

Abstract
Kisspeptin (Kiss1) and neurokinin B (NKB) (encoded by the Kiss1 and Tac2 genes, respectively) are indispensable for reproduction. In the female of many species, Kiss1 neurons in the arcuate nucleus (ARC) coexpress dynorphin A and NKB. Such cells have been termed Kiss1/NKB/Dynorphin (KNDy) neurons, which are thought to mediate the negative feedback regulation of GnRH/LH secretion by 17β-estradiol. However, we have less knowledge about the molecular physiology and regulation of Kiss1/Kiss1-expressing neurons in the ARC of the male. Our work focused on the adult male mouse, where we sought evidence for coexpression of these neuropeptides in cells in the ARC, assessed the role of Kiss1 neurons in negative feedback regulation of GnRH/LH secretion by testosterone (T), and investigated the action of NKB on KNDy and GnRH neurons. Results showed that 1) the mRNA encoding Kiss1, NKB, and dynorphin are coexpressed in neurons located in the ARC; 2) Kiss1 and dynorphin A mRNA are regulated by T through estrogen and androgen receptor-dependent pathways; 3) senktide, an agonist for the NKB receptor (neurokinin 3 receptor, encoded by Tacr3), stimulates gonadotropin secretion; 4) KNDy neurons express Tacr3, whereas GnRH neurons do not; and 5) senktide activates KNDy neurons but has no discernable effect on GnRH neurons. These observations corroborate the putative role for KNDy neurons in mediating the negative feedback effects of T on GnRH/LH secretion and provide evidence that NKB released from KNDy neurons is part of an auto-feedback loop that generates the pulsatile secretion of Kiss1 and GnRH in the male.
Neurokinin B (NKB) and its receptor, neurokinin 3 receptor (NK3R), play key roles in the regulation of reproduction in mammals. Humans bearing mutations in either the TAC3 or TACR3 gene (encoding NKB and NK3R, respectively) exhibit hypogonadotropic hypogonadism and infertility (1, 2). Although Tac2 (encoding NKB in rodents) and Tacr3 are expressed throughout the brain (3), they are expressed together in cells of the arcuate nucleus (ARC), a nodal point for the regulation of GnRH secretion (4). These cells are often called Kiss1/NKB/Dynorphin (KNDy) neurons, because they coexpress kisspeptin (Kiss1), NKB (Tac2), and dynorphin A (Pdyn) (5–7). Kiss1 secreted from KNDy cells acts on GnRH neurons, which express Kiss1 receptor (Kiss1r) (8–10). Moreover, KNDy neurons are thought to mediate the negative feedback control of GnRH secretion by sex steroids (6, 11). In the female mouse, 17β-estradiol (E2) inhibits the expression of Kiss1, Tac2, and Pdyn in KNDy neurons by acting through estrogen receptor (ER)α (6, 12). Likewise, in the male, testosterone (T) inhibits Kiss1 expression in the ARC, but T can act through either ERα (after aromatization to E2) or the androgen receptor (AR) (13). If and how T regulates the expression of Tac2, Tacr3, and Pdyn in KNDy neurons is unexplored, and addressing this problem was one of our experimental objectives.
Based on studies of KNDy neurons in the ARC, we and others have suggested that these cells constitute a pacemaker network that drives the ultradian secretion of GnRH (6, 14, 15). This system requires the stimulatory effect of NKB/NK3R signaling acting through recurrent collaterals to provide a feed-forward regenerative and synchronizing input to KNDy neurons. Operation of the KNDy pulse generator depends on phase-delayed inhibitory feedback, which would subsequently suppress volleys of action potentials after NKB stimulation, a function we originally attributed to dynorphin/κ opioid receptor (KOR) (encoded by Oprk1) signaling via recurrent collaterals (6, 15). Recent evidence supports the proposition that pulsatile secretion of Kiss1 drives the ultradian rhythm of GnRH secretion. First, studies in monkeys have shown that pulses of Kiss1 measured in the median eminence are temporally correlated with GnRH pulses (16). Second, volleys of multiunit activity recorded near KNDy neurons in goats are periodic and coordinated with LH pulses (15). To understand more about KNDy neurons, we investigated their characteristics in the male mouse by analyzing the coexpression of Kiss1, Tac2, Tacr3, Pdyn, and Oprk1 in the ARC and characterizing their regulation by T. Moreover, we evaluated the ability of NKB agonists to induce GnRH/LH secretion and investigated the effect of NK3R agonists and antagonists on the biophysical properties of Kiss1-expressing neurons in vivo.
Materials and Methods
Animals
Animals were housed at the University of Washington, and all procedures and surgeries conducted there were in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Except as noted, all procedures were approved by the Institutional Animal Care and Use Committee at the University of Washington School of Medicine in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Adult male C57BL/6J mice (8 wk old) were purchased from The Jackson Laboratory. Mice carrying an AR (Ar) gene null mutation on a mixed 129S4/SvJaeSor x FVB/NJ background were generated in the lab of R. E. Braun at The Jackson Laboratory. Artm1.1Reb mice lack Ar in all cells in all tissues. They were generated by mating 129S4/SvJaeSor;Artm1Reb/X mice (17), in which the first exon of the Ar gene is flanked by inverted loxP sites, with stimulated by retinoic acid gene 8 (Stra8) cre recombinase male transgenic mice, FVB/NJ;(Tg(Stra8-cre) (18), to generate Artm1.1Reb/+ female mice, in which the first exon of Ar is stably inverted. Inversion of the first exon of Ar generates a null allele (17). Artm1.1Reb/+ female mice were mated with wild-type (WT) 129S4/SvJaeSor males to generate Artm1.1Reb/Y males. Hereafter, the Artm1.1Reb/Y mice will be referred to as Ar knockout (KO) mutant mice to denote Ar mice with disruption of Ar in all cells. The Jackson Laboratory Animal Care and Use Committee approved all animal studies. Adult male C57BL/6J mice intended for hormonal measurements were bred in the vivarium of the University of Córdoba. Experimental procedures in these animals were approved by the Córdoba University Ethical Committee and conducted in accordance with the European Union normative for the use of experimental animals.
The Kiss1-CreGFP mice were produced by gene targeting technology. Briefly, a fragment that included approximately 5.0-kb DNA 5′ of the Kiss1 translation start site and approximately 8 kb on the 3′ side from a C57Bl/6 BAC clone was subcloned into Bluescript. The targeting construct contained a CreGFP fusion protein cassette inserted upstream of the start site for Kiss1. Mice were confirmed to have enhanced green fluorescence protein (GFP) expression profiles that were indistinguishable from the reported distribution of Kiss1-expressing neurons in the mouse, with specific labeling in the medial and caudal ARC, as well as the anteroventral periventricular nucleus (8, 12, 19). Kiss1-CreGFP animals were developed and bred at the University of Washington, then supplied to Oregon Health and Science University and Yale University.
Animal procedures at Oregon Health and Science University
Adult male Kiss1-CreGFP mice were maintained under constant temperature (21–23 C) and light (light on between 0600 and 1800 h local time), with food (standard chow) and water provided ad libitum and housed in groups of no more than five animals per cage. The animals were castrated under isofluorane anesthesia 7 d before experimentation and given a dose of 4 mg/kg carprofen (Rimadyl, Pfizer Animal Health, NY) immediately after surgery for analgesia. On the day of experimentation, the animal was given an ip dose of 15 mg of ketamine. The animal was then rapidly decapitated, its brain removed from the skull, and a diencephalic block was dissected. The resultant block was mounted on a cutting platform that was then secured in a vibratome well filled with ice-cold, oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid (aCSF) (208 mm sucrose, 2 mm KCl, 1 mm MgCl, 1.25 mm NaH2PO4, 10 mm HEPES, 26 mm NaHCO3, 10 mm dextrose, 2 mm MgSO4, and 1 mm CaCl2). Four coronal slices (240 μm) were cut through the hypothalamus. The slices were transferred to a multiwell auxiliary chamber containing oxygenated aCSF (124 mm NaCl, 5 mm KCl, 1.44 mm NaH2PO4, 5 mm HEPES, 10 mm Dextrose, 25.99 mm NaHCO3, and 2 mm CaCl2) and kept there until recovery, at which time the slices were prepared for cell harvesting as described below.
Animal procedures at Yale University
GnRH-GFP mice were kindly provided by Sue Moenter (University of Virginia, Charlottesville, VA) and housed at Yale University. Tissue was collected from Kiss1-CreGFP and GnRH-GFP mice according to the requirements of the Institutional Animal Care and Use Committee at Yale University. All animals were housed in groups of three to five, maintained on a 14-h light, 10-h dark cycle with lights on at 0400 h, and had access to standard rodent chow and water ad libitum.
Gonadectomy (GDX) and steroid replacement
Testes were removed through a single abdominal incision from adult male mice while they were under isoflurane inhalation anesthesia (Abbott Laboratory, North Chicago, IL), delivered by a vaporizer (Veterinary Anesthesia Systems, Bend, OR). Testicular vasculature was sutured, and wound clips were used to close the incision. Immediately after GDX, oil-filled capsules (sham), E2 + oil-filled capsules, or dihydrotestosterone (DHT) (5α-androstan-17β-ol-3-one) + oil-filled capsules were implanted sc via a small midscapular incision at the base of the neck; wound clips were used to close the incision. T (4-androsten-17β-ol-3-one), DHT, and E2 were purchased from Sigma (St. Louis, MO). For T implants, SILASTIC brand tubing (inner diameter, 1.47 mm and outer diameter, 1.95 mm; Dow Corning Corp., Midland, MI) was cut to 15 mm, one end sealed with silicone glue and allowed to cure overnight. T crystals were packed into the tube to a length of 10 mm, and the remaining length of the tube was occluded with silicone glue. Implants were left to cure overnight. These implants maintain normal physiological levels of T (13). Capsules containing DHT were made as described for T. Similar capsules have previously been shown to produce significant androgenic actions at target tissues, e.g. spermatogenesis in gonadotropin-deficient mice (20, 21). Implants that significantly elevate serum E2 levels were constructed by packing SILASTIC brand tubing with 4 mm of an E2-cholesterol mix (1:4) (22). The day before surgery, implants were washed with two changes of 100% ethanol (10 min each) and then placed in physiological saline overnight. All untreated animals received empty (sham) capsules.
Tissue preparation
Blood was spun in a centrifuge to separate serum, which was stored at −20 C until hormone measurements were performed. Brains were removed for in situ hybridization (ISH), frozen on dry ice, and then stored at −80 C until sectioned. Brains were cut on a cryostat into 20-μm sections in the coronal plane (from the diagonal band of Broca to the mammillary bodies). Sections were thaw mounted onto SuperFrost Plus slides (VWR Scientific, West Chester, PA) and stored at −80 C. A single set (from five total per brain) was used for each ISH procedure (with adjacent sections 100 μm apart).
Radioimmunoassay
Serum levels of LH in animals treated with steroid replacement were measured at Northwestern University (Evanston, IL). Reagents for the LH assay were obtained from the NIH. For LH, the antiserum used was anti-rLH-S-11, and the standard was rLH-RP3. The assay sensitivity was 0.2 ng/ml, and the intraassay coefficient of variation was 4%.
Serum LH and FSH levels in animals treated centrally with senktide (or the vehicle control) were measured at the University of Córdoba in a volume of 25–50 μl by a double-antibody method with RIA kits supplied by the NIH (A. F. Parlow, National Institute of Diabetes and Digestive and Kidney Diseases, National Hormone and Peptide Program, Torrance, CA). LH-I-10 and FSH-I-9 were labeled with 125I in IODO-GEN tubes, following the instructions of the manufacturer (Pierce, Rockford, IL). Hormone concentrations were expressed in terms of the reference preparations LH-RP-3 and FSH-RP-2 that were used as standards. Intra- and interassay coefficients of variation were less than 8 and 10% for LH and less than 6 and 9% for FSH. The sensitivity of the assays was 0.2 ng/ml for LH and 0.8 ng/ml for FSH. For each hormone determination, all samples were measured in the same assay. The accuracy of hormone measurements was confirmed by assessment of mouse serum in samples containing known hormone concentrations.
Detection of Kiss1 mRNA
The probe used for detection of Kiss1 mRNA was previously described in (8). The Kiss1-specific sequence of the probe spans bases 76–486 of the mouse cDNA sequence (GenBank accession no. AF472576). The procedure for ISH is described briefly below.
Detection of other mRNA
The probes used for detection of Pdyn, Oprk1, Tac2, Tacr3, and Gnrh1 mRNA have been described elsewhere (6, 8, 10).
Single-label ISH of Kiss1, Tac2, and Pdyn mRNA
Kiss1, Tac2, and Pdyn mRNA sense and antisense probes were transcribed with T7 or T3 polymerase (Fermentas, Glen Burnie, MD), as previously described (6). Radiolabeled probes were synthesized in vitro by inclusion of the following ingredients in a volume of 20 μl: 250 μCi 33P-UTP (PerkinElmer Life Sciences, Boston, MA); 1 μg of linearized DNA (or 1 μg of PCR product); 0.5 mm each ATP, CTP, and GTP; and 40 U of polymerase. Residual DNA was digested with 4 U of deoxyribonuclease (Ambion, Foster City, CA), and the deoxyribonuclease reaction was terminated by addition of 2 μl of 0.5 m EDTA (pH 8.0). The riboprobes were separated from unincorporated nucleotides with NucAway Spin Columns (Ambion). Slides containing mouse hypothalamic sections were then processed as previously described (8, 23).
Double-label ISH
Antisense mouse Kiss1 or GnRH probe was transcribed from linearized pAMP1 plasmid containing the mouse Kiss1 or GnRH insert with T7 polymerase (Fermentas) (8). The cDNA template for the Pdyn, Oprk1, Tac2, and Tacr3 riboprobes was generated by PCR with primers that were designed to contain promoters for T7 RNA polymerase in the antisense direction and T3 RNA polymerase in the sense direction. Radiolabeled riboprobes for Oprk1, Tac2, and Tacr3 were synthesized as described above. Digoxigenin (DIG)-labeled Kiss1 or Gnrh1 antisense riboprobes were synthesized with T7 RNA polymerase and DIG labeling mix (Roche, Indianapolis, IN) according to the manufacturer's instructions. Slides were processed for double-labeled ISH as described previously (10). Slides were stored at 4 C and developed 8–12 d later.
Detection, quantification, and analysis of Pdyn, Oprk1, Tac2, Tacr3, and Gnrh1 mRNA
The brain sections were analyzed bilaterally unless otherwise specified. Slides from all of the animals were assigned a random three-letter code, alphabetized, and read under dark-field illumination with custom-designed software designed to count the total number of cells and the number of silver grains (corresponding to radiolabeled Pdyn, Oprk1, Tac2, and Tacr3 over each cell) (24). Kiss1 or Gnrh1 mRNA-containing cells were visualized under fluorescent illumination, and custom software was used to count the number of silver grains over each DIG-labeled cell, depending on which radiolabeled probe was used. The number of cells reported for experiment 1 represents the number of cells within the coronal sections containing the ARC for each set, not the total number of cells in the ARC. We presented cell number for experiment 1, because we sought to compare the fraction of Kiss1-expressing cells that coexpress Tac2 and the fraction of Tac2-expressing cells that coexpress Kiss1. For experiment 3, the results are presented as total mRNA, which reflects the number of detectable cells (for each species of mRNA) times average grain counts/cell per animal (in arbitrary units). In all instances, the total mRNA paralleled cell counts, but only values for total mRNA are presented. The starting and ending point of quantification was determined according to Paxinos and Franklin (from Interaural, 2.58 mm/Bregma −1.22 mm to Interaural 1.00 mm/Bregma −2.80 mm) (25). Signal to background ratios for individual cells were calculated; an individual cell was considered to be double-labeled if it had an signal to background ratio of 3 or more. For each animal, the number of double-labeled cells was calculated as a percent of the total number of Kiss1 or Gnrh1 mRNA-positive cells, then averaged across animals to produce a mean ± sem.
Harvesting GFP-labeled cells in the ARC
The method used for neuronal harvesting was according to previously established procedures (26) with some modifications as follows. Briefly, the ARC was microdissected and exposed to protease and gentle tituration to disperse the neurons. The dispersed cells were visualized using a Leitz inverted microscope, patched, and then harvested with gentle suction using the XenoWorks microinjector system (Sutter Instrument Co., Novato, CA). The contents of the pipette were expelled into a test tube containing a solution with 1 μl of Invitrogen Superscript III 5× buffer (Invitrogen, Carlsbad, CA), 15 U of RNasin (Promega, Madison, WI) and 10 mm of dithiothreitol in a total of 5 μl. Each harvested cell was RT as described previously (26) with modifications in which both random primers (100 ng/cell; Promega) and anchored oligo(dT)20 primer (400 ng/cell; Invitrogen), and Superscript III reverse-transcriptase (100 U/cell; Invitrogen) were used. In addition, harvested aCSF in the vicinity of the dispersed cells also underwent RT and was used as a control. Cells and tissue RNA used as negative controls were processed as described above but without RT. Each cell was then analyzed for the expression of Kiss1 mRNA by PCR as described below.
Primer design and single cell RT-PCR
Primer pairs for single cell RT-PCR were developed using the mRNA sequence of the respective genes from the National Center for Biotechnology Information. Primers were designed using the Clone Manager software (Sci Ed Software, Cary, NC) based on the mRNA sequences. Primers were as follows: mouse Kiss1 (132-bp product) accession no. NM_178260, forward primer 64–80 bp, reverse primer 195–179 bp; mouse β-actin (110-bp product) accession no. NM_007393, forward primer 416–435 bp, reverse primer 505–525 bp. The primers were designed to cross an intron to eliminate any genomic DNA contamination, and the PCR product in single cells was sequenced to confirm the correct product. The PCR was performed using 2 μl of cDNA template from each RT reaction in a 20 μl of PCR mix. Forty to 50 cycles of amplification were performed using a Bio-Rad C1000 Thermal Cycler (Bio-Rad, Hercules, CA) according to established protocols (26, 27). PCR products were visualized with ethidium bromide on a 2% agarose gel. Analysis of 104 cells from three animals revealed that the vast majority (96.0 ± 4.1%) of GFP-labeled neurons tested positive for Kiss1 mRNA (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org), and Kiss1 mRNA was undetectable in non-GFP-labeled neurons (six cells from three animals). All Kiss1-negative cells were positive for β-actin, indicating that cellular RNA was preserved (data not shown).
Slice preparation and recordings from Kiss1 and GnRH neurons
Animals were anesthetized with chloral hydrate (400 mg/kg ip) and killed by decapitation between 1200 and 1900 h. After decapitation, brains were removed, placed in a Petri dish containing aCSF, and trimmed to yield a small block containing the region of interest. Coronal slices of thickness 300 μm were obtained with a Vibratome 1500 (Vibratome Co., St. Louis, MO) and transferred to a Plexiglass recording chamber (1.5 ml) on the fixed stage of an Olympus BX50WI scope for visualized whole-cell recording (Olympus, New York, NY). The chamber was perfused continuously with normal aCSF at a rate of 2–3 ml/min, with its temperature maintained at 33 ± 0.5 C. One to 2 h later, the slice was used for recording.
Whole-cell current and voltage-clamp recordings were performed through the use of previously described methods (28). The low resistance (2.5–3.5 mΩ) patch pipettes were filled with a solution containing 125 mm K gluconate, 10 mm HEPES, 5 mm [1,2-bis(o-aminophenoxy)ethane-N,N,N′N′-tetraacetic acid] K4, 2.38 mm CaCl2, 4 mm Mg-ATP, 10 mm Na phosphocreatine, and 0.3 mm Na2-GTP (pH 7.32–7.35). Data were acquired using an Axoclamp-2B and pClamp 9 (Axon Instruments, Foster City, CA). No correction was made for the calculated liquid junction potential of approximately 11 mV for the internal solution.
A medium of aCSF containing 128 mm NaCl, 3 mm KC, 1.25 mm NaH2PO4, 10 mm D-glucose, 26 mm NaHCO3, 2 mm CaCl2, and 2 mm MgCl2 (pH 7.35–7.38) was prepared and equilibrated with 95% O2-5% CO2. NKB, senktide, and SB2222000 (an NK3R antagonist, purchased from Tocris, Ellisville, MO) and kisspeptin-10 (Sigma-Aldrich, St. Louis, MO) were diluted in aCSF from previously prepared stock solutions that were stored at −20 C. Agonists were applied through a Y-tube (29), and the NK3R receptor antagonist, SB222200, was bath applied.
Statistical analysis
All data are expressed as the mean ± sem for each group. One- and two-way ANOVA followed by t test were used to assess variation among experimental groups and confirm statistical significance. Significance level was set at P < 0.05. All analyses were performed with GraphPad Prism (GraphPad, La Jolla, CA).
Experimental design
Experiment 1. Coexpression of Kiss1 mRNA with Pdyn, Oprk1, Tac2, and Tacr3 mRNA in the ARC
To determine whether Kiss1 neurons in the mouse male ARC coexpress Pdyn, Oprk1, Tac2, and Tacr3 mRNA, WT mice were divided into two groups (n = 5 per group): GDX + sham and GDX + T replacement. Seven days after GDX, at approximately 0900 h, mice were anesthetized with isoflurane, their blood was collected for LH RIA by retroorbital bleeding (to confirm GDX), and they were killed by decapitation. Their brains were collected and processed for double-label ISH, as described above.
Experiment 2. Coexpression of Gnrh1 mRNA with Tacr3 mRNA in the preoptic area (POA)
To determine whether GnRH neurons in the POA of the male mouse express Tacr3 mRNA, we used the same experimental design as in experiment 1 but here sought evidence for coexpression of Gnrh1 and Tacr3 mRNA by double-label ISH in the POA of the brain samples used in experiment 1.
Experiment 3. The mechanisms by which T regulates Kiss1, Tac2, and Pdyn expression
To determine whether the regulation of Kiss1, Tac2, and Pdyn mRNA by T in the ARC is mediated by AR and/or ER, nine adult male Ar KO mice and nine WT littermates were GDX and three from each group received DHT, E2, or sham replacement. Tissue was collected and prepared for ISH as described for experiment 1.
Experiment 4. Gonadotropin response to senktide
To assess the effect of the NK3R agonist senktide (Sigma-Aldrich) on the secretion of LH and FSH, two groups of adult (8 wk old) intact male WT mice (n = 7–10/group) received an intracerebroventricular (icv) injection (8) of either vehicle (NaCl 0.9%) or senktide (600 pmol/3 μl) as described (6, 30); senktide was used instead of NKB in these experiments, because senktide is more soluble in saline than NKB. Thirty minutes later, the animals were bled, and serum was collected as described for experiment 1.
Experiment 5. Electrophysiological responses of Kiss1 and GnRH neurons to an NK3R agonist and antagonist
In this experiment, the effects of NKB, senktide (an NK3R agonist), and SB2222000 (an NK3R antagonist) on the electrophysiological properties of Kiss1 (in the ARC) and GnRH neurons were examined. Brain slices were prepared from the coronal sections of intact and castrated adult male Kiss1-CreGFP and GnRH-GFP mice (31). Whole-cell patch clamp recordings were performed on Kiss1 and GnRH cells identified by GFP, as described above.
Results
Coexpression of Kiss1/Pdyn/Oprk1/Tac2/Tacr3 genes and their regulation by T
The percentage of neurons coexpressing Pdyn, Oprk1, Tac2, and Tacr3 mRNA with Kiss1 mRNA in GDX mice with and without T treatment was assessed by double-label ISH. In the ARC, most of the neurons that expressed Kiss1 mRNA also expressed Tac2 (94%) and Pdyn (86%) (Fig. 1, A and B). Many of these KNDy neurons also expressed Tacr3 (76%) mRNA (Fig. 1C), but only a few expressed detectable levels of Oprk1 mRNA (6%) (Fig. 1D). We also noted that although most of the Pdyn mRNA-containing neurons in the ARC appeared to coexpress Kiss1 mRNA, only about half of the Tac2-expressing neurons also expressed Kiss1 mRNA (Fig. 1B). The percentage of neurons that expressed both Kiss1 mRNA and any one of the other genes studied was not altered by T treatment. However, T significantly reduced the number of KNDy (Kiss1 expressing) cells and the total number of Tac2-expressing cells (P < 0.05), apparently including some of those that did not coexpress Kiss1 (Fig. 2).
Fig. 1.

A, Representative photomicrographs of coronal sections of the ARC showing coexpression of Kiss1 mRNA and Pdyn mRNA. B, Kiss1 mRNA and Tac2 mRNA. C, Kiss1 mRNA and Tacr3 mRNA. And D, Kiss1 mRNA and Oprk1 mRNA. Kiss1 mRNA-expressing cells are made fluorescent with Vector Red substrate, and clusters of white silver grains reflect the presence of Pdyn (A), Tac2 (B), Tacr3 (C), or Oprk1 (D) mRNA. Scale bars, 50 μm. In B, blue arrows represent single-labeled Kiss1 cells; yellow arrows represent single-labeled Tac2 cells; green arrows represent double-labeled Kiss1/Tac2 cells. Note, all animals are GDX. 3V, Third ventricle.
Fig. 2.
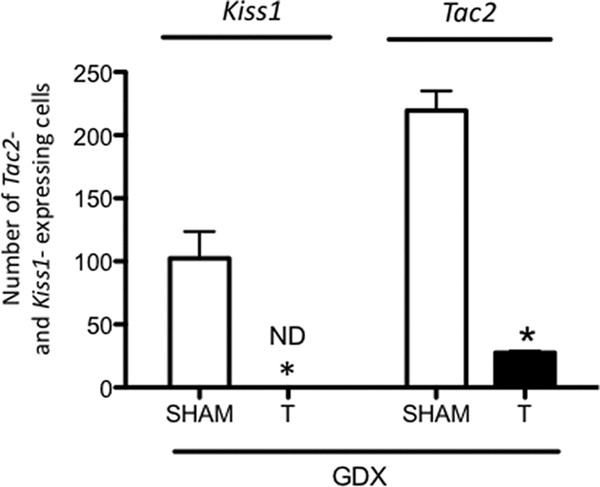
Effect of T replacement on the number of Kiss1 and Tac2-expressing cells in coronal sections of the ARC in castrated (GDX) WT mice. Data are presented as the mean ± sem. The asterisk indicates that the value depicted in the bar is significantly different from the sham-treated control group. ND, Nondetectable.
Coexpression of Gnrh1 and Tacr3 mRNA in the POA
To investigate whether KNDy neurons activate GnRH neurons through NKB → NK3R signaling, we used double-label ISH to determine whether GnRH neurons express Tacr3 mRNA. Although KNDy neurons expressed Tacr3 mRNA at high levels (see Fig. 1C), GnRH neurons showed no expression regardless of the sex steroid milieu (intact or GDX) (Fig. 3).
Fig. 3.

Representative photomicrograph showing no coexpression of Gnrh1 mRNA and Tacr3 in the POA of the male mouse. Gnrh1 mRNA-expressing cells (in red) are fluorescently labeled with Vector Red substrate, and the lack of clusters of white silver grains reflects the absence of Tacr3-expressing cells in this region of the brain. Scale bar, 50 μm.
Regulation of Kiss1, Tac2, and Pdyn mRNA through AR and ER
The biological effects of T on target tissues can be mediated by either a direct action of T on the AR or an indirect action involving aromatization of T to E2 and a subsequent action through the ER. To identify the pathway through which T acts to regulate the expression of Kiss1, Tac2, and Pdyn in the male, we analyzed and compared the effects of E2 and DHT (an androgen that cannot be aromatized) on the expression of these neuropeptides in GDX WT and Ar KO male mice. As expected, LH was suppressed by both DHT and E2 in WT males (P < 0.01) but only by E2 in Ar KO mice (P < 0.05) (Fig. 4). Likewise, Kiss1 and Pdyn mRNA levels were reduced by treatment with both DHT and E2 in GDX WT mice (Kiss1 WT: sham vs. E2, P < 0.05; sham vs. DHT, nondetectable Kiss1 cells in the DHT-treated group; Pdyn: sham vs. E2, P < 0.05; sham vs. DHT, P < 0.05). Tac2 mRNA was also significantly reduced by E2 in GDX WT animals (P < 0.05), although this reduction was not significant after DHT treatment. In GDX Ar KO mice, DHT had no detectable effect on the expression of Kiss1, Tac2, or Pdyn, whereas E2 inhibited the expression of all three (Kiss1 and Pdyn, P < 0.01; Tac2, P < 0.05) (Fig. 5). Of note, levels of Kiss1 and Tac2 mRNA in sham-treated Ar KO were markedly elevated compared with those of sham-treated WT mice (P < 0.05) (Fig. 5, A and B), suggesting that T-dependent AR signaling provides a constitutive restraint on the activity of Kiss1 neuronal activity, which is unmasked in animals bearing a congenital lack of AR.
Fig. 4.
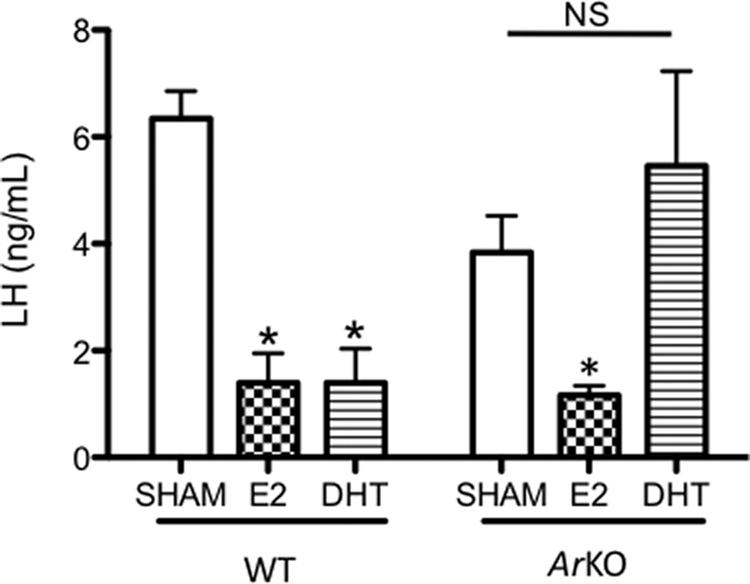
Serum LH levels in castrated (GDX) adult male WT and Ar KO mice, treated with capsules that were empty (sham) or filled with either E2 or DHT. Data are presented as the mean ± sem. Asterisks indicate either values that differ significantly from sham-treated controls (in the case of WT animals) or values that differ between bars (in the case of Ar KO). NS, Nonsignificant.
Fig. 5.
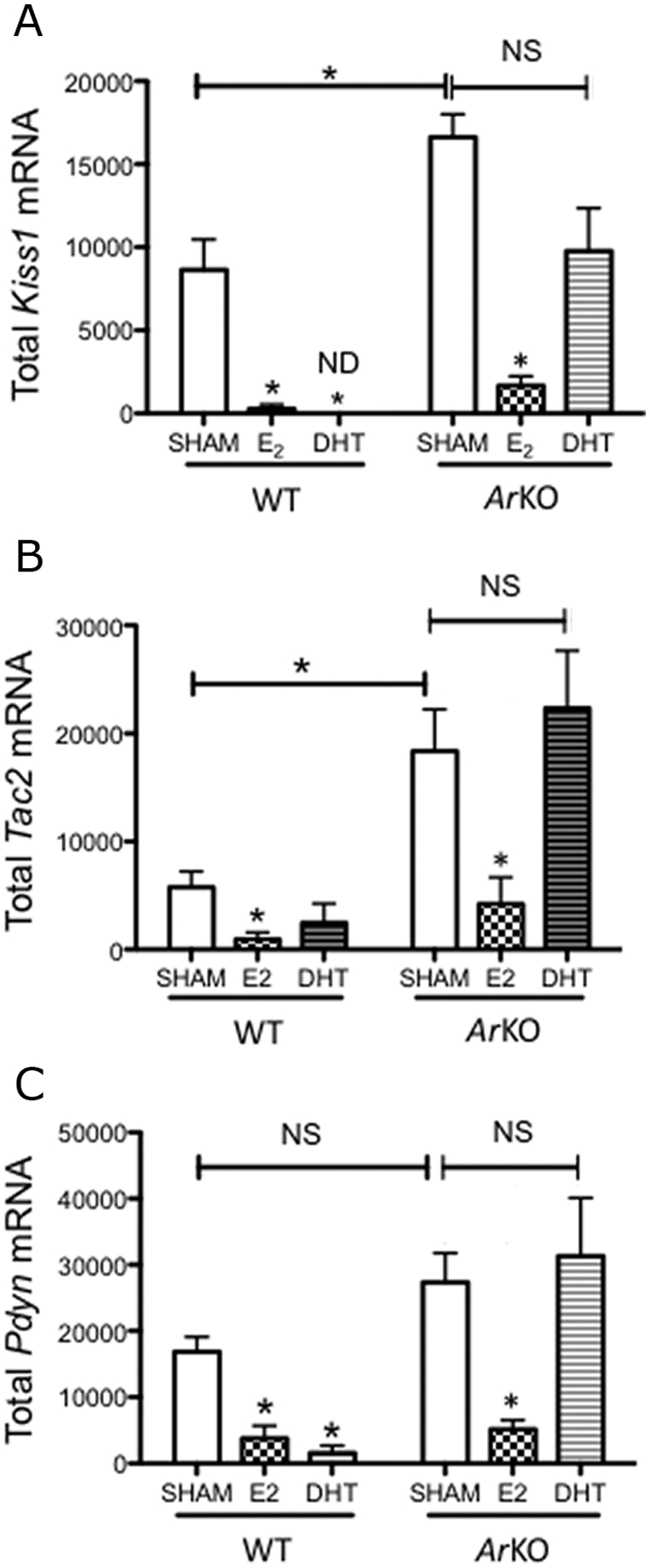
Effect of various treatments (sham/empty capsule, E2-filled capsule, or DHT-filled capsule) on the amount of Kiss1 (A), Tac2 (B), and Pdyn (C) mRNA (number of cells per cell content in grains) in the ARC of GDX WT and Ar KO mice. Data are presented as the mean ± sem. ND, Nondetectable; NS, nonsignificant.
Gonadotropin response to senktide
To investigate the role of NKB/NK3R signaling in the regulation of GnRH/LH/FSH secretion, we compared the effects of the NK3R agonist senktide with those of the vehicle alone in intact male mice. We observed a robust increase in serum levels of both LH and FSH after icv senktide administration (P < 0.01 and P < 0.05, respectively) (Fig. 6, A and B).
Fig. 6.
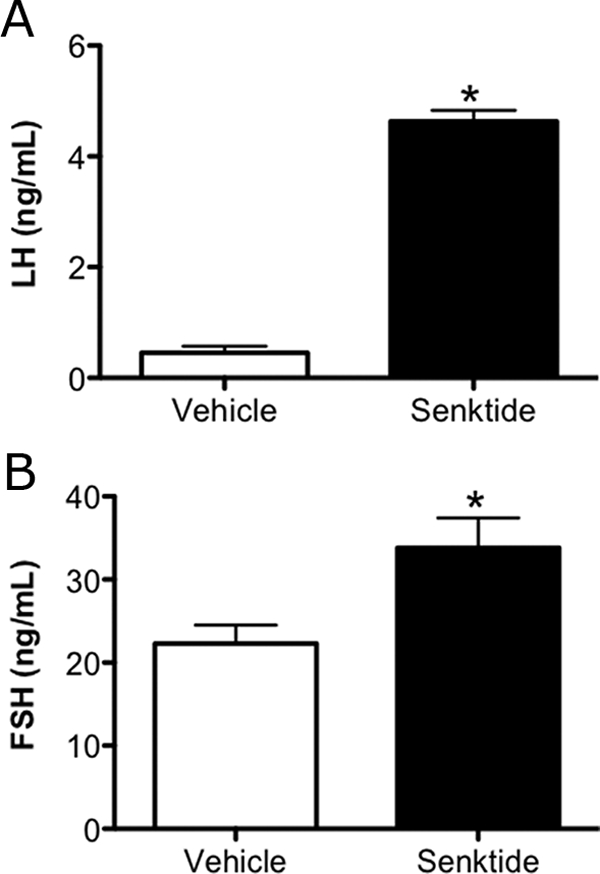
Serum levels of LH (A) and FSH (B) in adult males 30 min after treatment with senktide (600 pmol icv). Data are presented as the mean ± sem.
Electrophysiological responses of Kiss1 and GnRH neurons to NKB/senktide and Kiss1
In brain slices prepared from intact and GDX males, GFP-labeled Kiss1 neurons in the ARC were studied. Neurons in intact and GDX males had similar resting membrane potentials of −77 ± 2 mV (n = 14) and −75 ± 3 mV (n = 13), respectively. NKB (0.1–1 μm, 15 sec) activated 100% of Kiss1 neurons tested (n = 25), regardless of the gonadal status of the mouse from which slices were prepared. In current clamp studies, NKB activation was seen as a dramatic depolarization that drove volleys of action potentials in 85% of neurons tested. The magnitude of the depolarization in response to NKB (1 μm, 15 sec) was similar in neurons isolated from intact and GDX neurons (intact: 12.8 ± 3 mV, n = 9; GDX: 14.3 ± 2.8 mV; n = 6) (Fig. 7, A and C). The NKB response was reversible and had a mean duration of 7.7 ± 0.8 min (n = 8). In voltage-clamp experiments, NKB produced a strong inward current (26 ± 4.9 pA; n = 5; holding potential, −65 mV) (Fig. 7, B and C). A selective NK3R antagonist, SB222200 (1–3 μm, 15 min), reduced the NKB effect to less than 35% of control values (P < 0.05) (Fig. 7, D and E). Senktide (1 μm, 15 sec) mimicked the excitatory effect of NKB, producing a 16 ± 2.9 mV depolarization in the six neurons tested (Fig. 7F), implicating the NK3R in mediating effect of NKB on Kiss1 neurons.
Fig. 7.

A, Current-clamp recording from a quiescent GFP-labeled Kiss1 neuron, recorded from a brain slice of an adult male mouse with intact gonads. A 15-sec application of 0.1 μm NKB produced a 9-mV depolarization and induced action potentials. The NKB response lasted for more than 12 min in this cell. B, Voltage-clamp recording from another cell (holding potential, −65 mV) showing that NKB produced a 34-pA inward current, with an apparent decrease in membrane conductance. Downward vertical deflections are in response to −8-mV pulses delivered every 4 sec to monitor the input conductance of the recorded neuron. C, Left trace shows that 0.1 μm NKB produced a 35-mV depolarization and action potential firing in a quiescent GFP-labeled Kiss1 neuron recorded from a brain slice obtained from a GDX adult male mouse. The NKB response lasted for 270 sec in this cell. Also note that the NKB response was associated with an apparent increase in membrane resistance (as measured by applying repetitive 0.01-nA current pulses every 4 sec). Voltage-clamp recording from the same cell shows that NKB produced a 27-pA inward current, with an apparent decrease in membrane conductance. The time gap between the two applications was 13 min, illustrating the reproducibility of the response in the same cell. D, Traces showing an NKB-responsive cell from a GDX mouse cell, in which the NK3R antagonist, SB222200 (3 μm, 15 min), attenuated the response to a subsequent (at 22 min) application of NKB. E, Bar chart summarizing the antagonist effect of SB222200, which reduced the NKB response to less than 35% of control value in the cells tested. F, Trace from a cell that was strongly activated by senktide (1 μm, 15 sec), a selective NK3R receptor agonist, further confirming the presence of an NK3R-mediated activation of Kiss1 neurons in the ARC.
In contrast to Kiss1 neurons, GnRH neurons, whose cell bodies reside in the POA, were insensitive to NK3R receptor activation, regardless of the animal's gonadal status (intact, n = 9; GDX, n = 8). Despite prolonged applications of either NKB or senktide (1 μm, 15–120 sec), neither agonist had any apparent effect, which contrasts with the robust response of GnRH neurons to Kiss1 (Fig. 8A). NK3R agonists failed to induce a discernible effect, regardless of whether the agent was applied before or after exposure to Kiss1 (Fig. 8, A and B).
Fig. 8.

A, Current-clamp recording from a quiescent GnRH-GFP neuron in a brain slice obtained from an adult male mouse with intact gonads showing that senktide (1 μm, 120 sec) had no effect on its membrane potential, whereas a subsequent application of Kiss1 (0.1 μm, 15 sec) produced a strong excitatory effect. B, In a spontaneously firing cell, NKB applied after administration of Kiss1 also had no effect. C and D, Current-clamp recordings from two Kiss1-sensitive GnRH-GFP neurons, recorded from brain slices obtained from GDX mice, showing that NKB had no discernible effect, regardless of whether it was administered before or after Kiss1.
Discussion
We report that Kiss1-expressing cells in the ARC of male mice coexpress both Pdyn and Tac2 mRNA, as well as the mRNA for the NKB receptor (Tacr3), a property shared by females (6). Thus, although Kiss1-expressing neurons in the ARC of the rodent are sexually differentiated in other respects (32), they share this important feature. Furthermore, we report that virtually all Kiss1-expressing cells in the ARC coexpress Tac2, whereas only about half of the Tac2-expressing cell types in the ARC coexpress Kiss1. Differences in the threshold of mRNA detection in the double-labeling procedures could account for this observation. However, based on the fact that the radiolabeled and nonisotopically labeled probes (for Kiss1 and Tac2) worked well and that the differences in the coexpression phenomenon were large, it seems likely that there are at least two distinct populations of Tac2-expressing cells in the ARC (one that expresses Kiss1 and another that does not), each with its own unique physiological function. We also present evidence that T regulates the expression of Kiss1, Tac2, and Pdyn in KNDy neurons in the male by acting through AR- and ER-dependent mechanisms, whose actions contribute to the feedback regulation of gonadotropin secretion and could account for the sexual differentiation observed in KNDy neurons (32). Finally, we have demonstrated that Kiss1 neurons express the NKB receptor (NK3R) (Tacr3); moreover, NKB stimulates Kiss1 neurons, which show sustained volleys of action potentials in response to NKB, paralleled by robust LH and FSH secretory responses to the NKB agonist, senktide, in vivo. The prevailing steroidal milieu plays a critical role in determining the effect of NKB on LH secretion, because it has been shown that senktide inhibits LH secretion in agonadal animals (3, 6). On the other hand, NKB has no discernable effect on GnRH neurons, suggesting that an autosynaptic loop of NKB → NK3R signaling drives Kiss1 secretion (6, 15, 16, 33), which in turn governs pulsatile GnRH secretion.
NKB agonists induce LH release in the sheep, monkey, and rat (3, 7, 34). We have now shown this to be the case in the mouse and extend the downstream molecular targets to include FSH, as well as LH. We note that these results may be at variance with a report in the male rodent that NKB has no effect on gonadotropin secretion (35). This discordance may reflect that the former study used NKB (rather than the selective NK3R agonist senktide) that was administered in a diluent of water, in which NKB may not be readily soluble (7, 36). In any case, the fact that NKB is shown here to stimulate gonadotropin secretion in the mouse in vivo and to stimulate Kiss1 neurons in the mouse in vitro bolsters confidence in the conclusion that NKB is a potent secretagogue for Kiss1, GnRH, and gonadotropins in the mouse.
The mechanism by which NKB evokes gonadotropin secretion remains a matter of debate. One possibility is that NKB acts directly on GnRH neurons. In support of this contention is the observation that a low level of NK3R staining has been reported in GnRH somata and fibers in rats (37). Nevertheless, another report in the ewe asserts that NK3R is undetectable in GnRH neurons (34). Here, we report that senktide had no discernable effect on GnRH neurons in slice preparations, whereas NKB exerted a marked stimulatory effect on Kiss1 neurons under similar experimental conditions. Thus, we contend that NKB directly activates KNDy neurons, which subsequently drive GnRH neurons and thus gonadotropin secretion. This proposition is supported by the fact that NK3R is colocalized with NKB and dynorphin in the ARC of the rat (38) and mouse (6). Moreover, in the rat, senktide induces Cfos mRNA expression in Kiss1 neurons in the ARC (30) and stimulates multiple unit activity in the vicinity of KNDy neurons in the ARC of the goat (15). Finally, in the present study, we show that NKB induces trains of action potentials in Kiss1 neurons in vitro, an effect which is mimicked by senktide and can be blocked by NK3R antagonists. Together, these observations suggest that recurrent collaterals from KNDy neurons may help to perpetuate sustained firing from an interconnected network of cells that in turn drives the GnRH pulse generator.
Previous work in the female mouse suggested that KNDy neurons express low levels of the dynorphin receptor (KOR) (6). Based on this observation, we postulated that dynorphin might act through an autosynaptic loop to terminate the NKB-induced discharge of Kiss1 neurons (6). However, the results presented here demonstrate that KNDy neurons do not express detectable levels of KOR, at least not in males. This suggests that some other process, perhaps involving inhibitory interneurons, provides the inhibitory feedback required to establish and maintain oscillatory network behavior in the male, and quite possibly in the female as well.
We found that there are nearly twice as many Tac2-containing neurons in the ARC as there are Kiss1/Tac2-containing neurons. Although it is possible that this observation reflects a differential sensitivity of the radiolabeled and nonisotopically labeled probes used to identify Tac2 and Kiss1, it seems more probable that there are at least two populations of Tac2-expressing cells in the ARC, one that coexpresses Kiss1 and another that does not, which would imply that NKB serves multiple physiological functions in the ARC. Although compelling evidence suggests that the KNDy neurons drive basal GnRH release and mediate the negative feedback effects of gonadal steroids on GnRH secretion (14), little is known about the “other” Tac2-expressing neurons that express neither Kiss1 nor Pdyn mRNA, beyond the fact that they may be involved in the hypothalamic regulation of pituitary function (37, 39). One possibility is that neurons that express only Tac2 (and not Kiss1 or Pdyn) are part of the coupling mechanism that allows Kiss1 neurons to be synaptically linked to one another (through NKB → NK3R signaling) in a syncytium that allows maximum coordination of discharge among cells in the oscillator, this idea remains to be tested.
We and others have established that sex steroids inhibit the expression of Kiss1 in the ARC of several species (11). However, the effects of T on Tac2 and Pdyn expression have not been reported, nor has the mechanism by which T exerts its action. Here, we have confirmed our earlier finding that T acts through both ERα and AR to inhibit Kiss1 expression in KNDy neurons (13) and now extend these findings by showing that T works in a similar fashion to inhibit Pdyn expression in KNDy neurons. However, we could not establish for certain whether DHT exerts an effect on NKB, at least not in WT males. Despite the fact that levels of Tac2 mRNA were lower in the GDX DHT-treated groups compared with sham-treated controls, this difference was not statistically significant (perhaps reflecting the confounding effects of non-Kiss1 Tac2-expressing cells, which may or may not be equally responsive to DHT as KNDy neurons). However, we lack sufficient statistical power to rule out a possible effect. Moreover, E2 significantly decreases Tac2 expression, in agreement with our previous findings in female mice (6). The results gleaned in Ar KO mice suggest an additional role for AR in KNDy neurons, beyond that revealed in the study of steroid effects in WT animals. Indeed, the sham-treated Ar KO group had much greater levels of Kiss1, Tac2, and Pdyn mRNA in the ARC than their WT controls. This implies that AR is involved in the development of KNDy neurons in the ARC (40), possibly reflecting the action of AR on apoptosis in the ARC during development (41, 42).
In summary, our findings lend credence to the hypothesis that recurrent collaterals deliver NKB to KNDy neurons and provide a feed-forward signal that amplifies and synchronizes the activity of KNDy neurons, which in turn drives the GnRH pulse generator. This model is supported by the observations that 1) NKB activates Kiss1 neurons, 2) NKB induces LH and FSH secretion, 3) KNDy neurons express both NKB and NK3R, and 4) GnRH neurons do not respond to NKB nor do they express NK3R. Another aspect of the model contends that activation of KNDy neurons also triggers a delayed inhibitory signal that subsequently restrains the activity of KNDy neurons and thus accounts for the interpulse interval (6, 15). We had originally proposed that this is mediated by dynorphin delivered through recurrent axon collaterals to KNDy neurons. However, the present results indicate that KNDy neurons do not express Oprk1 mRNA (KOR), arguing that some other KOR-expressing interneurons (not KNDy neurons) mediate the action of Dyn. A multisynaptic circuit involving interneurons may help to explain how the interpulse interval is phase lagged from the pacemaker events that trigger activity in KNDy neurons. Finally, we show that T inhibits the expression of all three neurotransmitters expressed in KNDy neurons in the male, strengthening the proposition that such neurons play a vital role in the negative feedback regulation of GnRH/LH secretion in the male, as well as the female.
Supplementary Material
Acknowledgments
We thank Brigitte Mann, at Northwestern University (Evanston, IL), for performing some of the LH assays and Dr. Amy E. Oakley and Simina Popa for their assistance in the preparation of the manuscript.
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development/National Institutes of Health (NIH) through cooperative agreements U54 HD12629 (to the University of Washington Center for Research in Reproduction and Contraception); by NIH Grants R01 HD049651, R01 DA016898, RO1NS043330, and KO5 DA020570; by Spanish Research Grants BFU 2008-00984 (Ministerio de Ciencia e Innovación) and P08-CVI-00603 (Junta de Andalucía); by the State of Connecticut, Department of Mental Health and Addiction Services Grant MH61465; and by training grants awarded by the Marie Curie Outgoing International Fellowships from the 7th Framework Program of the European Union and the Mary Gates endowment for undergraduate research at the University of Washington.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- aCSF
- Artificial cerebrospinal fluid
- AR
- androgen receptor
- ARC
- arcuate nucleus
- DHT
- dihydrotestosterone
- DIG
- digoxigenin
- E2
- 17β-estradiol
- ER
- estrogen receptor
- GDX
- gonadectomy
- GFP
- green fluorescence protein
- icv
- intracerebroventricular
- ISH
- in situ hybridization
- Kiss1
- kisspeptin
- KNDy
- Kiss1/NKB/Dynorphin
- KO
- knockout
- KOR
- κ opioid receptor
- NIH
- National Institutes of Health
- NKB
- neurokinin B
- NK3R
- neurokinin 3 receptor
- Pdyn
- dynorphin A
- POA
- preoptic area
- T
- testosterone
- WT
- wild type.
References
- 1. Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, Serin A, Mungan NO, Cook JR, Ozbek MN, Imamoglu S, Akalin NS, Yuksel B, O'Rahilly S, Semple RK. 2009. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet 41:354–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Young J, Bouligand J, Francou B, Raffin-Sanson ML, Gaillez S, Jeanpierre M, Grynberg M, Kamenicky P, Chanson P, Brailly-Tabard S, Guiochon-Mantel A. 2010. TAC3 and TACR3 defects cause hypothalamic congenital hypogonadotropic hypogonadism in humans. J Clin Endocrinol Metab 95:2287–2295 [DOI] [PubMed] [Google Scholar]
- 3. Navarro VM, Castellano JM, McConkey SM, Pineda R, Ruiz-Pino F, Pinilla L, Clifton DK, Tena-Sempere M, Steiner RA. 2011. Interactions between kisspeptin and neurokinin B in the control of GnRH secretion in the female rat. Am J Physiol Endocrinol Metab 300:E202–E210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fink G. 2000. Neuroendocrine regulation of pituitary function: general principles. In: Conn PM, Freeman ME. eds. Neuroendocrinology in physiology and medicine. Totowa, NJ: Humana Press; 107–134 [Google Scholar]
- 5. Goodman RL, Lehman MN, Smith JT, Coolen LM, de Oliveira CV, Jafarzadehshirazi MR, Pereira A, Iqbal J, Caraty A, Ciofi P, Clarke IJ. 2007. Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology 148:5752–5760 [DOI] [PubMed] [Google Scholar]
- 6. Navarro VM, Gottsch ML, Chavkin C, Okamura H, Clifton DK, Steiner RA. 2009. Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/neurokinin B neurons in the arcuate nucleus of the mouse. J Neurosci 29:11859–11866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramaswamy S, Seminara SB, Ali B, Ciofi P, Amin NA, Plant TM. 2010. Neurokinin B stimulates GnRH release in the male monkey (Macaca mulatta) and is colocalized with kisspeptin in the arcuate nucleus. Endocrinology 151:4494–4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gottsch ML, Cunningham MJ, Smith JT, Popa SM, Acohido BV, Crowley WF, Seminara S, Clifton DK, Steiner RA. 2004. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology 145:4073–4077 [DOI] [PubMed] [Google Scholar]
- 9. Han SK, Gottsch ML, Lee KJ, Popa SM, Smith JT, Jakawich SK, Clifton DK, Steiner RA, Herbison AE. 2005. Activation of gonadotropin-releasing hormone neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J Neurosci 25:11349–11356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Irwig MS, Fraley GS, Smith JT, Acohido BV, Popa SM, Cunningham MJ, Gottsch ML, Clifton DK, Steiner RA. 2004. Kisspeptin activation of gonadotropin releasing hormone neurons and regulation of KiSS-1 mRNA in the male rat. Neuroendocrinology 80:264–272 [DOI] [PubMed] [Google Scholar]
- 11. Oakley AE, Clifton DK, Steiner RA. 2009. Kisspeptin signaling in the brain. Endocr Rev 30:713–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA. 2005. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology 146:3686–3692 [DOI] [PubMed] [Google Scholar]
- 13. Smith JT, Dungan HM, Stoll EA, Gottsch ML, Braun RE, Eacker SM, Clifton DK, Steiner RA. 2005. Differential regulation of KiSS-1 mRNA expression by sex steroids in the brain of the male mouse. Endocrinology 146:2976–2984 [DOI] [PubMed] [Google Scholar]
- 14. Lehman MN, Coolen LM, Goodman RL. 2010. Minireview: kisspeptin/neurokinin B/dynorphin (KNDy) cells of the arcuate nucleus: a central node in the control of gonadotropin-releasing hormone secretion. Endocrinology 151:3479–3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wakabayashi Y, Nakada T, Murata K, Ohkura S, Mogi K, Navarro VM, Clifton DK, Mori Y, Tsukamura H, Maeda K, Steiner RA, Okamura H. 2010. Neurokinin B and dynorphin A in kisspeptin neurons of the arcuate nucleus participate in generation of periodic oscillation of neural activity driving pulsatile gonadotropin-releasing hormone secretion in the goat. J Neurosci 30:3124–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Keen KL, Wegner FH, Bloom SR, Ghatei MA, Terasawa E. 2008. An increase in kisspeptin-54 release occurs with the pubertal increase in luteinizing hormone-releasing hormone-1 release in the stalk-median eminence of female rhesus monkeys in vivo. Endocrinology 149:4151–4157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holdcraft RW, Braun RE. 2004. Androgen receptor function is required in Sertoli cells for the terminal differentiation of haploid spermatids. Development 131:459–467 [DOI] [PubMed] [Google Scholar]
- 18. Sadate-Ngatchou PI, Payne CJ, Dearth AT, Braun RE. 2008. Cre recombinase activity specific to postnatal, premeiotic male germ cells in transgenic mice. Genesis 46:738–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gottsch M, Lawhorn J, Popa S, Oakley A, Alreja M, Navarro V, Mcclean M, Clifton D, Palmiter R, Steiner R, Generation and confirmation of a Kiss1 eGFP-Cre knock-in mouse. Society for Neuroscience 40th Annual Meeting San Diego, 2010. (Abstract P633.614) [Google Scholar]
- 20. Lindzey J, Wetsel WC, Couse JF, Stoker T, Cooper R, Korach KS. 1998. Effects of castration and chronic steroid treatments on hypothalamic gonadotropin-releasing hormone content and pituitary gonadotropins in male wild-type and estrogen receptor-α knockout mice. Endocrinology 139:4092–4101 [DOI] [PubMed] [Google Scholar]
- 21. Singh J, O'Neill C, Handelsman DJ. 1995. Induction of spermatogenesis by androgens in gonadotropin-deficient (hpg) mice. Endocrinology 136:5311–5321 [DOI] [PubMed] [Google Scholar]
- 22. Pak TR, Lynch GR, Ziegler DM, Lunden JB, Tsai PS. 2003. Disruption of pubertal onset by exogenous testosterone and estrogen in two species of rodents. Am J Physiol Endocrinol Metab 284:E206–E212 [DOI] [PubMed] [Google Scholar]
- 23. Cunningham MJ, Scarlett JM, Steiner RA. 2002. Cloning and distribution of galanin-like peptide mRNA in the hypothalamus and pituitary of the macaque. Endocrinology 143:755–763 [DOI] [PubMed] [Google Scholar]
- 24. Chowen JA, Argente J, Vician L, Clifton DK, Steiner RA. 1990. Pro-opiomelanocortin messenger RNA in hypothalamic neurons is increased by testosterone through aromatization to estradiol. Neuroendocrinology 52:581–588 [DOI] [PubMed] [Google Scholar]
- 25. Paxinos G, Franklin KBJ. 2001. The mouse brain in stereotaxic coordinates. San Diego: Academic Press [Google Scholar]
- 26. Zhang C, Bosch MA, Rick EA, Kelly MJ, Rønnekleiv OK. 2009. 17β-Estradiol regulation of T-type calcium channels in gonadotropin-releasing hormone neurons. J Neurosci 29:10552–10562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qiu J, Fang Y, Bosch MA, Rønnekleiv OK, Kelly MJ. 2011. Guinea pig kisspeptin neurons are depolarized by leptin via activation of TRPC channels. Endocrinology 152:1503–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu M, Hajszan T, Xu C, Leranth C, Alreja M. 2004. Group I metabotropic glutamate receptor activation produces a direct excitation of identified septohippocampal cholinergic neurons. J Neurophysiol 92:1216–1225 [DOI] [PubMed] [Google Scholar]
- 29. Wu M, Hajszan T, Leranth C, Alreja M. 2003. Nicotine recruits a local glutamatergic circuit to excite septohippocampal GABAergic neurons. Eur J Neurosci 18:1155–1168 [DOI] [PubMed] [Google Scholar]
- 30. Navarro VM, Tena-Sempere M. 2011. Kisspeptins and the neuroendocrine control of reproduction. Front Biosci 3:267–275 [DOI] [PubMed] [Google Scholar]
- 31. Spergel DJ, Krüth U, Hanley DF, Sprengel R, Seeburg PH. 1999. GABA- and glutamate-activated channels in green fluorescent protein-tagged gonadotropin-releasing hormone neurons in transgenic mice. J Neurosci 19:2037–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kauffman AS, Navarro VM, Kim J, Clifton D, Steiner RA. 2009. Sex differences in the regulation of Kiss1/NKB neurons in juvenile mice: implications for the timing of puberty. Am J Physiol Endocrinol Metab 297:E1212–E1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohkura S, Uenoyama Y, Yamada S, Homma T, Takase K, Inoue N, Maeda K, Tsukamura H. 2009. Physiological role of metastin/kisspeptin in regulating gonadotropin-releasing hormone (GnRH) secretion in female rats. Peptides 30:49–56 [DOI] [PubMed] [Google Scholar]
- 34. Billings HJ, Connors JM, Altman SN, Hileman SM, Holaskova I, Lehman MN, McManus CJ, Nestor CC, Jacobs BH, Goodman RL. 2010. Neurokinin B acts via the neurokinin-3 receptor in the retrochiasmatic area to stimulate luteinizing hormone secretion in sheep. Endocrinology 151:3836–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Corander MP, Challis BG, Thompson EL, Jovanovic Z, Loraine Tung YC, Rimmington D, Huhtaniemi IT, Murphy KG, Topaloglu AK, Yeo GS, O'Rahilly S, Dhillo WS, Semple RK, Coll AP. 2010. The effects of neurokinin B upon gonadotrophin release in male rodents. J Neuroendocrinol 22:181–187 [DOI] [PubMed] [Google Scholar]
- 36. Yang J, Dhawan V, Morrish DW, Kaufman S. 2007. Bimodal effects of chronically administered neurokinin B (NKB) on in vivo and in vitro cardiovascular responses in female rats. Regul Pept 143:136–142 [DOI] [PubMed] [Google Scholar]
- 37. Krajewski SJ, Anderson MJ, Iles-Shih L, Chen KJ, Urbanski HF, Rance NE. 2005. Morphologic evidence that neurokinin B modulates gonadotropin-releasing hormone secretion via neurokinin 3 receptors in the rat median eminence. J Comp Neurol 489:372–386 [DOI] [PubMed] [Google Scholar]
- 38. Burke MC, Letts PA, Krajewski SJ, Rance NE. 2006. Coexpression of dynorphin and neurokinin B immunoreactivity in the rat hypothalamus: morphologic evidence of interrelated function within the arcuate nucleus. J Comp Neurol 498:712–726 [DOI] [PubMed] [Google Scholar]
- 39. True C, Kirigiti M, Ciofi P, Grove KL, Smith MS. 2011. Characterisation of arcuate nucleus kisspeptin/neurokinin B neuronal projections and regulation during lactation in the rat. J Neuroendocrinol 23:52–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ciofi P, Lapirot OC, Tramu G. 2007. An androgen-dependent sexual dimorphism visible at puberty in the rat hypothalamus. Neuroscience 146:630–642 [DOI] [PubMed] [Google Scholar]
- 41. Estrada M, Varshney A, Ehrlich BE. 2006. Elevated testosterone induces apoptosis in neuronal cells. J Biol Chem 281:25492–25501 [DOI] [PubMed] [Google Scholar]
- 42. Semaan SJ, Murray EK, Poling MC, Dhamija S, Forger NG, Kauffman AS. 2010. BAX-dependent and BAX-independent regulation of Kiss1 neuron development in mice. Endocrinology 151:5807–5817 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.