

Abstract
The oncoprotein meningioma 1 (MN1) is overexpressed in several subtypes of acute myeloid leukemia (AML) and overexpression was associated with a poor response to chemotherapy. MN1 is a cofactor of retinoic acid receptor/retinoic x receptor (RAR/RXR)-mediated transcription and this study identified genes in the promonocytic cell line U937 that were regulated by MN1. We found that MN1 can both stimulate and inhibit transcription. Combining MN1 expression with all-trans retinoic acid (ATRA), the ligand of the RAR/RXR dimer, showed that MN1 could both enhance and repress ATRA effects. Many of the identified genes are key players in hematopoiesis and leukemogenesis (e.g. MEIS1 and BMI1). Another interesting target is DHRS9. DHRS9 is involved in the synthesis of ATRA from vitamin A. MN1 inhibited DHRS9 expression and completely abolished its induction by ATRA. MN1 is also the target of a rare AML-causing translocation encoding the MN1–TEL protein. MN1–TEL induces expression of only a few genes and its most pronounced effect is inhibition of a large group of ATRA-induced genes including DHRS9. In conclusion, both MN1 and MN1–TEL interfere with the ATRA pathway and this might explain the differentiation block in leukemias in which these genes are involved.
Introduction
Recently, meningioma 1 (MN1) was identified as an important player in myeloid leukemogenesis. Overexpression of MN1 is observed in acute myeloid leukemia (AML) specified by the chromosomal aberration inv(16) (1,2), in some AMLs overexpressing the transcription factor ecotropic viral integration (EVI1) (2) and in some adult AMLs with normal karyotype (3). A high level of MN1 expression is a predictor of poor prognosis in these latter patients and recently it has been shown that low MN1 expression can be used as a marker to predict sensitivity to all-trans retinoic acid (ATRA) treatment (4). By retroviral insertional mutagenesis, Slape et al. (5) have identified MN1 as a second hit that co-operates with the NUP98–HOXD13 fusion in causing myelodysplastic syndrome or AML in mice.
However, the involvement of MN1 in AML has already been known much longer. In 1995, we cloned the MN1 gene (6) and soon thereafter it was identified as a target gene of the recurrent t(12;22) found in AML patients (7). As a result, almost all the coding sequence of MN1 is fused to two-thirds of the coding sequence of TEL (ETV6). MN1 appeared to act as a transcriptional coactivator of retinoic acid receptor/retinoic x receptor (RAR/RXR) (8), but recent research (4) and also this paper show that MN1 has a dual function. Depending on the type of RAR/RXR target gene, MN1 can either stimulate or repress transcription, with or without collaboration with ATRA. TEL, the other fusion partner of the MN1–TEL fusion, is a member of the E26 transformation-specific (ETS) family of transcription factors and represses transcription by binding to ETS elements in promoters (9,10). MN1–TEL is thought to act as a novel transcription factor causing transcription deregulation of genes normally repressed by TEL (11). Recently, we have shown that MN1–TEL has an additional characteristic important for its pathogenic properties. By the use of a point mutant that cannot bind to ETS elements, we have shown that MN1–TEL represses RAR/RXR-mediated transcription (12).
Bone marrow (BM) transduction/transplantation experiments in mice have shown that both MN1–TEL and MN1 are hematopoietic oncogenes. MN1–TEL induces AML within 3 months after BM transplantation (13). MN1 overexpression in mouse BM causes myeloproliferative disease, a condition slightly different from AML. Mice died within 5–8 weeks after receiving transplants. Combined expression in mouse BM of MN1 and CBFb-MYH11, the product of inv(16), resulted in rapid development of AML strongly suggesting that overexpression of MN1 is indeed an important step in the development of inv(16) AML (14). Heuser et al. (4) have also shown that MN1 is a powerful oncogene in mouse BM and based on the rapid development of disease suggested that overexpression of MN1 alone might be sufficient to cause AML in mice. In agreement with the results of the in vivo studies, Mn1 expression is found in the granulocytes–monocytes progenitor fraction of hematopoietic cells in mice (14). Wagner et al. (15) detected MN1 in the slow dividing, primitive fraction of hematopoietic stem cells obtained from human umbilical cord blood.
Although all these reports document the importance of MN1 in different forms of leukemia, little is known about the downstream targets of MN1 expression, which severely hampers our understanding of its involvement in leukemogenic transformation. This study aimed to identify genes regulated by MN1 and MN1–TEL by expression profiling in the promonocytic cell line U937 (16) and the effect of expression of these genes in absence or presence of ATRA. The U937 cell line has a progenitor phenotype with features close to the granulocytes–monocytes progenitor stage of hematopoiesis. The cell line has extensively been employed to study myeloid differentiation induced by agents such as ATRA and dimethyl sulfoxide. We show that MN1, directly or indirectly, affects expression of many genes that play a role in hematopoiesis. Moreover, MN1 can both inhibit and stimulate expression of genes induced by RAR/RXR. MN1–TEL inhibits some genes that are also inhibited by MN1 but its greatest effect is in inhibiting RAR/RXR-induced genes.
Materials and methods
Origin of cell lines, cell culture, induction of gene expression and western blotting
The cell lines were based on U937T (17) [derived from cell line U937 (18)] and inducibly express MN1 or MN1–TEL using the tet-off system. They were generated as described previously (17), by selecting cells transfected with pUHD10S plasmid, containing complementary DNA (cDNA) of the gene of interest. The cell lines will be referred to as the MN1, MN1–TEL and UHD cell line. The UHD cell line is transfected with empty expression plasmid. The cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, 1000 U/ml penicillin, 1 mg/l streptomycin, 1 μg/ml tetracycline, 0.5 μg/ml puromycin and 100 μg/ml hygromycin. Gene expression was induced by withdrawal of tetracycline. Two separate induction experiments were performed a few weeks apart to serve as biological duplicates. For each time point (0, 16, 20, 24, 48 and 72 h), 3·× 106 cells of each cell line were exposed to four different medium conditions: absence of tetracyclin (−tet: induction of target gene); presence of tetracyclin (+tet: no induction of target gene) and both in absence or presence of 1 μM (Sigma–Aldrich, Zwijdrecht, The Netherlands). The cells were harvested and washed in phosphate-buffered saline. Dry cell pellets were stored at −80°C corresponding to a 1 ml portion (to be used later for RNA extraction) and two 100 μl portions of resuspended cells (each to be used for western blot analysis; enough for loading of four gels). The antibody against the N-terminal part of MN1 (2F2) has been described (11). The antibody against BMI1 was F6 (Upstate Lake Placid, NY), for YES1 #610375 (BD Biosciences, Breda, The Netherlands) and ID1 SC-488 (Santa Cruz, CA). The western blots were incubated with horseradish peroxidase-labeled secondary antibodies (P044701, Dako, Glostrup, Denmark) and IRDye 680 conjugated antimouse (LI-COR, Lincoln, NE). The latter was used to scan and quantify the protein signals on an Odyssey infrared imaging system (LI-COR). Protein levels were calculated relative to the amount of β-tubulin present in the sample. The beta-Tubulin antibody was from hybridoma E7.
RNA extraction and amplification of RNA
Total RNA was extracted from the frozen cell pellets by TRIzol reagent (Invitrogen, Carlsbad, CA) and further purified using RNeasy columns with on-column DNase treatment (Qiagen, Hilden, Germany). The concentration and purity of the RNA were measured on a NanoDrop ND-1000 UV-VIS spectrophotometer and the quality of the RNA was checked using the Agilent 2100 BioAnalyzer with an RNA 6000 Nano LabChip. Three microgram of total RNA was used for linear amplification using the Superscript III RNA Amplification kit (Invitrogen). The concentration and purity of the amplified RNA were measured on a NanoDrop instrument and the quality of the amplified RNA was checked by agarose gel electrophoresis.
Gene expression profiling with microarrays
Spotted oligo microarrays with the Operon V3.0 library (35K Human, http://omad.operon.com/humanV3) were obtained from the Netherlands Cancer Institute Central Microarray Facility. Protocols for sample preparation were taken from the Netherlands Cancer Institute Central Microarray Facility Web site (http://microarrays.nki.nl). In short, 1 μg of amplified RNA was labeled using the ULS™-Cy3/5 amplified RNA fluorescent labeling kit (Kreatech, Amsterdam, The Netherlands) and was used for hybridization on the same day. The labeling efficiency was checked on a NanoDrop instrument. The labeled amplified RNA was fragmented (RNA Fragmentation Reagents, Ambion, Austin, TX) and mixed with blocking solution containing Poly d(A), Cot-1 DNA and yeast tRNA (GE Healthcare, Zeist, The Netherlands and Roche, Basel, Switzerland). The arrays were hybridized overnight at 42°C on a Tecan HS4800 hybridization station, according to the M016 protocol developed by the Erasmus Center for Biomics (http://www.biomics.nl). Samples were cohybridized according to a multifactorial design scheme as shown in supplementary Figure 1 (available at Carcinogenesis Online), with each combination of samples in a straight and dye-swap fashion for each biological duplicate. The hybridized arrays were scanned on a Perkin Elmer ScanArray Express HT instrument. The measured fluorescence intensities were determined using ImaGene software version 6.0 (Biodiscovery, El Segundo, CA).
Microarray data analysis
The ImaGene data were uploaded into the CMF database (CMFdb, http://cmfdb.nki.nl) and normalized using the lowess subarray method (default settings, except for background correction). No background correction was applied, as overall background was very low and even (mean background 311 ± 75 for Cy5 and 446 ± 173 for Cy3, averaged overall arrays, versus a mean signal of 2376 ± 592 for Cy5 and 2927 ± 650 for Cy3, corresponding to 15%). Background correction would result in negative intensities for low-intensity spots, indicating that any background signal was not superimposed on the spots but rather present next to the spots. This was also indicated by the appearance of the blank spotting controls, which showed up as holes in the background. The normalized data were downloaded from the CMF database and further analyzed in R using limmaGUI (19–21). The data were analyzed with each time point separately as well as combined for each condition. Linear parametrizations were created including each of the comparisons of interest. Each separate comparison at each time point is composed of four arrays (supplementary Figure 1 is available at Carcinogenesis Online). In addition, each biological duplicate was analyzed separately to assess the biological variability. Gene lists were ranked based on the B-statistic (log odds of differential expression); P-values are also reported. Unsupervised hierarchical clustering was performed using Spotfire DecisionSite 9.0 (Tibco, Somerville, MA) with default settings. In the cluster analysis, log2 ratios at the individual time points with B-values <1 were set to 0 when the accompanying B-value of the combined time points of a certain reporter was also <1.
Reverse transcription–polymerase chain reaction and real-time quantitative polymerase chain reaction
Total RNA was converted into cDNA with the M-MLV reverse transcriptase system (Invitrogen), using 3 μg of total RNA and 750 ng of random primer (Invitrogen). Real-time quantitative polymerase chain reaction (qPCR) was performed using the amount of cDNA equivalent to 25 ng total RNA in a total volume of 25 μl containing 330 nM of primers each and 12.5 μl Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). The primers used for qPCR for MEIS1 and YES1 were taken from the RTprimerDB database (http://medgen.ugent.be/rtprimerdb/) (22) (IDs indicated). The primer sets for NDRG1 and POLR2A were obtained from fellow researchers. Whenever possible, intron-spanning primers were used. The sequences are shown in supplementary Table 1 (available at Carcinogenesis Online). Expression levels were determined relative to a standard curve for which standards were prepared by making 4-fold serial dilutions of a pooled sample. For each biological duplicate, qPCR was performed in duplicate and thus the results are represented as the average and standard deviation of four measurements. PCRs and real-time fluorescence measurements were done on an ABI 7700 Sequence Detection System (Applied Biosystems; annealing: 60°C, measurement: 72°C). The qPCR results were normalized using the messenger RNA levels of RNA polymerase 2A (POLR2A). The results are presented relative to the expression levels in the presence of tetracycline for each cell line.
Fluorescent in situ hybridization
The U937 cell line cultures were treated for 30 min with colcemid and fixed with ethanol:acetic acid (3:1). Nuclei were spotted onto slides and incubated with the nicktranslation-labeled plasmids (Bio-nicktranslation kit, Invitrogen) or BAC probes (Random Prime labellings system, Invitrogen). The amount of plasmid probes was 5 ng per slide and that of BAC probes 50–75 ng per slide. BAC clones were selected from the UCSC genome browser (UC Santa Cruz, Santa Cruz, CA) and purchased from BACPAC Resources (Oakland, CA). FISH slides were analyzed with an Axioplan 2 Imaging microscope (Zeiss, Göttingen, Germany) and images were captured using Isis software (MetaSystems, Altlussheim, Germany).
Results
Induction of expression of MN1 and MN1–TEL in U937T cells
Expression of MN1 and MN1–TEL was induced in U937T cells by tetracycline (tet) withdrawal for 16, 20, 24, 40, 48 and 72 h. A western blot visualizing the induction of MN1 or MN1–TEL protein is shown in Figure 1A. In the presence of tetracycline (0 h), there was no detectable expression of either protein but expression gradually increased from 16 h onward, as indicated by the appearance of single bands of the correct molecular weight. At induction times exceeding 24 h, additional bands appeared, possibly representing breakdown products of MN1 and MN1–TEL. The blots were stained with Coomassie blue to verify equal loading of the lanes (data not shown). In parallel, the messenger RNA expression levels were examined by qPCR analysis as shown in Figure 1B. Both cell lines contained a low, but clearly detectable level of MN1 or MN1–TEL transcripts even in the presence of tetracycline, whereas these products were not detected in the other inducible cell lines or the control cell line UHD. Tetracycline release resulted in a 10-fold increase in transcript levels within 16–24 h, increasing further with longer induction times. Interestingly, the presence of ATRA during tetracycline release led to elevated expression levels in both cases (25- to 30-fold for MN1 and 30- to 40-fold for MN1–TEL at 16–24 h). In pilot experiments, a similar effect of ATRA on induction of MN1 and MN1–TEL protein was observed (data not shown).
Fig. 1.

Induction of MN1 and MN1–TEL expression in U937T cells. The expression of MN1 and MN1–TEL was induced by withdrawal of tetracycline for 16, 20, 24, 40, 48 and 72 h, after which samples were collected for western blotting and real-time qPCR. The cells collected at 0 h were grown in the presence of tetracycline and without the addition of ATRA. (A) Assessment of the expression of MN1 or MN1–TEL by western blot, using an antibody raised against the N-terminal part of MN1 (2F2). (B) Assessment of the levels of MN1 and MN1–TEL messenger RNA expression by qPCR. The qPCR results were normalized, using the messenger RNA levels of POLR2A, and are presented relative to those in the presence of only the tet-repressor: white squares: +tet and +ATRA; black circles: −tet, no ATRA; gray triangles: −tet and +ATRA).
Alterations in gene expression mediated by MN1 and MN1–TEL and ATRA in U937 cells
To examine the effects of MN1 and MN1–TEL on the expression of genes and the influence of ATRA on these effects, oligonucleotide microarray analysis was performed following the scheme presented in supplementary Figure 1 (available at Carcinogenesis Online). Only samples taken after 16, 20 and 24 h of induction were used for microarray analysis because there is a clear expression of the genes at these time points and we want to detect early effects and not focus on possible secondary effects of expression of MN1 and MN1–TEL. For assessment of the effect of ATRA, only samples induced for 16 and 20 h were used. Each combination of samples was hybridized in a straight and dye-swap fashion for two biological duplicates, adding up to four arrays per combination. Both raw and normalized data have been deposited in NCBI’s Gene Expression Omnibus (23) and are accessible through GEO Series accession number GSE11441 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE11441).
On average, 29 129 ± 2699 unflagged spots were quantified per array (out of 37 632 on each of the 100 arrays in total). We identified 4468 spots showing statistically significant differences [B-values of 1 or higher when all the time points (16, 20 and 24 h) were combined for analysis] in mean expression levels under the conditions selected for supplementary Figure 2 (available at Carcinogenesis Online). The unsupervised hierarchical clustering (supplementary Figure 2 is available at Carcinogenesis Online) of these signals shows that time points of each different treatment cluster together, and that all the ATRA effects cluster together. It is obvious that addition of ATRA results in higher expression levels than those observed with MN1 or MN1–TEL expression alone. When analyzing the expression data, we realized that MN1, although present on the array, did not show up as one of the differentially expressed genes. A closer inspection of the signal showed that it was consistently low for both Cy5 and Cy3 in all arrays (514 ± 114 and 516 ± 115, respectively), suggesting that the oligo for MN1 most probably did not hybridize very well with the labeled amplified RNA.
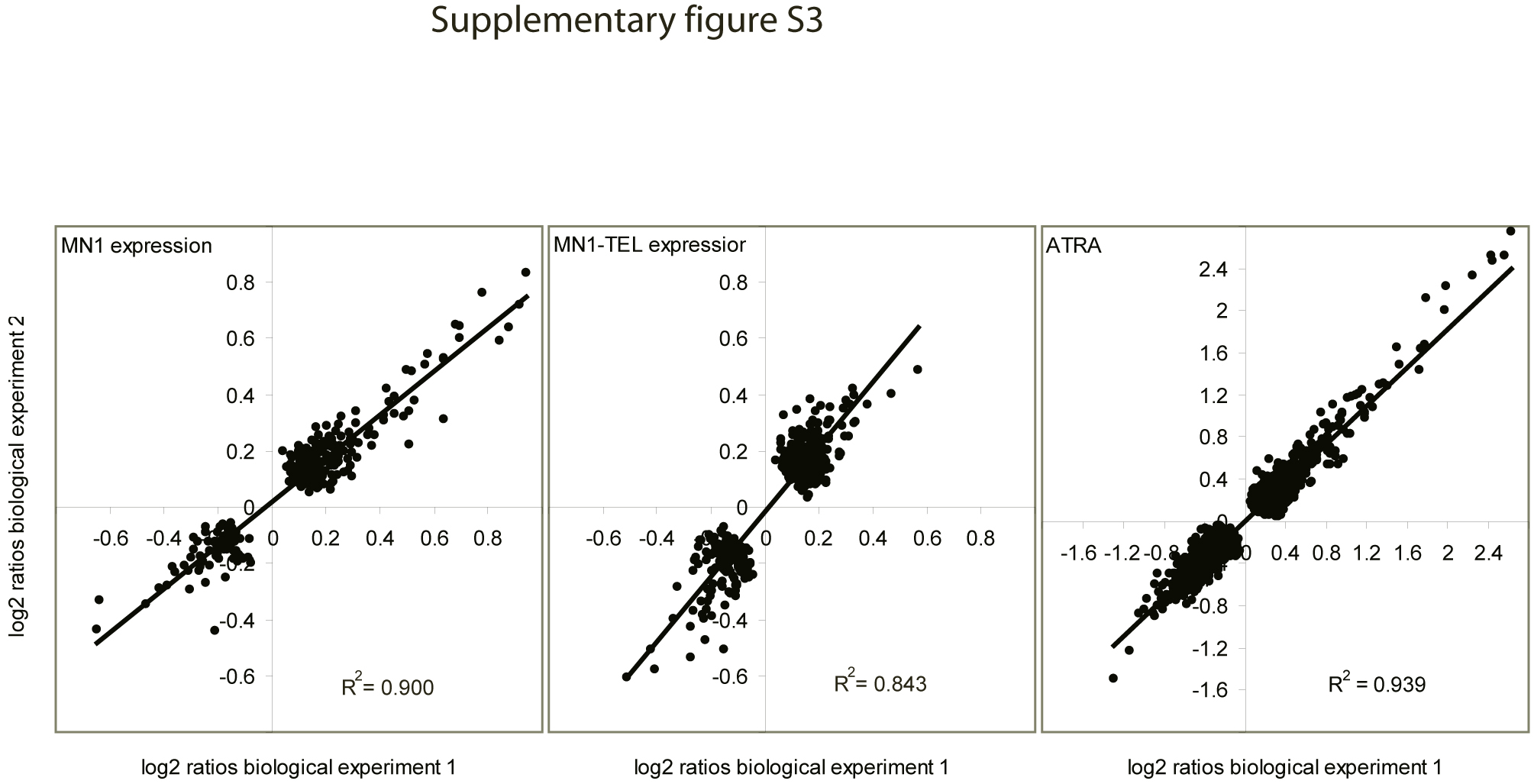
To assess the reproducibility between biological duplicates for the genes affected by MN1, MN1–TEL and ATRA, correlation plots for the resulting reporter sets are shown in supplementary Figure 3 (available at Carcinogenesis Online). For these, the microarrays were analyzed for each biological replicate separately. Correlation coefficients (R2) were obtained by linear regression of the log2 ratios in each biological duplicate plotted against each other. This resulted in correlation coefficients between the log2 ratios in each biological duplicate of 0.900 for genes affected by MN1, 0.843 for genes affected by MN1–TEL and 0.938 for genes affected by ATRA alone (all time points combined), indicating very similar behavior in both experiments. Even though most of the log2 ratios found were relatively low, just >0.2 (absolute value), with maximal values up to 1.0 for MN1 and up to 0.6 for MN1–TEL, these high correlation coefficients give us confidence that even low log2 ratios are meaningful.
Genes affected by MN1, MN1–TEL and ATRA
A list was assembled consisting of all genes influenced by MN1 or MN1–TEL expression with a cutoff of log2 ratios > 0.2 or <−0.2 (n = 85) supplemented with 60 genes whose expression was most significantly affected by ATRA treatment. A cutoff value of 0.2 corresponds to a 15% difference in expression between conditions. The list was supplemented with a set of genes that do not meet the above criteria, but of which the expression is influenced by ATRA treatment in combination with MN1/MN1–TEL (n = 28). Genes were removed from the lists if a similar significant change in expression was observed by tetracycline release of the control cell line as that invoked by MN1 or MN1–TEL expression. The final list, consisting of 165 reporters comprising 155 different genes, was used for cluster analysis. The results are visualized in Figure 2 and it is clear that there is clustering of the different time points of each different treatment and all the ATRA effects. Supplementary Table 2 (available at Carcinogenesis Online) contains all data including the extended gene names, Genbank accession numbers, combined log2 ratios, B-values and P-values. Figure 3 displays the genes that are regulated by MN1 or MN1–TEL (n = 85). The different time points are represented by cluster analysis and accompanied by the combined log2 ratios and P-values. It is obvious that MN1 expression affects many more genes than MN1–TEL expression. About 50% of the genes that are repressed by MN1 are also repressed by MN1–TEL (yellow), whereas MN1–TEL has no effect on most genes that are induced by MN1 (blue). This suggests that the transcription-stimulating properties of MN1 are lost in the MN1–TEL protein. In addition, we identified a small set of genes that is influenced by MN1–TEL and that does not respond to MN1.
Fig. 2.
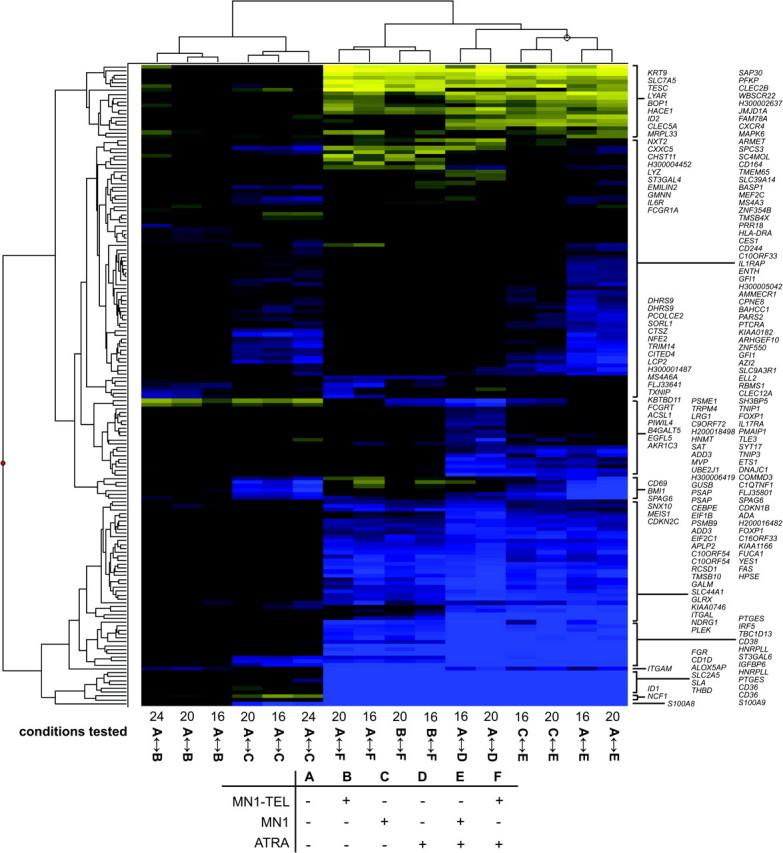
A selected set of 165 reporters cluster together for subsequent time points of MN1 or MN1–TEL expression and/or ATRA treatment in U937 cells. The log2 ratios of changes in expression between the conditions indicated were subjected to unsupervised hierarchical clustering. The conditions tested on the different arrays are indicated at the bottom with numbers (16, 20 or 24) and letter codes A–F. Numbers correspond to hours after induction of MN1 or MN1–TEL. The table underneath the figure explains the letter codes (+: presence of MN1 or MN1–TEL or ATRA; −: absence of MN1 or MN1–TEL or ATRA). For instance, A ↔ B means that a sample with induced MN1–TEL expression is hybridized against a sample without MN1–TEL expression. Blue color represents stimulation and yellow repression.
Fig. 3.

Genes regulated by MN1 and MN1–TEL. The clustering analysis was performed with expression data from the three different time points (16, 20 and 24 h after induction of MN1 or MN1–TEL). All genes listed show a significant change in expression (log2 ratio ≤0.2 or ≥0.2) for all three separate time points. The listed log2 ratios together with the P-value are calculated from combined time points. Genes indicated in bold are used for qPCR analyses (Figure 5 and 6 and supplementary Figure 5 is available at Carcinogenesis Online). Blue represents stimulation and yellow repression.
Figure 4 summarizes those genes that are differentially affected in the combination of MN1 or MN1–TEL with ATRA when compared with either treatment alone. Clearly, the differentials in expression in the presence of ATRA are in general larger than those of MN1 and MN1–TEL. An explanation for this striking difference might be that, in contrast to the effect of ATRA, which impacts all cells, only part of the cells are effectively expressing the gene of interest when released from tetracycline inhibition. Limitation of expression to a sub population of the culture is not uncommon in the expression system used in this study (Dr Judith Boer, Leiden University Medical Center, personal communication, 2006). Synergistic induction or repression of transcription is indicated in Figure 4 with a black or gray background, respectively. We concluded that there was synergy when the observed effect of ATRA in combination with MN1 or MN1–TEL was higher or lower than that expected from the sum of the observed separate effects (cutoff value log2 > 0.2). Figure 4A lists genes induced by ATRA. A small subset of ATRA-stimulated genes is synergistically stimulated by MN1 (indicated with a black box). These include the leukemogenic oncogene MEIS1. MN1–TEL either has no effect on these genes or inhibits the ATRA effect. Figure 4A also shows that expression of MN1 results in a synergistic inhibition of gene expression of about half of the ATRA-stimulated genes (indicated by the gray background) and an even larger group of genes is repressed by MN1–TEL. For instance, the induction of DHRS9, an enzyme involved in ATRA synthesis (24), by ATRA is completely annulled by both MN1 and MN1–TEL. The integrin family member ITGAM is the only gene whose expression is synergistically induced by MN1–TEL and ATRA. Figure 4B shows that MN1 or MN1–TEL can sometimes revert gene inhibition by ATRA. Most notable is the high induction of BMI1 by MN1, which is even further stimulated in the presence of ATRA. Figure 4C shows a selection of genes of which expression is not affected by ATRA alone, but that are affected by the combination of MN1 or MN1–TEL and ATRA. Of special interest are the genes SPAG6, DNAJC1 and COMMD3 that, together with BMI1 (Figure 4B), are similarly regulated by ATRA and MN1. These genes are all located on chromosome 10p12.31. None of these genes is upregulated in the control cell line or in the MN1–TEL cell line. BMI1 is a known common viral integration site for B-cell lymphomas in mice (25) and the possibility existed that integration of the MN1 expression plasmid in this region of the genome might have caused an artificial upregulation of the entire group of genes. We therefore performed fluorescent in situ hybridization on metaphase chromosomes using a plasmid containing the MN1 cDNA and a 10p12.31 specific BAC clone as probes. No integrations of the plasmid were detected in the 10p12.31 region of the genome (supplementary Figure 4 is available at Carcinogenesis Online). Thus, we conclude that the stimulation of these genes by MN1 is genuine. In conclusion, Figure 4 shows that both MN1 and MN1–TEL have considerable effects on RAR/RXR-mediated transcription.
Fig. 4.
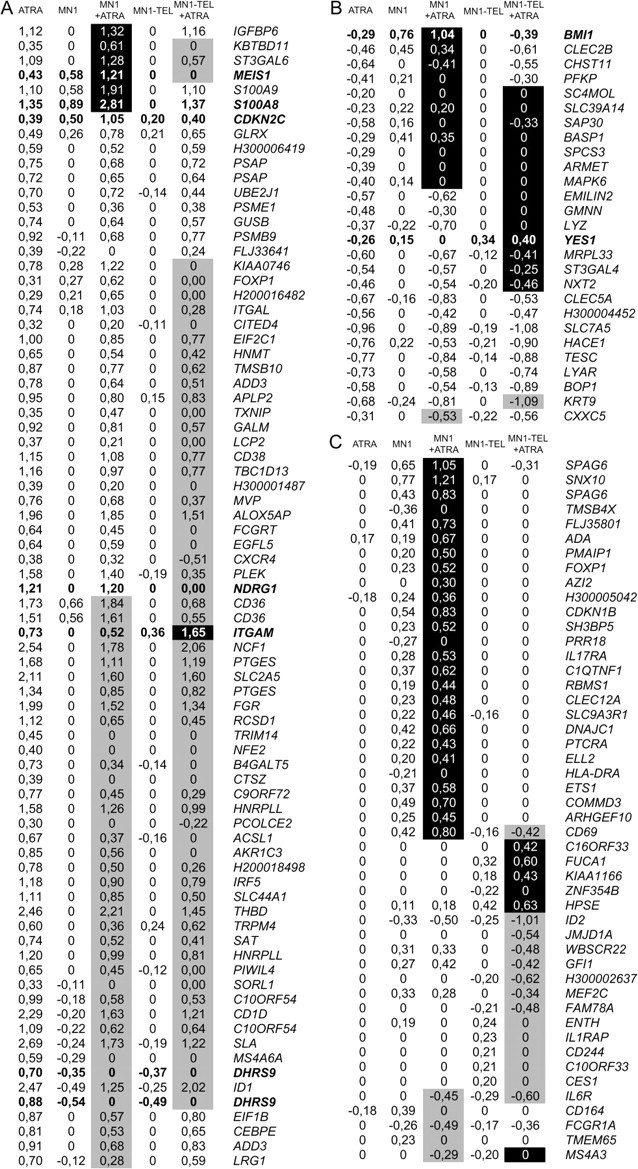
ATRA-regulated genes and the influence of MN1 and MN1–TEL on their expression. Log2 ratios of changes in expression are indicated for ATRA treatment, MN1 or MN1–TEL induction and the combination of MN1 or MN1–TEL induction and ATRA treatment. Black or gray background represents a synergistic induction or repression of ATRA effects (cutoff value log2 ratio 0.2). (A) ATRA-stimulated genes. (B) ATRA-repressed genes. (C) Genes not influenced by ATRA treatment alone, but affected by the combination of ATRA and MN1 or MN1–TEL. Gene names and Genbank number are shown in supplementary Table 2 (available at Carcinogenesis Online). Genes indicated in bold are used for qPCR analyses (Figures 5, 6 and supplementary Figure 5 is available at Carcinogenesis Online).
Validation and extension of microarray results by qPCR analysis
To validate the microarray results, we selected a number of genes for qPCR evaluation. Included were genes exhibiting interesting expression differences between MN1 and MN1–TEL or upon ATRA addition. The results were compared with the corresponding microarray data. This comparison is visually represented by heat maps in Figure 5. This shows that the qPCR results are very similar to those obtained by the microarray analysis, as indicated by the overall similarity in patterning of the heat maps. Notably, there is a 2-fold difference in magnitude in the color scale, which runs from −0.8 to 0.8 for the microarray data and from −1.5 to 1.5 for the qPCR data. This indicates that the microarray data give an ∼2-fold underestimation of the change in gene expression. RNA samples were not only collected for the time points used for microarray analysis but also at 0 h and at longer induction times of 40, 48 and 72 h. Therefore, we were able to follow the behavior of interesting candidate genes beyond the limited 24 h interval of the microarray analysis, and generally, the magnitude of the observed effects continued to increase. Some of the results for S100A8, NDRG1, DHRS9, BMI1, YES1 and ID1 are highlighted in Figure 6, and the full data set for these genes and for MEIS1, CDKN2C, ITGAM, ID1 and IGFBP6 is shown in supplementary Figure 5 (available at Carcinogenesis Online). Figure 6A shows that S100A8 expression after 72 h is 100-fold increased by ATRA, 600-fold by MN1 and 1800-fold by the combination of the two. On the contrary, MN1–TEL expression seems to negatively influence the ATRA effect (supplementary Figure 5B is available at Carcinogenesis Online). ATRA also stimulates NDRG1 expression that is completely abolished by MN1–TEL (Figure 6B). Both MN1 and MN1–TEL abolish the 8- to 10-fold induction by ATRA of DHRS9 expression (Figures 6C and D). Figure 6E shows that MN1 expression causes a considerable increase in BMI1 levels, regardless of the presence of ATRA. YES1 is a gene that is repressed by the addition of ATRA (supplementary Figure 5I is available at Carcinogenesis Online). Upon MN1 and MN1–TEL expression, there is an increase in YES1 expression that is similar in absence or presence of ATRA. The increase is most pronounced upon MN1–TEL expression at time points beyond 24 h (Figure 6F). ID1 expression is decreased upon MN1 induction, whereas the presence of ATRA stimulates (Figure 6G). Figure 6H shows the validation of the observed effects on the protein level for BMI1, ID1 and YES1. Together, the qPCR data and western blots confirm our microarray observations and show that many of the effects continue after the 24 h time point that was the end point for the microarray experiments.
Fig. 5.

Comparison of microarray and qPCR results. The RNA samples used for microarray analysis were also used for qPCR analysis. Expression levels were determined relative to a standard curve and used without further normalization. The results were compared with the corresponding microarray data by expressing the qPCR data as mean log2 ratios. (A) Unsupervised hierarchical clustering of the microarray data (log2 ratios) for the genes selected for qPCR validation. The conditions tested on the different arrays are indicated at the bottom with numbers (16, 20 or 24) and letter codes A–F. A detailed explanation is given in legend of Figure 2. (B) Heat map of the qPCR data in the order corresponding to the unsupervised hierarchical clustering of the microarray data. Blue color represents stimulation and yellow repression.
Fig. 6.
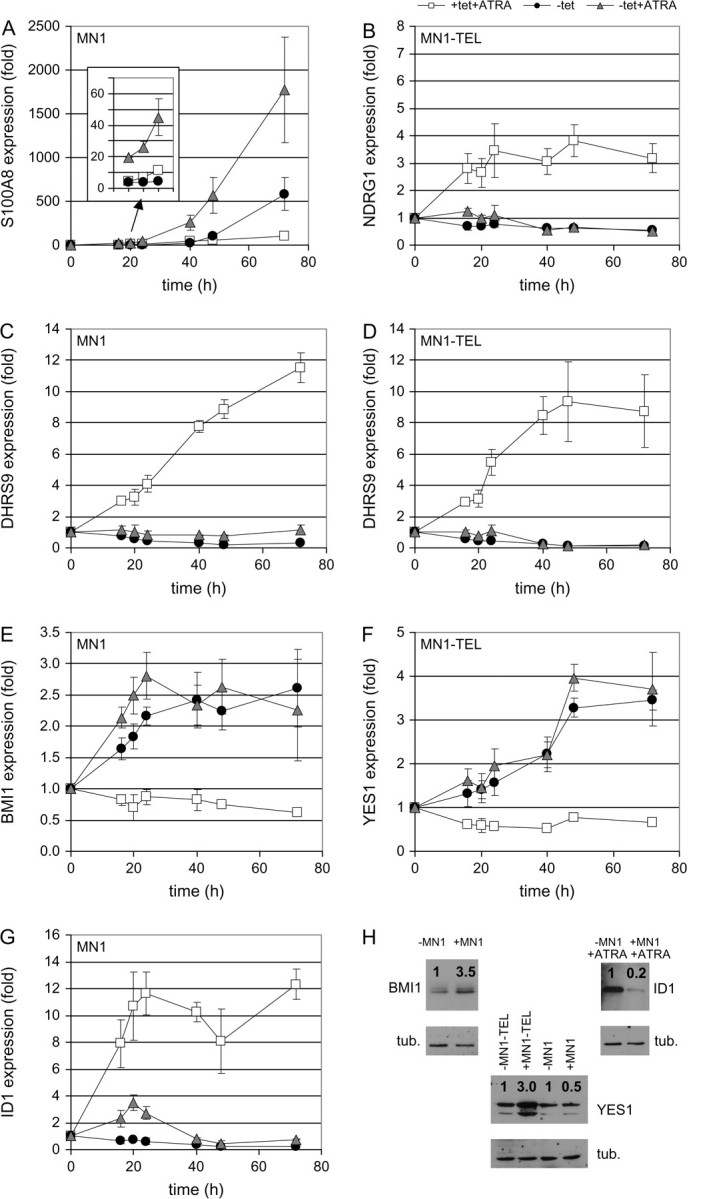
Time dependence of effects of MN1 and MN1–TEL on expression levels and validation of qPCR results by western blot. MN1 and MN1–TEL expression were induced by tetracycline release in the presence or absence of ATRA. In parallel with the RNA samples used for the microarray hybridizations (16, 20 and 24 h), samples were also collected and analyzed with qPCR for time point 0 h and longer incubation times of 40, 48 and 72 h. The qPCR results were normalized, using the messenger RNA levels of POLR2A, and are presented relative to those in the presence of only the tet repressor: white squares: +tet and +ATRA; black circles: −tet, no ATRA; gray triangles: −tet and +ATRA. The results are shown only for the cell line indicated in the upper left corner of each graph. (A) S100A8 (MN1 cell line), (B) NDRG1 (MN1–TEL cell line), (C) DHRS9 (MN1 cell line), (D) DHRS9 (MN1–TEL cell line), (E) BMI1 (MN1 cell line) and (F) YES1 (MN1–TEL cell line). Three of the target genes (BMI1, ID1 and YES1) were analyzed on the protein level. Time points after induction of MN1 or MN1–TEL expression and treatments were chosen (BMI1, 24 h; YES1, 48 h and ID1 72 h) and protein levels detected and quantified. Protein expression levels (indicated with a ratio above the signal) were calculated relative to the amount of β-tubulin present in the sample and the non-induced sample. tub. = tubulin.
Discussion
In this study, we have identified genes regulated by the oncoprotein MN1 and the fusion protein MN1–TEL in U937T cells. During the past year, MN1 and MN1–TEL have been identified as potent leukemogenic oncogenes in mice. In man, MN1 is overexpressed in different subgroups of AML, especially in inv(16) AML, whereas its overexpression in AML with a normal karyotype has been associated with a poor response to ATRA treatment. In the past, we and others showed that MN1 influences RAR/RXR-regulated gene expression and therefore we also studied the effect of MN1 expression on the activation of endogenously expressed retinoid receptors in the presence of ATRA.
The expression profiling resulted in a list of genes up- or downregulated by ATRA, MN1 or by the combination of MN1 and ATRA. We focused on direct effects by choosing the time points following induction of MN1 expression when, and shortly after, expression was visible. We, however, cannot rule out the possibility that some genes are indirectly affected by MN1 expression. For some genes such as MEIS1, CDKN2C, S100A8 and 9, the combination of MN1 and ATRA synergistically induced expression (Figure 4A). This regulation pattern is identical to the one we observed in transient transfection experiments using a mouse sarcoma virus long terminal repeat-driven reporter gene (8). In previous studies, we showed that the transactivating N-terminal domain of MN1, that binds co-activators such as p300 and RAC3, is responsible for this type of regulation. Here, we also show that MN1 can inhibit gene expression induced by ATRA, which is concordant with the data presented by Heuser et al. (4) who described that MN1 opposes the effects of ATRA and even conferred ATRA resistance to cells. At present, we do not know how MN1 exerts these effects. Besides the transactivating domain, not much is known about functional domains in the rest of this large protein. Finally, there are genes whose expression is minimally affected by ATRA alone but whose expression is induced to a higher level by the combination of MN1 and ATRA than by MN1 alone. Among these are BMI1, SPAG6, DNAJC1 and COMMD3, four genes that are located within 0.5 Mb of each other on chromosome 10p12. The joint regulation of these genes suggests that MN1 might be able to extend its influence over a larger genomic area. Interestingly, BMI1 and SPAG6 are also co-ordinately and highly expressed in inv(16) leukemias in which high expression of MN1 is thought to be obligatory (2).
It is obvious that many of the genes affected by MN1 play a role in hematopoiesis and leukemia. BMI1 is a member of the polycomb repression complex 1 and is required for the maintenance of adult hematopoietic stem cells. It functions by repressing genes that promote lineage specification and cell-cycle arrest, such as p16/Arf (26–28). SPAG6 was recently identified as a marker for minimal residual disease in AML patients (29) in cases where expression of SPAG6 is high. In case of a remission, the expression of SPAG6 drops, whereas a relapse is characterized by rising SPAG6 expression levels. The same is true for S100A8 and S100A9. The expression of these two calcium-binding proteins is upregulated by MN1 and they were recently described as genes upregulated in AML (30). The genes are downregulated in response to treatment with methotrexate. Meis1 gene is a common retroviral integration site and was shown to be important for the pathogenesis of AML in mice (31,32).
Overexpression of MN1 in mouse BM produces highly proliferating immortalized cell lines and mice receiving transplants with MN1 retrovirus-transduced cells that rapidly develop a lethal myeloproliferative disease/myeloid leukemia (4,14). Heuser et al. (4) showed that p21, p27 and PU.1 were repressed in these cells. We see no effect of MN1 and/or ATRA on p21 and PU.1 expression in U937 cells. However, we do see upregulation of the cyclin-dependent kinase inhibitors CDKN1B (p27) and CDKN2C (p18). A counterintuitive effect is that upregulation of these genes is expected to lead to cell-cycle arrest. On the other hand, it has been shown that CDKN2C is abundantly expressed in hematopoietic progenitors, AML and cell lines and its expression is downregulated along with myeloid differentiation (33,34).
The MN1–TEL fusion gene contains most of the coding sequence of MN1 and TEL (ETV6), thereby combining the transcription-activating domains of MN1 with the DNA-binding domain of TEL. The fusion protein is thought to act as a deregulated transcription factor possibly disturbing the function of both MN1 and TEL. We therefore expected a much more extensive list of genes whose expression would be affected by MN1–TEL. However, only few genes are induced or inhibited by MN1–TEL. Perhaps its most profound effects reside in the fact that it is unable to stimulate a large set of genes that are induced by MN1 (Figure 3). The other most noticeable activity of the fusion protein is its inhibition of the stimulatory effects of ATRA on a large set of genes (Figure 4A). Disturbing the function of TEL is most probably caused by the binding of MN1–TEL to ETS regulatory sequences using TEL’s DNA-binding domain. Further scrutiny of genes that are induced by MN1–TEL identified two genes, ITGAM and HPSE, that are indeed regulated by the ETS factors PU.1 and GABP (35–37). It is not known whether TEL also regulates these genes, but it has been described that ETS family members can bind to different ETS elements making it possible that MN1–TEL binds the ETS-responsive elements in these genes. In agreement with our qPCR and array data, there are retinoic acid-responsive elements in the promoter region of the ITGAM gene (37), explaining the increase in ITGAM expression in response to ATRA treatment.
The most interesting tumorigenesis-related genes that are influenced by MN1–TEL are NDRG1, ITGAM and DHRS9. The expression of NDRG1 is not directly influenced by the presence of MN1–TEL, but in combination with ATRA treatment it inhibits the ATRA effects on NDRG1 expression. NDRG1 is a metastasis suppressor, frequently downregulated in more advanced and poorly differentiated tumors (38). ITGAM, also known as CD11b or MAC-1, belongs to the family of integrins. CD11b/MAC-1 is expressed on mature monocytes and macrophages and is present on AML cells with a myelomonocytic differentiation. In fact, antibodies against CD11b/MAC-1 are widely used to sort and define the differentiation status of leukemic cells. High CD11b/MAC-1 expression was also seen in leukemias induced by expression of MN1–TEL in mouse BM cells (14). Overexpression of CD11b/MAC-1 has also been associated with an unfavorable prognosis in AML (39,40).
Finally, the effect of MN1 and MN1–TEL on DHRS9 is interesting in relation to the differentiation block observed in most leukemias. The gene encodes an enzyme that is essential for the synthesis of ATRA from vitamin A. We show that ATRA stimulates expression of this gene and may thus enhance its own synthesis in mammals. Both MN1 and MN1–TEL inhibit DHRS9 expression and completely abolish its induction by ATRA. Thus, this might inhibit the ATRA-induced differentiation in AML in man and of MN1- and MN1–TEL-induced leukemia in mice. One could hypothesize that AML with high MN1 expression would perhaps benefit from adjuvant treatment with ATRA. However, the contrary appears to be the case. AML patients who were treated with ATRA only benefited from this treatment when MN1 expression was low. High MN1 expression apparently conferred ATRA resistance to the leukemic cells (4). These findings suggest that perhaps other MN1 target genes play a more important role in defining the leukemia phenotype.
In conclusion, we have shown that MN1 and MN1–TEL, directly or indirectly, regulate expression of many genes that have been implicated in leukemogenesis. Both proteins are able to enhance or inhibit ATRA effects on gene expression. Further research is necessary to fully understand the pleiotropic effects executed by MN1 and MN1–TEL on gene expression.
Supplementary material
Supplementary Figures 1–5 and Tables 1 and 2 can be found at http://carcin.oxfordjournals.org/
Funding
The Dutch Cancer Society (2003-2869).
Supplementary Material
Acknowledgments
The E7 hybridoma against β-tubulin (M. Klymkowsky) was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA 52242.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- AML
acute myeloid leukemia
- ATRA
all-trans retinoic acid
- BM
bone marrow
- cDNA
complementary DNA
- ETS
E26 transformation-specific
- RAR/RXR
retinoic acid receptor/retinoic x receptor
- qPCR
quantitative polymerase chain reaction
References
- 1.Ross ME, et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104:3679–3687. doi: 10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- 2.Valk PJ, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N. Engl. J. Med. 2004;350:1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- 3.Heuser M, et al. High meningioma 1 (MN1) expression as a predictor for poor outcome in acute myeloid leukemia with normal cytogenetics. Blood. 2006;108:3898–3905. doi: 10.1182/blood-2006-04-014845. [DOI] [PubMed] [Google Scholar]
- 4.Heuser M, et al. MN1 overexpression induces acute myeloid leukemia in mice and predicts ATRA resistance in AML patients. Blood. 2007;110:1639–1647. doi: 10.1182/blood-2007-03-080523. [DOI] [PubMed] [Google Scholar]
- 5.Slape C, et al. Retroviral insertional mutagenesis identifies genes that collaborate with NUP98-HOXD13 during leukemic transformation. Cancer Res. 2007;67:5148–5155. doi: 10.1158/0008-5472.CAN-07-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lekanne Deprez RH, et al. Cloning and characterization of MN1, a gene from chromosome 22q11, which is disrupted by a balanced translocation in a meningioma. Oncogene. 1995;10:1521–1528. [PubMed] [Google Scholar]
- 7.Buijs A, et al. Translocation (12;22) (p13;q11) in myeloproliferative disorders results in fusion of the ETS-like TEL gene on 12p13 to the MN1 gene on 22q11 [published erratum appears in Oncogene (1995) 11, 809] Oncogene. 1995;10:1511–1519. [PubMed] [Google Scholar]
- 8.van Wely KH, et al. The MN1 oncoprotein synergizes with coactivators RAC3 and p300 in RAR-RXR-mediated transcription. Oncogene. 2003;22:699–709. doi: 10.1038/sj.onc.1206124. [DOI] [PubMed] [Google Scholar]
- 9.Chakrabarti SR, et al. The leukemia-associated gene TEL encodes a transcription repressor which associates with SMRT and mSin3A. Biochem. Biophys. Res. Commun. 1999;264:871–877. doi: 10.1006/bbrc.1999.1605. [DOI] [PubMed] [Google Scholar]
- 10.Lopez RG, et al. TEL is a sequence-specific transcriptional repressor. J. Biol. Chem. 1999;274:30132–30138. doi: 10.1074/jbc.274.42.30132. [DOI] [PubMed] [Google Scholar]
- 11.Buijs A, et al. The MN1-TEL fusion protein, encoded by the translocation (12;22)(p13;q11) in myeloid leukemia, is a transcription factor with transforming activity. Mol. Cell. Biol. 2000;20:9281–9293. doi: 10.1128/mcb.20.24.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Wely KH, et al. The MN1-TEL myeloid leukemia-associated fusion protein has a dominant-negative effect on RAR-RXR-mediated transcription. Oncogene. 2007;26:5733–5740. doi: 10.1038/sj.onc.1210382. [DOI] [PubMed] [Google Scholar]
- 13.Carella C, et al. MN1-TEL, the product of the t(12;22) in human myeloid leukemia, immortalizes murine myeloid cells and causes myeloid malignancy in mice. Leukemia. 2006;20:1582–1592. doi: 10.1038/sj.leu.2404298. [DOI] [PubMed] [Google Scholar]
- 14.Carella C, et al. MN1 overexpression is an important step in the development of inv(16) AML. Leukemia. 2007;21:1679–1690. doi: 10.1038/sj.leu.2404778. [DOI] [PubMed] [Google Scholar]
- 15.Wagner W, et al. Molecular evidence for stem cell function of the slow-dividing fraction among human hematopoietic progenitor cells by genome-wide analysis. Blood. 2004;104:675–686. doi: 10.1182/blood-2003-10-3423. [DOI] [PubMed] [Google Scholar]
- 16.Sundstrom C, et al. Establishment and characterization of a human histiocytic lymphoma cell line (U-937) Int. J. Cancer. 1976;17:565–577. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- 17.Boer J, et al. Overexpression of the nucleoporin CAN/NUP214 induces growth arrest, nucleocytoplasmic transport defects, and apoptosis. Mol. Cell. Biol. 1998;18:1236–1247. doi: 10.1128/mcb.18.3.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gossen M, et al. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 19.Wettenhall JM, et al. limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics. 2004;20:3705–3706. doi: 10.1093/bioinformatics/bth449. [DOI] [PubMed] [Google Scholar]
- 20.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 21.Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 22.Pattyn F, et al. RTPrimerDB: the real-time PCR primer and probe database, major update 2006. Nucleic Acids Res. 2006;34:D684–D8. doi: 10.1093/nar/gkj155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edgar R, et al. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones RJ, et al. Epstein-Barr virus lytic infection induces retinoic acid-responsive genes through induction of a retinol-metabolizing enzyme, DHRS9. J. Biol. Chem. 2007;282:8317–8324. doi: 10.1074/jbc.M608667200. [DOI] [PubMed] [Google Scholar]
- 25.van Lohuizen M, et al. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell. 1991;65:737–752. doi: 10.1016/0092-8674(91)90382-9. [DOI] [PubMed] [Google Scholar]
- 26.Sawa M, et al. BMI-1 is highly expressed in M0-subtype acute myeloid leukemia. Int. J. Hematol. 2005;82:42–47. doi: 10.1532/IJH97.05013. [DOI] [PubMed] [Google Scholar]
- 27.Raaphorst FM. Deregulated expression of Polycomb-group oncogenes in human malignant lymphomas and epithelial tumors. Hum. Mol. Genet. 2005;14:R93–R100. doi: 10.1093/hmg/ddi111. Spec No 1. [DOI] [PubMed] [Google Scholar]
- 28.Akala OO, et al. Hematopoietic stem cell self-renewal. Curr. Opin. Genet. Dev. 2006;16:496–501. doi: 10.1016/j.gde.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 29.Steinbach D, et al. Identification of a set of seven genes for the monitoring of minimal residual disease in pediatric acute myeloid leukemia. Clin. Cancer Res. 2006;12:2434–2441. doi: 10.1158/1078-0432.CCR-05-2552. [DOI] [PubMed] [Google Scholar]
- 30.Cheok MH, et al. Treatment-specific changes in gene expression discriminate in vivo drug response in human leukemia cells. Nat. Genet. 2003;34:85–90. doi: 10.1038/ng1151. [DOI] [PubMed] [Google Scholar]
- 31.Mamo A, et al. Molecular dissection of Meis1 reveals 2 domains required for leukemia induction and a key role for Hoxa gene activation. Blood. 2006;108:622–629. doi: 10.1182/blood-2005-06-2244. [DOI] [PubMed] [Google Scholar]
- 32.Rozovskaia T, et al. Upregulation of Meis1 and HoxA9 in acute lymphocytic leukemias with the t(4: 11) abnormality. Oncogene. 2001;20:874–878. doi: 10.1038/sj.onc.1204174. [DOI] [PubMed] [Google Scholar]
- 33.Nishimura N, et al. Suppression of ARG kinase activity by STI571 induces cell cycle arrest through up-regulation of CDK inhibitor p18/INK4c. Oncogene. 2003;22:4074–4082. doi: 10.1038/sj.onc.1206498. [DOI] [PubMed] [Google Scholar]
- 34.Schwaller J, et al. Expression and regulation of G1 cell-cycle inhibitors (p16INK4A, p15INK4B, p18INK4C, p19INK4D) in human acute myeloid leukemia and normal myeloid cells. Leukemia. 1997;11:54–63. doi: 10.1038/sj.leu.2400522. [DOI] [PubMed] [Google Scholar]
- 35.Lu WC, et al. Trans-activation of heparanase promoter by ETS transcription factors. Oncogene. 2003;22:919–923. doi: 10.1038/sj.onc.1206201. [DOI] [PubMed] [Google Scholar]
- 36.Jiang P, et al. Cloning and characterization of the human heparanase-1 (HPR1) gene promoter: role of GA-binding protein and Sp1 in regulating HPR1 basal promoter activity. J. Biol. Chem. 2002;277:8989–8998. doi: 10.1074/jbc.M105682200. [DOI] [PubMed] [Google Scholar]
- 37.Hickstein DD, et al. Identification of the promoter of the myelomonocytic leukocyte integrin CD11b. Proc. Natl Acad. Sci. USA. 1992;89:2105–2109. doi: 10.1073/pnas.89.6.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kovacevic Z, et al. The metastasis suppressor, Ndrg-1: a new ally in the fight against cancer. Carcinogenesis. 2006;27:2355–2366. doi: 10.1093/carcin/bgl146. [DOI] [PubMed] [Google Scholar]
- 39.Mason KD, et al. The immunophenotype of acute myeloid leukemia: is there a relationship with prognosis? Blood Rev. 2006;20:71–82. doi: 10.1016/j.blre.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 40.Graf M, et al. Expression of MAC-1 (CD11b) in acute myeloid leukemia (AML) is associated with an unfavorable prognosis. Am. J. Hematol. 2006;81:227–235. doi: 10.1002/ajh.20526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}