

Abstract
Interfaces are a most common motif in complex systems. To understand how the presence of interfaces affects hydrophobic phenomena, we use molecular simulations and theory to study hydration of solutes at interfaces. The solutes range in size from subnanometer to a few nanometers. The interfaces are self-assembled monolayers with a range of chemistries, from hydrophilic to hydrophobic. We show that the driving force for assembly in the vicinity of a hydrophobic surface is weaker than that in bulk water and decreases with increasing temperature, in contrast to that in the bulk. We explain these distinct features in terms of an interplay between interfacial fluctuations and excluded volume effects—the physics encoded in Lum–Chandler–Weeks theory [Lum K, Chandler D, Weeks JD (1999) J Phys Chem B 103:4570–4577]. Our results suggest a catalytic role for hydrophobic interfaces in the unfolding of proteins, for example, in the interior of chaperonins and in amyloid formation.
Keywords: binding, hydrophobicity, thermodynamics
Hydrophobic effects are ubiquitous and often the most significant forces of self-assembly and stability of nanoscale structures in liquid matter, from phenomena as simple as micelle formation to those as complex as protein folding and aggregation (1, 2). These effects depend importantly on length scale (3–5). Water molecules near small hydrophobic solutes do not sacrifice hydrogen bonds, but have fewer ways in which to form them, leading to a large negative entropy of hydration. In contrast, hydrogen bonds are broken in the hydration of large solutes, resulting in an enthalpic penalty. For hydrophobic solutes in bulk water at standard conditions, the cross-over from one regime to the other occurs at around 1 nm (3–6) and marks a change in the scaling of the solvation free energy from being linear with solute volume to being linear with exposed surface area. In bulk water, this cross-over provides a framework for understanding the assembly of small species into a large aggregate.
Typical biological systems contain a high density of interfaces, including those of membranes and proteins, spanning the entire spectrum from hydrophilic to hydrophobic. Whereas water near hydrophilic surfaces is bulk-like in many respects, water near hydrophobic surfaces is different, akin to that near a liquid–vapor interface (3–5, 7–9). Here, we consider how these interfaces alter hydrophobic effects. Specifically, to shed light on the thermodynamics of hydration at, binding to, and assembly at interfaces, we study solutes with a range of sizes at various self-assembled monolayer interfaces over a range of temperatures using molecular simulations and theory.
Our principal results are that, although the hydration thermodynamics of hydrophobic solutes at hydrophilic surfaces is similar to that in bulk, changing from entropic to enthalpic with increasing solute size, it is enthalpic for solutes of all length scales near hydrophobic surfaces. Further, the driving force for hydrophobically driven assembly in the vicinity of hydrophobic surfaces is weaker than that in bulk and decreases with increasing temperature, in contrast to that in bulk. These results suggest that hydrophobic surfaces will bind to and catalyze unfolding of proteins, which we predict is relevant in the formation of amyloids and the function of chaperonins.
Models
Molecular Simulations.
We simulate the solid–water interfaces of self-assembled monolayers (SAMs) of surfactants (Fig. 1A) with a range of head-group chemistries, from hydrophobic (-CH3) to hydrophilic (-OH) (8, 10). To study the size dependence of hydration at interfaces, we selected cuboid shaped (L × L × W) cavities, with thickness W = 0.3 nm, and side L, varying from small values comparable to the size of a water molecule to as large as 10 times that size. Thicker volumes will show qualitatively similar behavior, but will gradually sample the “bulk” region away from the interface. We model the monolayers and water with reasonably realistic force fields. But to focus specifically on solvent-induced hydrophobic interactions, we consider only idealized solutes—cavities that simply expel water from the volume they occupy. Solute–solvent and solute–solute interactions beyond those of excluded volume forces can further enrich our findings (5, 11, 12).
Fig. 1.

Size-dependent hydrophobic hydration at interfaces. (A) A schematic of a cuboidal cavity (green) at the SAM–water interface. The SAM head groups (black and white), alkane tails (gray), and water (red and white, partially cut out for clarity) are shown. (B) A typical configuration of the model membrane, color coded by its distance from the model surface (gray). (C) Important volumes in estimating the free energy, μex, of emptying the probe volume V (green) using the theoretical model. The region above the membrane is the volume B (blue), and the intersection of V and B is v (dark green). (D) Length-scale dependence of the cavity hydration free energy per unit area, μex/A, in bulk water and at interfaces, at T = 300 K, obtained from molecular dynamics simulations. (E) Theoretical model estimates of μex/A, near surfaces with different attraction strengths, η. (F) Connecting the microscopic binding free energy of a cavity to an interface, to the macroscopic surface wettability. The cos θ values were obtained from molecular dynamics simulations of a water droplet on SAM surfaces (10). Lines are predictions using Eq. 1 with size-dependent γLV taken from D.
Theoretical Model.
To rationalize the simulation results and obtain additional physical insights, we developed a model based on Lum–Chandler–Weeks (LCW) theory (4). LCW theory incorporates the interplay between the small length scale Gaussian density fluctuations and the physics of interface formation relevant at larger length scales, and captures the length scale dependence of hydrophobic hydration in bulk water. Near hydrophobic surfaces, LCW theory predicts the existence of a soft liquid–vapor-like interface, which has been confirmed by simulations (7–9).
We model this liquid–vapor-like interface near a hydrophobic surface, as an elastic membrane (Fig. 1B), whose energetics are governed by its interfacial tension and the attractive interactions with the surface. The free energy of cavity hydration, μex, is related to the probability of spontaneously emptying out a cavity-shaped volume, V. Such emptying can be conceptualized as a two-step process in which interfacial fluctuations of the membrane can empty out a large fraction of V in the first step, with the remaining volume v emptied out via a density fluctuation (Fig. 1C). When v is small, the probability that it contains N waters is well approximated by a Gaussian (8, 13, 14). The cost of emptying v can then be obtained from the average and the variance of the number of waters in v, which are evaluated by assuming that water density responds linearly to surface–water adhesive interactions.
We tune the strength of the model surface–water attraction, U(r), using a parameter η, where η ≈ 1 corresponds to a hydrophobic -CH3 SAM-like surface, with higher values representing increasingly hydrophilic surfaces. The representation of hydrophilic surfaces in our theoretical model lacks the specific details of hydrogen bonding interactions (e.g., between the hydrophilic -OH SAM surface and water), so comparisons between high-η model surfaces and hydrophilic SAM surfaces in simulations are qualitative in nature. Equations that put the above model on a quantitative footing are given in Appendix and the details of its exact implementation are included in SI Text.
Size-Dependent Hydrophobic Hydration at, and Binding to Interfaces.
Fig. 1D shows the excess free energy, μex, to solvate a cuboidal cavity at temperature T = 300 K, divided by its surface area (A = 2L2 + 4LW). The quantity μex/A can be thought of as an effective surface tension of the cavity–water interface. In bulk water, this value shows a gradual cross-over with increasing L, as expected (4, 6). Fig. 1D also shows the length-scale dependence of μex/A for solvating cavities in interfacial environments. Near the hydrophilic OH-terminated SAM, the behavior is similar to that in bulk water. However, with increasing hydrophobicity of the interface, the size dependence of μex/A becomes less pronounced and is essentially absent near the -CH3 surface, suggesting that hydration at hydrophobic surfaces is governed by interfacial physics at all length scales.
Fig. 1E shows the analogous solvation free energies predicted using the theoretical model. The essential features of solvation next to the SAM surfaces are captured well by this model, particularly for the hydrophobic surfaces (with η around one), where the potential U(r) closely mimics the effect of the real SAM on the adjacent water, and the agreement between theory and simulation is nearly quantitative. For the more hydrophilic SAMs, the comparison is qualitative, because the simple form for U(r) does not represent dipolar interactions well.
Fig. 1D also indicates that μex becomes favorable (smaller) with increasing surface hydrophobicity. The difference in μex at an interface and in the bulk,  , quantifies the hydration contribution to the experimentally measurable free energy of binding of solutes to interfaces. Because the solvation of large solutes is governed by the physics of interface formation, both in bulk and at the SAM surfaces, we can approximate Δμex = Ac(γSV - γSL - γLV), where Ac = L2 is the cross-sectional area, γ is the surface tension, and subscripts SV, SL, and LV, indicate solid–vapor, solid–liquid, and liquid–vapor interfaces, respectively. Using Young’s equation, γSV = γSL + γLV cos θ, we rewrite
, quantifies the hydration contribution to the experimentally measurable free energy of binding of solutes to interfaces. Because the solvation of large solutes is governed by the physics of interface formation, both in bulk and at the SAM surfaces, we can approximate Δμex = Ac(γSV - γSL - γLV), where Ac = L2 is the cross-sectional area, γ is the surface tension, and subscripts SV, SL, and LV, indicate solid–vapor, solid–liquid, and liquid–vapor interfaces, respectively. Using Young’s equation, γSV = γSL + γLV cos θ, we rewrite
| [1] |
where θ is the water droplet contact angle on the solid surface.
Although Eq. 1 is strictly valid only for macroscopic cavities, it can be applied to sufficiently large microscopic cavities with a length-scale dependent surface tension,  , which can be approximated by
, which can be approximated by  . Indeed, lines in Fig. 1F predicted using Eq. 1 are in excellent agreement with simulation data and indicate that the strength of binding increases with surface hydrophobicity, as well as with solute size. Hydrophobicity of flat surfaces is frequently characterized at the macroscale and even using molecular simulations (15) by measuring the contact angle of a water droplet placed on the surface. Such an approach is not feasible for protein surfaces, where topography and chemistry change over subnanometer length scales. Our demonstration of the connection between the macroscopic contact angle and the microscopic binding free energy suggests a molecular approach for characterizing the hydrophobicity of nanoscale surfaces (16, 17), for which the contact angle is ill-defined, but the binding free energy can be readily measured.
. Indeed, lines in Fig. 1F predicted using Eq. 1 are in excellent agreement with simulation data and indicate that the strength of binding increases with surface hydrophobicity, as well as with solute size. Hydrophobicity of flat surfaces is frequently characterized at the macroscale and even using molecular simulations (15) by measuring the contact angle of a water droplet placed on the surface. Such an approach is not feasible for protein surfaces, where topography and chemistry change over subnanometer length scales. Our demonstration of the connection between the macroscopic contact angle and the microscopic binding free energy suggests a molecular approach for characterizing the hydrophobicity of nanoscale surfaces (16, 17), for which the contact angle is ill-defined, but the binding free energy can be readily measured.
Temperature Dependence of Hydration at Interfaces.
The differences between cavity hydration at interfaces and in bulk are highlighted most clearly in the T dependence of μex, which characterizes the entropic and enthalpic contributions to the free energy. For small solutes in bulk, the entropy of hydration is known to be large and negative (18, 19), which reflects the reduced configurational space available to the surrounding water molecules. In contrast, for large solutes, the entropy of hydration is expected to be positive, consistent with the temperature dependence of the liquid–vapor surface tension (20). Fig. 2A shows that μex of large cuboidal cavities (L = 3 nm) in bulk water indeed decreases with increasing temperature, although the corresponding hydration entropy per unit surface area (25 J·mol-1·K-1·nm-2) is lower than that expected from the temperature derivative of surface tension of water (about 90 J·mol-1·K-1·nm-2; ref. 20). We note that solvation entropies calculated with the Simple Point Charge Extended (SPC/E) model of water are known to be smaller than experimental values by about 20% (21). Additionally, the cavity–water surface tension and its temperature derivative for these nanoscopic cavities are expected to be smaller than the corresponding macroscopic values (9).
Fig. 2.

Temperature dependence of μex in bulk water and at SAM–water interfaces for large (L = 3.0 nm) cavities (A and B) and for small (L = 0.5 nm, L = 0.75 nm) cavities (C and D), obtained from simulations (A, C) and from the model (B, D) of Eq. 5.
Fig. 2A also shows that, for large cuboidal cavities (L = 3 nm), μex decreases with increasing temperature not only in bulk water and near the hydrophilic (-OH) surface, but also near the hydrophobic (-CH3) surface, indicating a positive entropy of cavity formation. Thus, in all three systems, the thermodynamics of hydration of large cavities is governed by interfacial physics. Although the values of μex(L = 3 nm) at 300 K are rather large (573 kJ/mol in bulk water, 568 kJ/mol at the -OH interface, and 179 kJ/mol at the -CH3 interface), their variation with temperature, shown in Fig. 2A, is similar in bulk and at interfaces.
Fig. 2B shows that this same phenomenology is captured nearly quantitatively by the theoretical model. In the model, the cavity hydration free energies have large but athermal contributions from attractions between water and the model surfaces. The main temperature-dependent contribution to μex is the cost to deform the liquid–vapor-like interface near the surface to accommodate the large cavity. Because the necessary deformation is similar, regardless of the hydrophobicity of the surface, the variation of μex with temperature is similar as well.
Fig. 2C shows the temperature dependence of μex for small cavities (L = 0.5 nm) in bulk and at SAM–water surfaces. In bulk water, μex increases with temperature and yields an entropy of hydration of roughly -25 J·mol-1·K-1, characteristic of small length scale hydrophobic hydration. This negative value is consistent with those calculated for spherical solutes of a similar volume (18). With increasing hydrophobicity, the slope of the μex vs. T curve decreases and becomes negative, indicating a positive entropy of cavity formation near sufficiently hydrophobic surfaces. Near the most hydrophobic surface (-CH3), the entropy of hydration of this small cavity is +30 J·mol-1·K-1.
Fig. 2D shows that the same behavior is recovered by the theoretical model, though the correspondence is clearest at a slightly larger cavity size (L = 0.75 nm). Near hydrophilic model surfaces, the interface is pulled close to the surface by a strong attraction, so it is costly to deform it. As a result, small cavities are emptied through bulk-like spontaneous density fluctuations that result in a negative entropy of hydration. In contrast, near a hydrophobic surface, the interface is easy to deform, which provides an additional mechanism for creating cavities. In fact, this mechanism dominates near sufficiently hydrophobic surfaces, and because the surface tension of water decreases with increasing temperature, so does μex. Hence, even small cavities have a positive entropy of hydration near hydrophobic surfaces. The continuous spectrum of negative to positive solvation entropies observed in Fig. 2 C and D is thus revealed to be a direct consequence of the balance between bulk-like water density fluctuations and liquid–vapor-like interfacial fluctuations.
Fig. 3A shows that, near the hydrophobic CH3-terminated SAM, cavity hydration entropies per unit area, Sex/A, are positive and roughly constant (about 30 J·mol-1·K-1·nm-2) over a broad range of cavity sizes. In contrast, in bulk water, Sex/A depends on L, and changes from large negative to positive values with increasing L. The general trend is that, as the physics of interface formation becomes important (either in the vicinity of a hydrophobic surface or for increasing cavity sizes), Sex/A approaches a positive limiting value. This trend is already well known in the context of solvating hard spheres of various diameters in liquid water and other bulk solvents (22, 23). Here, however, we provide theoretical tools and results needed to establish how these trends play out in contexts of neighboring surfaces. The length scale at which entropy crosses zero, LS, can serve as a thermodynamic cross-over length. In bulk water, LS ≈ 1.8 ± 0.2 nm. The behavior of Sex/A is qualitatively similar at the -OH surface, with LS ≈ 1.3 ± 0.4 nm. Although the numerical value of LS may depend on the shape of the cavity and on solute–water attractions for nonidealized hydrophobes, the trend in entropy should not.
Fig. 3.

Length-scale dependence of the excess solvation entropy per unit surface area for (A) cavities in bulk water and at the -CH3 and -OH SAM–water interfaces, and (B) cavities in the model of Eq. 5 near surfaces of different attraction strengths, η.
Fig. 3B shows that our implementation of LCW ideas recovers many of the observed trends, with solvation entropy being everywhere positive for the smallest attraction strength η and a thermodynamic cross-over length of just under 1 nm emerging for the more hydrophilic model surfaces, similar to that in bulk water. Nevertheless, the agreement between Fig. 3 A and B is somewhat qualitative, mostly as a result of the crude form of U(r) used to model hydrophilic surfaces.
Thermodynamics of Binding to, and Assembly at, Hydrophobic Surfaces.
In the preceding sections, we have examined the hydration behavior of single, isolated, idealized cavities near flat surfaces and in the bulk. We now consider the consequences of our observations on hydrophobically driven binding and assembly, summarized schematically in Fig. 4.
Fig. 4.

Schematic illustrating the thermodynamics of binding and assembly. The points represent free energies of solvating small objects individually (Left) and in the assembled state (Right), in bulk (Top) and at a hydrophobic interface (Bottom), at a lower (blue, TL) and a higher (red, TH) temperature near ambient conditions. Assembly: The driving force for assembly at hydrophobic interfaces is smaller than that in bulk, and is enthalpic, decreasing with increasing temperature, unlike in bulk. Binding: The driving force for binding small objects to a hydrophobic surface increases with temperature, so it is entropic, whereas for large objects, it is enthalpic.
Fig. 4 indicates that, although the binding of both small and large solutes (or aggregates) to hydrophobic surfaces is highly favorable, their thermodynamic signatures are different. Binding of small solutes is entropic and becomes more favorable with increasing temperature, whereas binding of large solutes is enthalpic and depends only weakly on temperature. For example, for the L = 3 nm solute, the binding free energy is 386 kJ/mol at 280 K and increases to 397 kJ/mol at 320 K.
Fig. 4 also highlights the differences in the thermodynamics of hydrophobically driven assembly at interfaces and in bulk, inferred from our length-scale dependence studies. In bulk, the solvation of many small, isolated hydrophobes scales as their excluded volume. Accommodating small species inside the existing hydrogen-bonding network of water imposes an entropic cost, so the solvation free energy increases with increasing temperature. When several small hydrophobes come together, water instead hydrates the aggregate by surrounding it with a liquid–vapor-like interface. The corresponding solvation free energy scales as the surface area and decreases with increasing temperature.
Thus, the driving force for assembly of n small solutes (each of surface area A1, volume v1, and solvation free energy of  ) into a large aggregate (with surface area An and volume nv1) in bulk water is well approximated by
) into a large aggregate (with surface area An and volume nv1) in bulk water is well approximated by
| [2] |
where  and γbulk is a curvature-corrected effective surface tension (top curve of Fig. 1D). As the surface tension decreases with increasing temperature, so does the free energy to hydrate nanometer-sized aggregates (Fig. 2A). However, the free energy to individually hydrate the small solutes increases with temperature (Fig. 2C), resulting in a larger driving force for assembly. Conversely, although the driving force for assembly,
and γbulk is a curvature-corrected effective surface tension (top curve of Fig. 1D). As the surface tension decreases with increasing temperature, so does the free energy to hydrate nanometer-sized aggregates (Fig. 2A). However, the free energy to individually hydrate the small solutes increases with temperature (Fig. 2C), resulting in a larger driving force for assembly. Conversely, although the driving force for assembly,  , is large and negative (favorable) at ambient conditions, it decreases in magnitude with decreasing temperature (upper portion of Fig. 4) and can even change sign at a sufficiently low temperature. When adapted to particular systems, Eq. 2 can, with remarkable accuracy, explain complex solvation phenomena like the temperature-dependent aggregation behavior of micelles (24) and the cold denaturation of proteins (2).
, is large and negative (favorable) at ambient conditions, it decreases in magnitude with decreasing temperature (upper portion of Fig. 4) and can even change sign at a sufficiently low temperature. When adapted to particular systems, Eq. 2 can, with remarkable accuracy, explain complex solvation phenomena like the temperature-dependent aggregation behavior of micelles (24) and the cold denaturation of proteins (2).
In the presence of a hydrophobic surface, on the other hand, we have found that interfacial physics dominates at all length scales (Figs. 2 and 3). As a result, the driving force for assembly at interfaces,  , does not scale as in Eq. 2, but is instead given by
, does not scale as in Eq. 2, but is instead given by
| [3] |
where γint is the effective surface tension at the interface (the lower curves of Fig. 1 D and E). Because γint decreases with increasing temperature (Fig. 2), so does the hydration contribution to the driving force for assembly at a hydrophobic surface, in contrast to that in bulk.
The free energy barrier between disperse and assembled states is also expected to be very different in bulk and near hydrophobic surfaces. In bulk, the dispersed state has no liquid–vapor-like interface, whereas the assembled state does. The transition state is a critical nucleus of hydrophobic particles that nucleates the liquid–vapor-like interface. The nucleation barrier can be high and dominates the kinetics of hydrophobic collapse of idealized hydrophobic polymers (25–27) and plates (28). In contrast, at ambient conditions, we expect aggregation near hydrophobic surfaces to be nearly barrierless because an existing liquid–vapor-like interface is deformed continuously between the disperse and assembled states.
Finally, and most importantly, we find that, for large aggregates, the driving force of assembly is weaker near interfaces than in bulk. In the limit of large n, the terms  dominate both at interfaces and in bulk (Eqs. 2 and 3), and the results in Fig. 1D show that
dominate both at interfaces and in bulk (Eqs. 2 and 3), and the results in Fig. 1D show that  .
.
The nontrivial behavior of the driving forces and barriers to assembly at interfaces should be relevant in biological systems where hydrophobicity plays an important role. Experiments have shown that hydrophobic surfaces bind and facilitate the unfolding of proteins, including those that form amyloids (29–31). Our results shed light on these phenomena and suggest that large hydrophobic surfaces may generically serve as catalysts for unfolding proteins (32), via solvent-mediated interactions. Indeed, simulations show that the binding of model hydrophobic polymers to hydrophobic surfaces is accompanied by a conformational rearrangement from globular to pancake-like structures (33). Such conformations can further assemble into secondary structures, such as β-sheets (30–32, 34), and we predict that the solvent contribution to this assembly at the hydrophobic surface will be governed by interfacial physics. This behavior implies that manipulating the liquid-vapor surface tension, either by changing the temperature or by adding salts or cosolutes, will allow one to manipulate the driving force for assembly.
We further speculate that the catalysis of unfolding by hydrophobic surfaces may play a role in chaperonin function (35). The interior walls of chaperonins in the open conformation are hydrophobic and can bind misfolded proteins, whereupon their unfolding is catalyzed (36, 37). Subsequent ATP-driven conformational changes render the chaperonin walls hydrophilic (35, 36). As a result, the unfolded protein is released from the wall, as the free energy for a hydrophobe to bind to a hydrophilic surface is much lower than that to bind to a hydrophobic one (Fig. 1D).
Our results also provide insights into the interactions between biomolecules and nonbiological hydrophobic surfaces, such as those of graphite and of certain metals, which have been shown to bind and unfold proteins (38, 39). Such interactions are of interest in diverse applications including nanotoxicology (40) and biofouling (39).
Collectively, our findings highlight that the magnitude and temperature dependences of the driving forces for assembly near hydrophobic surfaces are different from that in bulk and near hydrophilic surfaces. Experimental measurements of the thermodynamics of protein folding have been performed primarily in bulk water (41). Although many experiments have probed how interfaces affect protein folding, structure, and function (29, 42), to the best of our knowledge, there are no temperature-dependent thermodynamic measurements of self-assembly at interfaces. Extensions of atomic force microscope experiments of pulling a hydrophobic polymer attached to a surface (43) have the potential to quantify the thermodynamics of hydrophobic interactions in interfacial environments described here. We hope that our results will motivate such measurements.
Appendix
Simulation Details.
Our simulation setup and force fields are similar to those described in refs. 8 and 10. Simulations were performed in the canonical (NVT) ensemble with a periodic box (7 × 7 × 9 nm) that has a buffering liquid–vapor interface at the top of the box, for reasons explained in ref. 9. It has been shown that free energies obtained in the above ensemble are indistinguishable from those obtained in the isothermal-isobaric ensemble at a pressure of 1 bar (44). We have chosen the SPC/E model of water (45) because it adequately captures experimentally known features of water, such as surface tension, compressibility, and local tetrahedral order, which play important roles in the hydrophobic effect (5). Electrostatic interactions were calculated using the particle mesh Ewald method (46), and bonds in water were constrained using SHAKE (47). Solvation free energies were calculated using test particle insertions (48) for smaller cavities (L < 1 nm) and the indirect umbrella sampling method (9, 44) for larger cavities.
Theoretical Model.
We model the liquid–vapor-like interface near hydrophobic surfaces as a periodic elastic membrane, z = h(x,y), with an associated Hamiltonian, H[h(x,y)]:
 |
[4] |
Here, γ is the experimental liquid–vapor surface tension of water, ρℓ is the bulk water density, and U(r) is the interaction potential between the model surface and a water molecule at position r = (x,y,z). The square-gradient term in Eq. 4 accurately captures the energetics of interfacial capillary waves only for wavelengths larger than atomic dimensions (Fig. 5), so we restrict h(x,y) to contain modes with wavevectors below 2π/9 Å. At any instant in time, part of V can be empty due to an interfacial fluctuation. The number of waters in the remaining volume, v, fluctuates, and we denote by Pv(N) the probability that v contains N waters. We thus estimate the free energy for emptying V completely to be
 |
[5] |
where  is the partition function of the membrane. The volume v depends on the interfacial configuration h(x,y)—i.e., v = v[h(x,y)].
is the partition function of the membrane. The volume v depends on the interfacial configuration h(x,y)—i.e., v = v[h(x,y)].
Fig. 5.

Power spectrum of the instantaneous liquid–vapor interface at T = 300 K. A liquid–vapor interface was simulated using a 24 × 24 × 3 nm3 slab of SPC/E water in a periodic box of size 24 × 24 × 9 nm3 and the instantaneous interface configuration, h(x,y), and its Fourier transform,  , were evaluated as in ref. 53. The power spectrum of our simulated instantaneous interface is in good agreement with the capillary-wave theory prediction (
, were evaluated as in ref. 53. The power spectrum of our simulated instantaneous interface is in good agreement with the capillary-wave theory prediction ( ) for wavevectors smaller than approximately 2π/9 Å. For larger wavevectors, the power spectrum is sensitive to molecular detail (i.e., the coarse-graining length, ξ, used to define the intrinsic interface), as expected (54). Fitting the ξ = 2.0 Å data in the range 0.01 Å-1 < k < 0.3 Å-1 yields γ = 62.0 ± 0.5 mJ/m2, in reasonable agreement with the experimental value of 72 mJ/m2 and some simulated values of the SPC/E surface tension (e.g., 63.6 ± 1.5 mJ/m2; ref. 55), but not others (e.g., 52.9 mJ/m2; ref. 54).
) for wavevectors smaller than approximately 2π/9 Å. For larger wavevectors, the power spectrum is sensitive to molecular detail (i.e., the coarse-graining length, ξ, used to define the intrinsic interface), as expected (54). Fitting the ξ = 2.0 Å data in the range 0.01 Å-1 < k < 0.3 Å-1 yields γ = 62.0 ± 0.5 mJ/m2, in reasonable agreement with the experimental value of 72 mJ/m2 and some simulated values of the SPC/E surface tension (e.g., 63.6 ± 1.5 mJ/m2; ref. 55), but not others (e.g., 52.9 mJ/m2; ref. 54).
It is known that Pv(N) is well approximated by a Gaussian when v is small (8, 13, 14). If water were far from liquid–vapor coexistence, then Pv(N) would also be close to Gaussian for arbitrarily large v. The fact that water at ambient conditions is near liquid–vapor coexistence, and that there is a liquid–vapor-like interface near the SAM, is captured by the additional interfacial energy factor Z-1 exp{-βH[h(x,y)]} in Eq. 5. The net result is that the thermal average of Eq. 5 is dominated by interface configurations where v is small, so that even at ambient conditions, we can approximate
where 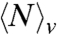 is the average number of waters in v and
is the average number of waters in v and  is the variance. We estimate these by noting that the solvent density responds linearly to the attractive potential, U(r), in the volume occupied by the water, B, depicted in Fig. 1C (13, 49, 50). Hence,
is the variance. We estimate these by noting that the solvent density responds linearly to the attractive potential, U(r), in the volume occupied by the water, B, depicted in Fig. 1C (13, 49, 50). Hence,
 |
Here, g(r) is the oxygen–oxygen radial distribution function of water (51).
The surface–water interaction is modeled by a potential, U(r), that closely mimics the attractive potential exerted by the -CH3 SAM on water:
The first term, Uwall(r), is a sharply repulsive potential in the region z < R0 that captures the hard-core exclusion of a plane of head groups at z = 0 with hard-sphere radius R0. The second term, Uhead(r), is scaled by η, and captures the head-group–water interaction, modeled as a plane of CH3 Lennard–Jones (LJ) interaction sites at z = 0 with an area density of μhead. The sites are parametrized with the Optimized Potential for Liquid Simulations United Atom (OPLS/UA) force field (52). The final term, Utail(r), similarly captures the alkane tail–water interaction, modeled as a uniform half-space of OPLS/UA CH2 LJ interaction sites of volume density ρtail at a distance ζ below the head groups. The parameters R0, ζ, μhead, and ρtail are dictated by the geometry of the SAM (see SI Text for details).
Supplementary Material
Acknowledgments.
The authors thank Steve Granick, Bruce Berne, and Frank Stillinger for providing helpful comments on an earlier draft. A.J.P. and P.V. were supported by National Institutes of Health Grant R01-GM078102-04. S.G. gratefully acknowledges partial financial support of the National Science Foundation (CBET-0933169, CBET-1134341, NSF-CBET-0967937) grants. D.C. was supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division and Chemical Sciences, Geosciences, and Biosciences Division of the US Department of Energy under Contract DE-AC02-05CH11231.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1110703108/-/DCSupplemental.
References
- 1.Tanford C. The Hydrophobic Effect—Formation of Micelles and Biological Membranes. New York: Wiley Interscience; 1973. [Google Scholar]
- 2.Kauzmann W. Some factors in the interpretation of protein denaturation. Adv Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 3.Stillinger FH. Structure in aqueous solutions of nonpolar solutes from the standpoint of scaled-particle theory. J Solution Chem. 1973;2:141–158. [Google Scholar]
- 4.Lum K, Chandler D, Weeks JD. Hydrophobicity at small and large length scales. J Phys Chem B. 1999;103:4570–4577. [Google Scholar]
- 5.Chandler D. Interfaces and the driving force of hydrophobic assembly. Nature. 2005;437:640–647. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 6.Rajamani S, Truskett TM, Garde S. Hydrophobic hydration from small to large lengthscales: Understanding and manipulating the crossover. Proc Natl Acad Sci USA. 2005;102:9475–9480. doi: 10.1073/pnas.0504089102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mittal J, Hummer G. Static and dynamic correlations in water at hydrophobic interfaces. Proc Natl Acad Sci USA. 2008;105:20130–20135. doi: 10.1073/pnas.0809029105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Godawat R, Jamadagni SN, Garde S. Characterizing hydrophobicity of interfaces by using cavity formation, solute binding, and water correlations. Proc Natl Acad Sci USA. 2009;106:15119–15124. doi: 10.1073/pnas.0902778106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel AJ, Varilly P, Chandler D. Fluctuations of water near extended hydrophobic and hydrophilic surfaces. J Phys Chem B. 2010;114:1632–1637. doi: 10.1021/jp909048f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shenogina N, Godawat R, Keblinski P, Garde S. How wetting and adhesion affect thermal conductance of a range of hydrophobic to hydrophilic aqueous interfaces. Phys Rev Lett. 2009;102:156101. doi: 10.1103/PhysRevLett.102.156101. [DOI] [PubMed] [Google Scholar]
- 11.Huang DM, Chandler D. The hydrophobic effect and the influence of solute-solvent attractions. J Phys Chem B. 2002;106:2047–2053. [Google Scholar]
- 12.Wallqvist A, Gallicchio E, Levy RM. A model for studying drying at hydrophobic interfaces: Structural and thermodynamic properties. J Phys Chem B. 2001;105:6745–6753. [Google Scholar]
- 13.Hummer G, Garde S, Garcia AE, Pohorille A, Pratt LR. An information theory model of hydrophobic interactions. Proc Natl Acad Sci USA. 1996;93:8951–8955. doi: 10.1073/pnas.93.17.8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crooks GE, Chandler D. Gaussian statistics of the hard-sphere fluid. Phys Rev E Stat Nonlin Soft Matter Phys. 1997;56:4217–4221. [Google Scholar]
- 15.Giovambattista N, Debenedetti PG, Rossky PJ. Effect of surface polarity on water contact angle and interfacial hydration structure. J Phys Chem B. 2007;111:9581–9587. doi: 10.1021/jp071957s. [DOI] [PubMed] [Google Scholar]
- 16.Acharya H, Vembanur S, Jamadagni SN, Garde S. Mapping hydrophobicity at the nanoscale: Applications to heterogeneous surfaces and proteins. Faraday Discuss. 2010;146:353–365. doi: 10.1039/b927019a. [DOI] [PubMed] [Google Scholar]
- 17.Giovambattista N, Lopez CF, Rossky PJ, Debenedetti PG. Hydrophobicity of protein surfaces: Separating geometry from chemistry. Proc Natl Acad Sci USA. 2008;105:2274–2279. doi: 10.1073/pnas.0708088105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garde S, Hummer G, Garcia AE, Paulaitis ME, Pratt LR. Origin of entropy convergence in hydrophobic hydration and protein folding. Phys Rev Lett. 1996;77:4966–4968. doi: 10.1103/PhysRevLett.77.4966. [DOI] [PubMed] [Google Scholar]
- 19.Huang DM, Chandler D. Temperature and length scale dependence of hydrophobic effects and their possible implications for protein folding. Proc Natl Acad Sci USA. 2000;97:8324–8327. doi: 10.1073/pnas.120176397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alejandre J, Tildesley DJ, Chapela GA. Molecular dynamics simulation of the orthobaric densities and surface tension of water. J Chem Phys. 1995;102:4574–4583. [Google Scholar]
- 21.Athawale MV, Sarupria S, Garde S. Enthalpy-entropy contributions to salt and osmolyte effects on molecular-scale hydrophobic hydration and interactions. J Phys Chem B. 2008;112:5661–5670. doi: 10.1021/jp073485n. [DOI] [PubMed] [Google Scholar]
- 22.Ashbaugh HS, Pratt LR. Colloquium: Scaled particle theory and the length scales of hydrophobicity. Rev Mod Phys. 2006;78:159–178. [Google Scholar]
- 23.Ashbaugh HS, Pratt LR. Contrasting nonaqueous against aqueous solvation on the basis of scaled-particle theory. J Phys Chem B. 2007;111:9330–9336. doi: 10.1021/jp071969d. [DOI] [PubMed] [Google Scholar]
- 24.Maibaum L, Dinner AR, Chandler D. Micelle formation and the hydrophobic effect. J Phys Chem B. 2004;108:6778–6781. [Google Scholar]
- 25.ten Wolde PR, Chandler D. Drying-induced hydrophobic polymer collapse. Proc Natl Acad Sci USA. 2002;99:6539–6543. doi: 10.1073/pnas.052153299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller T, Vanden-Eijnden E, Chandler D. Solvent coarse-graining and the string method applied to the hydrophobic collapse of a hydrated chain. Proc Natl Acad Sci USA. 2007;104:14559–14564. doi: 10.1073/pnas.0705830104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferguson AL, Debenedetti PG, Panagiotopoulos AZ. Solubility and molecular conformations of n-alkane chains in water. J Phys Chem B. 2009;113:6405–6414. doi: 10.1021/jp811229q. [DOI] [PubMed] [Google Scholar]
- 28.Huang X, Zhou R, Berne BJ. Drying and hydrophobic collapse of paraffin plates. J Phys Chem B. 2005;109:3546–3552. doi: 10.1021/jp045520l. [DOI] [PubMed] [Google Scholar]
- 29.Beverung CJ, Radke CJ, Blanch HW. Protein adsorption at the oil/water interface: Characterization of adsorption kinetics by dynamic interfacial tension measurements. Biophys Chem. 1999;81:59–80. doi: 10.1016/s0301-4622(99)00082-4. [DOI] [PubMed] [Google Scholar]
- 30.Sethuraman A, Vedantham G, Imoto T, Przybycien T, Belfort G. Protein unfolding at interfaces: Slow dynamics of alpha-helix to beta-sheet transition. Proteins. 2004;56:669–678. doi: 10.1002/prot.20183. [DOI] [PubMed] [Google Scholar]
- 31.Nikolic A, Baud S, Rauscher S, Pomes R. Molecular mechanism of beta-sheet self-organization at water-hydrophobic interfaces. Proteins. 2011;79:1–22. doi: 10.1002/prot.22854. [DOI] [PubMed] [Google Scholar]
- 32.Sharma S, Berne BJ, Kumar SK. Thermal and structural stability of adsorbed proteins. Biophys J. 2010;99:1157–1165. doi: 10.1016/j.bpj.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jamadagni SN, Godawat R, Dordick JS, Garde S. How interfaces affect hydrophobically driven polymer folding. J Phys Chem B. 2009;113:4093–4101. doi: 10.1021/jp806528m. [DOI] [PubMed] [Google Scholar]
- 34.Krone MG, et al. Role of water in mediating the assembly of alzheimer amyloid-beta abeta 16–22 protofilaments. J Am Chem Soc. 2008;130:11066–11072. doi: 10.1021/ja8017303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fenton W, Horwich A. Chaperonin-mediated protein folding: Fate of substrate polypeptide. Q Rev Biophys. 2003;36:229–256. doi: 10.1017/s0033583503003883. [DOI] [PubMed] [Google Scholar]
- 36.England J, Lucent D, Pande V. Rattling the cage: Computational models of chaperonin-mediated protein folding. Curr Opin Struct Biol. 2008;18:163–169. doi: 10.1016/j.sbi.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 37.Jewett A, Shea JE. Reconciling theories of chaperonin accelerated folding with experimental evidence. Cell Mol Life Sci. 2010;67:255–276. doi: 10.1007/s00018-009-0164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marchin KL, Berrie CL. Conformational changes in the plasma protein fibrinogen upon adsorption to graphite and mica investigated by atomic force microscopy. Langmuir. 2003;19:9883–9888. [Google Scholar]
- 39.Anand G, Zhang F, Linhardt RJ, Belfort G. Protein-associated water and secondary structure effect removal of blood proteins from metallic substrates. Langmuir. 2011;27:1830–1836. doi: 10.1021/la1041794. [DOI] [PubMed] [Google Scholar]
- 40.Tian F, Cui D, Schwarz H, Estrada GG, Kobayashi H. Cytotoxicity of single-wall carbon nanotubes on human fibroblasts. Toxicol in Vitro. 2006;20:1202–1212. doi: 10.1016/j.tiv.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Makhatadze G, Privalov P. Energetics of protein structure. Adv Protein Chem. 1995;47:307–425. doi: 10.1016/s0065-3233(08)60548-3. [DOI] [PubMed] [Google Scholar]
- 42.Karajanagi SS, Vertegel AA, Kane RS, Dordick JS. Structure and function of enzymes adsorbed onto single-walled carbon nanotubes. Langmuir. 2004;20:11594–11599. doi: 10.1021/la047994h. [DOI] [PubMed] [Google Scholar]
- 43.Li ITS, Walker GC. Interfacial free energy governs single polystyrene chain collapse in water and aqueous solutions. J Am Chem Soc. 2010;132:6530–6540. doi: 10.1021/ja101155h. [DOI] [PubMed] [Google Scholar]
- 44.Patel AJ, Varilly P, Chandler D, Garde S. Quantifying density fluctuations in volumes of all shapes and sizes using indirect umbrella sampling. J Stat Phys. 2011 doi: 10.1007/s10955-011-0269-9. 10.1007/s10955-011-0269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berendsen HJC, Grigera JR, Straatsma TP. The missing term in effective pair potentials. J Phys Chem. 1987;91:6269–6271. [Google Scholar]
- 46.Essmann U, et al. A smooth particle mesh ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 47.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 48.Widom B. Some topics in the theory of fluids. J Chem Phys. 1963;39:2808–2812. [Google Scholar]
- 49.Chandler D. Gaussian field model of fluids with an application to polymeric fluids. Phys Rev E Stat Nonlin Soft Matter Phys. 1993;48:2898–2905. doi: 10.1103/physreve.48.2898. [DOI] [PubMed] [Google Scholar]
- 50.Varilly P, Patel AJ, Chandler D. An improved coarse-grained model of solvation and the hydrophobic effect. J Chem Phys. 2011;134:074109. doi: 10.1063/1.3532939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Narten AH, Levy HA. Liquid water: Molecular correlation functions from X-ray diffraction. J Chem Phys. 1971;55:2263–2269. [Google Scholar]
- 52.Jorgensen WL, Madura JD, Swenson CJ. Optimized intermolecular potential functions for liquid hydrocarbons. J Am Chem Soc. 1984;106:6638–6646. [Google Scholar]
- 53.Willard AP, Chandler D. Instantaneous liquid interfaces. J Phys Chem B. 2010;114:1954–1958. doi: 10.1021/jp909219k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sedlmeier F, Horinek D, Netz RR. Nanoroughness, intrinsic density profile, and rigidity of the air-water interface. Phys Rev Lett. 2009;103:136102. doi: 10.1103/PhysRevLett.103.136102. [DOI] [PubMed] [Google Scholar]
- 55.Vega C, de Miguel E. Surface tension of the most popular models of water by using the test-area simulation method. J Chem Phys. 2007;126:154707. doi: 10.1063/1.2715577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.