

Abstract
A three-dimensional microfluidic channel was developed for high purity cell separations. This system featured high capture affinity using multiple vertical inlets to an affinity surface. In cell separations, positive selection (capture of the target cell) is usually employed. Negative enrichment, the capture of non-target cells and elution of target cells, has distinct advantages over positive selection. In negative enrichment, target cells are not labeled, and are not subjected to strenuous elution conditions or dilution. As a result, negative enrichment systems are amenable to multi-step processes in microfluidic systems. In previous work, we reported cell capture enhancement effects at vertical inlets to the affinity surface. In this study, we designed a chip that has multiple vertical and horizontal channels, forming a three-dimensional separation system. Enrichment of target cells showed separation purities of 92-96%, compared with straight-channel systems (77% purity). A parallelized chip was also developed for increased sample throughput. A two-channel showed similar separation purity with twice the sample flow rate. This microfluidic system, featuring high separation purity, ease of fabrication and use, is suitable for cell separations when subsequent analysis of target cells is required.
Introduction
The enrichment, isolation, and sorting of target cells from mixtures are important to both clinical diagnostics and basic research.(1-8) The preparation of a pure sample of target cells from a mixture of background cells is an enabling technology for genetic screening, immunology, and a host of other biomedical applications. Separation techniques can be based on a variety of approaches including magnetic separation (MACS) and fluorescence-activated cell sorting (FACS). With the development of micro total analysis systems,(9) lab-on-a-chip based devices have become an important platform for biomedical research in recent years.(10)
Most conventional cell separation techniques have been implemented in microfluidic systems.(11-12) The key advantage to miniaturizing traditional separations include low sample volume, flexible design, and the ability to customize separation parameters for a particular need.(13-19) However, the true potential of chip based separations is to utilize the microfluidic format to achieve separations that cannot be readily implemented in traditional separation methods. For example, most separation approaches isolate and purify a target cell based on positive selection. In this case the target cells is selected based on size, electrical properties, or a labeled surface antigen. While this approach works well in most situations, there are some inherent disadvantages to using the positive selection approach. First, if there is no singular parameter that distinguishes the target cell (i.e. a unique surface antigen), then isolation by positive enrichment is difficult or impossible. Second, the positive selection process in many cases leaves the cell labeled with an affinity ligand or bound to a separation surface. When subsequent analysis or culture of the target cell is needed, the label may have to be removed or the cells recovered from the affinity surface. Removal of the ligand or the release of the cells from a capture surface requires disruption of the affinity bond(s), which can damage the cell or compromise viability. In the case of affinity capture, elution of the cells may result in excess shear stress(20-21) or dilution of the target cells. Efforts to reduce shear stress during cell elution have resulted in gentler elution conditions, but with added complexity.(22) Bubble induced elution can be employed for efficiency removal of cells from the affinity surface, but this approach cannot be easily interfaced to other chip-based processes.(23-24) Nevertheless, positive selection methods will continue to play an important role in cell analysis.
In the cases where positive selection is not possible or not optimal, a strategy of negative selection can be employed. In negative selection, target cells pass throughout the separation process without label or capture. Background cells are depleted by affinity capture, leaving the eluted sample enriched with target cells. Negative enrichment has been reported using peptide- coated serpentine channels(25) and spiral channels.(26) However, capture efficiency in traditional microfluidic channels is limited under continuous flow conditions. To implement negative selection with high efficiency, new channel geometries must be used.
Recently, we reported the effects of inlet geometry on cell capture in microfluidic devices.(27) The use of a vertical inlet, where cells are loaded from the top of the chip into the separation channel, resulted in higher cell capture near the inlet itself when compared with the remainder of the affinity channel. This higher cell capture was found to result from the inlet geometry itself, where lower flow rates and trajectory toward the affinity surface resulted in higher cell capture. However, a single inlet became saturated with captured cells during chip operation, limiting the cell purity. We have now designed a new chip that creates multiple inlet regions using a three-dimensional microfluidic circuit (Figure 1) to overcome this limitation. The new chip design was used to successfully separated target cells from different cell mixtures with high purity and sample throughput.
Figure 1.

Schematic of the microfluidic device. a) Side view of the negative enrichment device. Cell mixtures are loaded into the chip, where background (non-target) cells are captured and target cells pass through for collection at the chip outlet. b) Image of the experimental device. Red food coloring was introduced into separation channels to help visualization (the channel is reflected in the bottom glass in this image).
Experimental Section
Reagents
Biotinylated bovine serum albumin was obtained from Sigma. Neutravidin was purchased from Pierce. Mouse anti-Human CD71 and anti-Human CD19 were purchased from BD Biosciences. SU-8 2015 photoresist and developer were purchased from Micro Chem. Dow Corning Sylgard 184 (PDMS) was purchased in kit form from Ellesworth Adhesives. Calcein-AM was purchased from Invitrogen. Phosphate Buffered Saline (PBS, pH 7.4) was purchased from VWR.
Cells and Cell Culture
HuT 78 human lymphocytes (ATCC #TIB-161), Ramos human lymphocytes (ATCC #CRL-1596), and C166-GFP mouse endothelial cells (ATCC #CRL-2583) were purchased from American Type Culture Collection (ATCC). HuT 78 lymphocytes are human CD71+CD19-. Ramos cells are human CD71+CD19+. C166-GFP cells are human CD71-CD19-. Cell antigen expression was confirmed by flow cytometry (see Supporting Information). The C166-GFP cells express green fluorescent protein, which was used to identify the cells during separations. All cell lines were maintained in an incubator at 37°C and 5% CO2. Cells were grown in RPMI 1640 medium (Hyclone) supplemented with 10% fetal bovine serum (Hyclone) and 20mL/L antibiotic-antifungal solution (penicillin-streptomycin stabilized solution, Sigma-Aldrich). Cells were subcultured twice a week. Cell concentrations were determined using a hemacytometer before each experiment, with concentrations ranging from 300-2000 cells/μL, depending on the experiment. Specific cell concentrations are detailed in the figure captions.
Microfluidic Device Fabrication
Soft photolithography (28) was used to create the channels in poly (dimethylsiloxane) (PDMS). The microfluidic device (Figure 1) was comprised of two layers of PDMS bonded to a glass slide to produce a three-dimension chip.(28) A high resolution mask (20,000 dpi laser printer transparency, CAD Art Services) was used to selectively expose a Si wafer coated with SU-8 2015 (Micro Chem). The wafer was subsequently developed and coated with perfluorooctyltrichlorosilane (Alfa Aesar). PDMS (mixed at 5:1 prepolymer:curing agent ratio for enhanced mechanical rigidity) was poured onto the Si wafer and baked at 100°C for one hour. The entire PDMS piece was removed from the wafer and cut into two pieces to form the top and bottom layer (see Supporting Information). A hole punch (18 gauge blunt needle) was used to create the vertical inlets in the bottom channel. The vertical channels were 600 μm in diameter, while the affinity channels were 40 μm tall and 1 mm wide. The punched, bottom PDMS layer was plasma treated for 1 minute and sealed to a glass slide to form the lower separation channels and vertical connections. After baking the sealed pieces at 100°C for 2 hours, the sealed pieces and top layer were plasma treated for two minutes and then sealed together. The top piece was carefully aligned so that the top affinity channels matched the punched holes, forming the three-dimensional chip structure. PTFE tubing (30 gauge, ID = 300 μm, Zeus) was connected to the chip inlet and sealed with a drop of uncured PDMS. The finished device was baked at 100°C for 5 minutes and stored until needed.
Affinity Surface Preparation
The affinity surface was prepared using established protocols(27, 29) with slight modifications. All surface coating reagents were loaded into the device after sealing; incubation of reagents occurred at 20°C. First, the channels were rinsed with deionized water. 10 μL of biotinylated bovine serum albumin (1mg/mL in 10 mM Tris-HCl, pH 8.0, 50 mM NaCl) solution was introduced to the device manually using a syringe. Biotin- BSA was incubated in the channel for 45 minutes. The device was then rinsed with 10 μL of 10 mM Tris buffer. Neutravidin (10 μL of 0.2 mg/mL solution in 10 mM Tris buffer) was loaded into the channels and incubated for 15 minute. Rinsing steps of 10 μL Tris buffer and 10 μLdeionized water completed the initial surface preparation. Chips were dried under air and stored at 4°C until the final conjugation step. Just before use, chips were removed from the refrigerator and 10 μL of biotinylated antibody (6.25 μg/mL) was added to complete the affinity surface conjugation.
Cell Separations
Flow control in this experiment was achieved using syringe pumps (KD Scientific). The syringe holding the cell mixture was connected to the buffer syringe (3% bovine serum albumin in PBS) using a t-junction.(27) For each experiment, the separation buffer was injected into the syringe first to ensure no bubbles were present in the chip. The cell mixture was then loaded at a flow rate of 0.5 mL/hr to rapidly fill the tubing and displace the buffer. The flow rate was then lowered to 0.05 mL/hr for cell separation.
Cell separations were monitored via fluorescence microscopy (IX71, Olympus). Green fluorescent protein-expressing or calcein-labeled cells were imaged using appropriate filters. Both fluorescence and white light images and videos were collected using a 0.3 NA, 10X objective. All images and videos were recorded using cooled CCD cameras coupled to the microscope. Blank (unstained) HuT 78 or Ramos cells were used to establish a threshold to identify fluorescent cells (either GFP expressing or calcein stained). A threshold of the mean background intensity plus three times the background cell standard deviation was used. Images and video were processed with ImageJ software (v. 1.43u, National Institutes of Health). All fluorescence images were background subtracted prior to data analysis.
Results and Discussion
Cell Capture at Vertical Inlets
Enhanced cell capture has been reported when a vertical channel meets a horizontal affinity surface.(27) Since the vertical channel in our device had a larger cross section area than the affinity channel, cell velocity was slower in the inlet area than in the affinity channel. This velocity decrease enhanced cell capture, since the fluid flow follows the mass conservation law (see Supporting Information, Video S1). As reported in our previous work, cell density at the vertical-horizontal channel interface was approximately 10 times higher than in the center of the affinity channel. In positive selection, this enhanced cell capture can actually be detrimental, as the surface can saturate and nonspecific binding of background cells occurs. In negative selection, however, nonspecific binding effects result in accidental capture of target cells, so purity is not affected while cell throughput decreases.
In any affinity cell separation system, there is a possibility that cell capture is not due to interaction between the capture ligand and a surface antigen. To confirm that cell capture was specific in our device, HuT 78 cells were tested in chips with and without anti-CD71 capture antibodies (Figure 2). In the case of chips without antibodies, cells capture was not observed when the antibody was absent (leaving a coating of Neutravidin only). When the antibody was present (Figure 2B), cell capture was observed. It should be noted that cracks in the vertical channels can be seen in the figure. These cracks were not observed to affect fluid flow during the lifetime of the devices.
Figure 2.

Comparison of cell capture with and without the capture antibody coating. A) The first vertical interface without anti-CD71 coating after 60 minutes of flowing HuT 78 cells (CD71+) through the device. Low non-specific capture was observed at all vertical interfaces. B) The first vertical interface with anti-CD71 capture antibodies. HuT 78 cells were observed to collect at the channel interfaces, indicating antibody capture was the primary mechanism of cell separation. Scale bar represents 100 μm.
Shear Stress During Separation
To study the shear stress effects on negative enrichment, C166-GFP mouse cells were used as a target with HuT 78 cells as a background with anti-CD71 modified surface. In general, lower shear stress increases the probability of cell capture. The force of cell capture is the product of the number of affinity bonds formed per cell and the force of a single affinity bond. The total force of cell capture must exceed the shear force in order for the cell to remain captured. However, low shear forces also promote nonspecific binding, so an optimum value must be reached. Since shear forces are directly proportional to volumetric flow rate, the flow of the system was varied and cell purity compared to optimize cell capture. As shown in Figure 3, sample purity of C166-GFP cells increases from 53 +/− 8% in the initial sample as the flow rate decreases. Flow rates below 0.1 mL/h enabled depletion of HuT 78 cells using anti-CD71 ligands. Under 0.05 mL/h, 50% initial C166-GFP cells were enriched to 96% after separation, showing better purity. Since affinity bond strength and the number of affinity bonds per cell will differ between cell types, it is necessary to optimize the flow rate for each set of experimental conditions.
Figure 3.

Flow rate effects on sample purity. The initial C166-GFP purity (53%±8%) increases as flow rate decreases, indicating the reduced shear stress and increased cell-surface interaction time improve depletion of background HuT 78 cells. Differences in cell purity at 0.5 and 0.1 mL/hr are statistically significant.
Negative Selection of Target Cells from Mixtures
Two test cases were established in this study. First, C166-GFP mouse cells were enriched from mixture of HuT 78 cells. In this case anti-CD71 was used as a capture molecule. In the second case, Ramos cells served as the background cell and HuT 78 cells were the target cell. For the second case anti-CD19 served as the capture molecule. The C166-GFP/HuT 78 experiments were used to test the performance of this device, while the HuT 78/Ramos experiments demonstrated separation of same-species cell types.
Fluorescence histograms of cells before and after separation are shown in Figure 4. The fluorescence intensity of the background cells was 7.8 counts/pixel with a standard deviation of 2.9 counts/pixel. For this particular experiment, the threshold for positive fluorescence identification was 16.5 counts/pixel. The threshold was calculated for each experiment. A mixture of C166-GFP and HuT 78 cells had an initial target cell concentration of 51.6%. After separation, 95.8% of the cells eluting from the chip were fluorescent and identified as mouse C166-GFP cells.
Figure 4.
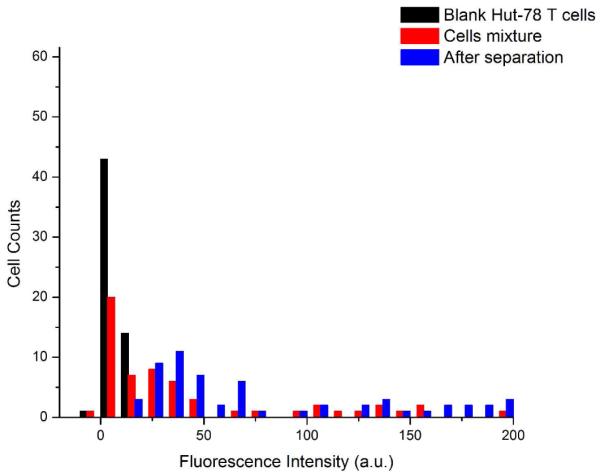
Fluorescence intensity histograms of cell mixtures and separated samples. Cell intensity (n = 59) was calculated for each experiment. Blank HuT 78 background cells (black) were used to set the fluorescence detection threshold. The cell mixture shows dim HuT 78 cells plus brighter C166-GFP cells expressing green fluorescent protein. The cells eluting from the chip (after separation) have the characteristically higher fluorescence of C166-GFP cells. C166- GFP cell concentration was 51.6% before separation and 95.8% after separation.
Since a higher flow rate was used to initially inject the sample, to cut down on loading time, the first five minutes of eluted sample were discarded since they may contain a large fraction of background cells. C166-GFP target cells were separated in mixtures containing 20- 50% target cells with total cell concentrations of 350-1800 cells/μL(See Figure 5A caption for specific cell concentrations). The overall purity in these experiments was 96 +/− 3%. A straight channel, two-dimensional chip was used with separation purities of 77%, indicating enhanced performance in the three-dimensional chip design. (Figure 5A). In previous work by Plouffe and coworkers(25) 96% depletion was achieved using a three-stage, serpentine channel (16 cm channel length in each stage). 9.31 and 3.43 μL min−1flow rate was applied to an eight parallel channels device. Sample purities in our case were comparable, but with shorter separation distances since the three-dimensional channel promotes cell capture. In another study(26), Green and coworkers used spiral-shaped two-dimensional channels to deplete cells, showing an enrichment of adipose-derived cells from 1.6% to 8.9% after negative selection with 7.54 μL min−1 flow rate. Our device can also be operated in serial circuits to deplete multiple cell types, and will be tested in the future to enrich a cell line out of a complex mixture.
Figure 5.

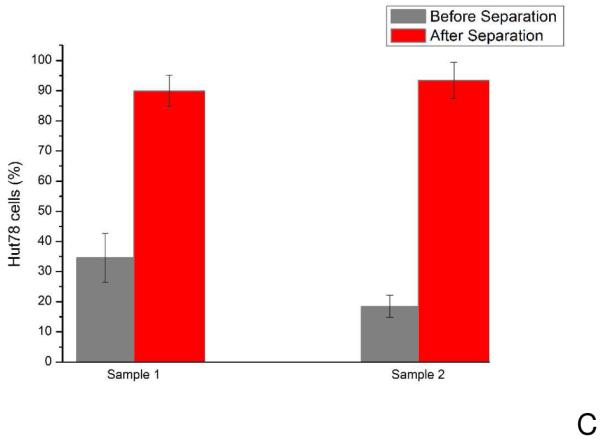
A) Enrichment of target cells (C166-GFP cells) of different initial mixture ratios. Sample 1 and 2 have 50% C166-GFP cells (HuT 78 cells were the background cells) with initial concentrations of 340±60 cells/μL and 1650±130 cells/μL, respectively. Sample 3 and 4 have approximately 30% C166-GFP cells in the initial mixture with total cell concentrations of 600±80 cells/μL and 1800±130 cells/μL, respectively. After collecting cells from outlet of the devices for 60 minutes, average C166-GFP purity of four samples was 96±3%. Straight channel chips operating under identical conditions (flow rate 0.05 mL/h) showed less enrichment. The initial C166-GFP ratio in the mixture was 60% with a total cell concentration of 720±80 cells/μL. Output of the straight channel chip was 77%, indicating the three-dimensional geometry enables superior cell separation. Error bars represent relative counting error (n=126, 193, 132, 106, 380, respectively). B) HuT 78 cell capture in the three-dimensional negative-selection device and a straight channel control. Samples containing 1100±164 cells/μL were introduced into both devices. The three-dimensional chip captured 5722 cells, compared with the straight channel chip, which captured 463 cells in the same experiment duration of 15 minutes. C) Enrichment of HuT 78 cells (target in this case) from Ramos cells using anti-CD19 chips. The HuT 78 cell percentage in Sample 1 was 35% with a total cell concentration of 1860±80 cells/μL. After separation the HuT 78 percentage was 90%. In Sample 2, the HuT 78 percentage was 18% with a total concentration of 3800±400 cells/μL. After separation the HuT 78 percentage was 93%. Error bars represent relative counting error (n=376 and 276 for samples 1 and 2, respectively).
The total number of captured cells in the three-dimensional chip was also compared to a straight channel chip. Samples containing only HuT 78 cells were divided in half and loaded into the two chip types. Both chip types were coated with anti-CD71, and the same flow rate was used for both chips (0.05 mL/h). A separation length of 2 cm was used for both channels. A total of 463 cells were captured in the straight channel chip, while 5722 cells were captured in the three-dimensional device (Figure 5b). The three dimensional channel geometry showed both higher purity and higher capture efficiency.
Separations of HuT 78 from Ramos background cells using anti-CD19 coated surfaces yielded similar results (Figure 5c). Ramos cells were pre stained with calcein-AM to distinguish them from HuT 78 cells. In this case a separation purity of 91% was achieved. To ensure that the transportation system (syringe and tubing) did not bias depletion of one cell type, cell throughput was observed for HuT 78/Ramos cell mixtures. Videos of cells passing through the device under constant flow were used to determine cell throughput. Two 30 second videos were recorded every 10 minutes at the first inlet under separation flow conditions (0.05 mL/hr, see Supporting Information for cell throughput figures). An uncoated device was used to avoid any cell capture. The initial percentage of HuT 78 cells was 38.0 +/− 5.1%. After 60 minutes of continuous operation, the HuT 78 cell percentage was 42.0 +/− 3.5%, which was not statistically different than the initial cell throughput. In contrast, a chip operated under identical conditions using anti-CD19 surfaces enriched a sample containing 18% HuT 78 cells to 93%. Therefore, affinity capture was the primary mechanism for cell enrichment.
Parallel Negative Selection Chips
It is possible to parallelize multiple channels in the same chip to increase throughput (See Supporting Information for figures of the chip). A two- channel chip, coated with anti-CD71, was used for negative selection of C166-GFP cells. A mixture containing 28% C166-GFP cells was enriched to 92% using this paralleled device (Figure 6). The performance of the left and right channels is not statistically different. The total sample flow rate was 0.1 mL/hr. The result is comparable in purity to single channel devices, indicating that parallel channels can achieve similar performance with higher sample throughput. We are currently working on a larger-scale parallelization for future studies.
Figure 6.
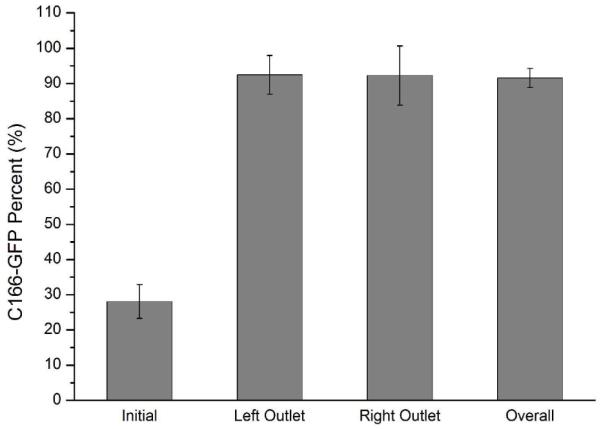
Enrichment of C166-GFP cells from Hut78 T cell using parallel separation channels at flow rate 0.1 mL/h. The initial sample concentration was 1900 ± 400 cells/μL and the percentage of C166-GFP target cells was 28%. Left and right outlets were monitored independently for target cell enrichment. The left and right channels performed similarly (no statistical difference) with an average purity of 92%. Error bars represent relative counting error (n=427, 332, 142, 1336, respectively).
Separation Conditions for Optimum Background Cell Depletion
It is expected that each additional vertical-horizontal channel interface will increase the capacity to deplete background cells. To verify this hypothesis, samples containing only the background cell (HuT 78 cells in this case) were passed through the device at the optimum flow rate of 0.05 mL/hr. Cell throughput (free moving cells) were measured at the output of the chip at 10 minute intervals. With an initial cell concentration of 660 HuT 78 cells/μL was introduced into the anti-CD71 chip, background cell output was 0.8 +/− 0.8 cells at the chip outlet in a 30-second video, indicating effective depletion of HuT 78 cells. Depletion of background cells was also conducted at higher concentrations (1330 cells/μL). In this case, cell throughput was monitored at each vertical channel interface (Figure 7).
Figure 7.

Depletion study of background HuT 78 cells. A) Cell throughput at different vertical channel interfaces (at bottom channel). Outlets 1,2,4,6 in the figure represent outlet area for bottom channel sections 1,2,4,6, respectively. Outlet 6 was the outlet of this device. Samples containing only Hut-78 T cells (1300±110 cells/μL) were introduced into anti CD71 coated devices. Every 10 minutes, a 30 second video was recorded for different outlets. Outlet 1 is seen to saturate at 30 minutes, however downstream outlets maintain efficient cell capture. B) Image of cell saturation on the surface at inlet 1 after 30minutes of separation. C) Comparison of cell throughout when target cells (C166-GFP, mouse endothelial cells (MEC)) are added to the sample. Initial C166-GFP cell concentration in Samples 1 and 2 were 540 cells/μL and 830 cells/μL, respectively. The output cell throughput increased according to C166-GFP initial concentration (Sample 1 = 29 cells, Sample 2 = 54 cells at 10 minutes). The output of outlet 6 was 0 cells. Exact cell counts are available in Supporting Information (Table S1 and S2). Examples of recorded videos are also available in supporting information (Video S2-S4).
The first vertical-horizontal channel interface showed the greatest degree of cell capture, but also saturated after 30 minutes of operation. Cell throughout at the first interface is seen to rise after 30 minutes as the affinity surface saturates with captured cells (Figure 7B). In addition, a single vertical interface cannot capture cells with 100% capture efficiency due to the random nature of cell capture. The additional vertical interfaces ensure efficient capture and serve as secondary capture surfaces as preceding interfaces become saturated. The use of multiple vertical interfaces prevents loss of system performance and allows cells to be depleted with high efficiency. In the case of a higher cell concentration, only 0.8 +/− 0.9 cells were observed to exit the chip at 60 minutes of separation. When C166-GFP cells were added to the HuT 78 sample, cell throughout increased (Figure 7C) as target cells passed through the device without capture.
Geometric Effects on Cell Velocity
Cell velocity plays a key role in cell capture. Cell velocity was measured using video data of cells moving though uncoated chips (to exclude capture effects). The three-dimensional chip design features a lower layer of channels connected to an upper layer via several (6) vertical channels, and cell velocity was found to vary depending on the location of the cell in the three-dimensional chip. Cells in the lower channels (glass bottom, coated with affinity ligands) had mean velocities of 0.11 +/− 0.04 mm/s as they entered the lower channel from the vertical inlet. Cells that were in the lower channel, after exiting the vertical channel interface region, accelerated to a velocity of 0.40 +/− 0.04 mm/s. Cells exiting the lower affinity channels have a mean velocity of 0.41 +/−0.06 mm/s. Cells exiting the top of the vertical channel and entering the upper horizontal channels had a mean velocity of 0.54 +/−0.09 mm/s, compared to a mean velocity of 0.42 +/− 0.05mm/s in the middle of the upper channels. Since cells at the inlet of each lower affinity channel have the lower velocity, the six bottom affinity channels are the major source of cell capture and negative cell depletion.
Conclusion
In this study, we reported enrichment of a target cell by negative selection of background cells. The microfluidic chip uses a three-dimensional architecture that results in efficient cell capture and low nonspecific binding. The device was capable of enriching target cells without labels or the need to elute captured cells. This approach is therefore directly amenable to applications where subsequent analyses of target cells are required, such as sub-culture, genetic analysis, or electron microscopy. The flexible nature of this approach enables parallelization. In future work, we will expand chip capabilities to simultaneously deplete multiple cell types.
The major limitation of this, and any, cell separation device is nonspecific binding when the target cell is rare (<1% of the total cell population). In these cases, new capture molecules can be developed to minimize nonspecific capture. In the case of negative enrichment, however, it is possible to separate rare target cells multiple times to reduce the number of background cells present in the sample. Our three-dimensional chip approach is an enabling tool for cell separations, and can enrich a target cell under continuous flow, with throughput scaling directly with the number of parallel separation channels.
Supplementary Material
Acknowledgements
Y.G. would like to acknowledge support by a Provost’s Fellowship. This work was supported by grants from the National Institutes of Health (Grant RR025782) and the Robert A. Welch Foundation (Grant D-1667).
References
- (1).Pappas D, Wang K. Anal. Chim. Acta. 2007;601:26–35. doi: 10.1016/j.aca.2007.08.033. [DOI] [PubMed] [Google Scholar]
- (2).Wysocki LJ, Sato VL. Proc. Natl. Acad. Sci. U. S. A. 1978;75:2844–2848. doi: 10.1073/pnas.75.6.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bonner WA, Hulett HR, Sweet RG, Herzenberg LA. Rev. Sci. Instrum. 1972;43:404–409. doi: 10.1063/1.1685647. [DOI] [PubMed] [Google Scholar]
- (4).Stefan M, Werner M, Walter W, Andreas R. Cytometry. 1990;11:231–238. [Google Scholar]
- (5).Springston SR, Myers MN, Giddings JC. Anal. Chem. 1987;59:344–350. [Google Scholar]
- (6).Hawkins BJ, Smith AE, Syed YA, Kirby BJ. Anal. Chem. 2007;79:7291–7300. doi: 10.1021/ac0707277. [DOI] [PubMed] [Google Scholar]
- (7).Vahey MD, Voldman J. Anal. Chem. 2008;80:3135–3143. doi: 10.1021/ac7020568. [DOI] [PubMed] [Google Scholar]
- (7).Hertz CM, Graves DJ, Lauffenburger DA, Serota FT. Biotechnol. Bioeng. 1985;27:603–612. doi: 10.1002/bit.260270509. [DOI] [PubMed] [Google Scholar]
- (8).Lara O, Tong X, Zborowski M, Farag SS, Chalmers JJ. Biotechnol. Bioeng. 2006;94:66–80. doi: 10.1002/bit.20807. [DOI] [PubMed] [Google Scholar]
- (9).Auroux P, Iossifidis D, Reyes DR, Manz A. Anal. Chem. 2002;74:2637–2652. doi: 10.1021/ac020239t. [DOI] [PubMed] [Google Scholar]
- (10).Salieb-Beugelaar GB, Simone G, Arora A, Philippi A, Manz A. Anal. Chem. 2010;82:4848–4864. doi: 10.1021/ac1009707. [DOI] [PubMed] [Google Scholar]
- (11).Didara TF, Tabrizian M. Lab Chip. 2010;10:3043–3053. doi: 10.1039/c0lc00130a. [DOI] [PubMed] [Google Scholar]
- (12).Lenshof A, Laurell T. Chem. Soc. Rev. 2010;39:1203–1217. doi: 10.1039/b915999c. [DOI] [PubMed] [Google Scholar]
- (13).Xu Y, Phillips JA, Yan JL, Li Q, Fan ZH, Tan W. Anal. Chem. 2009;81:7436–7442. doi: 10.1021/ac9012072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Phillips JA, Xu Y, Xia Z, Fan ZH, Tan W. Anal. Chem. 2009;81:1033–1039. doi: 10.1021/ac802092j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cheng X, Irimia D, Dixon M, Sekine K, Demirci U, Zamir L, Tompkins RG, Rodriguez W, Toner M. Lab Chip. 2007;7:170–178. doi: 10.1039/b612966h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dharmasiri U, Balamurugan S, Adams AA, Okagbare PI, Obubuafo A, Soper SA. Electrophoresis. 2009;30:3289–3300. doi: 10.1002/elps.200900141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Plouffe BD, Njoka DN, Harris J, Liao J, Horick NK, Radisic M, Murthy SK. Langmuir. 2007;23:5050–5055. doi: 10.1021/la0700220. [DOI] [PubMed] [Google Scholar]
- (18).Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Nature. 2007;450:1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sin A, Murthy SK, Revzin A, Tompkins RG, Toner M. Biotechnol. Bioeng. 2005;91:816–826. doi: 10.1002/bit.20556. [DOI] [PubMed] [Google Scholar]
- (20).Zhang X, Jonesa P, Haswell SJ. Chem. Eng. J. 2008;135:S82–S88. [Google Scholar]
- (21).Lu H, Koo LY, Wang WM, Lauffenburger DA, Griffith LG, Jensen KF. Anal. Chem. 2004;76:5257–5264. doi: 10.1021/ac049837t. [DOI] [PubMed] [Google Scholar]
- (22).Plouffe BD, Brown MA, Iyer RK, Radisic M, Murthy SK. Lab Chip. 2009;9:1507–1510. doi: 10.1039/b823523f. [DOI] [PubMed] [Google Scholar]
- (23).Wang K, Marshall MK, Garza G, Pappas D. Anal. Chem. 2008;80:2118–2124. doi: 10.1021/ac702553w. [DOI] [PubMed] [Google Scholar]
- (24).Cao X, Eisenthal R, Hubble J. Enzyme Microb. Technol. 2002;31:153–160. doi: 10.1016/s0141-0229(01)00329-5. [DOI] [PubMed] [Google Scholar]
- (25).Plouffe BD, Radisic M, Murthy SK. Lab Chip. 2008;8:462–472. doi: 10.1039/b715707j. [DOI] [PubMed] [Google Scholar]
- (26).Green JV, Murthy SK. Lab Chip. 2009;9:2245–2248. doi: 10.1039/b906768j. [DOI] [PubMed] [Google Scholar]
- (27).Li P, Tian Y, Pappas D. Anal. Chem. 2011;83:774–781. doi: 10.1021/ac102975g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Duffy DC, McDonald JC, Schueller OJA, Whitesides GM. Anal. Chem. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- (29).Pappas D. Practical Cell Analysis. Wiley-Blackwell; 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.