

Abstract
5-fluorouracil (5-FU) is widely used in chemotherapy for gastric and colorectal cancer, but gemcitabine (GEM), and not 5-FU, is approved as a standard drug for use in pancreatic cancer. Interindividual variation in the enzyme activity of the GEM metabolic pathway can affect the extent of GEM metabolism and the efficacy of GEM chemotherapy. Human equilibrative nucleoside transporter 1 (hENT1) is recognized as a major transporter of GEM into cells. In addition, a factor that activates hENT1 is the inhibition of thymidylate synthase (TS), one of the 5-FU metabolic enzymes; TS inhibition mediates depleting intracellular nucleotide pools, resulting in the activation of the salvage pathway mediated through hENT1. In this paper, the role of 5-FU in GEM-based chemotherapy for pancreatic cancer is discussed with special emphasis on enzymes involved in the 5-FU and GEM metabolic pathways and in the correlation between GEM responsiveness and the expression of 5-FU and GEM metabolic enzymes.
1. Introduction
Pancreatic cancer is one of the most life-threatening cancers; 35,240 deaths in Americans in 2009 (6% of all United States cancer deaths) make this cancer one of the leading causes of cancer-related death [1]. In spite of recent progress in surgical procedures, the operative resectability rate of pancreatic cancer remains unsatisfactory at 9% to 20% [2, 3]. Development of chemotherapeutic modalities has shifted from 5-fluorouracil (5-FU), one of the primary standard drugs used to treat solid cancers, to gemcitabine (GEM, Gemzar; Eli Lilly and Company, Indianapolis, Ind), the most anticipated agent for the treatment of this problematic disease [4]. However, treatment results and favorable outcomes with GEM remain variable; the response rate with GEM ranges from 5.4% to 16.7% [5, 6], and the median survival time (MST) of patients treated with 5FU of 4.2–4.5 months [5] is extended by GEM to 5.9–6.5 months [4, 6]. The concept of single-agent chemotherapy is clearly limited, and novel approaches to combination therapy should be considered. Here, we describe our recent challenges in pancreatic cancer and review the chemotherapeutic procedures currently available for the treatment of pancreatic cancer.
2. Application of Biological Study to Pancreatic Cancer Therapy
Recently, the study of the cell signaling pathway has been applied to the control of cancer proliferation, invasion, and metastasis as a molecular targeted therapy [7]. Among them, vitamin K3 (menadione), which induces cell apoptosis through activation of oxidative stress, has also been expected as a unique anticancer drug for pancreatic cancer [8, 9]. In the process of bringing experimental studies of such agents to the clinical trial stage, a drug delivery system is currently under consideration [10]. In parallel with the development of these future biochemical trials, recent studies based on standard chemotherapy with GEM, have also been developed.
Nucleoside transporters are commonly known to include two equilibrative nucleoside transporters (ENT1/2) and three concentrative nucleoside transporters (CNT1/2/3) [11]. Recent kinetic studies of human cell lines have shown the intercellular uptake of GEM to depend mainly on ENT1, which localizes in plasma and mitochondrial membranes [11–13]. ENT1 activity was reported to be a prerequisite for the occurrence of the growth inhibitory effect of GEM, because cells deficient in ENT1 activity were highly resistant to GEM; the rate of growth inhibition was increased 39- to 1800-fold in the presence of ENT1 [12]. Expression of mRNA and proteins was also evaluated as a favorable predictor of the effect of GEM clinically; MST was longer in patients with high versus low expression, 25.7 versus 8.5 months with mRNA expression (P = .001), and 13.0 versus 4.0 months with protein expression (P = .01) [14, 15]. These results suggest the action of ENT1 to be critical for GEM metabolism.
Intracellular enzymes deoxycytidine kinase (dCK), ribonucleotide reductase (RR), and 5′-nucleotidase (5′-NT) are also reported to be important in the conversion of GEM to its inactive form [16]. Some studies emphasizing the first step in limiting GEM phosphorylation by overexpression of dCK in tumor cells deficient in the enzyme have shown restoration of the response to GEM [17, 18]. RR is essential for DNA polymerization/repair [19] and consists of large and small dimerized subunits, M1 and M2, respectively. The M1 subunit possesses a binding site for enzyme regulation (regulatory subunit), and the M2 subunit is involved with RR activity (catalytic subunit) [20, 21]. Because 5′-NT reduces phosphorylated metabolites of GEM, the activity level of 5′-NT might also be a target for evaluation as the one factor most affecting the clinical outcome of GEM chemotherapy [22].
Although RRM1, dCK, and 5′-NT are useful predictors of GEM resistance [23], the individual actions of each of RR, dCK, and 5′-NT have not been reported as useful predictors of prognosis in pancreatic cancer chemotherapy. Namely, pancreatic cancer cells with a higher ratio of hENT1×dCK/RRM1×RRM2 showed higher cytotoxicity, and those cells with a lower ratio showed lower cytotoxicity [21]. Further studies are necessary to confirm the usefulness of these three factors as predictors of prognosis in pancreatic cancer.
3. The Role of One 5-FU Metabolic Enzyme, Thymidylate Synthase
Thymidylate synthase (TS) is generally known to be important in 5-FU metabolism [24, 25]. In solid-type carcinomas, TS expression was estimated for its ability to predict sensitivity to 5-FU; increase in the expression of 5-FU mRNA/protein resulted in resistance to 5-FU in colorectal cancer [26, 27]. Low expression of TS, as evaluated by immunohistochemistry and reverse-transcription PCR (RT-PCR), correlated with a favorable response to 5-FU-based therapy in colorectal cancer patients [25, 28, 29]. In pancreatic cancer, however, survival rate was better in the patients with high TS expression [30]. In contrast, expression of TS mRNA was found to correlate with patient survival; survival was longer in patients with low expression, but in patients with high TS expression, 5-FU-based chemotherapy showed favorable results [31]. Taken together, TS expression is related to 5-FU metabolism and its chemotherapeutic effect. In addition, Rauchwerger et al. previously reported that 5-FU itself plays a role in the inhibition of TS, and TS inhibitor modulates hENT1 [32]. On this basis, we investigated methods to improve pancreatic cancer therapy.
4. Challenges to the Better Treatment of Pancreatic Cancer
Expressed levels of 5-FU and/or GEM-related metabolic protein in seven independent pancreatic cancer cell lines (PANC-1, MIAPaCa-2, BxPC-3, Hs766T, Capan-2, AsPC-1, and CFPAC-1) were compared with the half maximal inhibitory concentrations (IC50s) of GEM or 5-FU, and only TS expression was found to correlate positively with drug-induced inhibitory effect on cell growth (P = .0169) [33]. hENT1 expression was found to be similar for each of these pancreatic cancer cell lines, whereas TS expression was found to be high in PANC-1, and moderate in MIAPaCa-2 and low in BxPC-3, as shown in (Figure 1). The relation between TS expression and GEM resistance was demonstrated by using these three pancreatic cancer cell lines (Figure 2). The inhibition of TS expression due to 5-FU (0.1 or 1.0 μM, doses having no cell-inhibitory or cell-death effects) was also shown to decrease GEM resistance in a dose-dependent manner. The inhibition of TS due to siRNA on PANC-1, which is the cell line most tolerant to GEM and which shows the highest expression of TS protein, also significantly decreased the resistance of PANC-1 to GEM, showing a decrease in IC50 from 77.0 ± 2.6 nM to 7.7 ± 1.1 nM (P = .0019). In addition, in our clinical study of patients treated with GEM after surgery for pancreatic cancer, low expression of TS protein evaluated in resected specimens by immunohistochemical techniques was found to correlate significantly with prolongation of disease-free survival (15.9 ± 12.4 versus 7.0 ± 3.5 months, P = .0256), as shown in (Figure 3).
Figure 1.
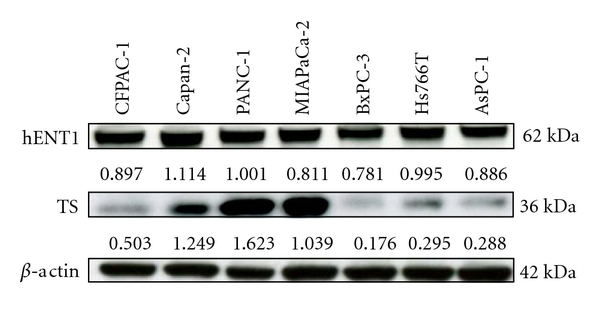
Cellular factor expression in several pancreatic cancer cell lines. Cellular factor-related metabolism for 5-FU or GEM was evaluated by Western blotting. Each value under the blotting band was obtained from comparison with the level of mouse monoclonal anti-beta-actin. hENT1, human equilibrative nucleoside transporter 1; TS, thymidylate synthase.
Figure 2.
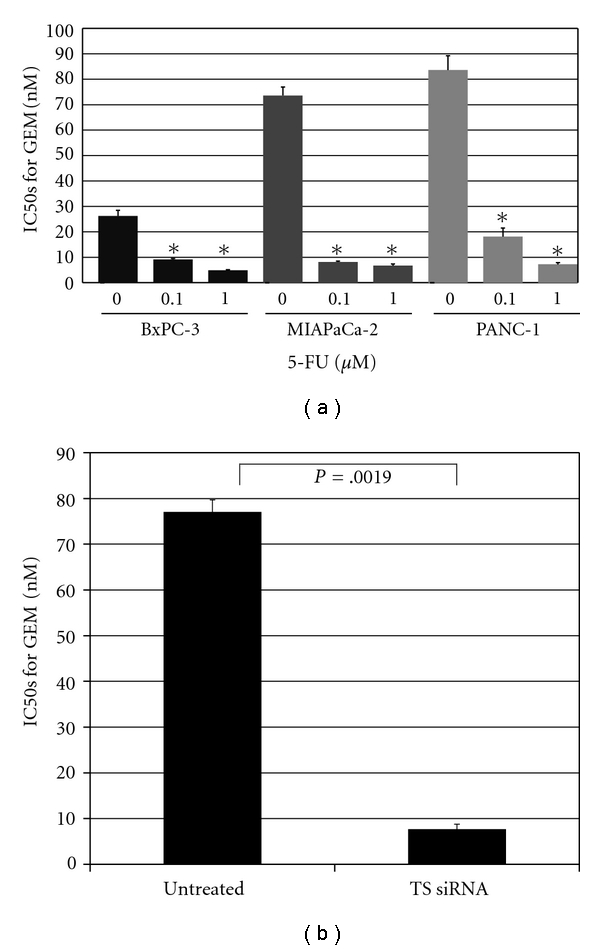
(a) Drug resistance of pancreatic cancer cells to 5-FU+GEM. Cultured cells (5 × 103 cells/well) were exposed to graded concentrations of gemcitabine (0, 10−2–104 nM) together with several 5-FU concentrations (0, 0.1, 1.0 μM) for 72 h. Drug resistance was expressed as the concentration of drug that inhibited colony formation by 50% (IC50). The control for each cell line (5-FU, 0 μM) showed statistical significance with each of the same cell lines (∗) (P < .01). (b) Effect of TS expression on GEM resistance. Knockout of TS expression significantly decreased resistance to GEM in pancreatic cancer cells as evidenced by the detected change in IC50s from 77.0 ± 2.6 nM to 7.7 ± 1.1 nM. Results are expressed as mean ± SD. Bar, SD; TS, thymidylate synthase.
Figure 3.
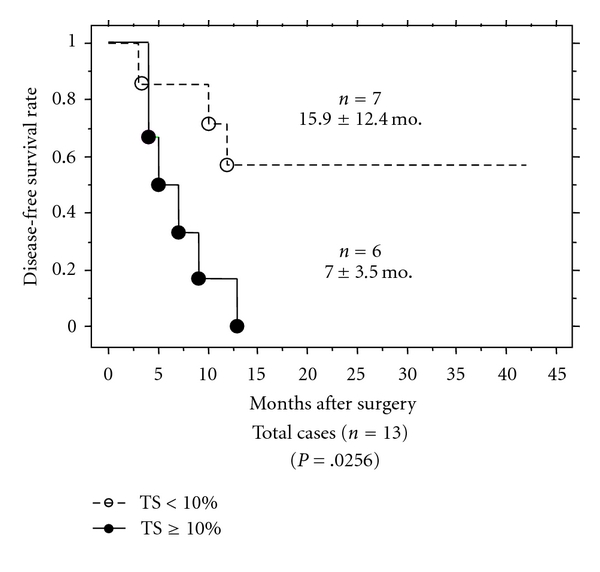
Comparison of patient disease-free survival rates. In patients receiving GEM treatment after surgery (n = 13), disease-free survival was compared according to percent of thymidylate synthase (TS) protein expression. Months after surgery; mean ± SD.
From our experience with pancreatic cancer cell lines, IC50s of 5-FU varied quite widely (9.0–1805.1 μM), but the IC50s of GEM were found to be stable (6.1–77.6 nM); therefore, GEM has been accepted as a first-line chemotherapeutic drug for the treatment of pancreatic cancer [4]. In contrast, according to these studies, 5FU could be useful not only to inhibit cell growth by its chemotherapeutic actions but also to reduce TS expression to improve sensitivity to GEM.
5. The Benefit of Combination Treatment of Gem with 5-FU
For GEM-induced intracellular changes, is hENT1 truly the most important factor? Although a previous report indicated that hENT1 is a useful predictor of GEM responsiveness or MST of pancreatic cancer patients treated with GEM, augmentation of hENT1 mRNA was detected after GEM treatment in the cells, whereas DNA synthesis inhibitor was found to increase the activity of some nucleoside transporters at the cell surface [34]. Resistance to GEM in pancreatic cancer cell lines does not appear to involve hENT1; inhibition of hENT1-mediated transport in pancreatic cell lines modifies GEM responsiveness either modestly or not at all [34]. In addition, we have previously reported on the contribution of TS to GEM chemotherapy [33]. Indeed, as shown in (Figure 1), the differences detected in the protein expression levels of TS (0.176–1.623), but not those of hENT1 (0.781–1.114), correlated with GEM responsiveness, suggesting that hENT1 itself has no close correlation to GEM resistance.
In the metabolic pathway of GEM, not only GEM metabolic enzymes but also TS has been shown to have a direct or indirect correlation with GEM metabolism: a TS inhibitor, such as 5-FU, in the de novo pathway mediates depleting intracellular nucleotide pools, resulting in activation of the salvage pathway and hENT1 as well [13, 35] (Figure 4). The process of activating hENT1 mediates the diffusion of nucleosides, including those associated with GEM, across plasma membranes in accordance with the concentration gradient [32]. Then, because hENT1 represents as a main GEM transporter, lower expression of TS might be related to transportation of GEM due to hENT1 activation. According to our experiments, TS inhibition itself is quite critical in GEM-mediated cancer cell death and also might be useful in GEM-resistant pancreatic cancer. Decreasing protein expression of TS with 5-FU probably induces a better prognosis for the patient undergoing GEM chemotherapy.
Figure 4.
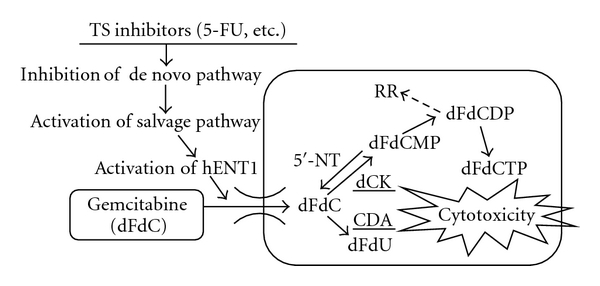
Contribution of thymidylate synthase (TS) in metabolic pathway of gemcitabine. For explanation of symbols and metabolic routes, see text. hENT1, human equilibrative nucleoside transporter; dCK, deoxycytidine kinase; RR, ribonucleotide reductase; 5′-NT, 5′-nucleotidase; CDA, cytidine deaminase.
6. Future Prospects
Although insufficient antitumor effect in pancreatic cancer has been detected with 5-FU alone, its modulating action in GEM treatment will be focused on in the future. In fact, the mid-trial report of the GEMSAP trial (Phase II) for Stage IV pancreatic cancer (Nakai et al.) in Japan revealed that chemotherapy with GEM plus S-1 (oral 5-FU prodrug, tegafur, gimeracil, and oteracil potassium) might be superior to chemotherapy with GEM alone (median progression-free survival, 5.4 versus 3.6 months, hazard ratio = 0.64 (95% confidence interval 0.42–0.97), P = .036; overall survival, 14.1 versus 8.7 months, P = .104) [36]. These experimental clinical results were shown by our recent study and suggested that TS and the dihydropyrimidine dehydrogenase (DPD) inhibitory effect of S-1 might produce additional effect on GEM [36]. In the future, the study of application timing and dosing of 5-FU, and S-1, that induce the most favorable outcome in regard to hENT1 should be continued. The challenge to provide better treatment against advanced pancreatic cancer is just beginning.
Financial Disclosure
There is no financial disclosure.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics. Cancer Journal for Clinicians. 2009;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Chua YJ, Cunningham D. Adjuvant treatment for resectable pancreatic cancer. Journal of Clinical Oncology. 2005;23(20):4532–4537. doi: 10.1200/JCO.2005.17.954. [DOI] [PubMed] [Google Scholar]
- 3.Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. Journal of the American Medical Association. 2007;297(3):267–277. doi: 10.1001/jama.297.3.267. [DOI] [PubMed] [Google Scholar]
- 4.Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. Journal of Clinical Oncology. 2007;25(15):1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 5.Burris HA, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first- line therapy for patients with advanced pancreas cancer: a randomized trial. Journal of Clinical Oncology. 1997;15(6):2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 6.Louvet C, Labianca R, Hammel P, et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. Journal of Clinical Oncology. 2005;23(15):3509–3516. doi: 10.1200/JCO.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 7.Osada S, Saji S. New approach to cancer therapy: the application of signal transduction to anti-cancer drug. Current Medicinal Chemistry. 2003;3(2):119–131. doi: 10.2174/1568011033353461. [DOI] [PubMed] [Google Scholar]
- 8.Osada S, Carr BI. Critical role of extracellular signal-regulated kinase (ERK) phosphorylation in novel vitamin K analog-induced cell death. Japanese Journal of Cancer Research. 2000;91(12):1250–1257. doi: 10.1111/j.1349-7006.2000.tb00911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osada S, Osada K, Carr BI. Tumor cell growth inhibition and extracellular signal-regulated kinase (ERK) phosphorylation by novel K vitamins. Journal of Molecular Biology. 2001;314(4):765–772. doi: 10.1006/jmbi.2001.5171. [DOI] [PubMed] [Google Scholar]
- 10.Osada S, Tomita H, Tanaka Y, et al. The utility of vitamin K3 (menadione) against pancreatic cancer. Anticancer Research. 2008;28(1A):45–50. [PubMed] [Google Scholar]
- 11.Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Annals of Oncology. 2006;17(5):v7–v12. doi: 10.1093/annonc/mdj941. [DOI] [PubMed] [Google Scholar]
- 12.Mackey JR, Mani RS, Selner M, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Research. 1998;58(19):4349–4357. [PubMed] [Google Scholar]
- 13.Damaraju VL, Damaraju S, Young JD, et al. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene. 2003;22(47):7524–7536. doi: 10.1038/sj.onc.1206952. [DOI] [PubMed] [Google Scholar]
- 14.Spratlin J, Sangha R, Glubrecht D, et al. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clinical Cancer Research. 2004;10(20):6956–6961. doi: 10.1158/1078-0432.CCR-04-0224. [DOI] [PubMed] [Google Scholar]
- 15.Giovannetti E, Mey V, Nannizzi S, Pasqualetti G, Del Tacca M, Danesi R. Pharmacogenetics of anticancer drug sensitivity in pancreatic cancer. Molecular Cancer Therapeutics. 2006;5(6):1387–1395. doi: 10.1158/1535-7163.MCT-06-0004. [DOI] [PubMed] [Google Scholar]
- 16.Kroep JR, Loves WJ, van der Wilt CL, et al. Pretreatment deoxycytidine kinase levels predict in vivo gemcitabine sensitivity. Molecular Cancer Therapeutics. 2002;1(6):371–376. [PubMed] [Google Scholar]
- 17.Bergman AM, Pinedo HM, Peters GJ. Determinants of resistance to 2′,2′-difluorodeoxycytidine (gemcitabine) Drug Resistance Updates. 2002;5(1):19–33. doi: 10.1016/s1368-7646(02)00002-x. [DOI] [PubMed] [Google Scholar]
- 18.Van der Wilt CL, Kroep JR, Bergman AM, et al. The role of deoxycytidine kinase in gemcitabine cytotoxicity. Advances in Experimental Medicine and Biology. 2000;486:287–290. doi: 10.1007/0-306-46843-3_56. [DOI] [PubMed] [Google Scholar]
- 19.Zhou BS, Tsai P, Ker R, et al. Overexpression of transfected human ribonucleotide reductase M2 subunit in human cancer cells enhances their invasive potential. Clinical and Experimental Metastasis. 1998;16(1):43–49. doi: 10.1023/a:1006559901771. [DOI] [PubMed] [Google Scholar]
- 20.Fan H, Villegas C, Wright JA. Ribonucleotide reductase R2 component is a novel malignancy determinant that cooperates with activated oncogenes to determine transformation and malignant potential. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(24):14036–14040. doi: 10.1073/pnas.93.24.14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakano Y, Tanno S, Koizumi K, et al. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. British Journal of Cancer. 2007;96(3):457–463. doi: 10.1038/sj.bjc.6603559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ueno H, Kiyosawa K, Kaniwa N. Pharmacogenomics of gemcitabine: can genetic studies lead to tailor-made therapy? British Journal of Cancer. 2007;97(2):145–151. doi: 10.1038/sj.bjc.6603860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bepler G, Kusmartseva I, Sharma S, et al. RRM1 modulated in vitro and in vivo efficacy of gemcitabine and platinum in non-small-cell lung cancer. Journal of Clinical Oncology. 2006;24(29):4731–4737. doi: 10.1200/JCO.2006.06.1101. [DOI] [PubMed] [Google Scholar]
- 24.Ulrich CM, Bigler J, Bostick R, Fosdick L, Potter JD. Thymidylate synthase promoter polymorphism, interaction with folate intake, and risk of colorectal adenomas. Cancer Research. 2002;62(12):3361–3364. [PubMed] [Google Scholar]
- 25.Garcia V, García JM, Peña C, et al. Thymidylate synthase messenger RNA expression in plasma from patients with colon cancer: prognostic potential. Clinical Cancer Research. 2006;12(7, part 1):2095–2100. doi: 10.1158/1078-0432.CCR-05-1644. [DOI] [PubMed] [Google Scholar]
- 26.Johnston PG, Drake JC, Trepel J, Allegra CJ. Immunological quantitation of thymidylate synthase using the monoclonal antibody TS 106 in 5-fluorouracil-sensitive and -resistant human cancer cell lines. Cancer Research. 1992;52(16):4306–4312. [PubMed] [Google Scholar]
- 27.Copur S, Aiba K, Drake JC, Allegra CJ, Chu E. Thymidylate synthase gene amplification in human colon cancer cell lines resistant to 5-fluorouracil. Biochemical Pharmacology. 1995;49(10):1419–1426. doi: 10.1016/0006-2952(95)00067-a. [DOI] [PubMed] [Google Scholar]
- 28.Ciaparrone M, Quirino M, Schinzari G, et al. Predictive role of thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase expression in colorectal cancer patients receiving adjuvant 5-fluorouracil. Oncology. 2006;70(5):366–377. doi: 10.1159/000098110. [DOI] [PubMed] [Google Scholar]
- 29.Kamoshida S, Matsuoka H, Ishikawa T, et al. Immunohistochemical evaluation of thymidylate synthase (TS) and p16 in advanced colorectal cancer: implication of TS expression in 5-FU-based adjuvant chemotherapy. Japanese Journal of Clinical Oncology. 2004;34(10):594–601. doi: 10.1093/jjco/hyh113. [DOI] [PubMed] [Google Scholar]
- 30.Takamura M, Nio Y, Yamasawa K, Dong M, Yamaguchi K, Itakura M. Implication of thymidylate synthase in the outcome of patients with invasive ductal carcinoma of the pancreas and efficacy of adjuvant chemotherapy using 5-fluorouracil or its derivatives. Anti-Cancer Drugs. 2002;13(1):75–85. doi: 10.1097/00001813-200201000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Hu YC, Komorowski RA, Graewin S, et al. Thymidylate synthase expression predicts the response to 5-fluorouracil-based adjuvant therapy in pancreatic cancer. Clinical Cancer Research. 2003;9(11):4165–4171. [PubMed] [Google Scholar]
- 32.Rauchwerger DR, Firby PS, Hedley DW, Moore MJ. Equilibrative-sensitive nucleoside transporter and its role in gemcitabine sensitivity. Cancer Research. 2000;60(21):6075–6079. [PubMed] [Google Scholar]
- 33.Komori S, Osada S, Mori R, et al. Contribution of thymidylate synthase to gemcitabine therapy for advanced pancreatic cancer. Pancreas. 2010;39(8):1284–1292. doi: 10.1097/MPA.0b013e3181dec17d. [DOI] [PubMed] [Google Scholar]
- 34.Réjiba S, Bigand C, Parmentier C, Hajri A. Gemcitabine-based chemogene therapy for pancreatic cancer using Ad-dCK::UMK GDEPT and TS/RR siRNA strategies. Neoplasia. 2009;11(7):637–650. doi: 10.1593/neo.81686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giovannetti E, Mey V, Nannizzi S, et al. Cellular and pharmacogenetics foundation of synergistic interaction of pemetrexed and gemcitabine in human non-small-cell lung cancer cells. Molecular Pharmacology. 2005;68(1):110–118. doi: 10.1124/mol.104.009373. [DOI] [PubMed] [Google Scholar]
- 36.Merl MY, Abdelghany O, Li J, Saif MW. First-line treatment of metastatic pancreatic adenocarcinoma: can we do better? Highlights from the “2010 ASCO Annual Meeting”. Chicago, IL, USA. June 4–8, 2010. Journal of the Pancreas. 2010;11(4):317–320. [PubMed] [Google Scholar]