1244, 1098, 1036, 818. 1H NMR (400 MHz, CDCl3) δ 7.26 (m, 2H), 6.86 (m, 2H), 4.45 (s, 2H), 3.80 (s, 3H), 3.54 (t, J=6.5 Hz, 2H), 2.97∼3.01 (m, 2H), 2.61 (s, 3H), 2.60 (s, 3H), 2.00∼2.07 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 159.1, 157.7, 155.1, 146.6, 130.8, 129.2, 129.2, 121.4, 121.0, 113.7, 113.7, 72.4, 69.5, 55.3, 34.7, 28.3, 25.6, 24.8. HRMS m/z calcd for C18H22Br2NO2 [M+H]+ = 442.0017, found 442.0003.
diisopropyl (Z)-1-decenylboronate (12)
To a solution of 1-decyne (1.38 g, 10 mmol) in acetone (50 mL) was added NBS (3.56 g, 20 mmol) and AgNO3 (170 mg, 1 mmol) at r.t. with protection from light. After 4h, the solvent was carefully removed under reduced pressure, diluted with hexane and filtered. The filtrate was concentrated and purified on silica gel (Hexane) to afford 1-bromo-1-decyne (1.84 g, 85%) as a colorless oil.
To a solution of 1-bromo-1-decyne (1.84 g, 8.49 mmol) in CH2Cl2 (10 mL) in a 100 mL round bottom flask surrounded by a water bath under argon, was slowly added a solution of BHBr2·Me2S (1M in CH2Cl2, 4.16 mL). After stirring of the reaction mixture overnight, pentane (4.16 mL) was added and the water bath was replaced with an ice-water bath. Isopropyl alcohol (16.62 mmol) was slowly introduced to the flask and stirring is continued for an additional 15 minutes. Then the mixture was concentrated to remove off CH2Cl2, the pentane layer was separated, the alcohol layer was extracted with pentane. The combined pentane extracts were concentrated to get diisopropyl (Z)-(1-bromo-1-decenyl) boronate.
To an ice-cooled solution of diisopropyl (Z)-(1-bromo-1-decenyl) boronate in THF (10 mL) in a 100 mL round bottom flask was added, with stirring, a solution of KIPBH (1M in THF, 8.49 mL) dropwise. The cold bath was then removed, and the stirring was continued at r.t. for 1h. Then the mixture was filtered and concentrated, the residue was purified on silica gel (Hexane-EtOAc, 4:1) to afford diisopropyl (Z)-1-decenyl boronate (1.31 g, 58%) as a colorless oil. The product was used for next step without further purification. IR (KBr, cm-1) vmax: 2923, 2854, 1621, 1418, 1337, 1246, 733. 1H NMR (400 MHz, CD3Cl3) δ 6.61 (dt, J = 7.5, 13.6 Hz, 1H), 5.42 (dt, J = 1.3, 13.6 Hz, 1H), 2.55 (m, 2H), 1.16∼1.43 (m, 26H), 0.88 (t, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CD3Cl3): δ 158.9, 151.1, 65.6, 60.4, 31.9, 31.9, 29.6, 29.5, 29.4, 29.3, 29.29, 29.27, 25.4, 24.6, 22.6, 14.1.
2-(3-(4-methoxybenzyloxy)propyl)-3,5-di((Z)-dec-1-enyl)-4,6-dimethylpyridine (13)
Pd(OAc)2 (9 mg, 0.04 mmol), PPh3 (32 mg, 0.12 mmol), and 11 (149 mg, 0.34 mmol) were stirred in toluene (1.36 mL) and aq Na2CO3 (0.68 mL, 2M), under a nitrogen atmosphere for 0.5h. To this solution was added a solution of 12 (361 mg, 1.35 mmol) in ethanol (0.68 mL). The solution was refluxed 6h, then diluted with EtOAc, filtered and concentrated, the residue was purified on silica gel (Hexane-EtOAc, 10:1) to afford the product (178 mg, 94%) as a pale yellow oil. IR (KBr, cm-1) vmax: 3000, 2923, 2852, 1613, 1512, 1246, 1099, 1038, 732. 1H NMR (400 MHz, CDCl3) 7.25 (m, 2H), 6.86 (m, 2H), 6.28 (d, J=11.8 Hz, 1H), 6.24 (d, J=11.8 Hz, 1H), 5.74∼5.81 (m, 2H), 4.43 (s, 2H), 3.80 (s, 3H), 3.50 (t, J=6.7 Hz, 2H), 2.75 (br s, 2H), 2.39 (s, 3H), 2.04 (s, 3H), 1.92∼2.00 (m, 2H), 1.74∼1.81 (m, 4H), 1.19∼1.34 (m, 24H), 0.86 (t, J=6.8 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 159.1, 156.4, 153.3, 134.7, 134.4, 131.0, 129.3, 129.2, 129.1, 129.1, 128.8, 125.9, 125.4, 113.9, 113.7, 72.3, 70.1, 55.3, 32.6, 31.9, 31.9, 29.4, 29.4, 29.4, 29.4, 29.3, 29.2, 29.2, 29.2, 28.9, 28.8, 28.7, 23.1, 22.7, 22.7, 17.5, 14.1, 14.1. HRMS m/z calcd for C38H60NO2 [M+H]+ = 562.4624, found 562.4621.
3-(3,5-di((Z)-dec-1-enyl)-4,6-dimethylpyridin-2-yl)propan-1-ol (14)
To a solution of 13 (178 mg, 0.32 mmol) in 7 mL of EtOH was added 1N HCl (3.5 mL). Then the mixture was refluxed for 3 hours. After cooled down, the mixture was concentrated to remove EtOH. The water layer was extracted with CH2Cl2 (3×30 mL) and the combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified on silica gel (DCM-MeOH, 20:1) to give 121 mg pale yellow oil in 80% yield as the hydrochloride salt. IR (KBr, cm-1) vmax: 3235, 2954, 2923, 2853, 1613, 1512, 1245, 1094, 1038, 722. 1H NMR (400 MHz, CDCl3) δ 6.26 (d, J=11.5 Hz, 1H), 6.23 (d, J=11.5 Hz, 1H), 5.76∼5.84 (m, 2H), 3.71 (t, J=5.5 Hz, 2H), 2.92 (br s, 2H), 2.38 (s, 3H), 2.06 (s, 3H), 1.89∼1.94 (m, 2H), 1.73∼1.80 (m, 4H), 1.19∼1.32 (m, 24H), 0.86 (t, J=6.8 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 155.6, 152.7, 144.5, 135.0, 134.9, 129.6, 129.2, 125.3, 125.2, 63.2, 33.7, 31.7, 31.7, 30.2, 29.4, 29.4, 29.4, 29.3, 29.2, 29.2, 28.9, 28.9, 28.78, 28.76, 22.6, 22.6, 22.6, 17.6, 14.1, 14.1. HRMS m/z calcd for C30H52NO [M+H]+ = 442.4049, found 442.4035.
Anibamine (1)
Methanesulfonyl chloride (53 mg, 0.46 mmol) was added to an ice-cooled solution of 14 (110 mg, 0.23 mmol) and triethylamine (70 mg, 0.69 mmol) in 6 mL CH2Cl2. The resulting mixture was allowed to warm to r.t. over 1h. The mixture was diluted with CH2Cl2 and washed with 1N HCl twice, dried over Na2SO4, filtered and concentrated. The residue was purified on silica gel (DCM-MeOH, 10:1) to afford the product (75 mg, 71%) as a pale yellow oil. IR (KBr, cm-1) vmax: 3382, 2955, 2923, 2853, 1605, 1466, 728. 1H NMR (400 MHz, CD3Cl3) δ 6.23 (d, J=11.2 Hz, 2H), 6.06 (dt, J=7.3 and 11.3 Hz, 1H), 6.03 (dt, J=7.4 and 11.3 Hz, 1H), 5.38 (br, 1H), 5.09 (br, 1H), 3.37 (m, 2H), 2.83 (s, 3H), 2.62 (m, 2H), 2.28 (s, 3H), 1.79∼1.84 (m, 4H), 1.22∼1.39 (m, 24H), 0.86 (t, J=6.8 Hz, 6H); 13C NMR (100 MHz, CD3Cl3): δ 154.6, 154.3, 148.8, 138.9, 138.8, 135.2, 131.3, 122.0, 121.0, 58.9, 32.6, 31.8, 31.8, 29.43, 29.40, 29.40, 29.33, 29.28, 29.2, 29.2, 29.2, 28.8, 28.6, 22.6, 22.6, 20.9, 18.8, 18.7, 14.1, 14.1. MS (ESI) m/z 424.6 (M+). (lit.10) HRMS m/z calcd for C30H50N [M]+ = 424.3943, found 424.3926.
(E)-dec-1-enylboronic acid (15)
A solution of 1-decyne (690 mg, 5 mmol) in anhydrous CH2Cl2 was cooled to 0°C, BHBr2·Me2S (1M in CH2Cl2, 5 mL) was slowly added. After 4h at r.t., the solution was carefully transferred into a cooled solution of 10% aqueous NaOH (10 mL) and the mixture was stirred at 0°C for 15 minutes. Saturated aqueous NH4Cl (17 mL) was added, and a voluminous white precipitate formed, CH2Cl2 was removed under reduced pressure. The (E)-decene-boronic acid was filtered and washed extensively with ice-water, afford (E)-dec-1-enylboronic acid (784 mg, 85%) as a white solid. The product was directly used for next step without any further purification. M.p. 52-53°C. IR (KBr, cm-1) vmax: 3443, 3322, 2921, 2848, 1634, 1346, 1151, 991. 1H NMR (400 MHz, CD3OD) δ 6.52 (dt, J = 6.6, 17.6 Hz, 1H), 5.55 (dt, J = 1.4, 17.6 Hz, 1H), 4.79 (s, 2H), 2.14 (m, 2H), 1.22∼1.43 (m, 12H), 0.90 (t, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CD3OD): δ 154.0, 152.3, 37.0, 33.0, 30.6, 30.4, 30.3, 29.7, 23.7, 14.4.
2-(3-(4-methoxybenzyloxy)propyl)-3,5-di((E)-dec-1-enyl)-4,6-dimethylpyridine (16)
Pd(OAc)2 (6 mg, 0.03 mmol), PPh3 (19 mg, 0.07 mmol), and 11 (88 mg, 0.2 mmol) were stirred in toluene (0.8 mL) and aqueous Na2CO3 (0.4 mL, 2M), under a nitrogen atmosphere for 0.5h. To this solution was added a solution of 15 (147 mg, 0.8 mmol) in ethanol (0.4 mL). The solution was refluxed for 6h, then diluted with EtOAc, filtered and concentrated, the residue was purified on silica gel (Hexane-EtOAc, 10:1) to afford the product (97 mg, 87%) as a pale yellow oil. IR (KBr, cm-1) vmax: 2954, 2922, 2852, 1613, 1512, 1246, 1098, 1038, 971. 1H NMR (400 MHz, CDCl3) δ 7.25 (m, 2H), 6.86 (m, 2H), 6.29 (d, J=15.9 Hz, 1H), 6.26 (d, J=16.0 Hz, 1H), 5.56∼5.67 (m, 2H), 4.43 (s, 2H), 3.79 (s, 3H), 3.50 (t, J=6.6 Hz, 2H), 2.79∼2.84 (m, 2H), 2.45 (s, 3H), 2.18∼2.26 (m, 4H), 2.18 (s, 3H), 1.93∼2.00 (m, 2H), 1.42∼1.50 (m, 4H), 1.28∼1.38 (m, 20H), 0.88 (t, J=6.5 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 159.1, 156.0, 152.9, 143.1, 137.0, 137.0, 131.0, 130.7, 130.6, 129.1, 129.1, 126.2, 125.8, 113.7, 113.7, 72.3, 70.0, 55.3, 33.4, 33.3, 32.6, 31.9, 29.6, 29.49, 29.47, 29.4, 29.4, 29.31, 29.31, 29.30, 29.30, 29.2, 23.7, 22.7, 22.7, 18.1, 14.1, 14.1. HRMS m/z calcd for C38H60NO2 [M+H]+ = 562.4624, found 562.4589.
3-(4,6-dimethyl-3,5-di((E)-prop-1-enyl)pyridin-2-yl)propan-1-ol (17)
The same procedure described for 14 was used and the residue was purified to give 75 mg pale yellow oil in 91% yield as the hydrochloride salt. IR (KBr, cm-1) vmax: 3242, 2922, 2852, 1552, 1454, 1245, 1059, 970. 1H NMR (400 MHz, CDCl3) δ 6.27 (d, J=16.1 Hz, 1H), 6.24 (d, J=16.1 Hz, 1H), 5.56∼5.68 (m, 2H), 3.70 (t, J=5.4 Hz, 2H), 2.96∼2.99 (m, 2H), 2.44 (s, 3H), 2.20∼2.26 (m, 4H), 2.19 (s, 3H), 1.88∼1.94 (m, 2H), 1.44∼1.51 (m, 4H), 1.24∼1.38 (m, 20H), 0.89 (t, J=6.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 155.2, 152.3, 144.0, 137.49, 137.46, 131.0, 131.0, 125.7, 125.6, 62.8, 33.6, 33.4, 33.3, 31.9, 31.9, 30.6, 29.4, 29.4, 29.3, 29.3, 29.3, 29.23, 29.23, 29.21, 23.1, 22.7, 22.7, 18.1, 14.1, 14.1. HRMS m/z calcd for C30H52NO [M+H]+ = 442.4049, found 442.4019.
(11E, 22E) isomer of Anibamine (2)
The same procedure described for 1 was used and the product was obtained as a pale yellow oil, yield: 51mg (71%). IR (KBr, cm-1) vmax: 3384, 2955, 2923, 2853, 1602, 1466, 976. 1H NMR (400 MHz, CD3Cl3) δ 6.26 (d, J=16.2 Hz, 1H), 6.23 (d, J=16.2 Hz, 1H), 5.98 (dt, J=6.8 and 16.2 Hz, 1H), 5.82 (dt, J=6.8 and 16.2 Hz, 1H), 5.18 (t, J=7.6 Hz, 2H), 3.52 (t, J=7.8 Hz, 2H), 2.85 (s, 3H), 2.53∼2.61 (m, 2H), 2.37 (s, 3H), 2.24∼2.31 (m, 4H), 1.45∼1.50 (m, 4H), 1.24∼1.32 (m, 20H), 0.89 (t, J=6.6 Hz, 6H); 13C NMR (100 MHz, CD3Cl3): δ 153.3, 153.1, 148.1, 142.1, 141.9, 136.7, 132.2, 122.4, 121.9, 58.8, 33.4, 33.23, 33.16, 31.9, 31.9, 29.38, 29.37, 29.27, 29.27, 29.25, 29.20, 28.84, 28.80, 22.7, 22.7, 21.2, 19.2, 19.0, 14.1, 14.1. MS (ESI) m/z 424.2 (M+). (lit.10) HRMS m/z: calcd for C30H50N [M]+ = 424.3943, found 424.3930.
2-(3-(4-methoxybenzyloxy)propyl)-5-bromo-3-((E)-dec-1-enyl)-4,6-dimethylpyridine (18) and 2-(3-(4-methoxybenzyloxy)propyl)-3-bromo-5-((E)-dec-1-enyl)-4,6-dimethylpyridine (19)
Pd(OAc)2 (11 mg, 0.05 mmol), PPh3 (37 mg, 0.14 mmol), and 11 (348 mg, 0.79 mmol) were stirred in toluene (1.6 mL) and aqueous Na2CO3 (0.79 mL, 2M), under a nitrogen atmosphere for 0.5h. To this solution was added a solution of (E)-dec-1-enylboronic acid (174 mg, 0.95 mmol) in ethanol (0.79 mL). The solution was then refluxed for 5h, diluted with EtOAc, filtered and concentrated, the residue was purified on silica gel (Hexane-EtOAc, 8:1) to afford a mixture (345 mg, 87%) as a pale yellow oil. 1H NMR indicated 1:1 (3E : 5E) ratio. The isomers were separated on preparative TLC plates with Hexane/EtOAc (4:1, v/v) as eluent after twice development give 18 as the top band a pale yellow oil (68 mg) and 19 as the lower band a pale yellow oil (70 mg).
Compound 18
IR (KBr, cm-1) vmax: 2953, 2923, 2852, 1512, 1246, 1100, 1038, 974, 820. 1H NMR (400 MHz, CDCl3) δ 7.23 (m, 2H), 6.86 (m, 2H), 6.28 (d, J=16.1 Hz, 1H), 5.61 (dt, J=6.9 and 16.1 Hz, 1H), 4.42 (s, 2H), 3.80 (s, 3H), 3.48 (t, J=6.6 Hz, 2H), 2.77∼2.81 (m, 2H), 2.62 (s, 3H), 2.36 (s, 3H), 2.18∼2.24 (m, 2H), 1.92∼1.99 (m, 2H), 1.42∼1.50 (m, 2H), 1.24∼1.36 (m, 10H), 0.89 (t, J=6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 159.1, 156.8, 154.2, 144.9, 138.2, 132.1, 130.8, 129.1, 129.1, 125.3, 122.2, 113.7, 113.7, 72.4, 69.8, 55.2, 33.2, 32.3, 31.9, 29.5, 29.32, 29.30, 29.2, 29.2, 25.9, 22.7, 21.3, 14.1. HRMS m/z calcd for C28H41BrNO2 [M+H]+ = 502.2321, found 502.2302.
Compound 19
IR (KBr, cm-1) vmax: 2954, 2924, 2853, 1512, 1248, 1101, 1037, 974, 820. 1H NMR (400 MHz, CDCl3) δ 7.27 (m, 2H), 6.87 (m, 2H), 6.26 (d, J=16.1 Hz, 1H), 5.64 (dt, J=6.9 and 16.1 Hz, 1H), 4.46 (s, 2H), 3.80 (s, 3H), 3.55 (t, J=6.6 Hz, 2H), 2.98∼3.02 (m, 2H), 2.41 (s, 3H), 2.37 (s, 3H), 2.22∼2.27 (m, 2H), 2.01∼2.08 (m, 2H), 1.44∼1.52 (m, 2H), 1.24∼1.38 (m, 10H), 0.89 (t, J=6.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 159.1, 156.7, 153.7, 144.8, 138.1, 132.3, 130.9, 129.2, 129.2, 125.8, 122.0, 113.7, 113.7, 72.4, 69.7, 55.3, 35.0, 32.2, 31.9, 29.4, 29.3, 29.2, 29.2, 28.6, 23.4, 22.7, 21.3, 14.1. HRMS m/z calcd for C28H41BrNO2 [M+H]+ = 502.2321, found 502.2295.
2-(3-(4-methoxybenzyloxy)propyl)-3-((E)-dec-1-enyl)-5-((Z)-dec-1-enyl)-4,6-dimethylpyridine (20)
Pd(OAc)2 (4 mg, 0.02 mmol), PPh3 (13 mg, 0.05 mmol), and 18 (65 mg, 0.13 mmol) were stirred in toluene (0.38 mL) and aqueous Na2CO3 (0.19 mL, 2M), under a nitrogen atmosphere for 0.5h. To this solution was added a solution of diisopropyl (Z)-1-decenylboronate (104 mg, 0.39 mmol) in ethanol (0.19 mL). The solution was refluxed 4h, then diluted with EtOAc, filtered and concentrated, the residue was purified on silica gel (Hexane-EtOAc, 8:1) to afford the product (68 mg, 93%) as a pale yellow oil. IR (KBr, cm-1) vmax: 2954, 2922, 2852, 1512, 1456, 1246, 1100, 1038, 971, 721. 1H NMR (400 MHz, CDCl3) δ 7.26 (m, 2H), 6.86 (m, 2H), 6.30 (d, J=16.1 Hz, 1H), 6.22 (d, J=11.2 Hz, 1H), 5.77 (dt, J=7.2 and 11.2 Hz, 1H), 5.62 (dt, J=6.9 and 16.1 Hz, 1H), 4.43 (s, 2H), 3.80 (s, 3H), 3.51 (t, J=6.7 Hz, 2H), 2.83 (t, J=7.8 Hz, 2H), 2.37 (s, 3H), 2.20∼2.24 (m, 2H), 2.11 (s, 3H), 1.95∼2.02 (m, 2H), 1.76∼1.82 (m, 2H), 1.43∼1.50 (m, 2H), 1.20∼1.31 (m, 22H), 0.88 (t, J=6.7 Hz, 3H), 0.86 (t, J=7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3)δ 159.0, 156.4, 152.9, 143.3, 137.0, 134.4, 130.9, 130.2, 129.4, 129.1, 129.1, 125.9, 125.7, 113.7, 113.7, 72.3, 70.0, 55.2, 33.4, 32.6, 31.9, 31.8, 30.9, 29.6, 29.5, 29.4, 29.34, 29.30, 29.26, 29.2, 28.9, 28.7, 23.2, 22.7, 22.6, 17.7, 14.11, 14.09. HRMS m/z calcd for C38H60NO2 [M+H]+ = 562.4624, found 562.4598.
3-(3-((E)-dec-1-enyl)-5-((Z)-dec-1-enyl)-4,6-dimethylpyridin-2-yl)propan-1-ol (21)
The same procedure described for 14 was used and the residue was purified to give 50 mg pale yellow oil in 90% yield as the hydrochloride salt. IR (KBr, cm-1) vmax: 3267, 2954, 2922, 2853, 1553, 1455, 1246, 1041, 970, 722. 1H NMR (400 MHz, CDCl3) δ 6.28 (d, J=16.1 Hz, 1H), 6.21 (d, J=11.2 Hz, 1H), 5.79 (dt, J=7.2 and 11.2 Hz, 1H), 5.62 (dt, J=6.8 and 16.1 Hz, 1H), 3.71 (t, J=5.3 Hz, 2H), 3.00 (t, J=6.0 Hz, 2H), 2.37 (s, 3H), 2.21∼2.27 (m, 2H), 2.13 (s, 3H), 1.92∼1.96 (m, 2H), 1.75∼1.80 (m, 2H), 1.45∼1.50 (m, 2H), 1.20∼1.33 (m, 22H), 0.89 (t, J=6.4 Hz, 3H), 0.86 (t, J=7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 155.5, 152.3, 144.3, 137.4, 134.9, 130.6, 129.7, 125.5, 125.4, 62.9, 33.6, 33.3, 31.9, 31.8, 30.6, 29.44, 29.38, 29.31, 29.28, 29.25, 29.23, 29.21, 29.21, 28.9, 28.7, 22.7, 22.6, 17.8, 14.10, 14.09. HRMS m/z calcd for C30H52NO [M+H]+ = 442.4049, found 442.4024.
(11Z, 22E) isomer of Anibamine (3)
The same procedure described for 1 was used. The product was obtained as yellow oil, yield: 44mg (94%). IR (KBr, cm-1) vmax: 3386, 2955, 2923, 2853, 1604, 1466, 1340, 978, 723. 1H NMR (400 MHz, CDCl3) δ 6.27 (d, J=16.2 Hz, 1H), 6.21 (d, J=11.4 Hz, 1H), 6.05 (dt, J=7.3 and 11.4Hz, 1H), 6.02 (dt, J=6.9 and 16.2Hz, 1H), 5.28 (br, 1H), 4.99 (br, 1H), 3.54 (m, 2H), 2.76 (s, 3H), 2.54∼2.63 (m, 2H), 2.33 (s, 3H), 2.25∼2.31 (m, 2H), 1.77∼1.83 (m, 2H), 1.46∼1.53 (m, 2H), 1.22∼1.35 (m, 22H), 0.89 (t, J=6.7 Hz, 3H), 0.86 (t, J=7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 153.8, 153.4, 148.0, 142.0, 138.8, 135.2, 132.2, 122.1, 121.8, 58.6, 33.5, 33.2, 31.9, 31.8, 29.4, 29.4, 29.25, 29.25, 29.22, 29.17, 29.10, 28.8, 28.6, 22.7, 22.6, 21.2, 18.7, 18.6, 14.11, 14.09. MS (ESI) m/z 424.3 (M+). HRMS m/z calcd for C30H50N [M]+ = 424.3943, found 424.3932.
2-(3-(4-methoxybenzyloxy)propyl)-5-((E)-dec-1-enyl)-3-((Z)-dec-1-enyl)-4,6-dimethylpyridine (22)
The same procedure described for 20 was used. The product was obtained as pale yellow oil, yield: 70mg (96%). IR (KBr, cm-1) vmax: 2954, 2922, 2852, 1612, 1512, 1455, 1246, 1100, 1038, 971, 722. 1H NMR (400 MHz, CDCl3) δ 7.25 (m, 2H), 6.86 (m, 2H), 6.27 (d, J=16.1 Hz, 1H), 6.26 (d, J=11.2 Hz, 1H), 5.77 (dt, J=7.2 and 11.2 Hz, 1H), 5.65 (dt, J=6.9 and 16.1 Hz, 1H), 4.42 (s, 2H), 3.80 (s, 3H), 3.49 (t, J=6.7 Hz, 2H), 2.74 (br s, 2H), 2.47 (s, 3H), 2.21∼2.26 (m, 2H), 2.12 (s, 3H), 1.91∼1.98 (m, 2H), 1.75∼1.81 (m, 2H), 1.45∼1.52 (m, 2H), 1.19∼1.39 (m, 22H), 0.89 (t, J=6.6 Hz, 3H), 0.86 (t, J=7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 159.0, 155.9, 153.3, 143.1, 136.9, 134.6, 131.0, 130.4, 129.1, 129.1, 129.1, 126.2, 125.5, 113.7, 113.7, 72.2, 70.1, 55.3, 33.4, 32.6, 31.88, 31.85, 29.45, 29.41, 29.35, 29.34, 29.30, 29.2, 29.2, 29.1, 28.9, 28.76, 23.75, 22.7, 22.6, 17.7, 14.09, 14.08. HRMS m/z calcd for C38H60NO2 [M+H]+ = 562.4624, found 562.4588.
3-(5-((E)-dec-1-enyl)-3-((Z)-dec-1-enyl)-4,6-dimethylpyridin-2-yl)propan-1-ol (23)
The same procedure described for 14 was used and the residue was purified to give 53 mg pale yellow oil in 91% yield as the hydrochloride salt. IR (KBr, cm-1) vmax: 3374, 2954, 2923, 2853, 1643, 1455, 1300, 1062, 973, 722. 1H NMR (400 MHz, CDCl3) δ 6.22 (m, 2H), 5.94 (dt, J=7.3 and 11.2 Hz, 1H), 5.76 (dt, J=6.9 and 16.2 Hz, 1H), 3.72 (t, J=5.5 Hz, 2H), 3.06 (t, J=6.6 Hz, 2H), 2.65 (s, 3H), 2.24∼2.30 (m, 2H), 2.24 (s, 3H), 1.88∼1.94 (m, 2H), 1.74∼1.80 (m, 2H), 1.46∼1.53 (m, 2H), 1.21∼1.35 (m, 22H), 0.89 (t, J=6.7 Hz, 3H), 0.87 (t, J=7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 154.6, 151.8, 146.3, 138.4, 135.8, 131.6, 130.2, 124.8, 124.4, 62.5, 33.4, 32.4, 31.85, 31.81, 30.5, 29.40, 29.36, 29.29, 29.27, 29.18, 29.17, 29.17, 29.17, 28.83, 28.80, 22.64, 22.61, 18.0, 14.06, 14.05. HRMS m/z calcd for C30H52NO [M+H]+ = 442.4049, found 442.4019.
(11E, 22Z) isomer of Anibamine (4)
The same procedure described for 1 was used. The product was obtained as yellow oil, yield: 46mg (91%). IR (KBr, cm-1) vmax: 3388, 2955, 2922, 2853, 1604, 1466, 978, 722. 1H NMR (400 MHz, CDCl3) δ 6.23 (d, J=16.2 Hz, 1H), 6.20 (d, J=11.3 Hz, 1H), 6.05 (dt, J=7.3 and 11.3 Hz, 1H), 6.02 (dt, J=6.9 and 16.2 Hz, 1H), 5.26 (t, J=7.3 Hz, 2H), 3.34 (t, J=7.6 Hz, 2H), 2.92 (s, 3H), 2.54∼2.62 (m, 2H), 2.31 (s, 3H), 2.26∼2.31 (m, 2H), 1.79∼1.84 (m, 2H), 1.47∼1.54 (m, 2H), 1.22∼1.38 (m, 22H), 0.89 (t, J=6.6 Hz, 3H), 0.87 (t, J=7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 154.0, 153.6, 148.8, 142.2, 138.9, 136.6, 131.2, 122.2, 121.0, 59.0, 33.2, 32.6, 31.84, 31.81, 29.4, 29.4, 29.4, 29.3, 29.23, 29.20, 29.16, 28.75, 28.74, 22.64, 22.61, 20.9, 19.2, 18.9, 14.09, 14.06. MS (ESI) m/z 424.3 (M+). HRMS m/z calcd for C30H50N [M]+ = 424.3943, found 424.3924.
Molecular modeling procedure
The structures of anibamine and its three isomers were built using Sybyl 8.1 with, unless specified, default parameters, followed by energy minimization (Tripos forcefield, Gasteiger-Hückel charges) with default termination at 0.05 kcal mol-1 Å-1. The energy minimized structures were then solvated in a water box (compound 1, periodic box size, 79,170.4 Å3, 42.76 Å each side, number of waters, 3474; compound 2, periodic box size, 119,018.2 Å3, 49.19 Å each side, number of waters, 5288; compound 3, periodic box size, 104,737.0 Å3, 46.99 Å each side, number of waters, 4609; compound 4, periodic box size, 95,426.2 Å3, 45.33 Å each side, number of waters, 4156). The isomers in these water boxes were again energy minimized (Tripos forcefield, Gasteiger-Hückel charges, constant dielectric = 80, termination 0.05 kcal mol-1 Å-1). Dynamic simulations were run on the four isomers within their respective water boxes for 200,000 fs. Average structures from the last 10,000 fs of the dynamic simulation run were generated and again energy minimized as above. Structures thus obtained were in the corresponding figures.
Calcium Assay Protocol
MOLT-4/CCR5 cells were plated in black 96-well plates with transparent bottom (Greinier Bio-one) at 100,000 cells per well in 50:1 HBSS:HEPES assay buffer. They were incubated for 1 hour at 37°C and 5% CO2 with control buffer or varying concentration of compound for a total volume of 130 μL per well. Cells were then incubated with 50 μL of Fluo-4-AM loading buffer (40 μL 2 μM Fluo-4 dye, 100 μL 2.5 mM probenacid, in 5 mL assay buffer) for an additional hour. Then 20 μL 200 nM RANTES solution in assay buffer or assay buffer alone were added to the wells right before changes in Ca2+ concentration were monitored by RFU for 90 seconds using a microplate reader (FlexStation3, Molecular Devices). Peak values were obtained using SoftMaxPro software (Molecular Devices) and non-linear regression curves were generated using Prism (GraphPad) to calculate IC50 values.
Supplementary Material
Acknowledgments
We are grateful for the funding support from US Army Prostate Cancer Research Program PC073739, and NIH/NIAID AI074461. The content of this report is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, nor the US Army Prostate Cancer Research Program. We thank the NIH AIDS Research and Reference Reagent Program for providing the MOLT-4/CCR5 cell line.
Footnotes
Supporting Information Available: Purity test results and copies of spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jayasuriya H, Herath KB, Ondeyka JG, Polishook JD, Bills GF, Dombrowski AW, Springer MS, Siciliano S, Malkowitz L, Sanchez M, Guan ZQ, Tiwari S, Stevenson DW, Borris RP, Singh SB. J Nat Prod. 2004;67:1036–1038. doi: 10.1021/np049974l. [DOI] [PubMed] [Google Scholar]
- 2.Klausmeyer P, Chmurny GN, McCloud TG, Tucker KD, Shoemaker RH. J Nat Prod. 2004;67:1732–1735. doi: 10.1021/np040114e. [DOI] [PubMed] [Google Scholar]
- 3.(a) Tagat JR, McCombie SW, Nazareno D, Labroli MA, Xiao Y, Steensma RW, Strizki JM, Baroudy BM, Cox K, Lachowicz J, Varty G, Watkins R. J Med Chem. 2004;47:2405–2408. doi: 10.1021/jm0304515. [DOI] [PubMed] [Google Scholar]; (b) Strizki JM, Tremblay C, Xu S, Wojcik L, Wagner N, Gonsiorek W, Hipkin RW, Chou CC, Pugliese-Sivo C, Xiao Y, Tagat JR, Cox K, Priestley T, Sorota S, Huang W, Hirsch M, Reyes GR, Baroudy BM. Antimicrob Agents Chemother. 2005;49:4911–4919. doi: 10.1128/AAC.49.12.4911-4919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M. Antimicrob Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baba M, Nishimura O, Kanzaki N, Okamoto M, Sawada H, Iizawa Y, Shiraishi M, Aramaki Y, Okonogi K, Ogawa Y, Meguro K, Fujino M. Proc Natl Acad Sci USA. 1999;86:5698–5703. doi: 10.1073/pnas.96.10.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maeda K, Nakata H, Koh Y, Miyakawa T, Ogata H, Takaoka Y, Shibayama S, Sagawa K, Fukushima D, Moravek J, Koyanagi Y, Mitsuya H. J Virology. 2004;78:8654–8662. doi: 10.1128/JVI.78.16.8654-8662.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]; (b) Littman DR. Cell. 1998;93:677–680. doi: 10.1016/s0092-8674(00)81429-4. [DOI] [PubMed] [Google Scholar]; (c) Howard OM, Oppenheim JJ, Wang JM. J Clin Immunol. 1999;19:280–292. doi: 10.1023/A:1020587407535. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schwarz MK, Wells TNC. Nat Rev Drug Discovery. 2002;1:347–357. doi: 10.1038/nrd795. [DOI] [PubMed] [Google Scholar]; (e) Lusso P. EMBO J. 2006;25:447–456. doi: 10.1038/sj.emboj.7600947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X, Haney KM, Richardson AC, Wilson E, Gewirtz DA, Ware JL, Zehner ZE, Zhang Y. Bioorg Med Chem Lett. 2010;20:4627–4630. doi: 10.1016/j.bmcl.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Coussens LM, Werb Z. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nelson WG, De Marzo AM, Isaacs WB. New Engl J Med. 2003;349:366–381. doi: 10.1056/NEJMra021562. [DOI] [PubMed] [Google Scholar]; (c) Robinson SC, Scott KA, Wilson JL, Thompson RG, Proudfoot AEI, Balkwill F. Cancer Res. 2003;63:8360–8365. [PubMed] [Google Scholar]; (d) Koenig JE, Senge T, Allhoff Ep, Koenig W. Prostate. 2004;58:121–129. doi: 10.1002/pros.10317. [DOI] [PubMed] [Google Scholar]
- 9.Vaday GG, Peehl DM, Kadam PA, Lawrence DM. Prostate. 2006;66:124–134. doi: 10.1002/pros.20306. [DOI] [PubMed] [Google Scholar]
- 10.Li G, Watson K, Buckheit RW, Zhang Y. Org Lett. 2007;9:2043–2046. doi: 10.1021/ol070748n. [DOI] [PubMed] [Google Scholar]
- 11.Mariella RP, Belcher EP. J Am Chem Soc. 1952;74:1916–1919. [Google Scholar]
- 12.Woon PB, Jung MK, Young SK, Cheol HY, Shin JK, Seok W. L U S Patent, 0092718 A1. 2004
- 13.Pitterna T, Renold P, Hueter OF, Maienfisch P, Zambach WWO. Patent, 109539 A2. 2009
- 14.(a) Siméon FG, Wendahl MT, Pike VW. J Org Chem. 2009;74:2578–2580. doi: 10.1021/jo802799c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bracher F, Daab J. Eur J Org Chem. 2002;14:2288–2291. [Google Scholar]
- 15.(a) Tilley JW, Zawoiski S. J Org Chem. 1988;53:386–390. [Google Scholar]; (b) Alami M, Linstrumelle G. Tetrahedron Lett. 1991;32:6109–6112. [Google Scholar]
- 16.Potts KT, Burton HR. J Org Chem. 1966;31:251–260. [Google Scholar]
- 17.(a) Brown HC, Imal T. Organometallics. 1984;3:1392–1395. [Google Scholar]; (b) Brown HC, Somayaji V. Synthesis. 1984:919–920. [Google Scholar]; (c) Miyaura N, Satoh M, Suzuki A. Tetrahedron Lett. 1986;27:3745–3748. [Google Scholar]; (d) Brown HC, Basavaiah D, Kulkarni SU, Bhat NG, Prasad JVNV. J Org Chem. 1988;53:239–246. [Google Scholar]
- 18.(a) Yanagi T, Oh-e T, Miyaura N, Suzuki A. Bull Chem Soc Jpn. 1989;62:3892–3895. [Google Scholar]; (b) Fairlamb IJS, Marrison LR, Dickinson JM, Lu FJ, Schmidt JP. Bioorg Med Chem. 2004;12:4285–4299. doi: 10.1016/j.bmc.2004.01.051. [DOI] [PubMed] [Google Scholar]
- 19.(a) Pietruszka J. J Org Chem. 1999;64:8287–8297. doi: 10.1021/jo9910278. [DOI] [PubMed] [Google Scholar]; (b) Molander GA, Bernardi CR. J Org Chem. 2002;67:8424–8429. doi: 10.1021/jo026236y. [DOI] [PubMed] [Google Scholar]
- 20.(a) Thoma G, Nuninger F, Schaefer M, Akyel KG, Albert R, Beerli C, Bruns C, Francotte E, Luyten M, MacKenzie D, Oberer L, Streiff MB, Wagner T, Walter H, Weckbecker G, Zerwes HG. J Med Chem. 2004;47:1939–55. doi: 10.1021/jm031046g. [DOI] [PubMed] [Google Scholar]; (b) Kaighn ME, Narayan S, Ohnuki Y, Lechner JF, Jones LW. Invest Urol. 1979;17:16–23. [PubMed] [Google Scholar]; (c) Haney Kendra M, Zhang Feng, Arnatt Christopher K, Yuan Yunyun, Li Guo, Ware Joy L, Gewirtz David A, Zhang Yan. Bioorg Med Chem Lett. 2011;21:5159–5163. doi: 10.1016/j.bmcl.2011.07.058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.