

Abstract
The following is a brief review of peroxidase structural biology since the initial structure determination of cytochrome c peroxidase (CCP) 30 years ago. An emphasis will be placed on what CCP has taught us about peroxidase mechanisms, especially Compound I formation and electron transfer.
Keywords: Crystallography, protein engineering, electron transfer, peroxidase
Introduction
Peroxidases hold a venerable position in the history of enzymology and especially in enzyme kinetics. Using horseradish peroxidase (HRP), rapid reaction methods were developed by Britton Chance in the 1940s [1]. The distinct spectroscopic characteristics of the famous Compounds I and II intermediates formed by HRP made it relatively straightforward to detect the green Compound I and red Compound II. Given the relative ease of preparing HRP and the extensive amount of kinetic information known, it may seem surprising that HRP was not an early target for crystallography once crystal structure determination became feasible. HRP was likely the victim of the “low lying fruit” dictum which governed the early days of protein crystallography. Not only was it necessary to be able to prepare large amounts of pure protein, but a critically important second criterion was that the protein must easily crystallize and diffract well. Here is where HRP fell short. In hindsight it appears that heterogeneity in carbohydrate content was a problem. Indeed, it was not until HRP was recombinantly expressed and purified free of carbohydrate that it became possible to grow decent crystals [2].
The initial breakthrough in peroxidase crystallography came with the early discovery [3] and subsequent characterization of yeast cytochrome c peroxidase (CCP) by Yonetani and colleagues [4]. CCP is simpler than HRP in having no carbohydrate, no S-S bonds, and no metal ions (other than the heme iron). CCP also exhibited the convenient property of crystallizing by dialysis against water [5]. With somewhat greater care it was possible to obtain diffraction quality crystals [6]. This ultimately resulted in the first heme peroxidase crystal structure published 30 years ago [7] (Fig. 1). At that time a total of 57 protein structures had been deposited in the PDB while as of today there are over 50,000. Of the 57 structures in the PDB in 1980, the known heme protein structures included myoglobin, hemoglobin, 2 eukaryotic cytochromes c (3CYT,1CYC), and one prokaryotic cytochrome c (155C). Therefore, the structure of CCP represented both the first peroxidase and heme enzyme structure to be solved. Twelve years later the second peroxidase structure was solved, myeloperoxidase [8], followed by lignin peroxidase [9], Coprinus peroxidase [10], ascorbate peroxidase [11] and plant peroxidases in 1996-1997 [2, 12]. Today there is at least one representative structure form each of the main 3 main classes, as defined by Welinder [13]. The primary reason for the long lapse between the first and second peroxidase structure followed by the relatively rapid solution of several other peroxidase structures in the mid 1990s was due to recombinant DNA technology. This also opened the way to using protein engineering to probe peroxidase structure and function and has been of enormous benefit in understanding peroxidase function.
Figure 1.
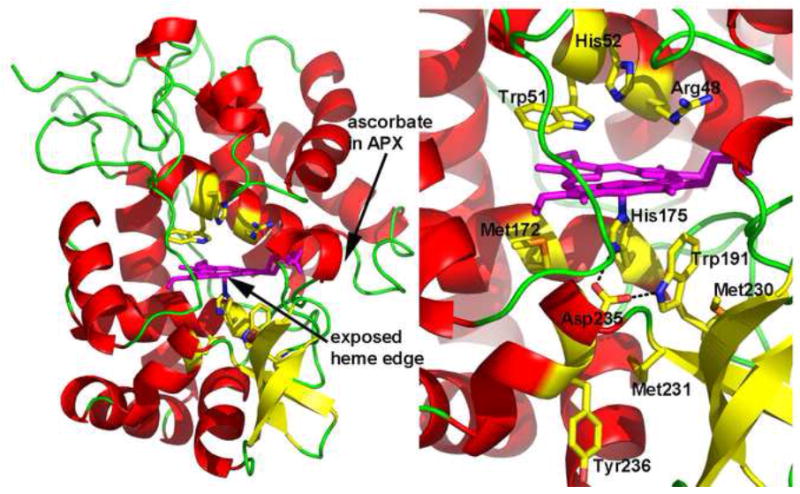
Crystal structure of yeast CCP. The location of the binding site for typical aromatic peroxidase substrates is indicated as the “exposed heme edge”.
The following is a brief review on what crystallography coupled with mutagenesis and other biophysical tools has taught us about peroxidase structure and function since solution of the CCP structure. Although there have been many elegant studies in this area on many peroxidase, here we focus primarily on yeast CCP not only because a majority of our lab’s peroxidase effort has been with CCP but because CCP was the first heme peroxidase where it became possible to probe structure function relationships using crystallography and protein engineering.
Solving the CCP Structure
The CCP structure was published in 1980 [7] although the crystallographic details were published in 1978 [14]. This was just prior to a renaissance in structural biology that began in the early 1980s when it became almost routine to produce proteins using recombinant DNA technology. Crystallographers now had the ability to produce large quantities of protein instead of relying on the natural host. Some companies in the newly emerging biotechnology industry recognized the importance of structural biology and began investing in protein crystallography. This growing interest provided the market driving force that helped spur the wider spread use of other major technological developments critical to protein crystallography. The first of these was relatively cheap computing which became more widespread in the 1980s with easy-to-use operating systems. The most noteworthy of these was the VMS (Virtual Memory System) developed by Digital Equipment Corp. and the famous VAX computers. This made programming much easier and computing more accessible which resulted in the rapid development of some key software packages for protein structure refinement, one of the most noteworthy being PROLSQ [15]. Computer graphics was also just starting to develop. Another important advance was the first area detector systems for rapid data collection which was first commercialized in the early 1980s. The CCP structure was solved just as these new technologies were being developed. What was not routinely available for several more years was synchrotron data collection.
During this transition point in the development of protein crystallography, the work on CCP was able to take full advantage of these advances. Most importantly was the strong commitment that the University of California at San Diego (UCSD, where the initial work on CCP was carried out) had made to structural biology especially considering that protein crystallography was still considered expensive, esoteric, and took way too long to solve structures. The UCSD Chemistry Department had cutting edge computational resources that were heavily used by local crystallographers and was one of the first universities to invest in the development of computer graphics for protein structure determination. Very importantly, the first area detectors for x-ray data collection were developed by Nh Xuong and colleagues at UCSD [16] (Fig. 2A) and, in fact, CCP was the second structure to be solved using the Xuong data collection system. The first electron density maps were displayed by stacking photocopies of electron density sections on to plastic sheets (Fig. 2C). The CCP project also was to be the first test case for building a structure using the new Evans and Sutherland Picture System together with locally developed software as opposed to the brass model optical comparators or “Richard’s box” used by most groups (Fig. 2B). The author was the guinea pig for this adventure but frustration soon set in with the cumbersome commands required to simply rotate around a bond and the frequent computer crashes. Therefore, the first CCP structure was constructed the old fashion way with an optical comparator. Thus, the CCP project had a bit of the new (data collection) and a bit of the old (model building) and was very likely the last structure built at UCSD using the brass model approach.
Figure 2.
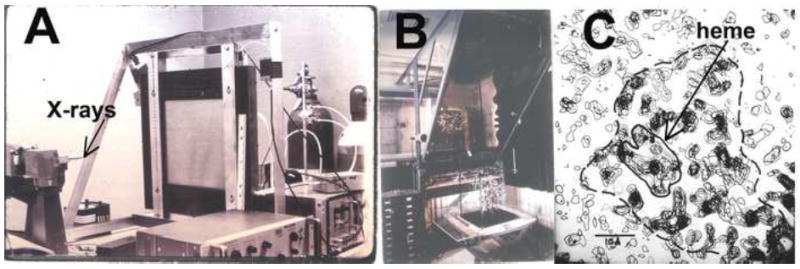
Some historical figures highlighting the state of crystallographic technology at the time the CCP structure was solved. A) One of the early area detectors developed by Xuong and colleagues at UCSD (cira mid-1970s). B) The type of optical comparator used to build the first CCP model. C) One of the early 2.6Å electron density maps of CCP. This is a 5Å thick section looking along the heme plane.
Compound I Formation
Prior to solution of the CCP structure, much was known about peroxidases, primarily through studies with HRP. The kinetics of Compounds I and II formation and reduction had been worked out in addition to many of their biophysical properties and several critical papers contributed to this work. However, a select few were particularly influential in developing the so-called Poulos-Kraut model of Compound I formation [17]. Using hydroperoxides to form Compound I, Schonbaum and Lo [18] found that there is a quantitative release of the alcohol, ROH. This meant that the O-O bond cleaved heterolytically leaving behind an oxygen atom with only 6 valence electrons, a potent oxidizing agent. The term “oxene” intermediate was adopted [19] in honor of the similarly reactive and better understood carbene. It also was known from magnetic susceptibility studies [20] and Mossbauer [21] that the redox state of the iron is Fe(IV). Taken together these data coupled with the finding that peroxidases and catalases form π cation radicals in Compound I [22], consistent with model heme studies [23], meant that the oxene O atom left behind after herterolysis of the O-O bond removes one electron from the iron and one from the porphyrin. This, then, provided the basis for a stereochemical mechanism for Compound I formation once the CCP structure was solved [17]. Acid base catalysis was anticipated in peroxidases and the location of the conserved distal side His52 was the obvious candidate (Fig. 3). What was unexpected was the active site Arg. In fact, the Arg was initially mis-interpreted as a Tyr (unpublished) for two reasons. First, the CCP sequence was not yet available when the first electron density maps were obtained. Second, since it was generally believed that active sites are usually hydrophobic it made no sense to have a large charged polar amino acid side chain situated directly over the heme in the active site so a Tyr was placed at this position even through the density was not consistent with Tyr. In those days one had to be a bit more creative in fitting electron density maps owing to the generally poor quality of heavy atom phases. It thus seemed more reasonable to place an aromatic group at this position despite the poor fit to the electron density map. Once the sequence became available [24] the active site structure was corrected in less than an hour.
Figure 3.
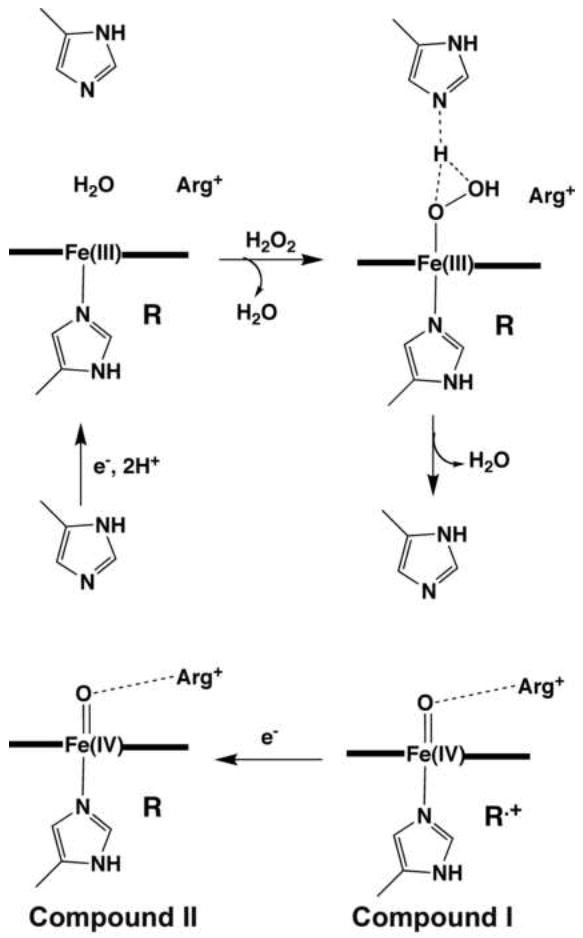
Peroxidase catalytic cycle. R is the site that forms a radical in Compound I which is the porphyrin in most peroxidases but Trp in CCP.
As might be expected, replacement of the distal side His has a dramatic effect on Compound I formation. Substituting the distal His52 with Leu lowers the rate of Compound I formation by 5 orders of magnitude [25] while replacing the active site Arg48 with Leu decreases the rate of Compound I formation by only 2 orders of magnitude [26]. Initially it was proposed that the active site Arg provides H-bonding stabilization to the transition state. This still may be the case but clearly such stabilization is not essential since even a 102 decrease in the rate of Compound I formation is still a decent rate. However, the stability of Compound I is substantially lower in the Arg48Leu mutant [26]. More recent crystal structures are relevant to this potential function of Arg48. The 1.2Å structure of CCP [27] clearly shows that Arg48 occupies two positions: one termed “out” and the other “in”. In the 1.3Å structure of Compound I Arg48 occupies only the “in” position where it can H-bond with the iron linked O atom (Fig. 4). The active site Arg is similarly positioned in HRP Compound I [28]. Therefore, the main role of Arg48 may be in stabilizing the ferryl center in Compound I. Such interactions should influence the reactivity of Compound I. Here it is useful to briefly mention cytochrome P450. It is normally assumed that P450s form a peroxidase-like Compound I although a true Compound I has never been experimentally observed in an O2 supported P450 catalytic cycle despite claims to the contrary [29]. Even so, if we assume P450s and peroxidases both utilize ferryl intermediates, we are left with the problem of why P450 Compound I is such a potent hydroxylating agent while peroxidase Compound I is not. Structural restrictions such as lack of substrate access to the iron-linked O atom in peroxidases is one reason. However, modulation of ferryl reactivity by the protein is likely to be the predominant control. A major difference is that P450s use a Cys axial ligand while peroxidases use His or Tyr, in the case of catalases [30]. The obvious experiment here is to switch ligands but the interpretation of such results can be problematic. Of particular importance is the significant steric differences between His and Cys and local H-bonding environments which are nearly impossible to engineer correctly. For example, His is simply longer than Cys so the ligand-Fe bond length will be extremely difficult to reproduce in the engineered protein. In addition, in P450s the Cys accepts a H-bond from a peptide NH while in peroxidase the His donates a H-bond so the local electrostatic environments are totally different. Sadly to say, swapping ligands cannot give us the answer. On the other hand, changing ligands has shown that the rate of Compound I formation is not so dependent on the nature of the axial ligand but that the reactivity and stability of Compound I is highly ligand dependent [31]. Thus the Cys vs. His difference in P450s and peroxidases no doubt plays a large role in controlling the reactivity of Compound I although it is very difficult to get an experimental handle to shed further light on this problem. Here is where computational biology can contribute and much insight has been provided by recent DFT calculations [32]. The most notable difference in the distal pockets is that peroxidases have a distinctly more polar distal environment than P450s owing to the His and Arg. This could well be a key factor in controlling Compound I reactivity. The ability of Arg to donate a H-bond should promote greater electronegative character to the ferryl O atom thus decreasing its electron deficiency which, in turn decreases its reactivity. Another important factor controlling ferryl reactivity that has come to light recently is the state of protonation of the iron linked oxygen. Green et al [33] have found that in some cases the ferryl center exhibits a high pKa so the predominate species is Fe(IV)-OH and not Fe(IV)=O. Strong electron donation by the Cys thiolate ligand in chloroperoxidase and P450s favors Fe(IV)-OH which also favors the radical rebound hydroxylation process in P450s as opposed to the one electron oxidations of the traditional peroxidase Fe(IV)=O.
Figure 4.
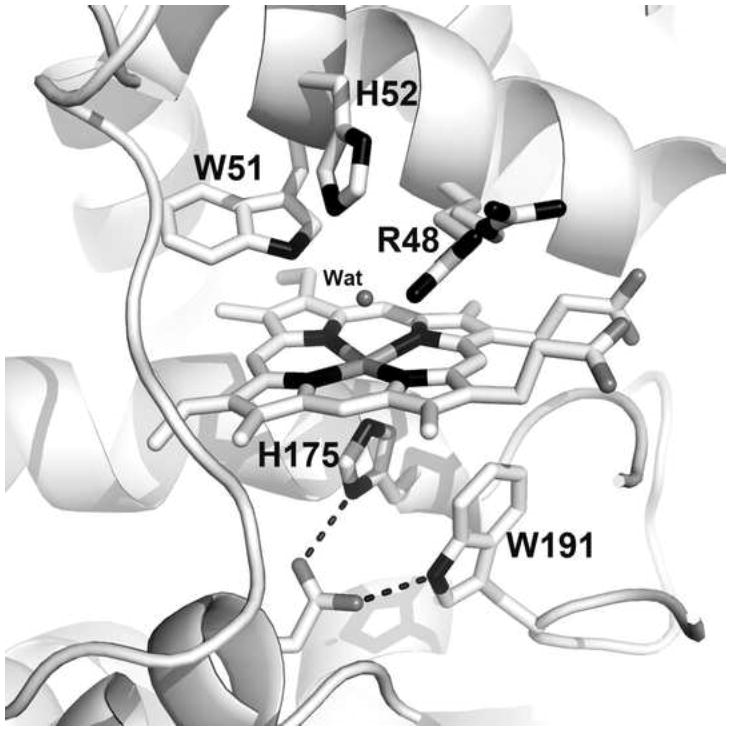
A close-up view of the CCP active site derived from the 1.2Å structure [27]. Note that Arg48 occupies two positions termed “in’ and “out”. In the “out” position, partially occupied water molecules occupy the space vacated by the Arg48 side chain. Conversely, when Arg48 occupies the “in” position, the vacated “out” space is occupied by water. In Compound I Arg48 is permanently “in” where it is able to H-bond with the ferryl O atom.
Chasing Down Radicals in Compound I
The structure of CCP also nicely explained a major difference between CCP and other peroxidases. CCP does not form a porphyrin radial but instead forms an amino acid free radical which generates a large and very stable EPR signal [34] and there was evidence that the location of the radical might be Trp [35]. The CCP structure has Trp51 directly adjacent to where peroxide will bind (Fig. 1) and from the limited sequence data available at the time [36] it was known that HRP has a Phe at this location. Since Trp is easier to oxidize than Phe it was no great leap in logic to conclude that Trp51 which must directly interact with the peroxide is the location of the Compound I free radical in CCP and that owing to the higher redox potential of Phe, the porphyrin is oxidized in HRP.
Even so a Trp radical was judged to be inconsistent with ENDOR data and instead a radical center consisting of at least one Met residue was proposed [37]. Remarkably enough the CCP structure shows three Met residues (172, 230, and 231) in the proximal heme pocket (Fig. 1) which are not present in HRP lending credence to the Met radical idea. However, the very first protein engineering studies of CCP proved that neither Trp51 nor Met172 are the free radical sites [38, 39]. The issue was finally settled by an elegant isotope substitution study [40] which showed that Trp191 is the radical site while mutagenesis showed that Trp191 also is essential for activity [41].
Although the location of the CCP radical moved over time before finally settling on Trp191, the simple idea that the relative ease of oxidation of Trp191 compared to the Phe at this position in most other peroxidases is why CCP forms a Trp191 radical and other peroxidases form a porphyrin radical remained intact. This idea was further strengthened by the observation that if Trp191 in CCP is replaced by Phe, a short lived porphyrin radical forms in the mutant Compound I [42]. This picture changed when ascorbate peroxidase (APX) was recombinantly expressed [43] thus providing sufficient material for detailed studies. Sequence comparisons showed that APX has both the distal and proximal side Trp residues [44] which later was substantiated by the APX crystal structure [45]. It was fully anticipated that APX would form a Trp radical but instead freeze-quench EPR [46] and stopped flow spectroscopy [47] showed that APX forms the traditional porphyrin radical. However, APX Compound I decays to a species that exhibits EPR properties consistent with a Trp radical similar to CCP [48]. It thus appears that the initial porphyrin radical formed in APX Compound I is able to oxidize the proximal Trp analogous to Trp191 CCP. This internal redox reactions requires about one minute indicating that the predominant form of Compound I under steady state conditions is the porphyrin radical
That APX forms a porphyrin radical, although unexpected, turned out to be a very useful result. Since APX and CCP are so similar in sequence and structure, it should be possible to work out precisely what structural variations are responsible for the differences in Compound I radical location and as such CCP and APX provided a means for probing in some detail how proteins stabilize radicals. That the Trp191 radical is a cation [49] helps a great deal since proteins are exquisite at electrostatic stabilization and modifying the local electrostatic environment can readily be achieved by mutagenesis. It thus seemed reasonable that CCP might be better able to stabilize the positive charge on the cationic Trp radical than APX by suitably positioned side chains that can stabilize a cation. Indeed, structural and small molecule binding data clearly showed that the proximal pocket surrounding Trp191 favors a positive charge. When Trp191 is converted to Gly the resulting cavity can be filled with various small molecules and those carrying a positive charge bind best [50]. The same mutant can also bind K+ in the engineered cavity [51]. It thus was clear that a cationic Trp191 radical would be electrostatically stabilized by the protein environment and subsequent computational approaches supported this view [52]. Not surprisingly it was shown that the buried and conserved Asp235 (Fig. 1) plays a major role in the binding of cations to the Trp191Gly mutant [53]. However, Asp235 cannot be the full story since APX also has this Asp but fails to form a stable Trp radical. Thus the answer must lie elsewhere.
One of the more obvious structural differences between CCP and APX that might influence cation radical stability is the proximal side metal ion binding site also found in plant peroxidases (Fig. 5) but not CCP. While in HRP this site is occupied by Ca2+ [2, 12], in APX this is a monovalent cation and was identified as K+ in the initial crystal structure [11]. The monovalent cation site in APX is about 8Å from the proximal Trp leading to the hypothesis that the presence of the K+ in APX would destabilize a Trp cationic radical. Engineering out the K+ site in APX proved difficult since in the K+ free mutant, the distal His drops down to coordinate the heme showing that the K+ helps to maintain the correct active site architecture [54]. The inverse experiment, however, worked and it was possible to engineer in the APX K+ site into CCP [55]. The characteristic intense EPR signal due to the Trp191 radical was absent in the K+ CCP mutant clearly showing that the cation site plays an important role in controlling radical stabilization. The mutant enzyme also exhibited very low activity [55]. Once again the picture appeared complete. Weather or not a Trp or porphyrin radical is formed depends on the relative stability of each and that in APX the K+ destabilizes the cationic Trp radical and thus, a porphyrin radical is formed. Further experimentation, however, showed that this picture also was a bit too simplistic. The K+ CCP mutant enzyme activity could be recovered depending on ionic strength and which cytc substrate is used. These differential effects of ionic strength and cytc are well known and associated with changes in the rate limiting step depending on the assay conditions [56]. This lead to the hypothesis that adding the K+ to CCP decreases the stability of the Trp191 radical but not its formation. That is, the initial reaction with H2O2 results in a full equivalent of Trp191 radical but that this rapidly decays [57], most likely to the nearby Tyr236 [58].
Figure 5.
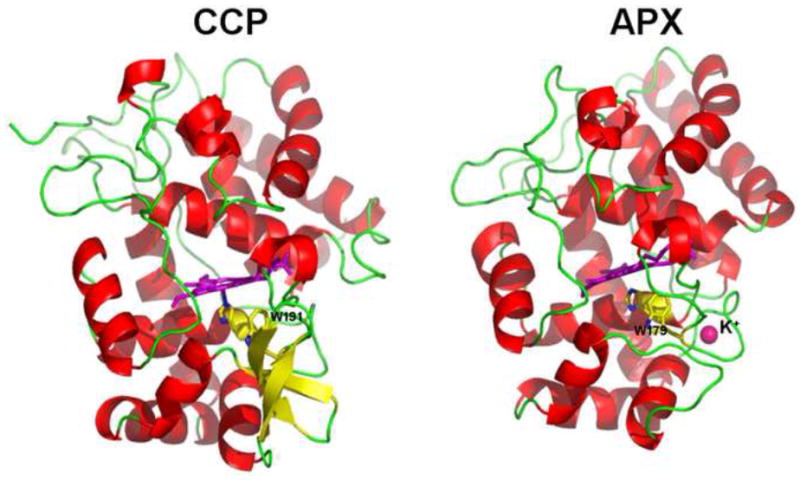
The CCP and APX structures showing the location of the K+ ion in APX relative to the proximal active site Trp.
Another important difference in the proximal pocket is the presence of Met230 and 231 in CCP which are Leu and Gln in APX. It was reasoned that the electronegative sulfur atoms of Met might also help to stabilize the Trp191 radical. It already was known that converting both Met residues to Leu perturbs the Trp191 EPR signal [59]. By both introducing the K+ site into CCP and mutating Met230 and 231 to the corresponding residues in APX, the Trp191 radical was eliminated [57]. The inverse experiment where the Leu and Gln in APX were both converted to Met resulted in formation of a Trp radical [60]. There is a third Met in the active site of CCP, Met172, which is Ser160 in APX. However, mutating APX Ser160 to Met causes some interesting modifications. This mutation results in the peroxide-dependent crosslinking of Met60 with a heme vinyl [61] and so this mutant was not included in our APX engineering work. These cumulative results indicate that, as anticipated, the unusual stability of the Trp191 radical can be fully explained by electrostatic stabilization. The one remaining problem is why none of these mutants form a porphyrin radical. Only when Trp191 is converted to Phe does CCP form a detectable porphyrin radical. One explanation for this apparent conundrum is that the initial preferred site of oxidation is always Trp191 no matter what mutants are introduced and that the key factor is stability of the Trp191 radical. It also is possible that the initial reactions results in a transient porphyrin radial too short lived to be observed which rapidly oxidizes Trp191. Thus changing the local electrostatic environment of the Trp191 radical does not prevent its initial formation but greatly effects its stability. One possibility for why the radical does not end up on the porphyrin in these mutants is the large number of alternate radical sites in CCP owing to the unusually high content of aromatic residues, 7 Trp and 14 Tyr, for a 30kDa protein. Indeed, when Compound I is allowed to decay CCP forms a small amount of intermolecular crosslinks in a complex and highly variable reaction via Tyr residues indicating that the radical site can migrate to surface Tyr residues [62]. Apparently oxidation of these sites is preferable to oxidation of the porphyrin. In the following scheme the radical migrates to other sites no matter where it initially forms.

In wild type CCP the radical eventually migrates to Tyr residues. Even though we appear to understand what controls Trp cation radical stability, what remains to be explained is why HRP forms such a stable porphyrin radical. Why doesn’t the porphyrin radical rapidly migrate to other sites as in CCP? Clearly the porphyrin radical is much more stable in HRP and understanding what structural features control this relative stability of porphyrin radicals in peroxidases remains an unsolved problem. However, recent computational studies [63] suggest that the heme propionates may play a role. The general idea is that a “naked” propionate with no H-bonding partners would be free to back-fill the positively charged hole of the porphyrin radical thus destabilizing a porphyrin cationic radical. Thus one might anticipate that peroxidases with strongly H-bonded propionates would form more stable porphyrin radicals. Indeed, a cursory examination of various peroxidase structures lends credence to this idea. CCP heme propionates are not H-bonded to either Arg or Lys while nearly all other heme peroxidases use Arg and/or Lys to balance the heme propionate charges and, unlike CCP, these peroxidases form porphyrin radicals. Even so, the limited mutagenesis data available does not support this notion. For example, introducing an Arg into CCP which forms a new charge pair with one heme propionate does nothing to help stabilize a porphyrin radical [64].
Non-Porphyrin Radicals in Other Peroxidases
CCP is not alone in producing amino acid centered radicals. One of the most notable examples is lignin peroxidase (LiP). The crystal structure of LiP showed a large lobe of electron density very near Cβ of Trp171 [65] (Fig. 6) which resulted in the finding that Trp171 is hydroxylated at Cβ during reactions with peroxides. Subsequent mutagenesis work showed that Trp171 is essential for activity in the oxidation of the natural substrate, veratryl alcohol [66]. It now is thought that veratryl alcohol undergoes oxidation near Trp171 and that Trp171 is involved in directing oxidizing equivalents to veratryl alcohol thus implicating Trp171 as forming at least a transient radical. Recently it has been possible to capture an EPR signal consistent with a Trp171 radical [67]. A second example is versatile peroxidase or VP. VP exhibits activities characteristic of both manganese and lignin perxoidase [68], hence the name “versatile”. Trp164 in VP is analogous to the redox active Trp171 in LIP (Fig. 6) and Trp164 parallels quite closely the behavior of Trp171 in LiP. EPR studies identified a radical centered on Trp164 [69] while mutagenesis showed that Trp164 is essential for the oxidation of high potential substrates like veratryl alcohol [70, 71].
Figure 6.
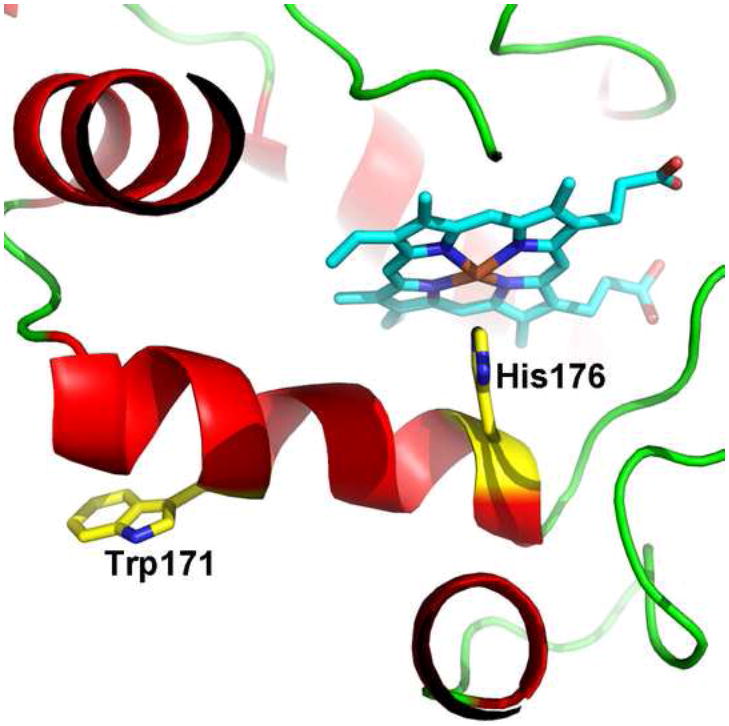
The structure of LiP showing the location of the redox active Trp171.
From the studies just discussed, it would appear that the location of the Compound I radical and location of the substrate binding site might be coupled. Multiple substrate binding sites also are implicated in other peroxidases with the earliest examples being CCP and APX. The most obvious place for small organic substrates to bind is near the exposed heme edge (Fig. 1). Chemical modification of the exposed heme edge of CCP blocks the ability of CCP to oxidize aromatic substrates but has little effect on the ability to oxidize cytc [72]. Likewise, the ascorbate binding site in APX contains the single Cys residue. Modification of this Cys blocks APX activity, presumably by preventing ascorbate from binding, but not the activity using aromatic substrates [73]. One final example is a newly characterized APX from Leishmania major (called LmAPX) which exhibits both APX and CCP activities [74] clearly implicating two substrate binding sites. LmAPX has a Trp208 which is analogous to Trp191 in CCP and Compound I of LmAPX shows no hint of the traditional peroxidase Compound I porphyrin radical. We have recently shown using EPR that LmAPX Compound I forms a very stable radical similar to Trp191 in CCP. Most interestingly, the W208F mutant in LmAPX exhibits very low cytc peroxidase activity but is fully active using ascorbate as the substrate [75].
Just like the corresponding mutant in CCP, the W208F mutant forms a transient porphyrin radical [75] . It thus appears that the path of electron transfer in LmAPX requires the redox active Trp208 for cytc oxidation but either the porphyrin or a Trp208 radical work fine in the oxidation of ascorbate.
Although the number of examples is limited, it does appear that the “generic” peroxidase substrate binding site is near the exposed heme edge while for the physiologically relevant substrates, Nature has evolved more specialized sites. In some cases, as in CCP, LiP, and VP, it also is necessary to provide redox active amino acid side chains as part of the electron transfer path.
Electron Transfer
CCP also has served as a model system for protein electron transfer. Early on CCP and cytc were one of the few redox pairs that could be prepared in sufficient quantities for detailed studies so here again CCP had a practical advantage in serving as a paradigm for inter-protein electron transfer reactions. Elegant chemical modification studies [76] showed that the necklace of Lys residues sprinkled over the surface near the exposed edge of cytc is critical in the reaction CCP. This strengthened the generally accepted picture that complementary electrostatic interactions are important in forming the redox complex. In the early days horse heart cytc was utilized for many studies with CCP and as expected, CCP activity decreased with increasing ionic strength presumably because at high ionic strength the complex is weakened. However, this simple picture became clouded with more detailed studies and it was found that with yeast cytc the activity actually increases with increasing ionic strength and only at moderately high salt concentrations does the activity decrease [77]. The reason for such behavior may be related to a change in rate limiting step as a function of ionic strength [56] and to best understand this explanation we refer to the following scheme fashioned after [78].
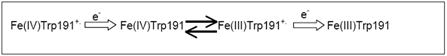
In this scenario the Trp radical is reduced by the first cytc electron but then there is an internal electron transfer from Trp191 to Fe(IV). The species that receives the second electron is Fe(III)Trp+. and thus, the Trp191 radical is the site of entry for both cytc electrons. At low ionic strength dissociation of cytc from CCP is rate limiting but at high ionic strength the off rate is fast and the redox equilibrium between the iron and Trp191 becomes limiting. The requirement of Trp191 in both electron transfer steps is consistent with the mutagenesis data [41]. The main problem with this mechanism is that the rate of intramolecular electron transfer from Trp191 to Fe(IV) is too slow to account for CCP turnover rates [78, 79]. There are two different scenarios to explain these apparent inconsistencies. First, CCP has a second site of electron transfer that directly reduces Fe(IV) which is consistent the work of Zhou&Hoffman [80] although why Trp191 would be needed for the second site model remains an open question. Second, the experimental conditions used to measure the Trp191/Fe(IV) internal electron transfer is far from what CCP experiences under steady state conditions and it could be that in the CCP-cytc complex the dynamics of the intramolecular electron transfer reaction favors rapid reduction of Fe(IV). This second explanation lacks direct experimental evidence for or against. Another interesting explanation [81] requires intermediate states of Compound I which, unfortunately, have not been detected but the general idea looks reasonable. However, the 1.3Å structure of peroxide-treated CCP does reveal some interesting structural changes that may be relevant to the problem of intramolecular electron transfer. Although there is some concern that the Compound I structure we reported a few years ago [27] may have been partially reduced in the x-ray beam, the structure revealed some clearly defined changes relative to the resting Fe(III) enzyme. First, the O atom is 1.87Å from the iron with strong continuous electron density between the two clearly indicating strong coordination. Second, the iron has moved into the plane of the porphyrin which is consistent in going from high-spin to low-spin. Third, Arg48 occupies only the “in” position and is ideally located to donate an H-bond to the iron-linked O atom. Fourth, two regions of the protein become more ordered in the peroxide-treated crystal. Met171 (see Fig. 1) is partially disordered in the Fe(III) enzyme as is a short section of polypeptide centered on Asn194. Asn194 is very near the CCP-cytc interface and also is quite close to Trp191. In the peroxide-treated structure these regions become ordered and effectively tighten the structure around Trp191 suggesting that the local environment adjusts upon formation of Compound I in order to stabilize the Trp191 radical. Early on it was suggested that one possible reason for why the observed intramolecular rate of electron transfer from Trp191 to Fe(IV) is so slow is that under normal steady state conditions the enzyme adopts a conformation [82] that favors rapid intramolecular electron transfer. The conformational changes we observe upon treating CCP crystals with peroxide may be related to the adjustments thought to be necessary of rapid internal electron transfer. Precisely how these changes alter the energetics in favor of electron transfer remains unknown but is a possible avenue for future studies.
Structure of the CCP-cytc Complex
Some of these issues were settled when the CCP-cytc structure was solved. The first attempt at solving the crystal structure of the CCP-cytc complex was reported in 1987 [83]. The complex crystallized as a dimer with only CCP clearly visible. Horse heart cytc, shown to be present by analysis of the crystals, occupied large open “solvent” regions in the crystal lattice but was not sufficiently ordered to contribute to the electron density. Given that cytc is a very stable well ordered molecule, the lack of cytc electron density must mean that the cytc was either orientationally disordered or experienced large rigid body motions around a favored orientation. This result was taken by many as evidence supporting the dynamic picture of protein-protein redox complexes. A few years later Pelletier and Kraut [84] succeeded in solving the CCP-horse heart cytc structure using the same crystal form reported in [83] but this time one cytc molecule was visible. Pelletier and Kraut [83] also obtained the CCP-yeast cytc. structure (Fig. 7) and since then, this model has been the paradigm for understanding CCP-cytc electron transfer. There are two important features of the structure worth emphasizing. First, there are no strong ionic interactions in the complex. This was somewhat unexpected given the strong ionic strength dependence of the CCP-cytc interaction and the well known requirement of surface Lys residues in cytc in forming redox complexes. Second, the heme of cytc. directly contacts the region of CCP involving Ala193 and Ala194. Therefore, Trp191 is only a few peptide bonds removed from the cytc. heme which could well provide a direct electron transfer path to the CCP Trp191 radical.
Figure 7.
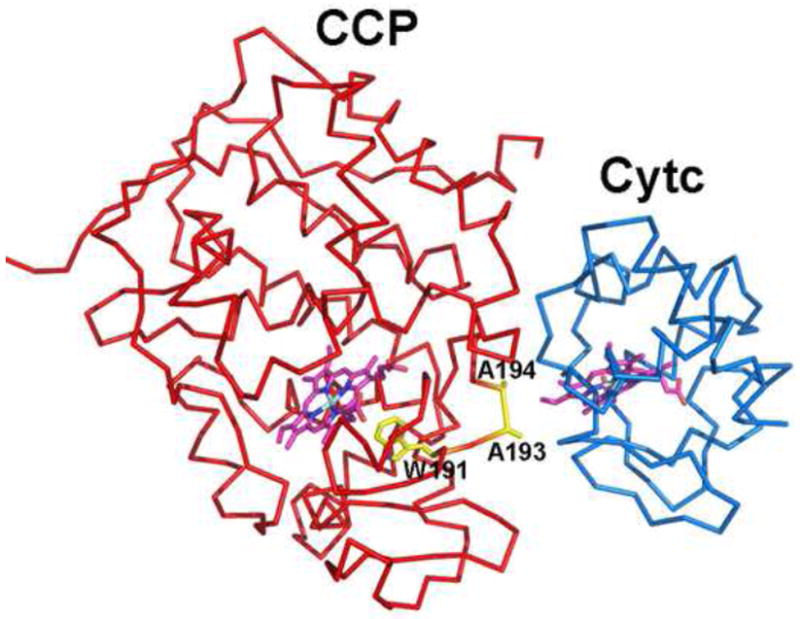
The CCP-cytc structure [84].
One further advance was made by crosslinking the CCP-cytc complex using engineered S-S bonds. The CCP-cytc. complex was used as a guide on where to engineer in Cys residues and this resulted in a very stable S-S linked complex that generated crystals that diffracted much better than the non-covalent complex [85]. The main advantage of this structure (Fig. 8) was a better picture of the CCP-cytc interface which showed no direct ionic interactions but several ordered water molecules. These water molecules bridge between polar groups. Ordered solvent serving as the main bridge between redox partners also was observed in the complex formed between P450BM3 and its redox partner [86]. One possible requirement for solvent at the interface is to “smooth” the interacting surfaces. The surfaces, while complementary in terms of charge, may not be quite complementary in terms of shape. Thus solvent serves to shape the surfaces for a better fit although the price paid is decreased affinity owing to solvent screening of complementary charges. This, however, may be an advantage since for efficient turnover the on and off rates must be balanced and dielectric solvent screening should help to provide such balance.
Figure 8.
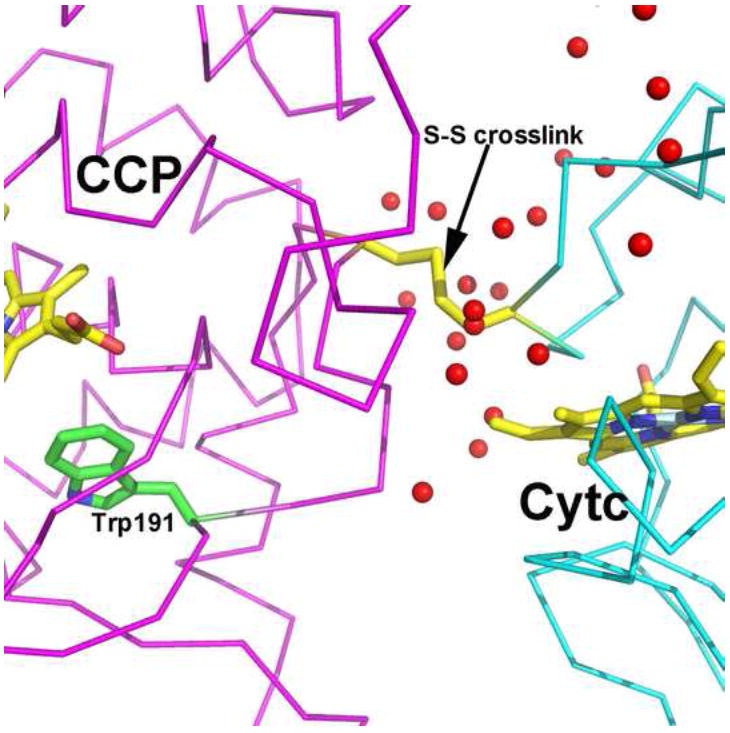
Crystal structure of the CCP-cytc crosslinked complex highlighting the ordered solvent molecules at the interface.
Although the CCP-cytc crystal structure now is generally accepted as a true representation of the physiologically important complex, the question remains on whether or not CCP has two different sites of electron transfer. Since the Trp191 radical is the point of entry for both cytc-derived electrons, it would appear that a single site would be favored. Even so it is well established that CCP does have a second, weak affinity site [80, 87, 88]. Theoretical approaches have been used to predict the location of alternate sites [89] and one such site was predicted to be near Asp148 of CCP. To test if this might be a functionally relevant region, cytc was selectively crosslinked to CCP in a region that will block access to the predicted second cytc binding site. To achieve selective crosslinking Lys149 in CCP and Lys79 of yeast cytc were converted to Cys, the two Cys residues crosslinked to give an intermolecular S-S bridge, and the complex purified and characterized [90]. As shown in Fig. 9 the crosslinking site is far removed from the location of cytc in the crystal structure. This complex exhibits near wild type levels of steady state activity which indicates that the cytc attached to CCP is not blocking a functionally important docking site. This initial effort has been followed by a more extensive analysis of various CCP-cytc crosslinked complexes [91, 92] that resulted in similar conclusions with all crosslinks giving near wild type activities except the one crosslink that mimics the crystal structure. An equally important observation from these studies was that in this covalent complex cytc reduces CCP Compound I very slowly, ≈1s-1, and that it is the Trp191 radical and not Fe(IV) that is reduced in a single turnover experiment [90]. Given the restrictions of the intermolecular S-S bond in this complex, there are only so many ways that cytc can be oriented relative in CCP and of the small number of possible orientations, the distance between hemes is shorter than the distance between the cytc heme and Trp191 yet Trp191 is reduced and not Fe(IV). It thus appears that independent from where the electron enters, it ends up reducing the Trp191 radical indicating that the Trp191 cation radical is the thermodynamically favored site of reduction.
Figure 9.
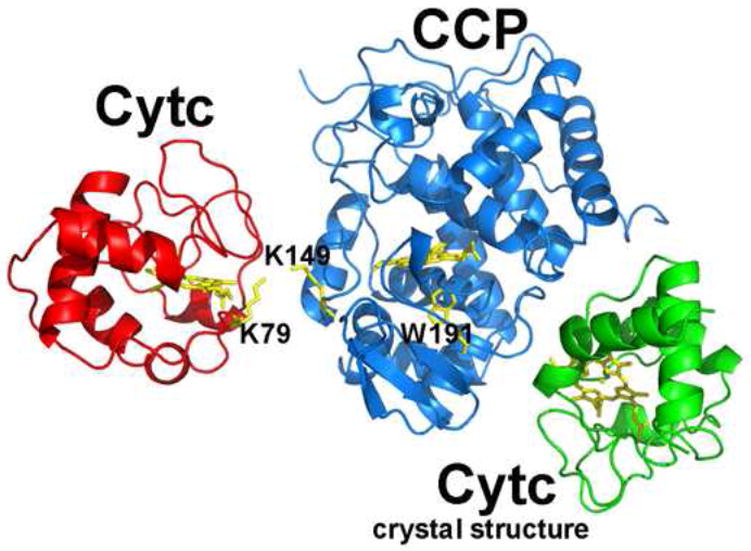
The red cytc indicates the location of cytc crosslinked S-S bond in the cytc K79C and CCP K149C mutant. The green cytc shows the location of the cytc binding site identified in the crystal structure.
The intermolecular S-S complex designed to mimic the crystal structure also provided some interesting kinetic results relevant to the one or two site model [85]. This complex exhibited about 7% wild type steady activity while intramolecular electron transfer from cytc to CCP Compound I in the covalent complex was too fast to measure by stopped flow methods. It later was shown that this 7% activity is due to a slight contamination of the complex with free CCP and that the highly purified complex exhibits <0.1% activity [91]. In a single turnover experiment about 10 min was required for Compound I in the complex to oxidize exogenously added reduced cytc. These data, coupled with mutagenesis/chemical modification experiments [93], point toward the CCP-cytc complex observed in the crystal structure as being the only complex relevant for electron transfer. However, it is quite possible that a second weaker site modulates the binding and/or affinity of the functionally important site. This would be consistent with the rather complex steady state kinetic behavior of CCP [56].
Conclusions and Future Prospects
Progress on peroxidase structure and function has been most impressive. A good deal of this is due the ability to clone and express a large number of peroxidases which has provided sufficient material for detailed biophysical investigations. The most immediate impact of peroxidase structural biology was unraveling the mechanism of Compounds I and II formation. This effort was greatly aided by the exceptional level of model heme chemistry prevalent in the 1970s and 1980s. The author was fortunate to have one of the leaders in this area, the late Teddy Traylor, close-by at UCSD so any naive notions about peroxidase function could be checked out with a real chemist who had a deep understanding of heme chemistry. With respect to CCP specifically, several problems can be considered solved. The acid base catalytic mechanism for Compound I formation is well understood and it now is clear that CCP has one predominant binding site for cytc. and that both electrons from cytc require Trp191. It also is most fortunate that CCP has proven to be such a forgiving protein since it is possible to make rather dramatic changes using mutagenesis yet the modified CCPs usually tolerate this abuse and crystallize. As a result CCP has provided an exceptional laboratory for some creative and novel engineering studies [94].
What remains unknown is why the porphyrin radical is so stable in some peroxidases and not in others. The simple electrostatic arguments employed to explain the stability of the CCP Trp191 radical are not sufficient. Part of the problem is the far more complex electronic structure of heme compared to a simple indole side chain. Moreover, as with much of biology, we are trying to explain fairly small differences. We often forget that activation free energy for reactions is related to the log of the rate so a difference of 1,000 fold in rate translates into only about 4kcal/mol which can be achieved by one good H-bond. Thus, subtle structural differences can result in large changes in rate. So far simple mutagenesis approaches have proven inadequate in understanding porphryin radical stability. This clearly is an area where some fresh approaches would benefit.
Peroxidases are not normally discussed in terms of biomedical relevance. However, peroxidases that have a very close structural and functional similarity to both CCP and APX now have been found in human pathogens. The most well studied of these is the Leishmania major LmAPX discussed earlier. Quite recently it has been found that over expression of LmAPX protects the organism against oxidative stress and associated protein damage [95, 96]. It remains to bee seen if LmAPX is essential for survival but it is clear that interfering with peroxidase activity should increase susceptibility to oxidative stress.
Peroxidases have long been of interest from a practical biotechnological perspective. The degradation of lignin is perhaps the premiere example. One very promising area is in altering substrate specificity. We now know that some peroxidases have specially designed binding sites for physiologically important substrates. Some key examples include APX, MnP, and probably LiP. Given the striking conservation in the three dimensional structure of peroxidases, it has been possible to swap side chains without effecting the overall fold and, indeed, there have been some recent successes in introducing new substrate sites into peroxidase (see Emma Raven’s chapter). It should then be possible to engineer sites that bind to interesting substrates for bioremediation or other useful purposes. We have all the tools in hand to accomplish this goal. Even so, the main issue will not be substrate binding but reactivity. Peroxidases catalyze electron/H atom transfer/abstraction reactions to generate substrate radicals. Here thermodynamic driving force is a key problem since the redox potential of Compounds I and II must be higher than the substrate. In this regard, peroxidases face a much different problem than P450s where the binding forces between substrate and protein hold the substrate in place near the iron-linked ferryl O atom for regio- and stereo-specific reaction so thermodynamic barriers to C-H bond rupture can be overcome by strong substrate-protein binding and orientation. Moreover, there is no need, as in peroxidases, to form a semi-stable Compound I so in P450s the “hot” ferryl center reacts as soon as it forms. This is why one has never observed a P450 Compound I in the normal catalytic cycle. In peroxidases, however, substrates are restricted from directly contacting the ferryl center so long-range electron transfer and driving force become key factors. To tackle the problem of novel substrate oxidation thus requires more than building in substrate binding sites but also requires a tuning of Compounds I and II reactivity in order to overcome thermodynamic barriers to electron transfer. We are, of course, limited by the redox potential of H2O2 itself. This presents a formidable problem. If Compound I reactivity is increased too much, then lifetime and stability become too short to enable substrates to bind, transfer an electron/H atom, and then dissociate. Increasing reactivity too much also could result in enzyme self-destruction. That protons and electrons are involved further complicates the chemistry. The rate limiting process, proton or electron transfer, may be different for different substrates. Thus to be able to exploit peroxidases for practical applications, we need a more complete understanding on how the enzyme tunes the reactivity and stability of Compounds I and II. With a recognition on the limits of what we can push peroxidases to do, the technologies and structural information are in hand to solve some of these remaining problems and more fully exploit peroxidases for practical applications.
Acknowledgments
I would like to thank Victoria S. Jasion for a careful proofreading job. Work on peroxidases has been supported by NIH grant GM42614
Abbreviations used
- CCP
cytochrome c peroxidase
- cytc
cytochrome c
- APX
ascorbate peroxidase
- LiP
lignin peroxidase
- VP
versatile peroxidase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chance B. J Biol Chem. 1943;151:553–577. [Google Scholar]
- 2.Gajhede M, Schuller DJ, Henriksen A, Smith AT, Poulos TL. Nature Structural Biology. 1997;4:1032–1038. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- 3.Altschul AM, Abrams R, Hogness TR. J Biol Chem. 1940;136:777–794. [Google Scholar]
- 4.Yonetani T. In: The Enzymes. Boyer B, editor. Academic Press; Orlandom Florida: 1976. pp. 345–361. [Google Scholar]
- 5.Yonetani T, Ray GS. J Biol Chem. 1965;240:4503–4508. [PubMed] [Google Scholar]
- 6.Larsson LO, Hagman LO, Kierkegaard P. J Biol Chem. 1970;245:902–903. [PubMed] [Google Scholar]
- 7.Poulos TL, Freer ST, Alden RA, Edwards SL, Skogland U, Takio K, Eriksson B, Xuong N, Yonetani T, Kraut J. J Biol Chem. 1980;255:575–580. [PubMed] [Google Scholar]
- 8.Zeng J, Fenna RE. J Mol Biol. 1992;226:185–207. doi: 10.1016/0022-2836(92)90133-5. [DOI] [PubMed] [Google Scholar]
- 9.Edwards SL, Raag R, Wariishi H, Gold MH, Poulos TL. Proc Natl Acad Sci USA. 1993:4429–4440. doi: 10.1073/pnas.90.2.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen JF, Kadziola A, Larsen S. FEBS letters. 1994;339:291–296. doi: 10.1016/0014-5793(94)80433-8. [DOI] [PubMed] [Google Scholar]
- 11.Patterson WR, Poulos TL. Biochemistry. 1995;34:4331–4341. doi: 10.1021/bi00013a023. [DOI] [PubMed] [Google Scholar]
- 12.Schuller DJ, Ban N, Huystee RB, McPherson A, Poulos TL. Structure. 1996;4:311–321. doi: 10.1016/s0969-2126(96)00035-4. [DOI] [PubMed] [Google Scholar]
- 13.Welinder KG. Current Biology. 1992;2:388–393. [Google Scholar]
- 14.Poulos TL, Freer ST, Alden RA, Xuong NH, Edwards SL, Hamlin RC, Kraut J. J Biol Chem. 1978;253:3730–3735. [PubMed] [Google Scholar]
- 15.Hendrickson WA, Konnert JH. Indian Inst. Sicnece; Bangalore, India: 1980. [Google Scholar]
- 16.Cork C, Hamlin RC, Vernon W, Xuong Nh. Acta Crystallogr. 1975;A31:702–703. [Google Scholar]
- 17.Poulos TL, Kraut J. J Biol Chem. 1980;255:8199–8205. [PubMed] [Google Scholar]
- 18.Schonbaum GR, Lo S. J Biol Chem. 1972;247:3353–3360. [PubMed] [Google Scholar]
- 19.Hamilton G. In: Chemical models and mechanisms for oxygenases. Hayaishi O, editor. Academic Press; New York: 1974. pp. 405–451. [Google Scholar]
- 20.Iizuka T, Kotani M, Yonetani T. J Biol Chem. 1971;246:4731–4736. [PubMed] [Google Scholar]
- 21.Lang G, Spartalian K, Yonetani T. Biochim et Biophys acta. 1976;451 doi: 10.1016/0304-4165(76)90275-0. [DOI] [PubMed] [Google Scholar]
- 22.Dolphin D, Forman A, Borg DC, Fajer J, Felton RH. Proc Natl Acad Sci USA. 1971;68:614–618. doi: 10.1073/pnas.68.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fajer J, Borg DC, Forman A, Dolphin D, Felton RH. J Amer Chem Soc. 1970;92:3451–3459. doi: 10.1021/ja00714a038. [DOI] [PubMed] [Google Scholar]
- 24.Takio K, Titani K, Ericsson LH, Yonetani T. Arch Biochem Biophys. 1980;203:615–629. doi: 10.1016/0003-9861(80)90219-2. [DOI] [PubMed] [Google Scholar]
- 25.Erman JE, Vitello LB, Miller MA, Shaw A, Brown KA, Kraut J. Biochemistry. 1993;32:9798–9806. doi: 10.1021/bi00088a035. [DOI] [PubMed] [Google Scholar]
- 26.Vitello LB, Erman JE, Miller MA, Wang J, Kraut J. Biochemistry. 1993;32:9807–9818. doi: 10.1021/bi00088a036. [DOI] [PubMed] [Google Scholar]
- 27.Bonagura CA, Bhaskar B, Shimizu H, Li H, Sundaramoorthy M, McRee D, Goodin DB, Poulos TL. Biochemistry. 2003;42:5600–5608. doi: 10.1021/bi034058c. [DOI] [PubMed] [Google Scholar]
- 28.Berglund GI, Carlsson GH, Smith AT, Szoke H, Henriksen A, Hajdu J. Nature. 2002;417:463–468. doi: 10.1038/417463a. [DOI] [PubMed] [Google Scholar]
- 29.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. Vol. 287. New York, N.Y: 2000. pp. 1615–1622. [DOI] [PubMed] [Google Scholar]
- 30.Murthy MR, Reid TJd, Sicignano A, Tanaka N, Rossmann MG. J Mol Biol. 1981;152:465–499. doi: 10.1016/0022-2836(81)90254-0. [DOI] [PubMed] [Google Scholar]
- 31.Choudhury K, Sundaramoorthy M, Hickman A, Yonetani T, Woehl E, Dunn MF, Poulos TL. J Biol Chem. 1994;269:20239–20249. [PubMed] [Google Scholar]
- 32.Kumar D, de Visser SP, Sharma PK, Hirao H, Shaik S. Biochemistry. 2005;44:8148–8158. doi: 10.1021/bi050348c. [DOI] [PubMed] [Google Scholar]
- 33.Green MT, Dawson JH, Gray HB. Science. Vol. 304. New York, N.Y: 2004. pp. 1653–1656. [DOI] [PubMed] [Google Scholar]
- 34.Yonetani T, Schleyer H, Eherenberg A. J Biol Chem. 1966;241:3240–3243. [PubMed] [Google Scholar]
- 35.Coulson AF, Yonetani T. Biochemical and biophysical research communications. 1972;49:391–398. doi: 10.1016/0006-291x(72)90423-8. [DOI] [PubMed] [Google Scholar]
- 36.Welinder KG. Eur J Biochrem. 1979;96:483–502. doi: 10.1111/j.1432-1033.1979.tb13061.x. [DOI] [PubMed] [Google Scholar]
- 37.Hoffman BM, Roberts JE, Brown TG, Kang CH, Margoliash E. Proc Natl Acad Sci USA. 1979;76:6132–6136. doi: 10.1073/pnas.76.12.6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fishel LA, Villafranca JE, Mauro JM, Kraut J. Biochemistry. 1987;27:351–360. doi: 10.1021/bi00376a004. [DOI] [PubMed] [Google Scholar]
- 39.Goodin DB, Mauk AG, Smith M. J Biol Chem. 1987;262:7719–7724. [PubMed] [Google Scholar]
- 40.Sivaraja M, Goodin DB, Smith M, Hoffman BM. Science. Vol. 245. New York, N.Y: 1989. pp. 738–740. [DOI] [PubMed] [Google Scholar]
- 41.Mauro JM, Fishel LA, Hazzard JT, Meyer TE, Tollin G, Cusanovich MA, Kraut J. Biochemistry. 1988;27:6243–6256. doi: 10.1021/bi00417a008. [DOI] [PubMed] [Google Scholar]
- 42.Erman JE, Vitello LB, Mauro JM, Kraut J. Biochemistry. 1989;28:7992–7995. doi: 10.1021/bi00446a004. [DOI] [PubMed] [Google Scholar]
- 43.Patterson WR, Poulos TL. J Biol Chem. 1994;269:17020–17024. [PubMed] [Google Scholar]
- 44.Mittler R, Zilinskas BA. FEBS letters. 1991;289:257–259. doi: 10.1016/0014-5793(91)81083-k. [DOI] [PubMed] [Google Scholar]
- 45.Patterson WR, Poulos TL. Biochemistry. 1995;34:4331–4341. doi: 10.1021/bi00013a023. [DOI] [PubMed] [Google Scholar]
- 46.Patterson WR, Poulos TL, Goodin DB. Biochemistry. 1995;34:4342–4345. doi: 10.1021/bi00013a024. [DOI] [PubMed] [Google Scholar]
- 47.Marquez LA, Quitoriano M, Zilinskas BA, Dunford HB. FEBS letters. 1996;389:153–156. doi: 10.1016/0014-5793(96)00562-5. [DOI] [PubMed] [Google Scholar]
- 48.Hiner AN, Martinez JI, Arnao MB, Acosta M, Turner DD, Lloyd Raven E, Rodriguez-Lopez JN. Eur J Biochem. 2001;268:3091–3098. doi: 10.1046/j.1432-1327.2001.02208.x. [DOI] [PubMed] [Google Scholar]
- 49.Huyett JE, Doan PE, Gurbriel R, Houseman ALP, Sivaraja M, Goodin DB, Hoffman BM. J Amer Chem Soc. 1995;117:9033–9041. [Google Scholar]
- 50.Fitzgerald MM, Churchill MJ, McRee DE, Goodin DB. Biochemistry. 1994;33:3807–3818. [PubMed] [Google Scholar]
- 51.Miller MA, Han GW, Kraut J. Proc Natl Acad Sci USA. 1994;91:11118–11122. doi: 10.1073/pnas.91.23.11118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jensen GM, Bunte SW, Warshel A, Goodin DB. J Phys Chem B. 1998;102:8221–8228. [Google Scholar]
- 53.Fitzgerald MM, Trester ML, Jensen GM, McRee DE, Goodin DB. Protein Sci. 1995;4:1844–1850. doi: 10.1002/pro.5560040919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheek J, Mandelman D, Poulos TL, Dawson JH. J Bio Inorg Chem. 1999;4:64–72. doi: 10.1007/s007750050290. [DOI] [PubMed] [Google Scholar]
- 55.Bonagura CA, Sundaramoorthy M, Pappa HS, Patterson WR, Poulos TL. Biochemistry. 1996;35:6107–6115. doi: 10.1021/bi960122x. [DOI] [PubMed] [Google Scholar]
- 56.Miller MA. Biochemistry. 1996;35:15791–15799. doi: 10.1021/bi961488c. [DOI] [PubMed] [Google Scholar]
- 57.Barrows TP, Bhaskar B, Poulos TL. Biochemistry. 2004;43:8826–8834. doi: 10.1021/bi049531g. [DOI] [PubMed] [Google Scholar]
- 58.Musah RA, Goodin DB. Biochemistry. 1997;36:11665–11674. doi: 10.1021/bi9708038. [DOI] [PubMed] [Google Scholar]
- 59.Fishel LA, Farnum MF, Mauro M, Miller MA, Kraut J, Liu YL, Scholes CP. Biochemistry. 1991;30:1986–1996. doi: 10.1021/bi00221a036. [DOI] [PubMed] [Google Scholar]
- 60.Barrows TP, Poulos TL. Biochemistry. 2005;44:14062–14068. doi: 10.1021/bi0507128. [DOI] [PubMed] [Google Scholar]
- 61.Metcalfe CL, Ott M, Patel N, Singh K, Mistry SC, Goff HM, Raven EL. J Amer Chem Soc. 2004;126:16242–16248. doi: 10.1021/ja048242c. [DOI] [PubMed] [Google Scholar]
- 62.Spangler BD, Erman JE. Biochim Biophys Acta. 1986;872:155–157. doi: 10.1016/0167-4838(86)90159-7. [DOI] [PubMed] [Google Scholar]
- 63.Guallar V, Olsen B. J Inorg Biochem. 2006;100:755–760. doi: 10.1016/j.jinorgbio.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 64.Meharenna YT, Oertel P, Bhaskar B, Poulos TL. Biochemistry. 2008;47:10324–10332. doi: 10.1021/bi8007565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blodig W, Doyle WA, Smith AT, Winterhalter K, Choinowski T, Piontek K. Biochemistry. 1998;37:8832–8838. doi: 10.1021/bi9727186. [DOI] [PubMed] [Google Scholar]
- 66.Doyle WA, Blodig W, Veitch NC, Piontek K, Smith AT. Biochemistry. 1998;37:15097–15105. doi: 10.1021/bi981633h. [DOI] [PubMed] [Google Scholar]
- 67.Smith AT, Doyle WA, Dorlet P, Ivancich A. Proc Natl Acad Sci USA. 2009;106:16084–16089. doi: 10.1073/pnas.0904535106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruiz-Duenas FJ, Camarero S, Perez-Boada M, Martinez MJ, Martinez AT. Biochem Soc Trans. 2001;29:116–122. doi: 10.1042/0300-5127:0290116. [DOI] [PubMed] [Google Scholar]
- 69.Pogni R, Baratto MC, Teutloff C, Giansanti S, Ruiz-Duenas FJ, Choinowski T, Piontek K, Martinez AT, Lendzian F, Basosi R. J Biol Chem. 2006;281:9517–9526. doi: 10.1074/jbc.M510424200. [DOI] [PubMed] [Google Scholar]
- 70.Ruiz-Duenas FJ, Pogni R, Morales M, Giansanti S, Mate MJ, Romero A, Martinez MJ, Basosi R, Martinez AT. J Biol Chem. 2009;284:7986–7994. doi: 10.1074/jbc.M808069200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ruiz-Duenas FJ, Morales M, Mate MJ, Romero A, Martinez MJ, Smith AT, Martinez AT. Biochemistry. 2008;47:1685–1695. doi: 10.1021/bi7020298. [DOI] [PubMed] [Google Scholar]
- 72.DePillis GD, Sishta BP, Mauk AG, Ortiz de Montellano PR. J Biol Chem. 1991;266:19334–19341. [PubMed] [Google Scholar]
- 73.Mandelman D, Jamal J, Poulos TL. Biochemistry. 1998;37:17610–17617. doi: 10.1021/bi981958y. [DOI] [PubMed] [Google Scholar]
- 74.Adak S, Datta AK. The Biochem J. 2005;390:465–474. doi: 10.1042/BJ20050311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yadav RK, Dolai S, Pal S, Adak S. Biochim Biophys acta. 2008;1784:863–871. doi: 10.1016/j.bbapap.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 76.Kang CH, Brautigan DL, Osheroff N, Margoliash E. J Biol Chem. 1978;253:6502–6510. [PubMed] [Google Scholar]
- 77.Kang CH, Ferguson-Miller S, Margoliash E. J Biol Chem. 1977;252:919–926. [PubMed] [Google Scholar]
- 78.Liu RQ, Miller MA, Han GW, Hahm S, Geren L, Hibdon S, Kraut J, Durham B, Millet F. Biochemistry. 1994;33:8678–8685. doi: 10.1021/bi00195a008. [DOI] [PubMed] [Google Scholar]
- 79.Ho PS, Hoffman BM, Solomon N, Kang CH, Margoliash E. Biochemistry. 1984;23:4122–4128. doi: 10.1021/bi00313a017. [DOI] [PubMed] [Google Scholar]
- 80.Zhou JS, Hoffman BM. Science. Vol. 265. New York, N.Y: 1994. pp. 1693–1696. [DOI] [PubMed] [Google Scholar]
- 81.Miller MA, Vitello L, Erman JE. Biochemistry. 1995;34:12048–12058. doi: 10.1021/bi00037a048. [DOI] [PubMed] [Google Scholar]
- 82.Ho PS, Hoffman BM, Kang CH, Margoliash E. J Biol Chem. 1983;258:4356–4363. [PubMed] [Google Scholar]
- 83.Poulos TL, Sheriff S, Howard AJ. J Biol Chem. 1987;262:13881–13884. [PubMed] [Google Scholar]
- 84.Pelletier H, Kraut J. Science. Vol. 258. New York, N.Y: 1992. pp. 1748–1755. [DOI] [PubMed] [Google Scholar]
- 85.Guo M, Bhaskar B, Li H, Barrows TP, Poulos TL. Proc Natl Acad Sci USA. 2004;101:5940–5945. doi: 10.1073/pnas.0306708101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sevrioukova IF, Li H, Zhang H, Peterson JA, Poulos TL. Proc Natl Acad Sci USA. 1999;96:1863–1868. doi: 10.1073/pnas.96.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mauk MR, Ferrer JC, Mauk AG. Biochemistry. 1994;33:12609–12614. doi: 10.1021/bi00208a011. [DOI] [PubMed] [Google Scholar]
- 88.Stemp ED, Hoffman BM. Biochemistry. 1993;32:10848–10865. doi: 10.1021/bi00091a041. [DOI] [PubMed] [Google Scholar]
- 89.Northrup SC, Boles JO, Reynolds JC. Science. Vol. 241. New York, N.Y: 1988. pp. 67–70. [DOI] [PubMed] [Google Scholar]
- 90.Pappa HS, Tajbaksh S, Saunders AJ, Pielak GJ, Poulos TL. Biochemistry. 1996;35:4837–4845. doi: 10.1021/bi952935b. [DOI] [PubMed] [Google Scholar]
- 91.Nakani S, Viriyakul T, Mitchell R, Vitello LB, Erman JE. Biochemistry. 2006;45:9887–9893. doi: 10.1021/bi060586n. [DOI] [PubMed] [Google Scholar]
- 92.Nakani S, Vitello LB, Erman JE. Biochemistry. 2006;45:14371–14378. doi: 10.1021/bi061662p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Miller MA, Geren L, Han GW, Saunders A, Beasley J, Pielak GJ, Durham B, Millett F, Kraut J. Biochemistry. 1996;35:667–673. doi: 10.1021/bi952557a. [DOI] [PubMed] [Google Scholar]
- 94.Hays Putnam AM, Lee YT, Goodin DB. Biochemistry. 2009;48:1–3. doi: 10.1021/bi8020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dolai S, Yadav RK, Pal S, Adak S. Eukaryotic cell. 2009;8:1721–1731. doi: 10.1128/EC.00198-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dolai S, Yadav RK, Pal S, Adak S. Free Rad Biol Med. 2008;45:1520–1529. doi: 10.1016/j.freeradbiomed.2008.08.029. [DOI] [PubMed] [Google Scholar]