

Abstract
The discovery of regulatory T cells almost 15 years ago initiated a new and exciting research area. The growing evidence for a critical role of these cells in controlling autoimmune responses has raised expectations for therapeutic application of regulatory T cells in patients with autoimmune arthritis. Here, we review recent studies investigating the presence, phenotype and function of these cells in patients with RA and juvenile idiopathic arthritis (JIA) and consider their therapeutic potential. Both direct and indirect methods to target these cells will be discussed. Arguably, a therapeutic approach that combines multiple regulatory T-cell-enhancing strategies could be most successful for clinical application.
Keywords: Rheumatoid arthritis, Juvenile idiopathic arthritis, Regulatory T cells, Treatment, Pro-inflammatory cytokines, Effector T cells, Combination therapy
Introduction
RA and juvenile idiopathic arthritis (JIA) are autoimmune diseases characterized by destructive joint inflammation. In the chronic phase of the diseases, a non-remitting activation of cells and expression of soluble mediators of especially the innate immune system dominates the inflammatory process. The resulting synovial inflammation is characterized by non-specific infiltration of both lymphocytes and innate immune cells, such as synoviocytes, macrophages and neutrophils. The importance of this innate immune activation in chronic arthritis is underscored by the success of interventions with biologicals that target non-specific effector mediators such as TNF-α. In contrast, interventions directed against CD4+ T cells have been disappointing. This has led to the assumption that T cells are of less importance in the chronic phase of RA and JIA [1].
Data obtained over the last years, however, have shed new light on the role of T cells in regulation of the inflammatory response. This line of research started almost 15 years ago with the discovery of so-called regulatory T cells (Treg) [2]. This exciting discovery raised expectations for novel ways of treating arthritis by targeting these Treg. Their presence and function in RA and JIA, and the questions still surrounding their potential therapeutic application will be discussed in this review.
Treg
Treg are capable of suppressing effector cell proliferation and cytokine production, and play an important role in immune homeostasis. Several subtypes of CD4+ Treg have been identified that can be either naturally occurring, derived from the thymus or induced in the periphery. These subtypes of Treg are depicted in Table 1 together with their supposed mechanism of action. Natural Treg constitutively express the IL-2 receptor (CD25) and require IL-2 for their survival and function [2, 3]. These cells are further characterized by the transcription factor FOXP3, which controls the development and suppressive function of the cells [4–6]. CD25+FOXP3+ Treg can suppress via multiple mechanisms, probably depending on the context in vivo [7]. These cells are critical in preventing autoimmune disease in animal models [3], which is confirmed in humans by the fact that patients with mutations in the FOXP3 gene suffer from immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX), characterized by autoimmune disease in multiple organs [8, 9]. More recently, it has been established in both mice and humans that Treg can also be induced in the periphery upon antigen encounter. These cells can be not only FOXP3+ [10–13], but also FOXP3−, such as T regulatory 1 (Tr1) cells that depend on IL-10 for their development and function [14, 15] and T helper 3 (Th3) cells, producing TGF-β [16]. CD25+FOXP3+ Treg are highly important in the control of autoimmune arthritis both in experimental models [17–19] and in human disease [20]. Therefore, we will further refer to this specific CD25+FOXP3+ subset by the term Treg and we will discuss the potential of these cells as a target for immune intervention in arthritis.
Table 1.
Subtypes of CD4+ Treg and supposed mechanism of action
| Term | Markers | Origin | Mechanism of action |
|---|---|---|---|
| Natural Treg | CD25 FOXP3 | Thymus | Cell–cell contact and suppressive cytokines |
| CTLA-4 GITR | |||
| Adaptive Treg | CD25 FOXP3 | Induced in the periphery | Cell–cell contact and suppressive cytokines |
| CTLA-4 GITR | |||
| Tr1 | – | Induced in the periphery | IL-10 production |
| Th3 | – | Induced in the periphery | TGF-β production |
Presence, phenotype and function of Treg in arthritis patients
Given the convincing evidence that Treg play a critical role in preventing experimental autoimmune arthritis, numerous groups have studied the presence, phenotype and function of Treg in patients with RA and JIA (summarized in Table 2) [20–28]. When analysing these data, it should be kept in mind that several studies were performed before FOXP3 was identified as a marker for Treg. In these studies, Treg were identified based on (high) CD25 expression, which is a less definitive marker for Treg compared with FOXP3. In addition, FOXP3 can also be up-regulated in effector cells during activation [29] and this makes it difficult to distinguish Treg from activated effector T cells in patients with ongoing autoimmune inflammation.
Table 2.
Presence, phenotype and function of Treg in arthritis
| Peripheral blood |
SF |
|||||||
|---|---|---|---|---|---|---|---|---|
| Disease | Markers used to identify Treg | Presence | Phenotype | Function | Presence | Phenotype | Function | Reference |
| JIA | CD4+CD25high | ↓ | FOXP3 mRNA = | NA | ↑⇑ | FOXP3 mRNA ↑⇑ HLA-DR MFI, CTLA-4 MFI, GITR MFI CD69+⇑ | ⇑ | [20] |
| JIA | CD4+CD25+a CD4+CD25+CD27+b | NA | NA | NA | ⇑ | FOXP3 mRNA⇑ | ⇑ | [25] |
| RA | CD4+CD25high | NA | NA | NA | ⇑ | NA | – | [21] |
| RA | CD4+CD25high | = | NA | ↓ | NA | NA | NA | [22] |
| RA | CD4+FOXP3higha CD4+CD25+CD127lowb | = | CTLA-4 MFI ↓ | ↓ | NA | NA | NA | [23] |
| RA | CD4+CD25high | Early active RA ↓ stable RA= | NA | = | ⇑ | NA | NA | [24] |
| RA | CD4+CD25+ | ↑ | CD69+ HLA-DR+ OX40+ GITR+ = CTLA-4 MFI | = | ↑⇑ | CD69+ HLA-DR+, OX40+ GITR+, CTLA-4 MFI⇑ | ⇑ | [26] |
| RA | CD4+CD25high | NA | FOXP3 mRNA ↓ TNFRII+ GITR+↑ CD69+ = | ↓ | NA | NA | NA | [27] |
| RA | CD4+CD25+ | = | NA | NA | ⇑ | CTLA-4+ GITR+, OX40+ CD69+↑⇑ | – | [28] |
aUsed for phenotyping. bUsed for functional assays. cSuppressive, but functionality not compared with peripheral blood. = : not changed; ↑: increased; ↓ : decreased,compared with peripheral blood of healthy controls; ⇑: increased compared with paired peripheral blood of patients; NA: not analysed; MFI: Mean Fluorescence Index; + : percentage of positive cells.
Nevertheless, the majority of studies suggest that Treg numbers in the periphery are not reduced in arthritis patients compared with healthy controls [22, 23, 26, 28]. Instead, Treg are enriched at the site of inflammation, since increased levels of these cells are found in the SF compared with peripheral blood [20, 21, 24–26, 28]. These SF-derived Treg show enhanced expression of FOXP3 mRNA, cytotoxic T lymphocyte antigen 4 (CTLA-4), glucocorticoid-induced tumor necrosis factor receptor (GITR), HLA-DR, CD69 and OX40 [20, 25, 26, 28] and are more efficient in inhibiting effector cell activation [20, 25, 26]. In contrast, reduced suppressive function has been reported for peripheral blood-derived Treg from RA patients in some [22, 23, 27], but not all studies [24, 26]. Thus, there is still conflicting evidence on the suppressive function of Treg in arthritis, which can result from the different test systems used to analyse the suppressive function of the cells.
For obvious technical reasons, all the above studies investigated Treg-mediated suppression in vitro. However, in vivo the local pro-inflammatory environment can interfere with the suppressive function of the cells. High levels of pro-inflammatory cytokines are present in the inflamed synovium of RA and JIA patients, including IL-6, IL-7, IL-15 and TNF-α [30–32]. In addition, human CD25hi cells express the TNF receptor, TNF receptor II (TNFRII) and expression of this receptor is up-regulated on cells from RA patients [27]. As a result, TNF-α can act directly on Treg and, in line with this, it was shown that pre-incubation of Treg with TNF-α reduces FOXP3 expression and abrogates suppression [27]. Other pro-inflammatory cytokines, IL-6, IL-7 and IL-15, can also interfere with Treg function [25, 33, 34], or even worse facilitate the conversion of Treg into IL-17 producing effector cells [35–37]. Finally, monocytes and dendritic cells from the site of inflammation express elevated levels of CD80, CD86 and CD40 [34, 38] and this enhanced expression of co-stimulatory molecules might also interfere with Treg-mediated suppression [34]. Thus, though Treg function in patients with RA and JIA is still incompletely understood, data from both animal models and human disease indicate that Treg play an important role in controlling autoimmune arthritis. As such, these cells form a promising treatment option for arthritis patients. Here, we will discuss several strategies to target these cells, both directly and indirectly.
Direct approaches to enhance Treg function
There are several methods available to directly target Treg for the treatment of autoimmune disease. These include expansion and induction of Treg in vitro followed by reinfusion into the patient, or in vivo by immunomodulatory compounds.
Ex vivo expansion of Treg
Treg can be isolated and expanded ex vivo by anti-CD3/anti-CD28 stimulation in the presence of IL-2 [39, 40]. With this protocol up to 3000-fold expansion can be reached without loss of suppressive function. Moreover, the cells have a higher inhibitory potential compared with directly isolated Treg, even in co-cultures with pre-activated effector cells [39]. Therefore, expanded Treg could have enhanced suppressive capacity in ongoing immune responses in vivo and be useful in the treatment of autoimmune disease. In favour of this argument, it has been shown that in vitro expanded Treg survive upon transfer in vivo and reverse pathology in new-onset diabetic mice [41]. Similarly, in experimental lupus, adoptive transfer of expanded Treg delayed the progression to severe renal disease, resulting in prolonged survival [42].
However, a potential hazard with expanding Treg for therapeutic purposes is the outgrowth of contaminating effector cells, since it is difficult to distinguish Treg from activated effector cells. This risk can be reduced by adding rapamycin to expansion cultures, which selectively allows for regulatory T-cell proliferation and survival, while depleting effector cells [43, 44]. Still, expanded Treg can also convert into effector cells themselves. Using the same protocol as described before, Hoffmann et al. [39], discovered that, although FOXP3 purity at the start of culture was almost 100%, subpopulations of Treg lost FOXP3 expression and suppressive capacity. Furthermore, these cells started to produce effector cytokines, such as IL-2 and IFN-γ. Only cells that co-expressed CCR7 and CD62L after expansion showed a stable Treg phenotype and these cells could be generated by selecting the CD45RA+CD4+CD25high subpopulation for Treg expansion [45]. In addition, Tran et al. [46] identified latency-associated peptide and IL-1 receptor type I/II (CD121a/CD121b) as markers to purify stable Treg after expansion in vitro. However, under certain circumstances, FOXP3+ Treg can also convert into effector cells, after transfer in vivo [47]. This could pose a risk for worsening, instead of dampening inflammation, especially in autoimmune disease [48]. Therefore, the stability of ex vivo expanded Treg should be further investigated and conditions reinforcing this stability need to be thoroughly determined. In addition, expansion of antigen-specific Treg can enhance efficacy [41] and reduce general immune suppression and this approach should, therefore, be explored in humans as well. Altogether, standardized protocols have to be developed to allow for reliable expansion of Treg with compliance to the Good Manufacturing Practices approved by the US Food and Drug Administration. Only then will clinical application in a large cohort of patients become feasible.
In vitro induction of Treg
In addition to expansion of already existing Treg, Treg can also be induced in vitro from non-Treg. This method circumvents the difficulty of obtaining high numbers of natural Treg required for expansion. Treg induction works well in mice in which CD4+CD25− cells activated in the presence of TGF-β develop into FOXP3-expressing cells with suppressive capacity that is maintained after transfer in vivo [11]. However, TCR stimulation of human CD4+CD25− cells can also result in transient expression of FOXP3 [49]. Furthermore, activation-induced expression of FOXP3 in humans does not confirm a regulatory phenotype and can even coincide with IL-2 and IFN-γ production [50]. Therefore, in vitro induction of Treg is far more complicated in humans compared with mice and it still needs to be established which culture conditions reinforce stable FOXP3 expression and suppressive function.
In vivo expansion and induction of Treg with immunomodulatory compounds
Next to expanding and inducing Treg in vitro, several immunoactive agents can be used to enhance Treg function in vivo. The most extensively studied Treg-enhancing agents will be discussed in this section and are summarized in Table 3.
Table 3.
Immunoactive compounds with Treg-enhancing capacity
| Compound | Effect on Treg | Results in experimental autoimmune disease | Results in human autoimmune disease |
|---|---|---|---|
| Anti-CD3 antibodies | Treg induction | Complete and sustained remission in recent-onset diabetic NOD mice [56] | Long-term improved insulin production in new-onset Type 1 diabetes [52, 53] |
| Short-term improvement in the number of inflamed joints in PsA [54] | |||
| Neuropeptides | Treg induction, expansion and enhanced function | VIP reduces development of CIA and established disease in DBA/1J mice [57, 63] | – |
| Urocortin reduces severity of established CIA in DBA/1J mice [64] | |||
| Retinoic acid | Treg induction | Reduced incidence of diabetes in NOD mice with established insulitis [70] | – |
| Improved bodyweight and reduced colon inflammation in TNBS-induced colitis [71] | |||
| Reduced severity and incidence of CIA in DBA/1J mice [72] | |||
| HDAC inhibitors | Enhancement and stabilization of FOXP3 expression leading to enhanced function | TSA and SAHA prevent bodyweight loss and histological damage in DSS-induced colitis [80, 81] | – |
| TSA reduces development of renal pathology in lupus-prone NZB/W F1 mice [82] | |||
| VPA reduces the incidence and severity of CIA in DBA/1J mice [83] |
NOD: non-obese diabetic; TNBS: trinitrobenzene sulfonic acid; DSS: dextran sodium sulphate.
Anti-CD3 antibodies
The immunosuppressive efficiency of mAbs against CD3 was initially established in the transplantation field, where they prevented allograft rejection. After humanizing the antibodies into non-Fc-receptor-binding antibodies that are not mitogenic, application in the treatment of autoimmune disease was tested as well [51]. In new-onset Type 1 diabetes patients, treatment with humanized CD3 antibodies led to preserved β-cell function and reduced insulin need [52, 53]. Also in rheumatic disease, efficacy of anti-CD3 treatment was confirmed: in a Phase I/II trial in patients with PsA, administration of huOKT3γ1 led to a 75% improvement in the number of inflamed joints in six out of seven patients [54]. Studies in experimental diabetes further elucidated the mechanisms involved in immune suppression by anti-CD3 antibodies. These studies revealed that short-term disease improvement was achieved by elimination of pathogenic effector cells and Th2 polarization. However, long-term beneficial effects depended on a non-depleting, Treg-inducing activity of the antibody [55, 56]. Thus, CD3-specific antibodies are capable of inducing Treg and have already been proved to be safe and effective in patients with autoimmune disease. As such, they may provide a valuable treatment option for RA and JIA as well, which should be further investigated.
Neuropeptides
Vasoactive intestinal peptide (VIP), an immune-regulatory neuropeptide, has been shown to have suppressive effects in experimental autoimmune disease, including CIA. This suppressive effect was accompanied by inhibition of pro-inflammatory cytokines and chemokines and immune deviation towards Th2 responses [57, 58]. However, similarly to the CD3-specific antibodies, it has become clear that VIP is capable of enhancing Treg numbers and suppressive function as well [59], presumably via the induction of tolerogenic dendritic cells [60–62]. In CIA, administration of VIP increased both the absolute number and percentage of Treg, leading to lower arthritis scores [63]. Another neuropeptide, urocortin, also reduced disease severity in this model via the induction of Treg [64]. Although clinical trials in human autoimmune disease are still awaiting, neuropeptides could be of therapeutic value, due to their Treg-enhancing capacity.
Retinoic acid
All-trans retinoic acid (ATRA) is an active metabolite of vitamin A that regulates various cellular functions, including lymphocyte proliferation and differentiation. Recently, several research groups have found that ATRA induces Treg, while simultaneously inhibiting Th17 development [65–68]. Therefore, ATRA might be able to restore the balance between Treg and pathogenic Th17 cells that is thought to be disturbed in autoimmune pathology [69]. In experimental models of diabetes [70] and colitis [71], ATRA treatment improved clinical outcome by inducing Treg. ATRA-mediated induction of Treg has not been investigated in arthritis models; however, ATRA administration has been shown to reduce severity and incidence of CIA. This beneficial effect was accompanied by a decrease in pro-inflammatory cytokines and collagen-specific antibodies [72]. Given the therapeutic effects of ATRA in experimental arthritis and its potent Treg-enhancing capacity, it would be valuable to further explore this mechanism for the treatment of arthritis. In addition, Treg induced in vitro in the presence of ATRA are resistant to conversion into FOXP3− cells [73] and Treg expanded in the presence of ATRA have enhanced suppressive capacity [74]. Therefore, ATRA can also be used to optimize protocols for the in vitro expansion and induction of Treg.
Histone deacetylase inhibitors
The FOXP3 gene is subject to epigenetic modifications, including acetylation mediated by histone acetyltransferases (HAT) that increases the negative charge of histones in the nucleosome. This leads to an open chromatin structure, allowing for gene transcription [75]. The described induction of Treg by ATRA probably depends on this modification, since acetylation of the FOXP3 promotor is enhanced in ATRA-treated cells [76]. Acetyl groups can also be removed by histone deacetylases (HDACs), introducing a positive charge that leads to tight DNA binding and reduced transcription [75]. In addition, FOXP3 can directly interact with HAT and HDAC at the protein level [77] and a very recent study shows that hyperacetylation of FOXP3, reciprocally controlled by the acetyltransferase p300 and the HDAC SIRT1, prevents poly-ubiquination and subsequent proteasomal degradation of the protein [78]. Agents counteracting HDAC activity, so-called HDAC inhibitors, can therefore both increase FOXP3 gene transcription and prevent protein degradation, thereby enhancing and stabilizing FOXP3 expression.
Two HDAC inhibitors, MS-275 and suberoylanilide hydroxamic acid (SAHA), have been shown to induce FOXP3 expression and suppressive function in human CD4+CD25− cells in vitro [79]. Exposure to another HDAC inhibitor, nicotinamide, increased the number of FOXP3+ cells in CD4+ cell cultures as well as the amount of FOXP3 per cell and the suppressive capacity of CD4+CD25+ cells [78]. Also in vivo, administration of HDAC inhibitors leads to increased numbers of FOXP3+ T cells with enhanced suppressive capacity. Moreover, treatment with HDAC inhibitors reduces pathology in dextran sodium sulphate-induced colitis [80, 81], lupus-prone mice [82] and experimental arthritis [83], by enhancing Treg function. Trichostatin-A (TSA) treatment even improved already established colitis and HDAC inhibitors have been shown to reduce the down-regulating effect of IL-6 on FOXP3 [84]. This makes them attractive candidates for the treatment of ongoing inflammation. Interestingly, several HDAC inhibitors are now being developed for application in human autoimmune disease based not on their capacity to enhance Treg function, but on their more familiar anti-inflammatory and immunosuppressive capacities. Among those agents is hydroxamic acid, which is being tested for therapeutic application in arthritis [85]. In addition, HDAC inhibitors can be used to stabilize FOXP3 expression in induced or expanded Treg since they prevent conversion of these cells into Th17 cells [35].
Antigen-specific induction of Treg by mucosal tolerization with self-antigen
The above-described methods are all based on enhancing the polyclonal Treg population. However, these non-specific approaches might lead to increased risk of infections and cancer, due to general immune suppression [86]. These unwanted side effects can be avoided by antigen-specific induction of Treg. This can be achieved by mucosal administration of self-antigen, which is a powerful way of inducing Treg towards a specific antigen [87]. Oral or nasal administration of self-antigens works well in animal models of arthritis, leading to delayed onset of disease and reduced severity [88–90], presumably via the induction of Treg [91–93]. Moreover, beneficial effects of oral antigen administration have also been described in already established disease, making a therapeutic application in humans feasible [94, 95].
However, results from animal models have been difficult to translate into humans. Clinical trials have shown that oral administration of antigen is safe [96–101]; however, in many cases only small improvements were found [97, 98], or only a minority of the patients responded to treatment [100]. These disappointing results are presumably caused by the fact that the disease-triggering antigen in humans is less clear and at the time of intervention multiple antigens are involved, due to epitope spreading [102]. Still, through bystander suppression, Treg specific for one antigen can also suppress immune responses towards other antigens that are presented in the same vicinity [103, 104]. This can be achieved by the production of non-specific, inhibitory cytokines, such as IL-10 and TGF-β by the induced Treg [88, 91, 92]. As a result, mucosal tolerization with self-antigen could work in human disease as well, as long as an immunogenic antigen is used that is presented at the same location as the self-antigens driving the immune response. A special class of proteins, termed heat shock proteins (HSPs), are promising antigens for this Treg induction via mucosal tolerization.
HSPs
HSPs are a set of evolutionarily conserved chaperones that are up-regulated under conditions of cellular stress, for instance during infection and inflammation [105]. As a result, they are abundantly present at the site of inflammation in RA and JIA [106, 107] and, because of their unique features HSPs are very immunogenic [108–110]. Therefore, these antigens are good candidates for mucosal tolerization in autoimmune disease, since they trigger T-cell responses and are highly present at the site of inflammation. Moreover, studies with cells from JIA patients suggest that HSPs might have a natural role in controlling inflammation via the induction of regulatory responses [111–114].
Several HSP family members have been shown to be protective upon mucosal administration in experimental arthritis, even in already established disease [115], probably via the induction of Treg [116, 117]. Moreover, nasal administration of a mycobacterial HSP peptide inhibited adjuvant arthritis, but also arthritis induced by an unrelated, non-microbial stimulus [118]. Thus, HSPs suppress experimental arthritis irrespective of the initial trigger and are effective in already established disease. This makes them suitable for therapeutic application in human arthritis. Studies with OM-89, an extract of Escherichia coli used for the treatment of RA, provide the first evidence that HSP could be effective in the treatment of human arthritis. Multicentre placebo-controlled trials with OM-89 showed that it ameliorates RA with few side effects [119, 120]. Later on, analysis of the OM-89 content revealed that it contained HSP [121] and oral administration in animal models led to HSP-directed T-cell responses [122]. Therefore, HSP is thought to be responsible for the therapeutic effect of OM-89 in arthritis. More direct evidence comes from a pilot Phase II trial with a peptide derived from E. coli HSP, dnaJP1. Oral administration of this peptide in RA patients was well tolerated and led to enhanced IL-4 and IL-10, and reduced TNF-α and IFN-γ production towards the peptide. Furthermore, dnaJP1-induced expression of FOXP3 in CD25bright cells was increased following treatment [123]. Subsequently, the clinical efficacy of this approach was studied in a placebo-controlled Phase II trial enrolling 160 patients with active RA. Again treatment was safe and well tolerated and reduced TNF-α responses towards dnaJP1 were found. Furthermore, a difference in the ACR20 and ACR50 score between treatment and placebo groups suggested clinical efficacy [124].
Indirect approaches to enhance Treg function
In addition to the above-described strategies that target the Treg population directly, indirect approaches can also be taken to enhance Treg function in patients with autoimmune disease. These include reducing the pro-inflammatory environment and enhancing responsiveness of effector cells to suppression.
Inhibition of pro-inflammatory cytokines
As described above, the in vivo pro-inflammatory environment at the site of inflammation in patients with autoimmune disease can have profound negative effects on Treg function. Therefore, dampening the ongoing inflammation, for instance by inhibiting pro-inflammatory cytokines, can indirectly lead to better Treg-mediated suppression. This is clearly shown by two studies that examined Treg function in RA patients before and after anti-TNF-α (infliximab) therapy. Both studies reported impaired Treg function before therapy, which was completely restored after infliximab treatment [22, 27]. Probably, neutralizing the high TNF-α levels in these patients directly reduced the down-regulating effect of TNF-α on Treg [27], thereby restoring their suppressive function. However, it is also possible that, instead of reconstituting the suppressive function of already existing Treg, anti-TNF-α therapy actually induced a new Treg population with enhanced regulatory potential [125].
Enhancing the responsiveness of effector cells to suppression
Indirect improvement of Treg function can also be achieved by enhancing responsiveness of effector cells to suppression. In Type 1 diabetes, inflammatory bowel disease and lupus, effector cells are refractory to inhibition by Treg [126–129]. Also in the SF of JIA and RA patients, effector cells appear to be less responsive to suppression compared with their peripheral blood counterparts [20, 26]. Elucidating the cause of this resistance to suppression and subsequent targeting will enhance Treg-mediated inhibition and restrict uncontrolled activation of effector cells. Several studies suggest that this can, at least partially, be achieved by blocking the production of pro-inflammatory cytokines. In experimental autoimmune encephalomyelitis (EAE), it was found that Treg isolated from the CNS could suppress effector cells from the spleen, but failed to inhibit effector cells isolated from the site of inflammation. When analysing these CNS effector cells, they were found to produce high levels of IL-6 and TNF-α. Furthermore, adding both these cytokines to naïve effector cells reversed their responsiveness to suppression. Thus, the increased resistance of effector cells at the site of inflammation in EAE mice is caused by TNF-α and IL-6 produced by these cells [130]. Another study describing the negative effects of IL-6 on Treg-mediated suppression also found that IL-6 acts on effector cells rather than on Treg [33]. Similarly IL-7, known to reduce Treg-mediated suppression, is expected to target effector cells as well [34], since expression of the IL-7 receptor (CD127) is low on Treg [131]. Therefore, blocking these pro-inflammatory cytokines will reduce the resistance of effector cells to suppression and thereby enhance control of inflammation by Treg.
Combination therapy
So far, we have described multiple approaches that can be taken to target Treg function in patients with autoimmune disease (Fig. 1), including direct, antigen-specific induction of Treg by tolerization with self-antigen. In addition, inhibition of the inflammatory response increases the responsiveness of effector cells to suppression and reduces the down-regulating effect of pro-inflammatory cytokines on Treg, thereby indirectly enhancing Treg function. It is therefore expected that clinical outcome can be enhanced by a combination of both these direct and indirect strategies. This is nicely illustrated by a study in which antigen-specific induction of Treg was combined with anti-TNF-α therapy in adjuvant arthritis. Both nasal administration of HSP60 peptide as well as a single dose of anti-TNF-α (etanercept) treatment, led to a small and insignificant reduction in arthritis scores. However, combining the two therapies resulted in a highly significant improvement of disease, as shown by lower arthritis scores and reduced joint destruction [132]. In addition, in several models of autoimmune diabetes, mucosal tolerization with islet antigen induced Treg and prevented development of disease, but was incapable of reversing established disease. Bresson et al. [133] now show that, when combined with a suboptimal dose of anti-CD3 therapy, intranasal administration of proinsulin peptide reverses recent-onset diabetes. Also in humans there is evidence for enhanced effectiveness of Treg induction, when combined with anti-inflammatory treatment. In the previously described trial with dnaJP1 in RA patients a synergistic clinical effect was found in patients receiving HCQ, a drug with potent anti-inflammatory properties [124]. Together, these data clearly demonstrate that combining Treg induction with anti-inflammatory treatment enhances clinical outcome. In addition to increased effectiveness, dampening the ongoing inflammation might also be crucial in preventing adverse effects, as it has been shown that in a pro-inflammatory environment TGF-β produced by Treg drives Th17 differentiation [37, 134] and Treg can convert into Th17 cells themselves [35–37].
Fig. 1.
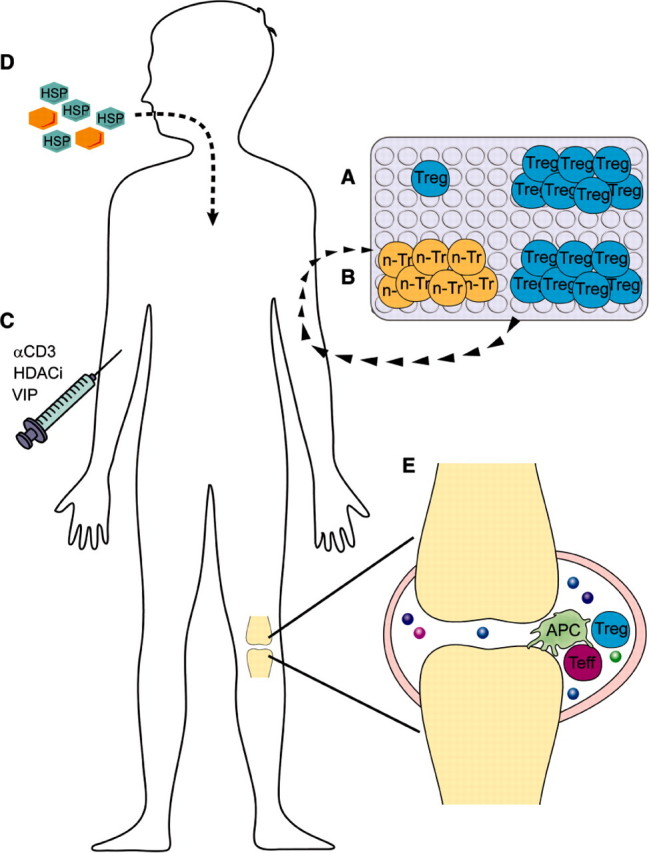
Methods of enhancing Treg function in arthritis patients. Treg can be enhanced in arthritis patients via different methods: (A) isolation and ex vivo expansion of natural Treg or (B) in vitro induction of Treg from non-Treg (n-Tr), followed by reinfusion into the patient; (C) in vivo induction and expansion of Treg by anti-CD3 antibodies (αCD3), HADC inhibitors (HADCi) and neuropetides, such as VIP; (D) mucosal tolerization with self-antigen, preferably HSP; (E) indirect improvement of Treg function by enhancing the responsiveness of effector cells to suppression and blocking pro-inflammatory cytokines.
Autologous bone marrow transplantation as a multifactorial therapeutic approach
One very powerful therapy applied for the treatment of refractory autoimmune disease is autologous bone marrow transplantation (aBMT). The idea of using aBMT in the treatment of autoimmunity stems from observed remission in patients transplanted for co-existing haematological malignancies and from efficacy in experimental models [135]. Initially, the mechanism of action was thought to depend on the elimination of autoreactive lymphocytes by intensive immune ablation, followed by the development of a new tolerant lymphocyte population after aBMT. However, more recently it has become clear that induction of Treg is also important in the clinical efficacy of aBMT [136]. aBMT has been used in the treatment of RA and systemic JIA patients who are unresponsive to other treatments [135, 137]. Especially in systemic JIA patients, this approach has been successful, leading to long-lasting, drug-free remission in 53% of the patients and a partial response in 18% of patients [137]. In a follow-up study of JIA patients receiving aBMT, it was demonstrated that in addition to a more tolerogenic response observed in effector T cells, Treg were affected as well. The low Treg levels before treatment were restored after aBMT and even after long-term follow-up the numbers of Treg were significantly increased compared with pre-treatment [138].
The importance of Treg in aBMT has also been investigated in experimental models of autoimmune disease. In EAE, pseudo-autologous BMT prevented relapses and resulted in increased levels of CD25bright cells and FOXP3 mRNA expression [139] and in CIA, co-transfer of purified Treg with the graft enhanced clinical outcome [18]. Furthermore, in proteoglycan-induced arthritis, it was found that depletion of CD25+ Treg after pseudo-autologous BMT abrogated disease remission induced by aBMT [140]. This last result clearly demonstrates a key role for Treg in the clinical efficacy of aBMT, next to elimination of autoreactive T cells and reduced inflammation, caused by immune suppression. aBMT is therefore a good example of how a multifactorial approach targeting Treg, effector T cells and ongoing inflammation is highly effective, even in the treatment of severe, systemic autoimmunity. It also shows that intensive immune ablation followed by aBMT provides an environment that is optimally suited for the development of Treg and might provide a window of opportunity for the induction of antigen-specific Treg.
Conclusion
Treg play a critical role in controlling autoimmune disease and several strategies are now being explored to target these cells for therapeutic purposes. For patients with RA and JIA, Treg provide a valuable new treatment option, since current therapies, such as anti-TNF-α therapy, cause a rather general immune suppression and do not induce sustained remission. As a result, side effects occur and life-long treatment is required. To enhance Treg function, the cells can be expanded and induced in vitro followed by adoptive transfer. However, these protocols have severe drawbacks, especially the risks associated with conversion of Treg into effector cells, and the costs and complexities associated with cellular therapy. Alternatively, Treg can be induced in vivo by immunomodulatory compounds and some of these agents have already been tested in patients.
Also, to avoid risks associated with general immune suppression, antigen-specific induction of Treg provides a potential safe and efficient approach, for which HSPs are promising candidate antigens. These proteins induce Treg that specifically recognize antigen at the site of inflammation, thereby avoiding systemic immune suppression. Clinical trials have shown that HSP treatment is safe and induces clinical improvement. Since the majority of studies indicate that Treg are not deficient in arthritis patients, but are functionally compromised by their pro-inflammatory environment, the efficacy of this approach can be optimized by inhibiting the ongoing inflammation in these patients. This is illustrated by a synergistic effect of Treg induction and anti-inflammatory treatment in both patients and experimental models. When combined with HSP treatment, only a single dose of anti-TNF-α therapy is sufficient to reduce pathology in experimental arthritis. The possibility of lowering the dose of anti-inflammatory treatment will have great impact on patient care, since it reduces the side effects associated with life-long drug administration. Therefore, Treg targeted approaches may significantly add to therapies that are in the clinic for arthritis today and deserve thorough future investigation.

Disclosure statement: F.vW. is supported by a Veni grant from the Netherlands Organisation for Scientific Research (NWO). E.J.W. is financially supported by Top Institute Pharma. All other authors have declared no conflicts of interest.
References
- 1.Strand V, Kimberly R, Isaacs JD. Biologic therapies in rheumatology: lessons learned, future directions. Nat Rev Drug Discov. 2007;6:75–92. doi: 10.1038/nrd2196. [DOI] [PubMed] [Google Scholar]
- 2.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. [PubMed] [Google Scholar]
- 3.Sakaguchi S. Naturally arising FOXP3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–52. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 4.Fontenot JD, Gavin MA, Rudensky AY. FOXP3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 5.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 6.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–42. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 7.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–32. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 9.Wildin RS, Ramsdell F, Peake J, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 10.Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. 2004;199:1401–8. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor FOXP3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+ J Immunol. 2004;172:5149–53. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 13.Yamagiwa S, Gray JD, Hashimoto S, Horwitz DA. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol. 2001;166:7282–9. doi: 10.4049/jimmunol.166.12.7282. [DOI] [PubMed] [Google Scholar]
- 14.Groux H, O’Garra A, Bigler M, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–42. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 15.Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev. 2006;212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- 16.Weiner HL. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol Rev. 2001;182:207–14. doi: 10.1034/j.1600-065x.2001.1820117.x. [DOI] [PubMed] [Google Scholar]
- 17.Morgan ME, Sutmuller RP, Witteveen HJ, et al. CD25+ cell depletion hastens the onset of severe disease in collagen-induced arthritis. Arthritis Rheum. 2003;48:1452–60. doi: 10.1002/art.11063. [DOI] [PubMed] [Google Scholar]
- 18.Morgan ME, Flierman R, van Duivenvoorde LM, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum. 2005;52:2212–21. doi: 10.1002/art.21195. [DOI] [PubMed] [Google Scholar]
- 19.Frey O, Petrow PK, Gajda M, et al. The role of regulatory T cells in antigen-induced arthritis: aggravation of arthritis after depletion and amelioration after transfer of CD4+CD25+ T cells. Arthritis Res Ther. 2005;7:R291–301. doi: 10.1186/ar1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Kleer I, Wedderburn LR, Taams LS, et al. CD4+CD25bright regulatory T cells actively regulate inflammation in the joints of patients with the remitting form of juvenile idiopathic arthritis. J Immunol. 2004;172:6435–43. doi: 10.4049/jimmunol.172.10.6435. [DOI] [PubMed] [Google Scholar]
- 21.Cao D, van Vollenhoven R, Klareskog L, Trollmo C, Malmstrom V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res Ther. 2004;6:R335–46. doi: 10.1186/ar1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ehrenstein MR, Evans JG, Singh A, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–85. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flores-Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci USA. 2008;105:19396–401. doi: 10.1073/pnas.0806855105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawson CA, Brown AK, Bejarano V, et al. Early rheumatoid arthritis is associated with a deficit in the CD4+CD25 high regulatory T cell population in peripheral blood. Rheumatology. 2006;45:1210–7. doi: 10.1093/rheumatology/kel089. [DOI] [PubMed] [Google Scholar]
- 25.Ruprecht CR, Gattorno M, Ferlito F, et al. Coexpression of CD25 and CD27 identifies FOXP3+ regulatory T cells in inflamed synovia. J Exp Med. 2005;201:1793–803. doi: 10.1084/jem.20050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. 2004;50:2775–85. doi: 10.1002/art.20499. [DOI] [PubMed] [Google Scholar]
- 27.Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108:253–61. doi: 10.1182/blood-2005-11-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mottonen M, Heikkinen J, Mustonen L, Isomaki P, Luukkainen R, Lassila O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol. 2005;140:360–7. doi: 10.1111/j.1365-2249.2005.02754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allan SE, Crome SQ, Crellin NK, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. 2007;19:345–54. doi: 10.1093/intimm/dxm014. [DOI] [PubMed] [Google Scholar]
- 30.de Jager W, Hoppenreijs EP, Wulffraat NM, Wedderburn LR, Kuis W, Prakken BJ. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross-sectional study. Ann Rheum Dis. 2007;66:589–98. doi: 10.1136/ard.2006.061853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harada S, Yamamura M, Okamoto H, et al. Production of interleukin-7 and interleukin-15 by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:1508–16. doi: 10.1002/1529-0131(199907)42:7<1508::AID-ANR26>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 32.Steiner G, Tohidast-Akrad M, Witzmann G, et al. Cytokine production by synovial T cells in rheumatoid arthritis. Rheumatology. 1999;38:202–13. doi: 10.1093/rheumatology/38.3.202. [DOI] [PubMed] [Google Scholar]
- 33.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 34.van Amelsfort JM, van Roon JA, Noordegraaf M, et al. Proinflammatory mediator-induced reversal of CD4+, CD25+ regulatory T cell-mediated suppression in rheumatoid arthritis. Arthritis Rheum. 2007;56:732–42. doi: 10.1002/art.22414. [DOI] [PubMed] [Google Scholar]
- 35.Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFOXP3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008;112:2340–52. doi: 10.1182/blood-2008-01-133967. [DOI] [PubMed] [Google Scholar]
- 36.Radhakrishnan S, Cabrera R, Schenk EL, et al. Reprogrammed FoxP3+ T regulatory cells become IL-17+ antigen-specific autoimmune effectors in vitro and in vivo. J Immunol. 2008;181:3137–47. doi: 10.4049/jimmunol.181.5.3137. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–9. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 38.Smolewska E, Stanczyk J, Brozik H, et al. Distribution and clinical significance of blood dendritic cells in children with juvenile idiopathic arthritis. Ann Rheum Dis. 2008;67:762–8. doi: 10.1136/ard.2007.077669. [DOI] [PubMed] [Google Scholar]
- 39.Hoffmann P, Eder R, Kunz-Schughart LA, Andreesen R, Edinger M. Large-scale in vitro expansion of polyclonal human CD4(+)CD25high regulatory T cells. Blood. 2004;104:895–903. doi: 10.1182/blood-2004-01-0086. [DOI] [PubMed] [Google Scholar]
- 40.Levings MK, Sangregorio R, Roncarolo MG. Human CD25(+)CD4(+) t regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med. 2001;193:1295–302. doi: 10.1084/jem.193.11.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang Q, Henriksen KJ, Bi M, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–65. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI. Suppression of disease in New Zealand Black/New Zealand White lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. J Immunol. 2006;177:1451–9. doi: 10.4049/jimmunol.177.3.1451. [DOI] [PubMed] [Google Scholar]
- 43.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743–8. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 44.Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177:8338–47. doi: 10.4049/jimmunol.177.12.8338. [DOI] [PubMed] [Google Scholar]
- 45.Hoffmann P, Eder R, Boeld TJ, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood. 2006;108:4260–7. doi: 10.1182/blood-2006-06-027409. [DOI] [PubMed] [Google Scholar]
- 46.Tran DQ, Andersson J, Hardwick D, Bebris L, Illei GG, Shevach EM. Selective expression of latency-associated peptide (LAP) and IL-1 receptor type I/II (CD121a/CD121b) on activated human FOXP3+ regulatory T cells allows for their purification from expansion cultures. Blood. 2009;113:5125–33. doi: 10.1182/blood-2009-01-199950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural FOXP3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci USA. 2009;106:1903–8. doi: 10.1073/pnas.0811556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou X, Bailey-Bucktrout SL, Jeker LT, et al. Instability of the transcription factor FOXP3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–7. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol. 2007;37:129–38. doi: 10.1002/eji.200636435. [DOI] [PubMed] [Google Scholar]
- 50.Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–90. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chatenoud L. CD3-specific antibody-induced active tolerance: from bench to bedside. Nat Rev Immunol. 2003;3:123–32. doi: 10.1038/nri1000. [DOI] [PubMed] [Google Scholar]
- 52.Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–8. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 53.Keymeulen B, Vandemeulebroucke E, Ziegler AG, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 54.Utset TO, Auger JA, Peace D, et al. Modified anti-CD3 therapy in psoriatic arthritis: a phase I/II clinical trial. J Rheumatol. 2002;29:1907–13. [PubMed] [Google Scholar]
- 55.Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–8. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- 56.Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol. 1997;158:2947–54. [PubMed] [Google Scholar]
- 57.Delgado M, Abad C, Martinez C, Leceta J, Gomariz RP. Vasoactive intestinal peptide prevents experimental arthritis by downregulating both autoimmune and inflammatory components of the disease. Nat Med. 2001;7:563–8. doi: 10.1038/87887. [DOI] [PubMed] [Google Scholar]
- 58.Juarranz Y, Abad C, Martinez C, et al. Protective effect of vasoactive intestinal peptide on bone destruction in the collagen-induced arthritis model of rheumatoid arthritis. Arthritis Res Ther. 2005;7:R1034–45. doi: 10.1186/ar1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delgado M, Chorny A, Gonzalez-Rey E, Ganea D. Vasoactive intestinal peptide generates CD4+CD25+ regulatory T cells in vivo. J Leukoc Biol. 2005;78:1327–38. doi: 10.1189/jlb.0605299. [DOI] [PubMed] [Google Scholar]
- 60.Chorny A, Gonzalez-Rey E, Fernandez-Martin A, Pozo D, Ganea D, Delgado M. Vasoactive intestinal peptide induces regulatory dendritic cells with therapeutic effects on autoimmune disorders. Proc Natl Acad Sci USA. 2005;102:13562–7. doi: 10.1073/pnas.0504484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delgado M, Gonzalez-Rey E, Ganea D. The neuropeptide vasoactive intestinal peptide generates tolerogenic dendritic cells. J Immunol. 2005;175:7311–24. doi: 10.4049/jimmunol.175.11.7311. [DOI] [PubMed] [Google Scholar]
- 62.Gonzalez-Rey E, Chorny A, Fernandez-Martin A, Ganea D, Delgado M. Vasoactive intestinal peptide generates human tolerogenic dendritic cells that induce CD4 and CD8 regulatory T cells. Blood. 2006;107:3632–8. doi: 10.1182/blood-2005-11-4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonzalez-Rey E, Fernandez-Martin A, Chorny A, Delgado M. Vasoactive intestinal peptide induces CD4+, CD25+ T regulatory cells with therapeutic effect in collagen-induced arthritis. Arthritis Rheum. 2006;54:864–76. doi: 10.1002/art.21652. [DOI] [PubMed] [Google Scholar]
- 64.Gonzalez-Rey E, Chorny A, Varela N, O’Valle F, Delgado M. Therapeutic effect of urocortin on collagen-induced arthritis by down-regulation of inflammatory and Th1 responses and induction of regulatory T cells. Arthritis Rheum. 2007;56:531–43. doi: 10.1002/art.22394. [DOI] [PubMed] [Google Scholar]
- 65.Elias KM, Laurence A, Davidson TS, et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–20. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mucida D, Park Y, Kim G, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–60. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 67.Schambach F, Schupp M, Lazar MA, Reiner SL. Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur J Immunol. 2007;37:2396–9. doi: 10.1002/eji.200737621. [DOI] [PubMed] [Google Scholar]
- 68.Pino-Lagos K, Benson MJ, Noelle RJ. Retinoic acid in the immune system. Ann N Y Acad Sci. 2008;1143:170–87. doi: 10.1196/annals.1443.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nistala K, Wedderburn LR. Th17 and regulatory T cells: rebalancing pro- and anti-inflammatory forces in autoimmune arthritis. Rheumatology. 2009;48:602–6. doi: 10.1093/rheumatology/kep028. [DOI] [PubMed] [Google Scholar]
- 70.Van YH, Lee WH, Ortiz S, Lee MH, Qin HJ, Liu CP. All-trans retinoic acid inhibits type 1 diabetes by T regulatory (Treg)-dependent suppression of interferon-gamma-producing T-cells without affecting Th17 cells. Diabetes. 2009;58:146–55. doi: 10.2337/db08-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bai A, Lu N, Guo Y, Liu Z, Chen J, Peng Z. All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. J Leukoc Biol. 2009;86:959–69. doi: 10.1189/jlb.0109006. [DOI] [PubMed] [Google Scholar]
- 72.Nozaki Y, Yamagata T, Sugiyama M, Ikoma S, Kinoshita K, Funauchi M. Anti-inflammatory effect of all-trans-retinoic acid in inflammatory arthritis. Clin Immunol. 2006;119:272–9. doi: 10.1016/j.clim.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 73.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–74. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang J, Huizinga TW, Toes RE. De novo generation and enhanced suppression of human CD4+CD25+ regulatory T cells by retinoic acid. J Immunol. 2009;183:4119–26. doi: 10.4049/jimmunol.0901065. [DOI] [PubMed] [Google Scholar]
- 75.Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood. 2009;144:3727–35. doi: 10.1182/blood-2009-05-219584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kang SG, Lim HW, Andrisani OM, Broxmeyer HE, Kim CH. Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. J Immunol. 2007;179:3724–33. doi: 10.4049/jimmunol.179.6.3724. [DOI] [PubMed] [Google Scholar]
- 77.Li B, Samanta A, Song X, et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci USA. 2007;104:4571–6. doi: 10.1073/pnas.0700298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Loosdregt J, Vercoulen Y, Guichelaar T, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood. 2009;115:965–74. doi: 10.1182/blood-2009-02-207118. [DOI] [PubMed] [Google Scholar]
- 79.Lucas JL, Mirshahpanah P, Haas-Stapleton E, Asadullah K, Zollner TM, Numerof RP. Induction of Foxp3+ regulatory T cells with histone deacetylase inhibitors. Cell Immunol. 2009;257:97–104. doi: 10.1016/j.cellimm.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 80.Tao R, de Zoeten EF, Ozkaynak E, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 81.de Zoeten EF, Wang L, Sai H, Dillmann WH, Hancock WW. Expression of HDAC9 by T regulatory cells prevents colitis in mice. Gastroenterology. 2009;138:583–94. doi: 10.1053/j.gastro.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Reilly CM, Thomas M, Gogal R, Jr, et al. The histone deacetylase inhibitor trichostatin A upregulates regulatory T cells and modulates autoimmunity in NZB/W F1 mice. J Autoimmun. 2008;31:123–30. doi: 10.1016/j.jaut.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 83.Saouaf SJ, Li B, Zhang G, et al. Deacetylase inhibition increases regulatory T cell function and decreases incidence and severity of collagen-induced arthritis. Exp Mol Pathol. 2009;87:99–104. doi: 10.1016/j.yexmp.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Samanta A, Li B, Song X, et al. TGF-beta and IL-6 signals modulate chromatin binding and promoter occupancy by acetylated FOXP3. Proc Natl Acad Sci USA. 2008;105:14023–7. doi: 10.1073/pnas.0806726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang L, de Zoeten EF, Greene MI, Hancock WW. Immunomodulatory effects of deacetylase inhibitors: therapeutic targeting of FOXP3+ regulatory T cells. Nat Rev Drug Discov. 2009;8:969–81. doi: 10.1038/nrd3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bluestone JA. Regulatory T-cell therapy: is it ready for the clinic? Nat Rev Immunol. 2005;5:343–9. doi: 10.1038/nri1574. [DOI] [PubMed] [Google Scholar]
- 87.Zhang X, Izikson L, Liu L, Weiner HL. Activation of CD25(+)CD4(+) regulatory T cells by oral antigen administration? J Immunol. 2001;167:4245–53. doi: 10.4049/jimmunol.167.8.4245. [DOI] [PubMed] [Google Scholar]
- 88.Garcia G, Komagata Y, Slavin AJ, Maron R, Weiner HL. Suppression of collagen-induced arthritis by oral or nasal administration of type II collagen. J Autoimmun. 1999;13:315–24. doi: 10.1006/jaut.1999.0320. [DOI] [PubMed] [Google Scholar]
- 89.Nagler-Anderson C, Bober LA, Robinson ME, Siskind GW, Thorbecke GJ. Suppression of type II collagen-induced arthritis by intragastric administration of soluble type II collagen. Proc Natl Acad Sci USA. 1986;83:7443–6. doi: 10.1073/pnas.83.19.7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Staines NA, Harper N, Ward FJ, Malmstrom V, Holmdahl R, Bansal S. Mucosal tolerance and suppression of collagen-induced arthritis (CIA) induced by nasal inhalation of synthetic peptide 184–198 of bovine type II collagen (CII) expressing a dominant T cell epitope. Clin Exp Immunol. 1996;103:368–75. doi: 10.1111/j.1365-2249.1996.tb08289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Min SY, Hwang SY, Park KS, et al. Induction of IL-10-producing CD4+CD25+ T cells in animal model of collagen-induced arthritis by oral administration of type II collagen. Arthritis Res Ther. 2004;6:R213–9. doi: 10.1186/ar1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Broere F, Wieten L, Klein Koerkamp EI, et al. Oral or nasal antigen induces regulatory T cells that suppress arthritis and proliferation of arthritogenic T cells in joint draining lymph nodes. J Immunol. 2008;181:899–906. doi: 10.4049/jimmunol.181.2.899. [DOI] [PubMed] [Google Scholar]
- 93.Park MJ, Min SY, Park KS, et al. Indoleamine 2,3-dioxygenase-expressing dendritic cells are involved in the generation of CD4+CD25+ regulatory T cells in Peyer's patches in an orally tolerized, collagen-induced arthritis mouse model. Arthritis Res Ther. 2008;10:R11. doi: 10.1186/ar2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Khare SD, Krco CJ, Griffiths MM, Luthra HS, David CS. Oral administration of an immunodominant human collagen peptide modulates collagen-induced arthritis. J Immunol. 1995;155:3653–9. [PubMed] [Google Scholar]
- 95.Zhang ZY, Lee CS, Lider O, Weiner HL. Suppression of adjuvant arthritis in Lewis rats by oral administration of type II collagen. J Immunol. 1990;145:2489–93. [PubMed] [Google Scholar]
- 96.Barnett ML, Combitchi D, Trentham DE. A pilot trial of oral type II collagen in the treatment of juvenile rheumatoid arthritis. Arthritis Rheum. 1996;39:623–8. doi: 10.1002/art.1780390413. [DOI] [PubMed] [Google Scholar]
- 97.Barnett ML, Kremer JM, St Clair EW, et al. Treatment of rheumatoid arthritis with oral type II collagen. Results of a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 1998;41:290–7. doi: 10.1002/1529-0131(199802)41:2<290::AID-ART13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 98.Choy EH, Scott DL, Kingsley GH, et al. Control of rheumatoid arthritis by oral tolerance. Arthritis Rheum. 2001;44:1993–7. doi: 10.1002/1529-0131(200109)44:9<1993::AID-ART347>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 99.Myers LK, Higgins GC, Finkel TH, et al. Juvenile arthritis and autoimmunity to type II collagen. Arthritis Rheum. 2001;44:1775–81. doi: 10.1002/1529-0131(200108)44:8<1775::AID-ART313>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 100.Sieper J, Kary S, Sorensen H, et al. Oral type II collagen treatment in early rheumatoid arthritis. A double-blind, placebo-controlled, randomized trial. Arthritis Rheum. 1996;39:41–51. doi: 10.1002/art.1780390106. [DOI] [PubMed] [Google Scholar]
- 101.Trentham DE, Dynesius-Trentham RA, Orav EJ, et al. Effects of oral administration of type II collagen on rheumatoid arthritis. Science. 1993;261:1727–30. doi: 10.1126/science.8378772. [DOI] [PubMed] [Google Scholar]
- 102.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–7. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- 103.Miller A, Lider O, Weiner HL. Antigen-driven bystander suppression after oral administration of antigens. J Exp Med. 1991;174:791–8. doi: 10.1084/jem.174.4.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yoshino S, Quattrocchi E, Weiner HL. Suppression of antigen-induced arthritis in Lewis rats by oral administration of type II collagen. Arthritis Rheum. 1995;38:1092–6. doi: 10.1002/art.1780380811. [DOI] [PubMed] [Google Scholar]
- 105.van Eden W, van der ZR, Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol. 2005;5:318–30. doi: 10.1038/nri1593. [DOI] [PubMed] [Google Scholar]
- 106.Boog CJ, de Graeff-Meeder ER, Lucassen MA, et al. Two monoclonal antibodies generated against human hsp60 show reactivity with synovial membranes of patients with juvenile chronic arthritis. J Exp Med. 1992;175:1805–10. doi: 10.1084/jem.175.6.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Albani S, Keystone EC, Nelson JL, et al. Positive selection in autoimmunity: abnormal immune responses to a bacterial dnaJ antigenic determinant in patients with early rheumatoid arthritis. Nat Med. 1995;1:448–52. doi: 10.1038/nm0595-448. [DOI] [PubMed] [Google Scholar]
- 108.Cohen IR. Autoimmunity to chaperonins in the pathogenesis of arthritis and diabetes. Annu Rev Immunol. 1991;9:567–89. doi: 10.1146/annurev.iy.09.040191.003031. [DOI] [PubMed] [Google Scholar]
- 109.Munk ME, Schoel B, Modrow S, Karr RW, Young RA, Kaufmann SH. T lymphocytes from healthy individuals with specificity to self-epitopes shared by the mycobacterial and human 65-kilodalton heat shock protein. J Immunol. 1989;143:2844–9. [PubMed] [Google Scholar]
- 110.Zugel U, Kaufmann SH. Immune response against heat shock proteins in infectious diseases. Immunobiology. 1999;201:22–35. doi: 10.1016/s0171-2985(99)80044-8. [DOI] [PubMed] [Google Scholar]
- 111.de Kleer I, Kamphuis SM, Rijkers GT, et al. The spontaneous remission of juvenile idiopathic arthritis is characterized by CD30+ T cells directed to human heat-shock protein 60 capable of producing the regulatory cytokine interleukin-10. Arthritis Rheum. 2003;48:2001–10. doi: 10.1002/art.11174. [DOI] [PubMed] [Google Scholar]
- 112.Kamphuis S, Kuis W, de Jager W, et al. Tolerogenic immune responses to novel T-cell epitopes from heat-shock protein 60 in juvenile idiopathic arthritis. Lancet. 2005;366:50–6. doi: 10.1016/S0140-6736(05)66827-4. [DOI] [PubMed] [Google Scholar]
- 113.de Graeff-Meeder ER, van Eden W, Rijkers GT, et al. Juvenile chronic arthritis: T cell reactivity to human HSP60 in patients with a favorable course of arthritis. J Clin Invest. 1995;95:934–40. doi: 10.1172/JCI117801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Massa M, Passalia M, Manzoni SM, et al. Differential recognition of heat-shock protein dnaJ-derived epitopes by effector and Treg cells leads to modulation of inflammation in juvenile idiopathic arthritis. Arthritis Rheum. 2007;56:1648–57. doi: 10.1002/art.22567. [DOI] [PubMed] [Google Scholar]
- 115.Cobelens PM, Heijnen CJ, Nieuwenhuis EE, et al. Treatment of adjuvant-induced arthritis by oral administration of mycobacterial Hsp65 during disease. Arthritis Rheum. 2000;43:2694–702. doi: 10.1002/1529-0131(200012)43:12<2694::AID-ANR9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 116.Haque MA, Yoshino S, Inada S, Nomaguchi H, Tokunaga O, Kohashi O. Suppression of adjuvant arthritis in rats by induction of oral tolerance to mycobacterial 65-kDa heat shock protein. Eur J Immunol. 1996;26:2650–6. doi: 10.1002/eji.1830261116. [DOI] [PubMed] [Google Scholar]
- 117.Wendling U, Paul L, van der Zee R, Prakken B, Singh M, van Eden WA. Conserved mycobacterial heat shock protein (hsp) 70 sequence prevents adjuvant arthritis upon nasal administration and induces IL-10-producing T cells that cross-react with the mammalian self-hsp70 homologue. J Immunol. 2000;164:2711–7. doi: 10.4049/jimmunol.164.5.2711. [DOI] [PubMed] [Google Scholar]
- 118.Prakken BJ, van der Zee R, Anderton SM, van Kooten PJ, Kuis W, van Eden W. Peptide-induced nasal tolerance for a mycobacterial heat shock protein 60 T cell epitope in rats suppresses both adjuvant arthritis and nonmicrobially induced experimental arthritis. Proc Natl Acad Sci USA. 1997;94:3284–9. doi: 10.1073/pnas.94.7.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rosenthal M, Bahous I, Ambrosini G. Longterm treatment of rheumatoid arthritis with OM-8980. A retrospective study. J Rheumatol. 1991;18:1790–3. [PubMed] [Google Scholar]
- 120.Vischer TL. Follow-up with OM-8980 after a double-blind study of OM-8980 and auranofin in rheumatoid arthritis. Clin Rheumatol. 1990;9:356–61. doi: 10.1007/BF02114396. [DOI] [PubMed] [Google Scholar]
- 121.Polla BS, Baladi S, Fuller K, Rook G. Presence of hsp65 in bacterial extracts (OM-89): a possible mediator of orally-induced tolerance? Experientia. 1995;51:775–9. doi: 10.1007/BF01922429. [DOI] [PubMed] [Google Scholar]
- 122.Bloemendal A, van der Zee R, Rutten VP, van Kooten PJ, Farine JC, van Eden W. Experimental immunization with anti-rheumatic bacterial extract OM-89 induces T cell responses to heat shock protein (hsp)60 and hsp70; modulation of peripheral immunological tolerance as its possible mode of action in the treatment of rheumatoid arthritis (RA)? Clin Exp Immunol. 1997;110:72–8. doi: 10.1046/j.1365-2249.1997.4841378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Prakken BJ, Samodal R, Le TD, et al. Epitope-specific immunotherapy induces immune deviation of proinflammatory T cells in rheumatoid arthritis. Proc Natl Acad Sci USA. 2004;101:4228–33. doi: 10.1073/pnas.0400061101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Koffeman EC, Genovese M, Amox D, et al. Epitope-specific immunotherapy of rheumatoid arthritis: clinical responsiveness occurs with immune deviation and relies on the expression of a cluster of molecules associated with T cell tolerance in a double-blind, placebo-controlled, pilot phase II trial. Arthritis Rheum. 2009;60:3207–16. doi: 10.1002/art.24916. [DOI] [PubMed] [Google Scholar]
- 125.Nadkarni S, Mauri C, Ehrenstein MR. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med. 2007;204:33–9. doi: 10.1084/jem.20061531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.D’Alise AM, Auyeung V, Feuerer M, et al. The defect in T-cell regulation in NOD mice is an effect on the T-cell effectors. Proc Natl Acad Sci USA. 2008;105:19857–62. doi: 10.1073/pnas.0810713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fantini MC, Rizzo A, Fina D, et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology. 2008;136:1308–16. doi: 10.1053/j.gastro.2008.12.053. [DOI] [PubMed] [Google Scholar]
- 128.Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol. 2008;181:7350–5. doi: 10.4049/jimmunol.181.10.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Venigalla RK, Tretter T, Krienke S, et al. Reduced CD4+,CD25− T cell sensitivity to the suppressive function of CD4+,CD25high,CD127low/− regulatory T cells in patients with active systemic lupus erythematosus. Arthritis Rheum. 2008;58:2120–30. doi: 10.1002/art.23556. [DOI] [PubMed] [Google Scholar]
- 130.Korn T, Reddy J, Gao W, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–31. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Banham AH. Cell-surface IL-7 receptor expression facilitates the purification of FOXP3(+) regulatory T cells. Trends Immunol. 2006;27:541–4. doi: 10.1016/j.it.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 132.Roord ST, Zonneveld-Huijssoon E, Le T, et al. Modulation of T cell function by combination of epitope specific and low dose anticytokine therapy controls autoimmune arthritis. PLoS ONE. 2006;1:e87. doi: 10.1371/journal.pone.0000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bresson D, Togher L, Rodrigo E, et al. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006;116:1371–81. doi: 10.1172/JCI27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 135.van Laar JM, Tyndall A. Adult stem cells in the treatment of autoimmune diseases. Rheumatology. 2006;45:1187–93. doi: 10.1093/rheumatology/kel158. [DOI] [PubMed] [Google Scholar]
- 136.van Wijk F, Roord ST, Vastert B, de Kleer I, Wulffraat N, Prakken BJ. Regulatory T cells in autologous stem cell transplantation for autoimmune disease. Autoimmunity. 2008;41:585–91. doi: 10.1080/08916930802200182. [DOI] [PubMed] [Google Scholar]
- 137.de Kleer I, Brinkman DM, Ferster A, et al. Autologous stem cell transplantation for refractory juvenile idiopathic arthritis: analysis of clinical effects, mortality, and transplant related morbidity. Ann Rheum Dis. 2004;63:1318–26. doi: 10.1136/ard.2003.017798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.de Kleer I, Vastert B, Klein M, et al. Autologous stem cell transplantation for autoimmunity induces immunologic self-tolerance by reprogramming autoreactive T cells and restoring the CD4+CD25+ immune regulatory network. Blood. 2006;107:1696–702. doi: 10.1182/blood-2005-07-2800. [DOI] [PubMed] [Google Scholar]
- 139.Herrmann MM, Gaertner S, Stadelmann C, et al. Tolerance induction by bone marrow transplantation in a multiple sclerosis model. Blood. 2005;106:1875–83. doi: 10.1182/blood-2004-12-4607. [DOI] [PubMed] [Google Scholar]
- 140.Roord ST, de Jager W, Boon L, et al. Autologous bone marrow transplantation in autoimmune arthritis restores immune homeostasis through CD4+CD25+Foxp3+ regulatory T cells. Blood. 2008;111:5233–41. doi: 10.1182/blood-2007-12-128488. [DOI] [PubMed] [Google Scholar]