

Abstract
We collected a massive and heterogeneous dataset of 20 255 gene expression profiles (GEPs) from a variety of human samples and experimental conditions, as well as 8895 GEPs from mouse samples. We developed a mutual information (MI) reverse-engineering approach to quantify the extent to which the mRNA levels of two genes are related to each other across the dataset. The resulting networks consist of 4 817 629 connections among 20 255 transcripts in human and 14 461 095 connections among 45 101 transcripts in mouse, with a inter-species conservation of 12%. The inferred connections were compared against known interactions to assess their biological significance. We experimentally validated a subset of not previously described protein–protein interactions. We discovered co-expressed modules within the networks, consisting of genes strongly connected to each other, which carry out specific biological functions, and tend to be in physical proximity at the chromatin level in the nucleus. We show that the network can be used to predict the biological function and subcellular localization of a protein, and to elucidate the function of a disease gene. We experimentally verified that granulin precursor (GRN) gene, whose mutations cause frontotemporal lobar degeneration, is involved in lysosome function. We have developed an online tool to explore the human and mouse gene networks.
INTRODUCTION
Tens of thousands of protein–protein, protein–DNA and protein–RNA interactions have been experimentally identified in mammalian organisms (1,2). However, they constitute only a small part of the cell regulatory network. Efforts have been made to infer transcriptional gene networks directly from gene expression profiles, using a variety of ‘reverse-engineering’ algorithms (3–9). Among the plethora of different approaches to reverse engineering, only information-theoretic approaches are applicable at the genome scale (10). In these approaches, the network among genes is reconstructed by considering pairs of genes and checking whether the two genes in each pair are significantly co-regulated across the experimental dataset by mutual information (MI), a probabilistic measure of relatedness (11). Significant co-regulations among genes are then represented as a network, by connecting two genes with an edge if their pairwise MI is significant.
Since MI measures statistical dependencies between two variables, an edge in the network implies a coordinated response between the two connected genes, but does not necessarily imply causality. Hence, a gene–gene ‘connection’ is not necessarily a direct physical interaction between the protein products of the two genes, or a transcription factor (TF)–target gene interaction, but can also imply a functional, but indirect, regulation, through one or more intermediaries.
In order to eliminate indirect interactions, the final network is usually pruned by removing edges which have a higher probability of representing indirect relationships, using a variety of techniques (4,5). The pruned network can then be used to discover TF–target-gene interactions (4,5).
Another popular way to measure relatedness between two genes is correlation that measures co-expression between two genes. A limitation of correlation is its ability to measure only linear relationships between genes (i.e. gene A increases/decreases linearly with gene B). However, it fails when relationships are more complex (saturation, hysteresys, etc.), whereas MI is not affected at all by non-linearities (12).
Reverse engineering becomes much more powerful as the number of gene expression profiles (GEPs) used to infer the network increases (10,13). However, the requirement of using homogeneous GEPs (i.e. from a specific cell type, tissue or condition) typically limits their number to the order of hundreds. There has been a multitude of approaches towards integrating heterogeneous gene expression profiles from multiple experiments (14–16). Two main strategies can be recognized: (i) a ‘pluribus unum’ approach, where the different GEPs within each experiment are processed as if they were part of a single massive experiment. The disadvantage of this approach is that normalization of large heterogeneous datasets forces expression values to be comparable across conditions even if they are not; moreover, only around half of expression datasets in public repositories contain unprocessed data (e.g. Affymetrix CEL file), which are indeed needed for normalization; (ii) a ‘divide and conquer’ approach, where each experiment is used independently to compute a measure of co-regulation among genes of interest. This measure is then averaged out across the different experiments. The disadvantages are 2-fold: a loss of information, since experiments may differ considerably in the number of expression profiles, thus leading to discard some experiments due the paucity of samples; and a loss in the precision of the computed ‘co-regulation’ measure, due to the fragmentation of the dataset.
An example of the ‘pluribus unum’ approach can be found in Ref. (14), where a collection of 5372 microarrays from different tissues and conditions was simultaneously normalized together using standard procedures. This is considered a significant achievement due to the large number of samples analysed. The results were used to relate genes to the conditions in which they are over- or under-expressed. Examples of the ‘divide and conquer’ approach, are found in Refs (15,16) where the Pearson correlation coefficient is measured independently in each experiment for each gene. In the study by Lukk et al. (15), a final list of genes co-expressed with a gene of interest is obtained via rank aggregation methods, whereas in Ref. (16) an averaged correlation value across experiments is attributed to each gene pair.
Our starting hypothesis was that, despite the extreme heterogeneity of gene expression profiles coming from different cell types, tissues and conditions, it is indeed possible to infer a meaningful ‘consensus’ gene network from tens of thousands of GEPs, which can then be used to investigate cell transcriptome organization and gene function. Towards this end, we collected a massive and heterogeneous dataset of >20 000 GEPs measured in human samples from almost 600 different experiments, as well as a similar number of GEPs in mouse samples. About a third of these, contained only normalized GEPs that cannot be directly compared across different experiments.
To infer a gene network from this massive dataset, we needed to overcome current limitations of multi-experiment intergration approaches and of MI-based reverse-engineering algorithms, which have been applied at most to hundreds of GEPs from the same tissue, or cell type, and which require normalized GEPs across the whole dataset. We, therefore, developed and applied a simple but powerful approach to recover a comprehensive gene network among most of the known genes from this massive dataset. We decided not to apply a ‘network pruning’ step, since we wanted to keep as many meaningful gene–gene connections as possible, even if these are not direct TF–target gene interactions. Indeed, we show that the resulting network is a powerful resource that can be used to discover new protein–protein interactions, to gain insight into the cell transcriptome organization, and to make hypothesis on the biological function of a gene.
We have developed an online tool (http://netview.tigem.it) for querying and exploring the gene network, for both human and mouse species.
MATERIALS AND METHODS
Gene expression profile dataset, gene network inference and computation of pairwise MI
We collected, from ArrayExpress (17), 20 255 GEPs (591 experiments) in human samples measured with the Affymetrix HG-U133A microarray, and 8895 GEPs (614 experiments) in mouse samples, with the Affymetrix Mouse430A_2 (Supplementary Table S1). Normalized GEPs within each experiment were retrieved from the database. The two mammalian dataset were analysed separately. The expression values of all GEPs within an experiment were discretized into a predetermined number n of bins (n = 3), with equal number of values in each bin. The bins are determined using the n quantiles of the normalized expression values as cut points. Each expression value is then replaced by an integer value corresponding to the bin it falls into (see also Supplementary Data).
In this way, we could use all the available experiments even when unprocessed data were not available in the public repository (∼30% of the experiments have no unprocessed data). Moreover, this simple technique works even when different normalization algorithms are used. The discretized values of gene expression are used to compute a ‘co-occurency’ matrix between each pair of probe-sets in each experiment, which is then used to estimate the joint probability distribution across the experiments.
Specifically, we considered two discrete random variables I and J assuming values in the set {1, 2, 3} describing the discretized expression values of two probe-sets I and J. In this context, MI can be defined as:
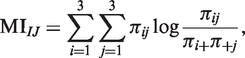 |
(1) |
where πij represents the joint probability P(I = i, J = j) with (i, j) ∈ {1, 2, 3} × {1, 2, 3} and πi+ = ∑jπij and π+j = ∑iπij are, respectively, their marginal probabilities P(I = i) and P(J = j).
In order to estimate the joint probability πij, we used a simple frequentist approach: let 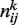 be the counts of the outcomes (I = i, J = j) across the nk GEPs of experiment k, then the frequency
be the counts of the outcomes (I = i, J = j) across the nk GEPs of experiment k, then the frequency 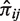 can be estimated jointly from the all the K experiments:
can be estimated jointly from the all the K experiments:
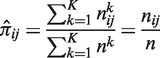 |
(2) |
This leads to a point estimate of MI equal to
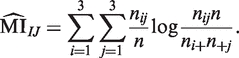 |
(3) |
The computational complexity of our algorithm is o(N2·n), where N is the number of probe-sets and n is the total number of GEPs.
The choice of a different number of discretization bins does not considerably affects the results (Supplementary Data and Supplementary Figure S4). The MI significant threshold was obtained by fitting a Gamma distribution to the values of the MI across all the probes' pairs (18) and selecting only MI with a P < 0.01.
Yeast two hybrid assays
The Yeast two Hybrid (Y2H) kit ‘ProQuest Two-Hybrid System’ (Invitrogen) included the Saccharomyces cerevisiae MAV 203 strain (MATα, leu2-3,112, trp1-901, his3Δ200, ade2-101, gal4Δ, gal80Δ, SPAL10::URA3, GAL1::lacZ, HIS3UAS GAL1::HIS3@LYS2, can1R, cyh2R), the bait vector pDEST32 and the prey vector pDEST22. The ‘Ultimate ORF’ (Invitrogen) of the genes of interest were used to generate prey and bait plasmids using the GateWay technology and protein–protein interaction assays were performed according to the manufacturer the instructions, along with the appropriate positive and negative control (Supplementary Table S5 and Supplementary Data).
Cell culture and transfection
For GRN functional assays, HeLa cells were cultured in DMEM supplemented with 10% FBS and treated for 96 h in the presence of sucrose to a final concentration of 100 mM with daily changes of medium. Cells were collected and analysed by immunofluorescence and RealTime PCR (Supplementary Data). For TFEB or GRN over-expression 500 000 HeLa cells were transfected with 4 μg of DNA expressing the human TFEB or GRN using lipofectamine transfection reagent (Invitrogen), and collected after 48 h (Supplementary Data).
Electron microscopy
GRN or EGFP over-expressing HeLa cells were fixed in 1% glutaraldehyde in 0.2 M HEPES buffer and further incubated in uranyl acetate and in OsO4. After dehydration through a graded series of ethanol, the cells were cleared in propylene oxide, embedded in the Epoxy medium (Epon 812) and polymerized at 60°C for 72 h. From each sample, thin sections were cut with a Leica EM UC6 ultramicrotome. Electron microscopic images were acquired from thin sections using an FEI Tecnai-12 electron microscope equipped with an ULTRA VIEW CCD digital camera (FEI, Einhoven, The Netherlands). Quantification of the lysosome-like organelle dimensions was performed using the AnalySIS software (Soft Imaging Systems GmbH, Munster, Germany).
RESULTS
We collected 20255 GEPs from 591 different experiments (Supplementary Table S1) performed in a variety of human tissues, cell types and conditions from public microarray repositories. A total of 22 283 different transcripts were measured, corresponding to the probe sets in the Affymetrix HG-U133A microarray. Our aim was to reverse engineer a consensus transcriptional gene network among the 22 283 transcripts from these multiple experiments. However, only ∼70% of these experiments contained unprocessed data (CEL file), whereas the remaining 30% contained just normalized GEPs.
We could not apply ‘off-the-shelf’ (Supplementary Data) state-of-the-art approaches to reverse engineering (5,19), because gene expression profiles are not comparable to each other across experiments in different tissues, cell types or conditions. For example, one of the most successful reverse-engineering algorithms based on MI [ARACNE (5)] is limited by a considerable loss of precision, if the ‘divide and conquer’ approach to multiple experiments is used (Supplementary Data).
To overcome these problems, we developed a simple MI reverse-engineering approach, which works in two steps: (i) by discretizing normalized gene expression values within each experiment and computing for each pair of genes a ‘co-occurrency’ matrix, which summarizes the coherence in gene expression changes across the samples in the same experiment; (ii) by estimating MI from the whole dataset at once, summing up the co-occurrency matrices across all the experiments, thus avoiding loss in precision due to dataset fragmentation (Supplementary Data). We then generated a network by connecting two genes in the network, if the associated MI was greater than a significance threshold (P < 0.01) obtained by fitting a Gamma distribution to the MI values, which has been shown to be a good approximation under the null hypothesis of statistical independence (18).
Our approach can in principle be applied also to GEPs obtained from RNA-seq technology, but at the moment their number is much smaller than the GEPs obtained from microarrays.
The inferred network consists of 4 817 629 connections among 22 283 transcripts (probe-set on the microarray). To our knowledge, this is the largest and most comprehensive dataset ever used to reverse engineer a transcriptional gene network.
Connections in the gene network are biologically relevant
To validate at least a subset of predictions, we generated a reference human interactome, Golden Standard (GoS), consisting of 105 588 experimentally verified interactions including protein–protein (2), TF–target gene and metabolic interactions (1).
Since the GoS interactome is at the gene/protein level, whereas the network we inferred is at the ‘probe-set’ level, we first needed to transform probe-sets to the corresponding genes.
In order to reduce as much as possible cross-hybridization issues known to affect microarray technology (20), we first removed 18.8% (4187) of the probe-sets that could not be reliably mapped to the coding genome, or that mapped to multiple genes (20).
After this step, only probes associated to a single gene were retained. However, some genes can be associated to more than one probe-set. We verified that 35% (4200/11978) of the genes represented on the microarray are indeed associated to multiple probe-sets (Supplementary Table S2). We checked for the consistency between probe-sets associated to the same gene by verifying that these probe-sets were indeed connected in the probe-set level network. We found that 69% (2884/4200) of the genes mapped by multiple probes are consistent (Supplementary Table S3). Out of the remaining 31% of the genes, 14% (596/4200) are associated to probe-sets that target alternative transcripts of the same gene, which may not be well co-regulated, and hence were retained in the analyis. The remaining 1243 of the probe-sets corresponding to 540 genes were removed from further analysis.
We then generated a gene-level network from the probe-set level network by connecting two genes with an edge, if the corresponding probe-sets were connected in the probe-set level network. When one, or both genes were associated to more than one probe-set, we required that at least one probe-set pair was connected in the probe-set level network, and assigned as the MI value of the gene pair, the maximum MI among the corresponding probe-set pairs (21).
Supplementary Figure S1a shows the percentage of inferred connections that were confirmed by the GoS interactome. The network reaches a maximum of 90% of correct predictions, with an average precision of 32%. We estimated the percentage of correct connections, had these been randomly guessed, to be equal to 0.0028%, as described in Ref. (10). The GoS interactome includes only a subset of the interactions occurring in a cell, because only a small subset of these have been experimentally verified. Moreover, a high MI does not necessarily imply physical interactions. Nevertheless, the GoS interactome provides evidence of the biological significance of the inferred connections.
For comparison, Supplementary Figure S1a also shows the percentage of correctly predicted connections by CoexpresDB (16), a database of human co-expressed genes measured using a classic Pearson Correlation Coefficient (PCC) from multiple experiments using the ‘divide and conquer’ strategy.
Connections tend to be conserved across species
To understand whether, and to what extent, connections among genes are conserved across human and mouse species, we collected 8895 gene expression profiles from 614 experiments in mouse, each measuring 45 101 transcripts, corresponding to the probe-sets in the Affymetrix Mouse430A_2 microarray. We then applied our approach to this massive dataset to reconstruct the network. Out of all the possible 1 017 027 550 gene–gene connections, only 14 461 095 connections among 45 101 transcripts were deemed significant (P < 0.01). The difference in the number of inferred connections between the human and mouse network is due to the different number of probe sets between the two microarray models analysed.
In order to compare the two networks, we first removed from the human network those genes without an ortholog in mouse, resulting in a ‘reduced’ network of 11 318 genes. We then found that 218 700 connections (12%) were conserved in mouse (Supplementary Figure S1b). This percentage is in line with previous studies involving a limited number of known protein–protein or protein–DNA interactions. In yeast, it has been reported that between 10% (22) and 30% (23) of protein–protein interactions occurring during the cell cycle of S. pombe (fission yeast) and S. cerevisiae (budding yeast) are conserved; another cross-species (fly and yeast) protein interaction study (24) resulted in a ratio of conservation ranging from 6% to 15%; in Ref. (25), the authors report a database of protein–protein interactions occurring among transcription factors in human and mouse, where the percentage of effective conserved interactions is ∼16% (the estimated range is 34–64% when taking into account the false positive rate of the experimental technique). A recent genome-wide analysis (26) integrating heterogeneous sets of experimental data (including 338 expression profiles in human and 1048 in mouse) showed a conservation of 15% of interactions between the two mammalian species.
Prediction of new protein–protein interactions
We investigated the identity of the top one thousand connections with the highest MI in the network (Figure 1, Supplementary Figure S2, and Supplementary Table S4). Forty per cent of these connections, involving a total of 302 genes, were confirmed by the GoS interactome. An additional 13% of the connections were predicted among genes in the same gene-family, which, therefore, may well be functionally related, although not physically interacting.
Figure 1.

Subnetworks obtained by collecting the top 1000 connections with the highest MI within the network. Subnetwork (a) contains genes that codify for the ‘ribosomal protein complex’, (‘Translation’, P < 1.0−113). Subnetwork (b) is enriched for genes involved in the ‘spindle checkpoint’ (‘Nuclear division’ P < 3.1−7), for clarity only a subset of interactions are shown. Subnetwork (c) is enriched for ‘metallothionein’ genes, a family of low molecular weight, heavy metal binding proteins. Subnetwork (d) contains major histocompatibility complex proteins (Antigen processing and presentation, P < 7.5−16). Pairs of genes are connected if their MI (probabilistic measure of relatedness) confirms a significant co-regulation.
In order to test the predictive ability of the network, we focused on the subnetwork (b) in Figure 1 consisting of 12 genes most of which (CENPF, NUSAP1, KIF2C, BUB1B, ASPM, ZWINT and CCNB2) involved in mitotic spindle checkpoint, chromosome motility and mitotic progression (27–30).
According to the GoS interactome, three protein–protein interactions were known to occur among the genes in subnetwork (b), therefore we decided to verify whether the predicted connections, could be yet undiscovered protein–protein interactions. We selected only the subset of seven genes (NUSAP1, KIF2C, BUB1B, ASPM, ZWINT, KIAA0101 and RRM2), which according to our network formed a tight cluster of genes all connected to each other. We performed a series of yeast two hybrid (Y2H) assays to test a total of 21 connections (i.e. all the possible interactions among seven proteins). According to the Y2H assay, 20 of these were positive. Since Y2H are known to be prone to false positive detections, we also experimentally estimated the precision (true positives over true positives plus false positives) of the Y2H assay by using appropriate positive and negative controls (Supplementary Data). The estimated precision resulted to be equal to 77%, hence, at least 15 (=77% of 20) of the experimentally identified interactions should be true positive predictions. This means that we can predict new protein–protein interactions with a precision of 75% (15 out of 21, Supplementary Table S5).
The modular structure of the network
The structure of the network has a typical exponential degree distribution (31,32) consisting of a large number of genes with very few connections, and very few genes with a large number of connections, termed hubs. We observed that, as the number of connections of a gene increases, so does its average expression level (Supplementary Figure S3a,b and Supplementary Data); in contrast, the intrinsic protein disorder (33) of its protein product significantly decreases (P = 0.009) (Supplementary Figure S3c,d and Supplementary Data). Protein disorder, defined as the length of the unstructured part of a protein, is an important determinant of gene dosage sensitivity (34).
A cell is able to regulate its complex behaviour thanks to sets of genes that perform different but coordinated functions. We asked whether we could find such functional modules within the network, which could reveal how the cell transcriptome is organized. We searched the network for modules, which are defined as ‘communities’ and ‘rich-clubs’ in network theory. A community is a group of genes highly inter-connected to each other, but with few connections to genes outside the group. A ‘rich-club’ can be defined as a ‘community of communities’, i.e. a group of closely inter-connected communities.
In order to identify communities, we represented the network as a matrix, as shown in Figure 2. The matrix was obtained by defining each entry mij as the value of the MI between gene i (in the i-th row) and gene j (in the j-th column). We observed that genes lying within the same chromosome (squares in Figure 2a) tend to be connected to each other more often than what would be expected by chance. We then applied a hierarchical clustering algorithm to the matrix representing the network, and thus identified 393 communities with more than 4 genes (Supplementary Table S6 and Supplementary Data).
Figure 2.

Modular structure of the network. Adjacency matrix of the network before (a) and after (b) the hierarchical clustering procedure used to identify communities. Each dot represents a connection among two genes, that is a matrix entry, whose MI is greater than the significance threshold. (a) Genes are sorted according to their chromosomal location. Numbers on the x and y axes indicate chromosomes. (b) Genes are sorted according the community they belong to. Square dimensions are proportional to the number of genes in each community. The inset shows an enlargement of an area of the adjacency matrix where single dots are visible.
Figure 2 shows the matrix representing the network before (a) and after (b) the hierarchical clustering procedure. Communities appear as dark squares, with genes belonging to the same community grouped together, giving a striking check-board pattern.
We assessed that 36% of these communities are enriched for a specific biological function by Gene Ontology analysis (Supplementary Table S7). This percentage increases up to 47%, when considering only communities composed by >10 genes. We also found that 6 out of 393 communities of the network are significantly enriched for disease genes (P < 0.05, Gene Set Enrichment Analysis, in Supplementary Data): community number 1, 11, 22, 40, 54 and 96. The most significant community, number 40, is composed by genes whose protein products localize to the ‘lysosome’, and is highly enriched for disease-genes involved in lysosomal storage disorders. Other examples include community 11, whose genes are related to cell adhesion and extracellular matrix organization, and include disease-genes causing developmental or cardiovascular defects; community 22, related to the immune system and including genes causing related disorders; community 54, composed by genes involved in oxygen transport and enriched for genes involved in haematological disorders. These ‘disease communities’ could contain other yet unknown disease-related genes, and could be helpful in identifying candidate genes in disease-related loci.
We observed that communities interact with each other; for example community 1, enriched for transmembrane receptor activity (P = 2.01 × 10−35) interacts with community 3, enriched for ‘extracellular region’ (P = 7.33 × 10−06, Figure 2b, arrow), but not to community 2 involved in ‘RNA processing’.
In order to better elucidate community function and interactions among them, we defined the interaction strength (IS) between two communities as the number of connections occurring among genes belonging to the two different communities, divided by the expected number of connections. The IS is equal to 0 if no connections exist among genes belonging to the two different communities. We computed the IS between all the pairs of 393 communities for a total of 77 028. Only 5074 pairs of communities had an IS >0. Similarly to the gene-wise network, also the community-wise network can be represented as a matrix. We can, therefore, apply a clustering procedure to group the communities into sets of highly interconnected communities (‘rich-clubs’) (35) (Supplementary Data).
We thus obtained 58 rich-clubs. Each rich-club has a representative community termed ‘exemplar’, to which all of the other communities in the rich-club are connected. The community-wise network is shown in Figure 3.
Figure 3.

Community-wise network. Each node is a community. A color and a number identify each rich-club (i.e. a group of highly interconnected communities). The width of each edge reflects the IS between communities. ‘Exemplars’ are indicated by triangles. Examples of rich clubs: (a) communities of genes involved in ‘intracellular trafficking’; (b) communities involved in the ‘extracellular matrix maintenance, and cell mobility’; (c) communities involved in ‘immune response’; (d) communities of genes involved in house-keeping functions: ‘gene expression’ (rich-club 197), ‘translation’ (rich-club 246), ‘RNA processing’ (rich-club 2) and ‘cell cycle’ (rich-club 7).
Communities in our network can be considered as functional modules consisting of genes whose expression is coordinated, and that carry out specific biological functions. We asked whether these communities could have a ‘physical counterpart’ in the cell. We investigated whether genes that are connected to each other according to our network, were also physically close to each other at the chromatin level, in the cell nucleus. We used a recent comprehensive 3D topological mapping of chromosomal loci physical interactions using an innovative ‘Hi-C’ chromatin capture technology (36).
This map of physical interactions can be represented as a matrix (Mp), where each entry mij reports the probability of the i-th Mb of the genome to be in physical contact with the j-th Mb, according to ‘Hi-C’ experimental results. Figure 4b is a graphical representation of this map for chromosome 19.
Figure 4.

Genes that are co-regulated tend to be physically close at the 3D chromatin level. (a) Connection tendency matrix of chromosome 19. Grey stripes highlight regions with no probes designed for the microarray model HG-U133A. A red color indicates two different 1 Mb loci whose genes are strongly connected to each other. (b) Physical contact matrix of chromosome 19. Grey stripes highlights chromosomal regions where centromeres are located, plus unalignable regions. A red color indicates two different 1 Mb loci that are physically close to each at the chromatin level. Physically close regions may also contain genes that are not co-expressed and vice versa: region (I) in (a) has an opposite tendency with respect to the corresponding region (I) in (b). This means that regions that are not in physical contact may contain genes that are co-expressed. The opposite can also be true, for example region (II) shows that loci physically interacting with each other do not necessarily contain genes that are co-expressed.
In order to compare our network with the physical contact probability map, we generated a ‘connection tendency matrix’ (Mc) at 1 Mb resolution from the network. Each entry mij of this matrix is simply the number of connections occurring among genes found within the i-th and j-th Mb of the genome, divided by the expected number of connections. Figure 4a is a graphical representation of the network at 1 Mb resolution, where red color represents two chromosomal regions which contain more connections than expected among the genes they harbour.
In both matrices (Figure 4a and b), there is a clear ‘plaid pattern’ highlighting chromosomal regions whose genes are strongly connected to each other (red in Figure 4a) and regions that are physically close to each other at the chromatin level (red in Figure 4b). These regions have a striking overlap (correlation=0.4, P = 7.3 × 10−123) especially the p-arm of chromosome 19 (upper left square in both matrices), revealing that genes that are physically close to each other at the chromatin level tend to be ‘co-regulated’ (i.e. have a significant MI) and vice versa.
By extending this analysis to all the chromosomes, we found a significant overlap (correlation significance: P < 0.01) for all but three chromosomes (9, 20 and 21). The significance is also present at the whole genome level, when considering also inter-chromosomal interactions.
Elucidating gene function
We exploited the information embedded in the network to identify gene function, or protein subcellular localization, via a guilty-by-association analysis. It consists in assigning a function to a gene (or a localization to the encoded protein) by checking whether there is a shared function among the genes connected to it (or a shared localization of their protein products). In what follows, we termed ‘gene neighbours’ the set of genes connected to a gene of interest.
We performed a Gene Ontology Enrichment Analysis (GOEA) on the set of gene neighbours for each gene (http://netview.tigem.it). We then selected, as a test set, 18 141 transcripts for which the function/localization was known according to their Gene Ontology classification. The percentage of correct predictions for each of the three GO classes (Biological Process, Molecular Function and Cellular Localization) ranged from 59% to 71%, respectively.
We next asked whether the guilty-by-association approach could be helpful in generating hypotheses on the function of genes involved in genetic diseases. We used our approach to identify human disease genes that may have a yet undiscovered role in lysosome function and organization.
To this end, we ranked, by their P-value from the GOEA, all the genes predicted to be lysosomal, or associated with lysosome organization, by our guilty-by-association analysis (http://netview.tigem.it and Supplementary Table S8). The top ranked genes included both lysosomal enzymes and other genes involved in lysosomal function. Among these, at position one, NPC2 (Niemann-Pick disease, type C2) disease-gene, is known to be an intralysosomal gene (37); at position two, we found another disease-gene, GRN. Both genes are also members of community 40, which is enriched both in disease genes and in lysosomal genes.
Despite extensive studies, the role of GRN is far from being understood and it has not been directly linked to lysosomal function. GRN is a highly conserved gene bearing multiple copies of the cysteine-rich granulin motifs. Proteolytic cleavage of the precursor protein by extracellular proteases, gives rise to smaller peptide fragments termed granulins which have been linked to a range of biological functions including ‘cell division’, ‘survival’, and ‘migration’ (38). Mutations in GRN cause frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions (FTLD-U) (39,40). Of note, mutations in NPC2 results in a wide spectrum of clinical phenotypes including a form of frontal lobe atrophy (41).
To investigate whether GRN was indeed related to lysosomal function, we first evaluated GRN expression levels following sucrose treatment, a known inducer of lysosomal biogenesis (42,43). Following sucrose treatment, we observed a 2-fold increase over baseline in GRN mRNA levels, along with a 3-fold increase in Cathepsin D (CTSD), a lysosomal enzyme used as positive control (Figure 5a). We also found an increased GRN immunostaining using a specific anti-GRN antibody in sucrose-treated cells (data not shown).
Figure 5.

GRN is involved in lysosomal function. (a) GRN and CTSD increase in expression level in HeLa following sucrose treatment, a known inducer of lysosomes, as measured by realtime qPCR. (b) Expression level of GRN and of CTSD increase in HeLa cells following TFEB over-expression, a transcription factor known to regulate lysosome biogenesis. (c) Immunofluorescence with antibody anti-LAMP1 and anti-LAMP2, used as a lysosomal marker, of transfected HeLa cells over-expressing GRN, or EGFP and in HeLa cells grown in medium collected after GRN, or EGFP over-expression.
The transcription factor EB (TFEB) has been recently identified as the transcription factor controlling most of the known lysosomal genes via direct binding to their proximal promoter (44), and therefore we next asked whether GRN is regulated by this transcription factor. We first identified, by bioinformatics analysis, two TFEB binding sites upstream of the GRN coding sequence (Supplementary Data). We then over-expressed TFEB in human cell lines and detected a 3-fold increase in GRN mRNA levels, along with a 3-fold increase in CTSD, a known target of TFEB, used as a positive control (Figure 5b).
We next over-expressed GRN in HeLa cells to investigate if this had any effect on lysosomes; as shown in Figure 5c, the LAMP1 and LAMP2 signal significantly increased when compared to a mock control, or over-expression of EGFP, as assessed by immunofluorescence (Supplementary Data). Electron microscopy in HeLa overexpressing GRN showed that the increase in LAMP1 and LAMP2 is likely due to an increase in the size of lysosomes but not in their number (Figure 6).
Figure 6.

Electron microscopy of HeLa cells overexpressing GRN reveals increase in lysosomes size. Electron microscopy of Hela cells after GRN, or EGFP over-expression. Lysosomes (indicated in EM images by arrows) appear to be larger in cells that overexpress GRN. Morphometric analysis of lysosome diameter (meanSD; n = 0 cells) confirms the increase in the lysosome size in GRN-expressing cells.
The effect of GRN on lysosomes was observed on the great majority of cells, despite transfection efficiency being not as extensive. Therefore, we hypothesized that GRN-transfected cells could secrete a factor inducing lysosome biogenesis in neighbouring cells. To test this hypothesis, we collected the medium from HeLa transfected cells over-expressing GRN and confirmed GRN protein expression in the medium by western blot analysis (data now shown). We then used this medium to grow untransfected wild-type cells. This resulted in a consistent increase (P < 0.003) in LAMP1 and LAMP2 signal compared to control as shown in Figure 5c (medium).
DISCUSSION
We inferred a network of co-regulated genes from a massive gene expression dataset in both human and mouse species. We showed that, when properly handled, this massive dataset, yields a powerful resource to identify both functional and physical interactions among genes, and to make hypotheses on transcriptome organization and gene function.
A further advantage of our resource is that it can automatically integrate gene expression measurements performed with different technologies, such as microarrays and RNA-seq from next-generation sequencing, since normalization is required only within each experiments and not across experiments.
Genes that are co-regulated in human tend to be co-regulated in mouse more than what is expected by chance; however, this is not the general trend, since only 12% of connections are conserved. Our observation adds weight to the hypothesis that regulation of gene expression may be different between species, even if they share a similar proteome.
The gene network structure is typical of complex networks with the presence of hub genes with a large number of connections. Our findings suggest that hub genes are highly expressed and may have been selected to be less dosage sensitive, i.e. not pathological when their expression is increased, as confirmed by their tendency to have a lower protein disorder.
We identified biologically relevant functional modules within the network, thus providing a modular view of the wiring diagram of a cell. Genes connected within functional modules in the network tend to have a ‘physical counterpart’ in the 3D conformation of the chromatin. We observed a striking similarity between genes that appear to be connected, and, therefore, are co-regulated, and their physical proximity at the 3D chromatin level. This suggests that regulation of coordinated gene expression is ‘hard-wired’ in the physical arrangement of the chromatin within the nucleus.
We have shown how the network can be used to identify new protein–protein interactions and to investigate the function of a disease-gene.
Upregulation of GRN by known inducers of lysosomal biogenesis and function, together with the increase in the LAMP1 and LAMP2 signal following GRN over-expression, or treatment with medium from GRN over-expressing cells, clearly supports a role of GRN in lysosome biology. This finding is also supported by previous evidence indicating that GRN colocalizes with lysosome-associated CD68 antigen in activated macrophages and microglia (45) and is overexpressed in the cerebral cortex of MPSIIIB and MPSI mice (46). Although GRN may bind the mannose 6-phosphate receptor (47,48), more recently it was demonstrated that GRN is endocytosed by sortilin and rapidly delivered to lysosomes (49). In addition, it has also been shown that inhibition of vacuolar ATPase increases intracellular and secreted GRN, which again support a lysosome involvement (50).
The approach we developed has some intrinsic limitations, since the MI measure does not allow to recover causal relationships between genes, hence we cannot distinguish direct from indirect regulation. To overcome this limitation, it should be possible to extend the approach to infer n-way dependencies, by computing for example conditional MI, in order to eliminate indirect interations. Another limitation is due to the fact that we used a variety of tissue and cell types in order to increase the statistical power of the inference method; however in so doing, we lost the capability of identifying tissue-specific (or cell-type specific) co-regulation among genes. Such information could be retrieved by first dividing the expression dataset according to the sample of origin, and then applying a Bayesian framework to determine the probability of each connection of occuring in a specific tissue or cell type.
We have made our resource publicly available as an online tool (http://netview.tigem.it). The gene network can be easily searched for a gene of interest, queried with a Gene Ontology term to detect all the genes with that predicted function, or searched with a list of genes to identify one or more common regulators, i.e. genes that are significantly co-regulated with most of the genes in the query list.
Additional studies can be easily conducted using our online resource. For example, we predicted for each gene its function. Specifically, it is possible to search for a function of interest, or a cellular compartment and identify the TFs predicted to be involved in that function, and thus possibly acting as master regulators. This analysis could help in hyoptheses generation, which should then be followed by ad hoc experimental investigation.
We believe our resource will provide a valuable tool to the research community.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Fondazione Telethon (Italy) (TCBP37TELC to N.B.-P., TDDP51TELC to D.d.B.); Italian Ministry of Research (MIUR-ITALBIONET to D.d.B.); Italian Association for Cancer Research (AIRC, Milan, Italy), EC (FP7 LipidomicNet) to N.B.-P. for financial support. Funding for open access charge: Fondazione Telethon (Italy).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
Authors are grateful to A. Carissimo for image quantification analysis, to M. Sardiello for TFEB binding site identification, and to G. Diez-Roux for critical reading the manuscript, to E. Polishchuk and A. Egorova for EM specimen preparation and morphometric analysis, to the Telethon Electron Microscopy Core Facility (TeEMCoF, IBP, CNR, Naples; Telethon project #GTF08001) and Integrated Microscopy Facility (IGB, CNR, Naples) for EM support.
REFERENCES
- 1.Joshi-Tope G, Gillespie M, Vastrik I, D'Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L, et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33 doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu X, Jiang R, Zhang MQ, Li S. Network-based global inference of human disease genes. Mol. Syst. Biol. 2008;4 doi: 10.1038/msb.2008.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2009;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nat. Genet. 2005;37:382–390. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- 6.Bansal M, Gatta GD, di Bernardo D. Inference of gene regulatory networks and compound mode of action from time course gene expression profiles. Bioinformatics. 2006;22:815–822. doi: 10.1093/bioinformatics/btl003. [DOI] [PubMed] [Google Scholar]
- 7.Della Gatta G, Bansal M, Ambesi-Impiombato A, Antonini D, Missero C, di Bernardo D. Direct targets of the TRP63 transcription factor revealed by a combination of gene expression profiling and reverse engineering. Genome Res. 2008;6:939–948. doi: 10.1101/gr.073601.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner T, di Bernardo D, Lorenz D, Collins J. Inferring genetic networks and identifying compound mode of action via expression profiling. Science. 2003;301:102–105. doi: 10.1126/science.1081900. [DOI] [PubMed] [Google Scholar]
- 9.di Bernardo D, Thompson M, Gardner T, Chobot S, Eastwood E, Wojtovich A, Elliott S, Schaus S, Collins J. Chemogenomic profiling on a genome-wide scale using reverse-engineered gene networks. Nat. Biotechnol. 2005;23:377–383. doi: 10.1038/nbt1075. [DOI] [PubMed] [Google Scholar]
- 10.Bansal M, Belcastro V, Ambesi-Impiombato A, di Bernardo D. How to infer gene networks from expression profiles. Mol. Syst. Biol. 2007;3 doi: 10.1038/msb4100120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butte AJ, Kohane IS. Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. Pac. Symp. Biocomput. 2000:418–429. doi: 10.1142/9789814447331_0040. [DOI] [PubMed] [Google Scholar]
- 12.Cover TM, Thomas JA. Elements of Information Theory. New York: Wiley-Interscience; 1991. [Google Scholar]
- 13.Marbach D, Prill RJ, Schaffter T, Mattiussi C, Floreano D, Stolovitzky G. Revealing strengths and weaknesses of methods for gene network inference. Proc. Natl Acad. Sci. USA. 2010;107:6286–6291. doi: 10.1073/pnas.0913357107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adler P, Kolde R, Kull M, Tkachenko A, Peterson H, Reimand J, Vilo J. Mining for coexpression across hundreds of datasets using novel rank aggregation and visualization methods. Genome Biol. 2009;10:R139–R150. doi: 10.1186/gb-2009-10-12-r139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lukk M, Kapushesky M, Nikkila J, Parkinson H, Goncalves A, Huber W, Ukkonen E, Brazma A. A global map of human gene expression. Nat. Biotechnol. 2010;28:322–324. doi: 10.1038/nbt0410-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Obayashi T, Hayashi S, Shibaoka M, Saeki M, Ohta H, Kinoshita K. COXPRESdb: a database of coexpressed gene networks in mammals. Nucleic Acids Res. 2008;36 doi: 10.1093/nar/gkm840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parkinson H, Kapushesky M, Shojatalab M, Abeygunawardena N, Coulson R, Farne A, Holloway E, Kolesnykov N, Lilja P, Lukk M, et al. ArrayExpress–a public database of microarray experiments and gene expression profiles. Nucleic Acids Res. 2007;35 doi: 10.1093/nar/gkl995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goebel B, Dawy Z, Hagenauer J, Mueller J. 2005. An approximation to the distribution of finite sample size mutual information estimates. In IEEE International Conference on Communications, Vol. 2, pp. 1102–1106. [Google Scholar]
- 19.Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8–e20. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ballester B, Johnson N, Proctor G, Flicek P. Consistent annotation of gene expression arrays. BMC genomics. 2010;11:294–308. doi: 10.1186/1471-2164-11-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu H, Wang F, Tu K, Xie L, Li Y-Y, Li Y-X. Transcript-level annotation of Affymetrix probesets improves the interpretation of gene expression data. BMC Bioinformatics. 2007;8:194–208. doi: 10.1186/1471-2105-8-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rustici G, Mata J, Kivinen K, Lio P, Penkett C, Burns G, Hayles J, Brazma A, Nurse P, Bahler J. Periodic gene expression program of the fission yeast cell cycle. Nat. Genet. 2004;36:809–817. doi: 10.1038/ng1377. [DOI] [PubMed] [Google Scholar]
- 23.Oliva A, Rosebrock A, Ferrezuelo F, Pyne S, Chen H, Skiena S, Futcher B, Leatherwood J. The cell cycle-regulated genes of Schizosaccharomyces pombe. PLoS Biol. 2005;3 doi: 10.1371/journal.pbio.0030225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bandyopadhyay S, Sharan R, Ideker T. Systematic identification of functional orthologs based on protein network comparison. Genome Res. 2006;16:428–435. doi: 10.1101/gr.4526006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, Tan K, Akalin A, Schmeier S, Kanamori-Katayama M, Bertin N, et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell. 2010;140:744–752. doi: 10.1016/j.cell.2010.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexeyenko A, Sonnhammer EL. Global networks of functional coupling in eukaryotes from comprehensive data integration. Genome Res. 2009;19:1107–1116. doi: 10.1101/gr.087528.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson VL, Scott MIF, Holt SV, Hussein D, Taylor SS. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J. Cell Sci. 2004;117(Pt 8):1577–1589. doi: 10.1242/jcs.01006. [DOI] [PubMed] [Google Scholar]
- 28.Manning AL, Ganem NJ, Bakhoum SF, Wagenbach M, Wordeman L, Compton DA. The kinesin-13 proteins Kif2a, Kif2b, and Kif2c/MCAK have distinct roles during mitosis in human cells. Mol. Biol. Cell. 2007;18:2970–2979. doi: 10.1091/mbc.E07-02-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raemaekers T, Ribbeck K, Beaudouin J, Annaert W, Van Camp M, Stockmans I, Smets N, Bouillon R, Ellenberg J, Carmeliet G. NuSAP, a novel microtubule-associated protein involved in mitotic spindle organization. J. Cell Biol. 2003;162:1017–1029. doi: 10.1083/jcb.200302129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turchi L, Fareh M, Aberdam E, Kitajima S, Simpson F, Wicking C, Aberdam D, Virolle T. ATF3 and p15PAF are novel gatekeepers of genomic integrity upon UV stress. Cell Death Diff. 2009;16:728–737. doi: 10.1038/cdd.2009.2. [DOI] [PubMed] [Google Scholar]
- 31.Newman MEJ. The structure and function of complex networks. SIAM Rev. 2003;45:167–256. [Google Scholar]
- 32.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 33.Linding R, Russell RB, Neduva V, Gibson TJ. GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003;31:3701–3708. doi: 10.1093/nar/gkg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vavouri T, Semple JI, Garcia-Verdugo R, Lehner B. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell. 2009;138:198–208. doi: 10.1016/j.cell.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 35.Frey BJJ, Dueck D. Clustering by passing messages between data points. Science. 2007;315 doi: 10.1126/science.1136800. [DOI] [PubMed] [Google Scholar]
- 36.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Z, Farver W, Kodukula S, Storch J. Regulation of Sterol Transport between Membranes and NPC2. Biochemistry. 2008;47:11134–11143. doi: 10.1021/bi801328u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eriksen JL, Mackenzie IRa. Progranulin: normal function and role in neurodegeneration. J. Neurochemi. 2008;104:287–297. doi: 10.1111/j.1471-4159.2007.04968.x. [DOI] [PubMed] [Google Scholar]
- 39.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 40.Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin J-J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 41.Klünemann HH, Elleder M, Kaminski WE, Snow K, Peyser JM, O'Brien JF, Munoz D, Schmitz G, Klein HE, Pendlebury WW. Frontal lobe atrophy due to a mutation in the cholesterol binding protein HE1/NPC2. Ann. Neurol. 2002;52:743–749. doi: 10.1002/ana.10366. [DOI] [PubMed] [Google Scholar]
- 42.Karageorgos LE, Isaac EL, Brooks DA, Ravenscroft EM, Davey R, Hopwood JJ, Meikle PJ. Lysosomal biogenesis in lysosomal storage disorders. Exp. Cell Res. 1997;234:85–97. doi: 10.1006/excr.1997.3581. [DOI] [PubMed] [Google Scholar]
- 43.Helip-Wooley A, Thoene JG. Sucrose-induced vacuolation results in increased expression of cholesterol biosynthesis and lysosomal genes. Exp. Cell Res. 2004;292:89–100. doi: 10.1016/j.yexcr.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 44.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 45.Naphade S, Kigerl K, Jakeman L, Kostyk S, Popovich P, Kuret J. Progranulin expression is upregulated after spinal contusion in mice. Acta Neuropathol. 2009;119 doi: 10.1007/s00401-009-0616-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl Acad. Sci. USA. 2003;100:1902–1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kollmann K, Mutenda KE, Balleininger M, Eckermann E, von Figura K, Schmidt B, Lübke T. Identification of novel lysosomal matrix proteins by proteome analysis. Proteomics. 2005;5:3966–3978. doi: 10.1002/pmic.200401247. [DOI] [PubMed] [Google Scholar]
- 48.Qian M, Sleat D, Zheng H, Moore D, Lobel P. Proteomics analysis of serum from mutant mice reveals lysosomal proteins selectively transported by each of the two mannose 6-phosphate receptors. Mol. Cell. Proteomics. 2007;7:58–70. doi: 10.1074/mcp.M700217-MCP200. [DOI] [PubMed] [Google Scholar]
- 49.Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Capell A, Liebscher S, Fellerer K, Brouwers N, Willem M, Lammich S, Gijselinck I, Bittner T, Carlson AM, Sasse F, et al. Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J. Neurosci. 2011;31:1885–1894. doi: 10.1523/JNEUROSCI.5757-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.