

Abstract
OBJECTIVE
Glucagon-like peptide 1 (GLP-1) is involved in the central regulation of food intake. It is produced within the brain by preproglucagon (PPG) neurons, which are located primarily within the brain stem. These neurons project widely throughout the brain, including to the appetite centers in the hypothalamus, and are believed to convey signals related to satiety. Previous work demonstrated that they are directly activated by leptin and electrical activity of the afferent vagus. Another satiety hormone, cholecystokinin (CCK), has also been linked to activation of brain stem neurons, suggesting that it might act partially via centrally projecting neurons from the nucleus tractus solitarius (NTS). The aim of this study was to investigate the neuronal circuitry linking CCK to the population of NTS-PPG neurons.
RESEARCH DESIGN AND METHODS
Transgenic mice expressing yellow fluorescent protein (Venus) under the control of the PPG promoter were used to identify PPG neurons in vitro and to record their electrical and pharmacological profile.
RESULTS
PPG neurons in the NTS were excited by CCK and epinephrine, but not by the melanocortin receptor agonist melanotan II. Both CCK and epinephrine acted to increase glutamatergic transmission to the PPG neurons, and this involved activation of α1-adrenergic receptors. Inhibition of adrenergic signaling abolished the excitatory action of CCK.
CONCLUSIONS
CCK activates NTS-PPG cells by a circuit involving adrenergic and glutamatergic neurons. NTS-PPG neurons integrate a variety of peripheral signals that indicate both long-term energy balance and short-term nutritional and digestional status to produce an output signal to feeding and autonomic circuits.
Glucagon-like peptide 1 (GLP-1) is a hormone produced by specialized endocrine cells in the intestinal epithelium (1) and a population of preproglucagon (PPG) neurons in the nucleus tractus solitarius (NTS) (2–6). Its physiological effects include the modulation of gastric emptying, glucose homeostasis, and appetite control, with both central and peripheral mechanisms likely contributing to its satiety evoking effects (7–15). Activation of central GLP-1 receptors seems likely to require release of GLP-1 from PPG neurons. In support of this hypothesis, a number of findings have suggested the involvement of the brain stem PPG neurons in appetite control. The immediate early gene cFOS, for example, is activated in PPG cells by peripheral satiety signals, such as gastric distension or systemically administered leptin (16,17).
The recent development of transgenic mice expressing eYFP (Venus) under the control of the PPG promoter (18) has enabled identification of this cell population in brain slice preparations and allowed the first characterization of PPG neuron activity in vitro (19). PPG neurons were shown to be directly regulated by leptin but were not affected by GLP-1 or peptide YY, two hormones that are released from enteroendocrine L-cells after a meal and have been shown to act as peripheral satiety signals.
Another peripheral satiety signal that might affect activity of PPG neurons is cholecystokinin (CCK). CCK, which is released postprandially from enteroendocrine L-cells, inhibits food intake and was actually the first gut-derived satiety hormone to be identified (20,21). It is well established that CCK acts on receptors in the periphery located on vagal afferent neurons that project to the nucleus of the solitary tract (22–24). Intraperitoneal application of CCK-8 induces cFOS immunoreactivity in pro-opiomelanocortin (POMC), catecholaminergic, and GLP-1-producing neurons in the vagal complex (25,26). However, recent studies on brain slice preparations have established that there are also direct effects of CCK within the lower brain stem (27). Although those experiments did not address the question of whether central CCK is released in a postprandial fashion, it is clear that microinjection of CCK-8 directly into the NTS suppresses food intake (28), thus suggesting that CCK acts locally within the NTS on neurons that integrate and relay satiety signals.
In this study, we examined the effect of CCK on the activity of PPG neurons, identified using the PPG-eYFP mouse strain (18). CCK-triggered activity was observed to be indirect, involving a complex neuronal network of catecholaminergic and glutamatergic signaling.
MATERIALS AND METHODS
Transgenic animals.
Transgenic mice were used that expressed a modified yellow fluorescent protein (YFP; Venus) under the control of the PPG promoter (18). Two founder strains, mGLU-V23-124 and mGlu-V50-144, created using mouse bacterial artificial chromosomes, were used interchangeably, since we observed no difference in the pattern of YFP expression in the brain stem. Animals were bred as heterozygotes on a C57/Bl6 background and were genotyped as described previously (18) before experimental use. All experiments were carried out in accordance with the U.K. Animals (Scientific Procedures) Act, 1986, with appropriate ethical approval.
Single-cell RT-PCR.
Samples for RT-PCR and single-cell RT-PCR were harvested and amplified in a multiplex and nested PCR protocol as described previously (29), using primers listed in Table 1. Reverse-transcribed samples were split in half, with one half used to test for PPG and the other half used to test for CCKA receptor (CCKAR) and CCKB receptor (CCKBR) in a multiplex first PCR and individual nested PCRs. The first PCR reaction product (5 μL) was used as a template for the nested PCR. As negative controls for single-cell RT-PCR, reactions were performed with solution from pipettes that were inserted into the slice without recording from a cell and with samples that were not reverse-transcribed. For positive controls, PCRs were performed on a 1:100 dilution of mouse brain stem cDNA.
TABLE 1.
PCR primers
| Primer | PPG | CCKA receptor | CCKB receptor |
|---|---|---|---|
| Outer primer | |||
| Forward | ATGAAGACCGTTTACATCGTGGC | TCAGTGTGCTGGGGAACACGCT | TCCTGGGACTGAGCCGACGC |
| Reverse | CTGGTGGCAAGGTTATCGAGA | ACCGTGTCATATGCCCGCCAG | GAAGGCACGCCACGTGTTGG |
| Inner primer | |||
| Forward | ACCAAGAGGAACCGGAAC | TCTTCGGAAGTGCCGTGTGCAA | GCCATCTGCCGACCACTGCAA |
| Reverse | CCAAGTTCCTCAGCTATGGCG | CACTTGGCAACAGGAAGCGGC | CCCCCTTGGTTTCGGACCCG |
| Product size (bp) | |||
| Outer | 492 | 886 | 860 |
| Inner | 186 | 285 | 341 |
Electrophysiology.
Coronal (200 μm) brain stem slices were obtained from adult (>8 weeks) transgenic mice of either sex after halothane anesthesia and dissection in ice-cold low Na+ solution containing (in mmol/L): 200 sucrose, 2.5 KCl, 28 NaHCO3, 1.25 NaH2PO4, 3 pyruvate, 7 MgCl2, 0.5 CaCl2, and 7 glucose (pH 7.4). After recovery at 34°C for 30 min in a solution containing (in mmol/L) 118 NaCl, 3 KCl, 25 NaHCO3, 1.2 NaH2PO4, 7 MgCl2, 0.5 CaCl2, and 2.5 glucose (pH 7.4), slices were kept at 34°C in artificial cerebrospinal fluid (ACSF) of the following composition (in mmol/L): 118 NaCl, 3 KCl, 25 NaHCO3, 1 MgCl2, 2 CaCl2, and 10 glucose (pH 7.4). Patch pipettes were pulled from thin-walled borosilicate capillaries (3–6 MΩ; Clark Electromedical Instruments, Pangbourne, U.K.) with a horizontal puller (Zeitz, Germany). Electrodes were filled with (in mmol/L) 120 potassium gluconate, 5 HEPES, 5 BAPTA, 1 NaCl, 1 MgCl2, 1 CaCl2, and 2 K2ATP (pH 7.2). For perforated-patch whole-cell recording, solubilized amphotericin B (Sigma, Gillingham, U.K.) was added to the pipette solution (final concentration ∼137.5 μg/mL).
Recordings were carried out in ACSF at 32°C. Experimental solutions were constantly bubbled with 95% O2-5% CO2. Most drugs were directly added to the ACSF. Epinephrine and norepinephrine were prepared as a 10 mmol/L stock solution in 0.5 mol/L HCl. 6,7-Dinitroquinoxaline-2,3-dione (DNQX) was prepared as a stock solution in DMSO. Tetrodotoxin (TTX) was prepared as a 1 mmol/L stock solution in Na-citrate buffer. The recording chamber (volume 2 mL) was perfused with ACSF at a rate of 4–5 mL/min. Drugs were either added to the ACSF or applied locally via pressure ejection from a glass pipette (opening diameter 5–10 μm) facing the recorded cell positioned at a distance of ∼100 μm. CCK-8s, DNQX, ICI118,551 hydrochloride, and TTX were obtained from Tocris Bioscience (Bristol, U.K.). Melanotan II was obtained from Phoenix Pharmaceuticals (Karlsruhe, Germany), and all other drugs, including clonidine and phenylephrine, were obtained from Sigma.
Recordings were performed in both voltage-clamp and current-clamp mode using an EPC-9 amplifier and Pulse/Pulsefit software (Heka Elektronik, Lambrecht, Germany). Currents or membrane potentials were filtered at 1 kHz and digitized at 4 kHz. Membrane resistance was monitored with 200 ms current or voltage pulses every 20 s.
Compensation for the junction potential (+10 mV for the potassium gluconate pipette solution) was performed offline. Recordings displayed in figures are adjusted for the junction potential. Action potentials were counted in 10-s bins to determine frequency. Mean action potential frequency was determined by taking the average frequency over a period of 3 min directly before drug application (control) or 3 min directly before washout of the drug. Voltage-clamp recordings of spontaneous excitatory postsynaptic currents (sEPSC) activity were analyzed with the Strathclyde Electrophysiology Software package (WinEDR/WinWCP; J. Dempster, University of Strathclyde, Glasgow, U.K.). Data are given as mean ± one S.E.M. Whether a cell responded to a drug was determined with unpaired t test by comparing the inter-event interval over 3 min under control conditions with that in the presence of the drug. A value of P < 0.05 was taken as positive response.
Statistical significance between groups of data was tested using one-way ANOVA followed by the post hoc Tukey test, unless stated otherwise. P values < 0.05 (*) and < 0.01 (**) were taken to indicate that the data were significantly different.
RESULTS
PPG neurons in the NTS were identified by their YFP fluorescence, and electrophysiological recordings were established under differential interference contrast optics as described previously (19). All experiments for this study were performed on the spontaneously active population of PPG neurons only. Any burst-firing cells were discarded (19). In current clamp (perforated-patch configuration) spontaneously active cells had a resting potential of −47 ± 1 mV (n = 29) and fired action potentials at a frequency of 1.5 ± 0.2 Hz (n = 28). Bath application of 100 nmol/L CCK octapeptide, sulfated (CCK-8s), had no significant effect on the resting membrane potential or the input resistance (data not shown) but led to a reversible increase in action potential frequency by 104 ± 34%, in 7 of 15 cells tested (Fig. 1), whereas no changes were observed in the remaining cells. This CCK-8s–induced increase in firing rate was abolished by bath application of 20 μmol/L DNQX, a non-N-methyl-d-aspartate (NMDA) glutamate receptor antagonist, in 5 out of 5 cells tested, suggesting that CCK-8s activated PPG cells via an increase in glutamatergic synaptic activity, rather than directly by activation of postsynaptic CCK receptors on PPG neurons. In agreement with these results, single-cell RT-PCR analysis demonstrated that m-RNA for the CCK receptors CCKAR and CCKBR is absent from PPG neurons (n = 8) but present in cDNA from mouse brain stem (Fig. 1E). These results indicated the presence of central CCK receptors, which might reside on either vagal afferent terminals or another cell population within the brain stem, but not on PPG neurons.
FIG. 1.
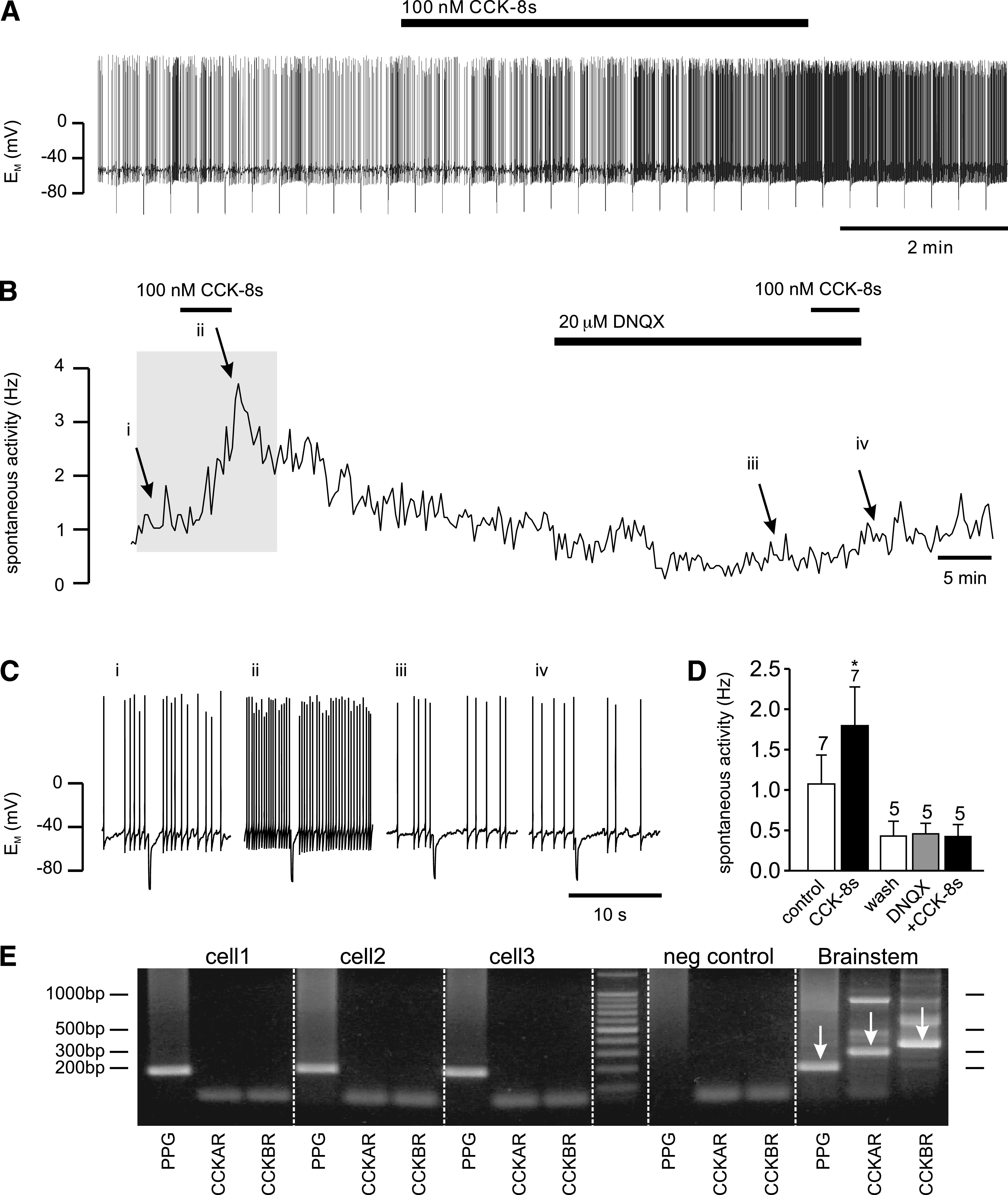
CCK increases spontaneous activity of PPG neurons. A: Current-clamp recording demonstrating that bath application of 100 nmol/L CCK-8s led to an increase in spontaneous action potential firing frequency of this PPG neuron. B: A plot of the firing frequency for the recording shown in A (the part of the recording shown in A is indicated by gray background). C: Short segments of the original current-clamp recording from B at time points indicated by i, ii, iii, and iv. D: Mean data for firing frequency from experiments as depicted in A and B. CCK-8 (100 nmol/L) significantly increased firing rate. This effect of CCK-8s was occluded in the presence of the non-NMDA glutamate receptor antagonist DNQX. Number of recordings for each condition is given above the bars. *P < 0.05. E: Typical single-cell RT-PCR analysis for PPG and the CCK receptors (CCKAR, CCKBR) for three PPG neurons and controls. Agarose gel (2%) demonstrating that the 186-bp PCR product for PPG, the 285-bp PCR product for CCKAR, and the 341-bp product for CCKBR can be obtained from brain stem cDNA (1:100 dilution; positive control; indicated by arrows) with the primers specified in Table 1. In contrast, cytoplasm extracted from single cells showing eYFP fluorescence (cell1, cell2, cell3) was only positive for PPG, but not CCKAR or CCKBR (only bands for primers visible). Negative (neg) control: pipette solution without cytoplasm extracted from cell. Molecular weight ladder shows bands at 100-bp intervals.
CCK modulation of spontaneous EPSC activity.
The previous results suggested that CCK might exert its effects via enhancement of glutamatergic synaptic inputs. To explore this possibility further, voltage-clamp recordings were performed to isolate EPSCs. EPSCs had a frequency of 2.3 ± 0.3 Hz (n = 53) and a mean amplitude of 25 ± 2 pA. These were predominantly glutamatergic, since DNQX (10 μmol/L) inhibited spontaneous synaptic activity by 91 ± 5% (Fig. 2; n = 6). Application of 100 nmol/L CCK-8s caused a reversible increase in sEPSC frequency in ∼60% PPG neurons tested (18 of 29; Fig. 2). The remaining cells showed no response.
FIG. 2.
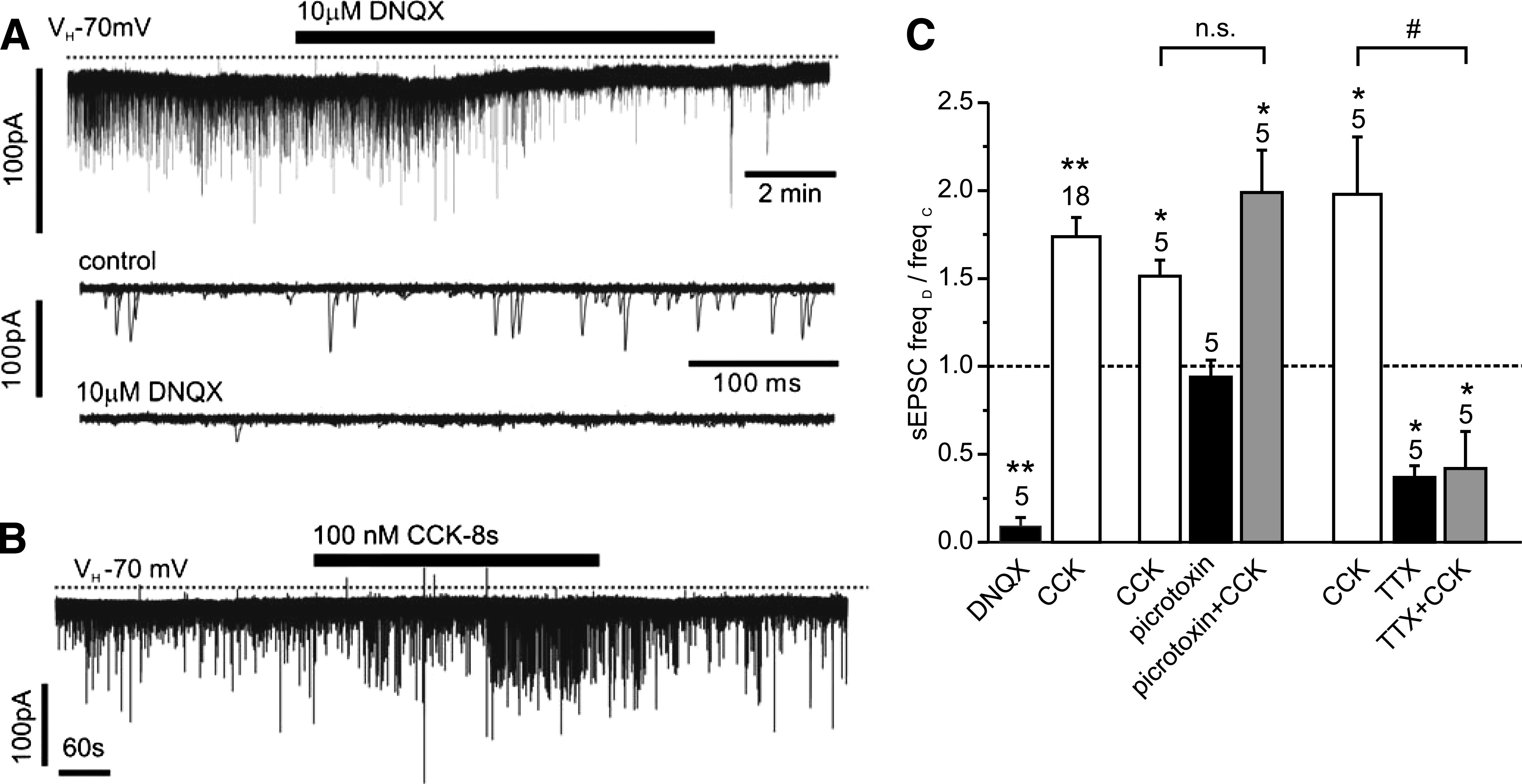
CCK stimulation of sEPSCs is sensitive to TTX but not picrotoxin. A: The vast majority of sEPSCs in PPG neurons are glutamatergic, as demonstrated by their inhibition by 10 μmol/L DNQX in this voltage-clamp recording at a holding potential of −70 mV. Bottom traces: Overlay of 15 consecutive 500 ms traces from the recording above under control conditions (top) and in the presence of DNQX (bottom). B: CCK-8s, bath-applied at 100 nmol/L, led to an increase in sEPSC frequency. C: Mean normalized effects of CCK on sEPSC frequency in the presence or absence of various drugs. The mean sEPSC frequency in presence of the drug (freqD) as a fraction of the frequency in the absence of any drug (freqC) is plotted. The excitatory CCK effect is not reduced by the GABA and glycine receptor antagonist picrotoxin (30 μmol/L) but is prevented by TTX (0.5 μM). *P < 0.05 compared with control; **P < 0.01 compared with control; #P < 0.05 compared with CCK. Numbers of cells tested are given above the bars.
Suppression of food intake by CCK-8s has previously been reported to be modulated by picrotoxin (30), and additionally CCK-8s has been shown to modulate γ-aminobutyric acid (GABA)ergic activity in the rat NTS (31). To verify that CCK activation of PPG cell activity is not because of the inhibition of an ongoing inhibitory input from GABAergic or glycinergic neurons, the effects of the GABAA and glycine receptor antagonist picrotoxin were tested on sEPSC frequency. Bath application of 30 μmol/L picrotoxin under control conditions had no significant effect on sEPSC frequency, thus demonstrating the absence of a tonic inhibition of glutamatergic input (n = 5). Furthermore, picrotoxin failed to affect the stimulatory effect of 100 nmol/L CCK-8s on EPSC frequency (Fig. 2; n = 5), thus precluding the involvement of inhibitory interneurons at any stage of the CCK pathway of PPG cell activation.
We previously demonstrated that the majority of NTS PPG neurons receive direct (monosynaptic) glutamatergic input from vagal afferent fibers (19). As the cell bodies of primary afferent neurons in the solitary tract are lost in the slice preparation, their nerve terminals should not fire action potentials; if they were responsible for the tonic glutamatergic tone on PPG neurons, this should be TTX independent. However, bath application of 0.5 μmol/L TTX reduced the frequency of EPSCs by 63 ± 7% (n = 5; Fig. 2), thus indicating that sEPSC activity is partially a result of action potential–dependent electrical activity within the in vitro brain stem slice preparation. Preincubation with 0.5 μmol/L TTX also completely prevented the stimulatory effect of CCK-8 on sEPSC frequency (n = 5; Fig. 2).
Postsynaptic effects of melanocortin receptor agonist on PPG neurons.
POMC neurons within the NTS were shown previously to be activated by peripherally administered CCK, as determined by cFOS staining (25,32). Furthermore, CCK administration failed to suppress food intake in the hyperphagic melanocortin receptor 4 (MC-4) knock-out mouse model (25). Consequently, we tested whether PPG neurons are electrically stimulated by MC-4 receptor activation. Bath application of the melanocortin receptor agonist MT-II (100 nmol/L) had no significant effect on firing rate (1.5 ± 0.7 Hz vs. 1.7 ± 0.8 Hz; Fig. 3), membrane potential (−52 ± 1 mV vs. −52 ± 2 mV), or input resistance of PPG neurons (n = 7), thus making it unlikely that the observed effects of CCK on PPG cells are mediated via NTS POMC neurons.
FIG. 3.

Epinephrine stimulation of firing frequency of PPG neurons is occluded by DNQX. A: Current-clamp recording showing the effect of bath application of 100 μmol/L norepinephrine on firing frequency of PPG neuron. B: Bath application of 10 μmol/L epinephrine leads to an increase in spontaneous action potential firing frequency of PPG neurons. This effect of epinephrine is prevented by the non-NMDA glutamate receptor antagonist DNQX (10 μmol/L). Top: instantaneous firing frequency; bottom: segments of the original current-clamp recording at time points i, ii, iii, and iv, indicated by arrows. C: Mean data for the change in firing frequency (FR) during experiments as shown in A. Epinephrine (10 μmol/L) and 100 μmol/L norepinephrine, but not 100 nmol/L Melanotan II (MT-II), 10 μmol/L norepinephrine, or 10 μmol/L dopamine significantly increased firing rate. The effect of 10 μmol/L epinephrine is occluded by 10 μmol/L DNQX. Number of recordings for each condition is given above the bars. *P < 0.05, **P < 0.01, compared with control; ##P < 0.01 compared with epinephrine.
Postsynaptic effects of catecholamines on PPG neurons.
It has been shown previously that systemically applied CCK activates noradrenergic cells in the NTS (33). To investigate the hypothesis that these neurons might mediate the effects of CCK on PPG cells, catecholamines were applied to PPG neurons in current-clamp recordings. Epinephrine (10 μmol/L) reversibly increased the firing rate from 1.6 ± 0.3 to 2.6 ± 0.6 Hz in five of six PPG neurons tested (Fig. 3) and had no effect on the remaining cell. Similarly, norepinephrine at a concentration of 100 μmol/L, but not 10 μmol/L, increased the action potential frequency by 85 ± 37% in six of eight cells tested (Fig. 3). Dopamine (10 μmol/L), by contrast, had no effect on firing rate (control 2.2 ± 0.4 Hz; dopamine 2.1 ± 0.5 Hz; n = 5). None of the catecholamines tested had a significant effect on membrane potential or input resistance, suggesting that the action of epinephrine and norepinephrine was presynaptic. In support of this notion, the effect of 10 μmol/L epinephrine on action potential frequency was suppressed by 10 μmol/L DNQX (Fig. 3). Thus, similar to CCK, epinephrine and norepinephrine appear to activate PPG neurons by increasing glutamatergic input.
Epinephrine increases spontaneous synaptic transmission.
The ionotropic glutamate receptor antagonist kynurenic acid (1 mmol/L) reduced the frequency of synaptic events by 93 ± 4% (n = 5; Fig. 4), whereas epinephrine (10 μmol/L; n = 12) significantly increased the frequency of EPSCs (Fig. 4). However, in the presence of 1 mmol/L kynurenic acid the effect of epinephrine was occluded (n = 4), thus verifying that epinephrine acted by modulating the glutamatergic input onto the PPG cells (Fig. 4). The α2-adrenoceptor agonists clonidine (10 μmol/L) and dexmedetomidine (1 μmol/L) had no significant effect on EPSC frequency, whereas the α1-receptor agonist phenylephrine (50 μmol/L) significantly increased EPSC frequency (Fig. 4).
FIG. 4.

Epinephrine acts on α1-adrenoreceptors and increases the frequency of spontaneous glutamatergic EPSCs. A: Voltage-clamp recording from a PPG neuron at a holding potential (VH) of −70 mV demonstrating the effects of epinephrine on sEPSCs. B: Overlay of 15 consecutive 500 ms traces from the recording shown in A under control conditions (top) and in the presence of epinephrine (bottom). C: Overlay of 15 consecutive 500 ms traces under control conditions and in the presence of phenylephrine or clonidine, respectively, as indicated above each overlay. Phenylephrine, but not clonidine, led to an increase in sEPSC frequency. D: Mean normalized effects of epinephrine and selective α1- (phenylephrine) and α2- (clonidine, dexmedetomidine) adrenoreceptor agonists on sEPSC frequency. The mean sEPSC frequency in presence of the drug (freqD) as a fraction of the frequency in the absence of any drug (freqC) is plotted. The effect of epinephrine is blocked by the glutamate receptor antagonist kynurenic acid (Kyn) and by the α-adrenergic receptor antagonist yohimbine. Yohimbine also blocked the effect of phenylephrine. Kyn itself blocked the majority of sEPSCs. *P < 0.05, **P < 0.01, compared with control. Numbers of cells tested are given above the bars.
Yohimbine (10 μmol/L), an α-receptor antagonist, had no significant effect on sEPSC frequency by itself (n = 5) but prevented the stimulatory effect of epinephrine on EPSC frequency (Fig. 4; n = 5). Similarly, yohimbine prevented the increase in EPSC frequency triggered by 100 nmol/L CCK-8s (n = 4; Fig. 5). By contrast, the β-adrenoreceptor antagonist ICI-118,551 (10 μmol/L) failed to reduce the stimulatory effect of CCK-8s on EPSC frequency (n = 5; Fig. 5). These results suggest that CCK acts on PPG neurons via modulation of adrenergic inputs.
FIG. 5.
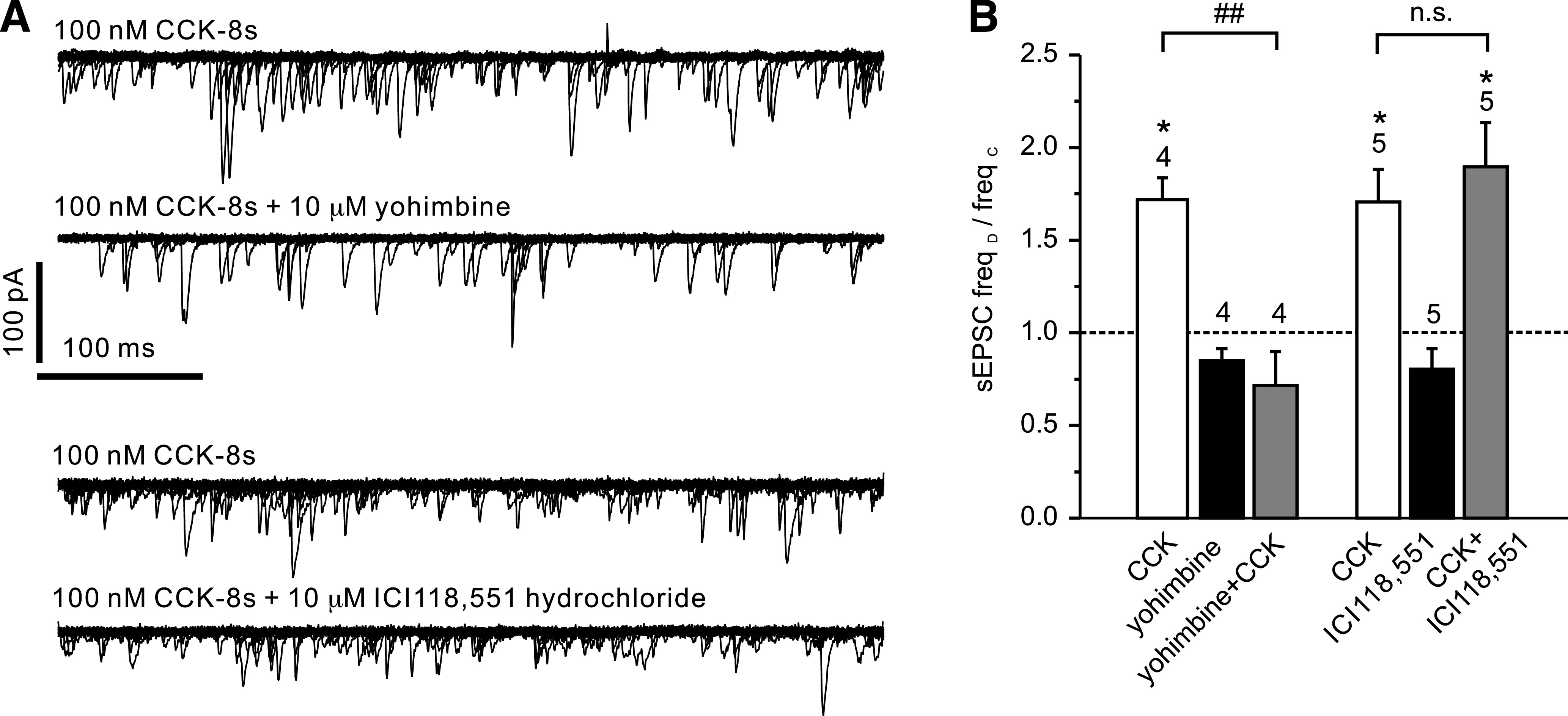
CCK stimulation of spontaneous EPSCs is sensitive to yohimbine. A: Overlay of 15 consecutive 500 ms traces from a recording like that shown in Fig. 2B in the presence of CCK (top) and CCK after preincubation with yohimbine (second from top). CCK-8s, bath-applied at 100 nmol/L, led to an increase in sEPSC frequency. This effect could be blocked by 10 μmol/L yohimbine. Bottom two traces show overlays from a recording where CCK was first applied alone (top trace) and then in the presence of the β-adrenoreceptor antagonist ICI118,551 hydrochloride (10 μmol/L; bottom trace). The CCK effect was not reduced by the β-adrenoreceptor antagonist. B: Mean normalized effects of CCK in the presence or absence of various drugs on sEPSC frequency from recordings as depicted in A. The mean sEPSC frequency in presence of the drug (freqD) as a fraction of the frequency in the absence of any drug (freqC) is plotted. The effect of CCK is blocked by yohimbine (10 μmol/L) but not ICI118,551. *P < 0.05 compared with control; ##P < 0.01 compared with CCK. Numbers of cells tested are given above the bars.
DISCUSSION
Application of CCK led to a significant increase in the frequency of spontaneous glutamatergic EPSCs in ∼50% of PPG neurons tested. This effect was sufficient to increase the firing frequency of these neurons and is therefore likely to be of physiological relevance. It is not clear whether the lack of response in the remaining PPG cells was caused by severed connections in the acute slice preparation or whether it reflects the existence of different subpopulations of PPG neurons that differ in their responsiveness to CCK. Similarly, ∼20% PPG neurons failed to respond to epinephrine or norepinephrine with a change in electrical activity.
It is well established that CCK activates vagal primary afferents, interacting with CCK receptors in the periphery (22,23,34,35), but potentially also directly within the dorsal vagal complex (27,31,36). It has been shown that neurons of the NTS centralis were stimulated by CCK-8 via an increase in spontaneous glutamatergic but not GABAergic synaptic transmission (37), similar to the observation made here. In contrast with our current observations, however, Baptista et al. (37) found that the effect of CCK persisted in the presence of TTX, suggesting a direct presynaptic effect of CCK. In their study, ∼50% of those cells that responded to CCK were tyrosine hydroxylase immunoreactive and thus catecholaminergic. Catecholaminergic NTS neurons tend to be second order neurons (38), but a follow-up study by Baptista et al. (27) showed that the CCK response persisted in vagally deafferented rats, thus suggesting that CCK does not necessarily act on vagal terminals within the NTS. Our present study also indicates that although PPG neurons are mainly second order neurons (19), CCK-8 has no effect on the vagal afferents impinging directly onto these cells, but likely involves the activation of catecholaminergic cells, which modulate the release of glutamate onto the PPG neurons (Fig. 6).
FIG. 6.
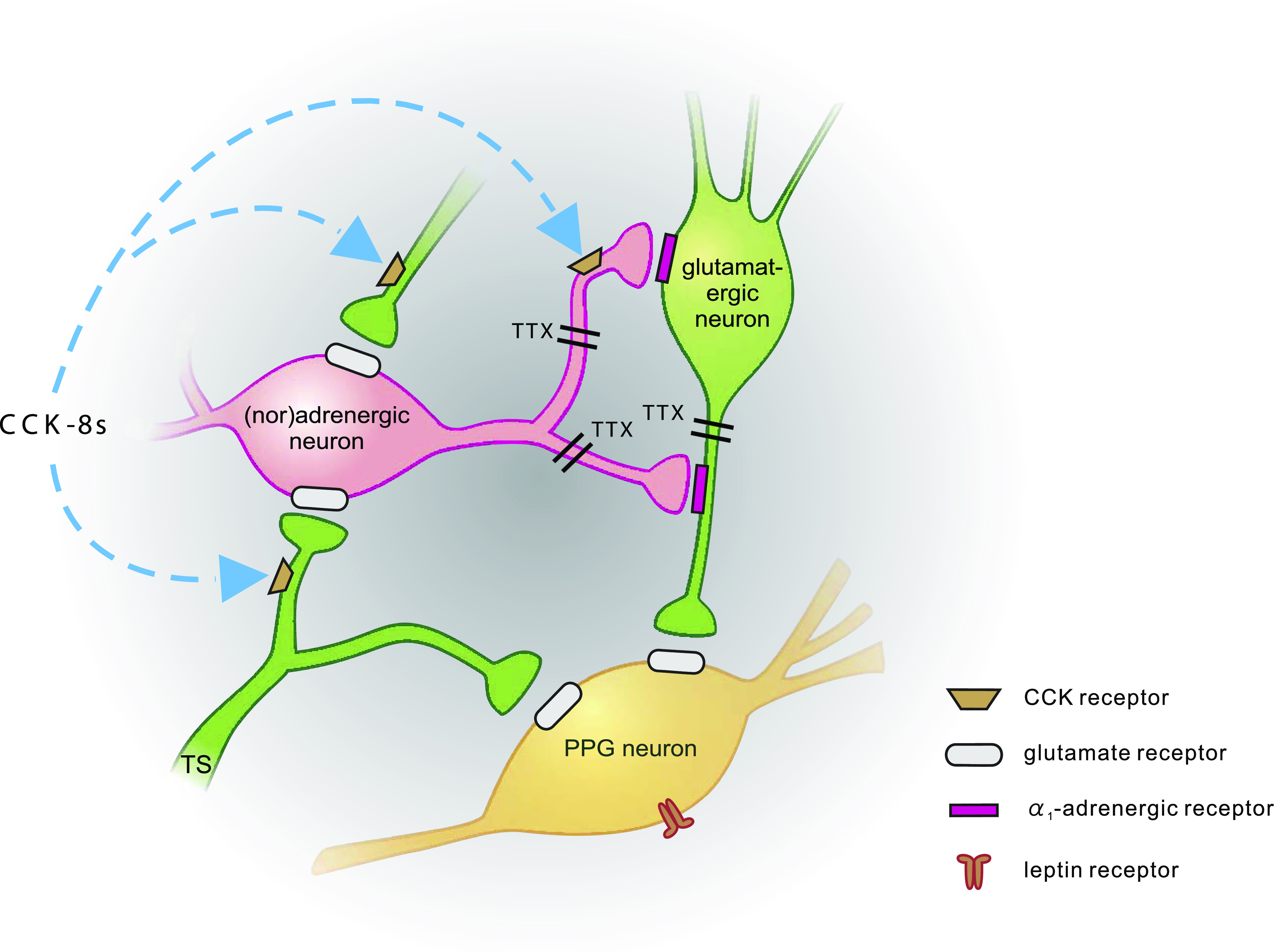
Schematic representation of synaptic inputs to PPG neurons. PPG neurons (yellow) express leptin receptors (Hisadome et al. [19]) and receive direct glutamatergic input both from the tractus solitarius (TS; TTX-insensitive; Hisadome et al. [(19)]) and local glutamatergic neurons (green; TTX-sensitive). Input from adrenergic/noradrenergic neurons (pink) is indirect via α1-adrenergic receptors, activation of which enhances glutamatergic input to PPG neurons. CCK enhances the activity of PPG neurons. Its effect is occluded by either α-adrenergic receptor antagonists or non-NMDA glutamate receptor antagonists. Thus, it acts either on (nor)adrenergic cells or presynaptic from those, as suggested by Baptista et al. (37).
Although CCK-8 also activates POMC neurons in the NTS (25,32), our finding that the melanocortin receptor agonist Melanotan II had no effect on PPG neuron activity suggests that CCK stimulation of PPG neurons does not involve activation of MC-3 or MC-4 receptors.
α-Adrenergic modulation of PPG cell activity.
In the hypothalamus, norepinephrine action can elicit either eating or satiety, mirroring the receptor type involved (39). Within the paraventricular nucleus, activation of α1-adrenoceptors seems to elicit satiety (40) and activation of α2-adrenoceptors causes hyperphagia (41,42). The findings that the satiety factor CCK activates adrenergic/noradrenergic (A2) NTS neurons (37,43) suggest that these transmitters act as anorectic signals at the level of the NTS. This is supported by our finding that epinephrine/norepinephrine, like CCK and like leptin (19), have an excitatory effect on PPG neurons.
Resembling what is seen with CCK, epinephrine, or norepinephrine had no direct effect on PPG neurons, but modulated glutamatergic synaptic input to cause an increase in firing frequency. Similar to the findings in the paraventricular nucleus, we observed that PPG neurons, which are likely to convey anorectic signals, were activated by the α1-receptor agonist phenylephrine but not by the α2-receptor agonists clonidine and dexmedetomidine. Intraperitoneal application of yohimbine, which is widely used as an α2-adrenoreceptor antagonist has, however, been reported to suppress feeding in rats (44). The underlying mechanism was postulated to involve enhanced release of norepinephrine as a result of blockade of α2-autoreceptors, resulting in activation of catecholaminergic A2/C2 neurons in the NTS and A1/C1 neurons in the ventrolateral medulla. In the current study application of yohimbine had no effect on spontaneous EPSCs on PPG neurons. This might suggest that there is no input from A1/C1 or A2/C2 neurons onto the glutamatergic synapses on the PPG neurons or that the anorexigenic effect of systemic yohimbine is independent of PPG-neuronal activity. In our brain stem slices, however, yohimbine suppressed the effect of epinephrine on EPSC frequency in PPG neurons, suggesting it acts on postsynaptic receptors. In fact, it blocked the response to the α1-receptor agonist phenylephrine, indicating that at a concentration of 10 μmol/L yohimbine is not selective for α2-receptors in the NTS and that systemically administered yohimbine might affect feeding independently of α2-mediated disinhibition of catecholamine release.
Physiological relevance.
In addition to the A1/C1 and A2/C2 neurons in ventrolateral medulla and NTS, respectively, catecholaminergic neurons have been found in the area postrema of rat. These neurons have been shown to express GLP-1 receptors and have been hypothesized to act as a link between circulating GLP-1 and activation of hypothalamus-projecting NTS PPG neurons (45). Our results presented here, however, indicate that there is no direct catecholaminergic input onto PPG neurons and instead that these cells are excited indirectly by adrenoreceptor activation. Although we showed previously that GLP-1 had no effect on PPG cell activity in the same brain stem slice configuration (19), our results are not compatible with the idea that the adrenergic population found here to modulate PPG neurons are themselves GLP-1 responsive. Because we recently demonstrated that NTS-PPG neurons send varicose axons into the area postrema (5), an alternative explanation for the GLP-1 receptor–positive catecholaminergic cells in the area postrema might be that they are themselves responsive to GLP-1 released from NTS-PPG neurons.
Although it is clear that gut-derived, as well as centrally injected, CCK causes satiety (20,28); at present, we do not know whether CCK receptors within the lower brain stem would be activated by peripherally released CCK that crosses the blood brain barrier or whether CCK released as a transmitter from central neurons or from vagal afferents serves these receptors. A number of studies have demonstrated CCK-immunoreactive fibers and even cell bodies in the caudal NTS of rodents (46–48). It has been suggested that a large fraction of these cell bodies are catecholaminergic (48), but it is unclear whether the CCK-immunoreactive fibers found in the NTS are of local origin (47) or represent vagal afferent fibers, as suggested by Palkovits et al. (46). In any case, it remains to be established whether the release of CCK within the NTS is in fact linked to food ingestion. It is, however, clear that CCK receptor activation within the NTS causes a reduction in food intake (28). Furthermore, intraperitoneal application of CCK-8 induces c-FOS immunoreactivity in the vagal complex, and immunohistochemical characterization of these c-FOS–positive cells revealed that they include POMC, catecholaminergic, and GLP-1-producing neurons (25,26).
A physiological role for PPG neurons in conveying anorectic signals is supported by the findings that they exhibit c-Fos immunoreactivity in response to leptin, CCK, and gastric distension (16,17,26) and are activated directly by leptin (19) and indirectly by CCK and epinephrine/norepinephrine in vitro. This suggests that NTS-PPG neurons integrate a variety of peripheral signals that indicate both long-term energy balance and short-term nutritional and digestional status to produce an output signal to feeding and autonomic circuits to optimize digestion and assimilation of nutrients and regulate calorific intake.
ACKNOWLEDGMENTS
This study was supported by the Medical Research Council, U.K. (Ref: G0600928 to K.H. and S.T.), the Wellcome Trust (Ref: 088357 to F.M.G. and Ref: 084210 to F.R.), and a Japan Society for the Promotion of Science International Collaboration Program grant (to K.H.).
No potential conflicts of interest relevant to this article were reported.
K.H. researched data and edited the manuscript. F.R. and F.M.G. contributed to the study design and edited the manuscript. S.T. designed the study and wrote the manuscript.
REFERENCES
- 1.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev 2007;87:1409–1439 [DOI] [PubMed] [Google Scholar]
- 2.Trapp S, Hisadome K. Glucagon-like peptide 1 and the brain: central actions-central sources? Auton Neurosci 2011;161:14–19 [DOI] [PubMed] [Google Scholar]
- 3.Merchenthaler I, Lane M, Shughrue P. Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J Comp Neurol 1999;403:261–280 [DOI] [PubMed] [Google Scholar]
- 4.Jin SL, Han VK, Simmons JG, Towle AC, Lauder JM, Lund PK. Distribution of glucagonlike peptide I (GLP-I), glucagon, and glicentin in the rat brain: an immunocytochemical study. J Comp Neurol 1988;271:519–532 [DOI] [PubMed] [Google Scholar]
- 5.Llewellyn-Smith IJ, Reimann F, Gribble FM, Trapp S. Preproglucagon neurons project widely to autonomic control areas in the mouse brain. Neuroscience 2011;180:111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larsen PJ, Tang-Christensen M, Holst JJ, Orskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience 1997;77:257–270 [DOI] [PubMed] [Google Scholar]
- 7.Hayes MR, Bradley L, Grill HJ. Endogenous hindbrain glucagon-like peptide-1 receptor activation contributes to the control of food intake by mediating gastric satiation signaling. Endocrinology 2009;150:2654–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest 1998;101:515–5209449682 [Google Scholar]
- 9.Williams DL, Baskin DG, Schwartz MW. Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology 2009;150:1680–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chelikani PK, Haver AC, Reidelberger RD. Intravenous infusion of glucagon-like peptide-1 potently inhibits food intake, sham feeding, and gastric emptying in rats. Am J Physiol Regul Integr Comp Physiol 2005;288:R1695–R1706 [DOI] [PubMed] [Google Scholar]
- 11.Baggio L, Kieffer TJ, Drucker DJ. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, regulates fasting glycemia and nonenteral glucose clearance in mice. Endocrinology 2000;141:3703–3709 [DOI] [PubMed] [Google Scholar]
- 12.Kinzig KP, D’Alessio DA, Seeley RJ. The diverse roles of specific GLP-1 receptors in the control of food intake and the response to visceral illness. J Neurosci 2002;22:10470–10476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang-Christensen M, Larsen PJ, Göke R, et al. Central administration of GLP-1-(7-36) amide inhibits food and water intake in rats. Am J Physiol 1996;271:R848–R856 [DOI] [PubMed] [Google Scholar]
- 14.Turton MD, O’Shea D, Gunn I, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 1996;379:69–72 [DOI] [PubMed] [Google Scholar]
- 15.Abbott CR, Monteiro M, Small CJ, et al. The inhibitory effects of peripheral administration of peptide YY(3-36) and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal-brainstem-hypothalamic pathway. Brain Res 2005;1044:127–131 [DOI] [PubMed] [Google Scholar]
- 16.Elias CF, Kelly JF, Lee CE, et al. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol 2000;423:261–281 [PubMed] [Google Scholar]
- 17.Vrang N, Phifer CB, Corkern MM, Berthoud HR. Gastric distension induces c-Fos in medullary GLP-1/2-containing neurons. Am J Physiol Regul Integr Comp Physiol 2003;285:R470–R478 [DOI] [PubMed] [Google Scholar]
- 18.Reimann F, Habib AM, Tolhurst G, Parker HE, Rogers GJ, Gribble FM. Glucose sensing in L cells: a primary cell study. Cell Metab 2008;8:532–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hisadome K, Reimann F, Gribble FM, Trapp S. Leptin directly depolarizes preproglucagon neurons in the nucleus tractus solitarius: electrical properties of glucagon-like Peptide 1 neurons. Diabetes 2010;59:1890–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaudhri O, Small C, Bloom S. Gastrointestinal hormones regulating appetite. Philos Trans R Soc Lond B Biol Sci 2006;361:1187–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibbs J, Young RC, Smith GP. Cholecystokinin decreases food intake in rats. J Comp Physiol Psychol 1973;84:488–495 [DOI] [PubMed] [Google Scholar]
- 22.Raybould HE, Gayton RJ, Dockray GJ. Mechanisms of action of peripherally administered cholecystokinin octapeptide on brain stem neurons in the rat. J Neurosci 1988;8:3018–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith GP, Jerome C, Cushin BJ, Eterno R, Simansky KJ. Abdominal vagotomy blocks the satiety effect of cholecystokinin in the rat. Science 1981;213:1036–1037 [DOI] [PubMed] [Google Scholar]
- 24.Dockray GJ. Cholecystokinin and gut-brain signalling. Regul Pept 2009;155:6–10 [DOI] [PubMed] [Google Scholar]
- 25.Fan W, Ellacott KL, Halatchev IG, Takahashi K, Yu P, Cone RD. Cholecystokinin-mediated suppression of feeding involves the brainstem melanocortin system. Nat Neurosci 2004;7:335–336 [DOI] [PubMed] [Google Scholar]
- 26.Rinaman L. Interoceptive stress activates glucagon-like peptide-1 neurons that project to the hypothalamus. Am J Physiol 1999;277:R582–R590 [DOI] [PubMed] [Google Scholar]
- 27.Baptista V, Browning KN, Travagli RA. Effects of cholecystokinin-8s in the nucleus tractus solitarius of vagally deafferented rats. Am J Physiol Regul Integr Comp Physiol 2007;292:R1092–R1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blevins JE, Stanley BG, Reidelberger RD. Brain regions where cholecystokinin suppresses feeding in rats. Brain Res 2000;860:1–10 [DOI] [PubMed] [Google Scholar]
- 29.Balfour RH, Hansen AM, Trapp S. Neuronal responses to transient hypoglycaemia in the dorsal vagal complex of the rat brainstem. J Physiol 2006;570:469–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson MC, Denson D, Bedford JA, Hunsinger RN. Pharmacological manipulation of sincalide (CCK-8)-induced suppression of feeding. Peptides 1983;4:351–357 [DOI] [PubMed] [Google Scholar]
- 31.Branchereau P, Champagnat J, Roques BP, Denavit-Saubie M. CCK modulates inhibitory synaptic transmission in the solitary complex through CCKB sites. Neuroreport 1992;3:909–912 [DOI] [PubMed] [Google Scholar]
- 32.Appleyard SM, Bailey TW, Doyle MW, et al. Proopiomelanocortin neurons in nucleus tractus solitarius are activated by visceral afferents: regulation by cholecystokinin and opioids. J Neurosci 2005;25:3578–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rinaman L, Hoffman GE, Dohanics J, Le WW, Stricker EM, Verbalis JG. Cholecystokinin activates catecholaminergic neurons in the caudal medulla that innervate the paraventricular nucleus of the hypothalamus in rats. J Comp Neurol 1995;360:246–256 [DOI] [PubMed] [Google Scholar]
- 34.Dockray GJ. The versatility of the vagus. Physiol Behav 2009;97:531–536 [DOI] [PubMed] [Google Scholar]
- 35.Moran TH, Baldessarini AR, Salorio CF, Lowery T, Schwartz GJ. Vagal afferent and efferent contributions to the inhibition of food intake by cholecystokinin. Am J Physiol 1997;272:R1245–R1251 [DOI] [PubMed] [Google Scholar]
- 36.Holmes GM, Tong M, Travagli RA. Effects of brain stem cholecystokinin-8s on gastric tone and esophageal-gastric reflex. Am J Physiol Gastrointest Liver Physiol 2009;296:G621–G631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baptista V, Zheng ZL, Coleman FH, Rogers RC, Travagli RA. Cholecystokinin octapeptide increases spontaneous glutamatergic synaptic transmission to neurons of the nucleus tractus solitarius centralis. J Neurophysiol 2005;94:2763–2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Appleyard SM, Marks D, Kobayashi K, Okano H, Low MJ, Andresen MC. Visceral afferents directly activate catecholamine neurons in the solitary tract nucleus. J Neurosci 2007;27:13292–13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wellman PJ. Norepinephrine and the control of food intake. Nutrition 2000;16:837–842 [DOI] [PubMed] [Google Scholar]
- 40.Wellman PJ, Davies BT. Reversal of phenylpropanolamine anorexia in rats by the alpha-1 receptor antagonist benoxathian. Pharmacol Biochem Behav 1991;38:905–908 [DOI] [PubMed] [Google Scholar]
- 41.Goldman CK, Marino L, Leibowitz SF. Postsynaptic alpha 2-noradrenergic receptors mediate feeding induced by paraventricular nucleus injection of norepinephrine and clonidine. Eur J Pharmacol 1985;115:11–19 [DOI] [PubMed] [Google Scholar]
- 42.Leibowitz SF. Hypothalamic paraventricular nucleus: interaction between alpha 2-noradrenergic system and circulating hormones and nutrients in relation to energy balance. Neurosci Biobehav Rev 1988;12:101–109 [DOI] [PubMed] [Google Scholar]
- 43.Rinaman L. Hindbrain noradrenergic lesions attenuate anorexia and alter central cFos expression in rats after gastric viscerosensory stimulation. J Neurosci 2003;23:10084–10092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Myers EA, Banihashemi L, Rinaman L. The anxiogenic drug yohimbine activates central viscerosensory circuits in rats. J Comp Neurol 2005;492:426–441 [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto H, Kishi T, Lee CE, et al. Glucagon-like peptide-1-responsive catecholamine neurons in the area postrema link peripheral glucagon-like peptide-1 with central autonomic control sites. J Neurosci 2003;23:2939–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palkovits M, Kiss JZ, Beinfeld MC, Williams TH. Cholecystokinin in the nucleus of the solitary tract of the rat: evidence for its vagal origin. Brain Res 1982;252:386–390 [DOI] [PubMed] [Google Scholar]
- 47.Takagi H, Kubota Y, Mori S, Tateishi K, Hamaoka T, Tohyama M. Fine structural studies of cholecystokinin-8-like immunoreactive neurons and axon terminals in the nucleus of tractus solitarius of the rat. J Comp Neurol 1984;227:369–379 [DOI] [PubMed] [Google Scholar]
- 48.Kawai Y, Takagi H, Tohyama M. Co-localization of neurotensin- and cholecystokinin-like immunoreactivities in catecholamine neurons in the rat dorsomedial medulla. Neuroscience 1988;24:227–236 [DOI] [PubMed] [Google Scholar]