

Summary
Inclusions comprised of α-synuclein (α-syn), i.e. Lewy bodies (LBs) and Lewy neurites (LNs), define synucleinopathies including Parkinson’s Disease (PD) and dementia with Lewy Bodies (DLB). Here, we demonstrate that pre-formed fibrils generated from full length and truncated recombinant α-syn enter primary neurons, likely by adsorptive-mediated endocytosis and promote recruitment of soluble endogenous α-syn into insoluble PD-like LBs and LNs. Remarkably, endogenous α-syn was sufficient for formation of these aggregates, and overexpression of wild type or mutant α-syn was not required. LN-like pathology first developed in axons and propagated to form LB-like inclusions in perikarya. Accumulation of pathologic α-syn led to selective decreases in synaptic proteins, progressive impairments in neuronal excitability and connectivity, and eventually, neuron death. Thus, our data contribute important insights into the etiology and pathogenesis of PD-like α-syn inclusions, their impact on neuronal functions, and provide a model for discovering therapeutics targeting pathologic α-syn- mediated neurodegeneration.
Introduction
Aggregates of amyloid proteins characterize many neurodegenerative disorders including Alzheimer’s (AD) and Parkinson’s (PD) disease. Formation of pathological inclusions occurs by a multi-step process including the misfolding of normal soluble proteins, their association into higher order oligomers, followed by their assembly into amyloid fibrils that form disease specific inclusions (Conway et al., 2000; Uversky et al., 2001). Recent evidence indicates that proteinaceous aggregates comprised of tau and α-synuclein (α-syn), which are characteristic lesions of AD and PD respectively, can induce pathology in healthy cells (Clavaguera et al., 2009; Desplats et al., 2009; Frost et al., 2009; Guo and Lee, 2011; Luk et al., 2009). This process is hypothesized to occur via uptake of misfolded polymers which can propagate by recruiting their endogenously expressed counterparts, followed by their spread to induce pathology throughout the nervous system (Aguzzi and Rajendran, 2009).
Support for this concept of transmissibility comes from studies showing that tau and α-syn pathology spread in a stereotypical temporal and topological manner (Braak and Braak, 1991; Braak et al., 2003). Furthermore, fetal mesencephalic grafts in the striatum of PD patients eventually show evidence of Lewy Bodies (LB), suggesting that pathologic α-syn could be transmitted from diseased striatal neurons to young grafted neurons (Kordower et al., 2008a; Kordower et al., 2008b; Li et al., 2008). However, these studies cannot determine if the LB-like inclusions were formed by the spread of α-syn fibrils, or if some other toxic effect of the neighboring diseased neurons induced α-syn inclusions.
Although previous studies in model systems demonstrate that exogenous amyloid fibrils can seed recruitment of intracellular soluble proteins into inclusions, (Clavaguera et al., 2009; Desplats et al., 2009; Frost et al., 2009; Guo and Lee, 2011; Hansen et al. 2011; Luk et al., 2009), they either employed additional factors to assist the entry of the fibrils into cells, or they utilized cell extracts containing disease proteins in which other components that contribute to development of pathology may exist. Also, all of these models rely on the overexpression of human wild type (WT) or mutant proteins. This contrasts with the majority of neurodegenerative diseases which are sporadic and express normal levels of the WT proteins that are the building blocks of the fibrillar inclusions in these disorders. Thus, it is crucial to determine whether formation of PD-like LBs and Lewy neurites (LNs) in cultured neurons can be facilitated under physiological conditions using pure WT α-syn preformed fibrils (pffs) and endogenous levels of α-syn expression.
Thus, we asked if α-syn pffs, formed from purified recombinant human WT α-syn (α-syn-hWT), recruit endogenous α-syn into pathologic, insoluble inclusions. We show that α-syn pffs are internalized and induce endogenous α-syn expressed in primary neurons to aggregate into inclusions resembling LBs and LNs in human PD brains. LN-like accumulations are initially detected in axons and α-syn pathology then propagates to the cell body where LB-like inclusions develop. Formation of these PD-like α-syn LNs and LBs causes selective reductions in synaptic proteins, and progressive impairments in neuronal network function and excitability that culminate in neuron death.
Results
Induction of insoluble α-syn aggregates in primary neurons by α-syn pffs
To determine if exogenous human α-syn pffs can seed recruitment of endogenously expressed mouse α-syn into insoluble LB-like and LN-like fibrillar aggregates, we added α-syn pffs generated from full length recombinant α-syn-hWT to primary hippocampal neurons derived from WT C57BL6 mice after culturing them for 5–6 days in vitro (DIV). These neurons were examined 2 weeks after the addition of α-syn-hWT pffs, when synapses are mature, and α-syn is normally localized to presynaptic terminals (Murphy et al., 2000). In PBS treated hippocampal neurons, endogenous mouse α-syn localized to presynaptic puncta as visualized using monoclonal antibody (mAB) Syn202, a pan-synuclein antibody (Giasson et al., 2000) (Figure 1A, top panels). In contrast, in α-syn-hWT pff-treated neurons, α-syn did not localize to the presynaptic terminal (Figure 1A), but instead formed fibrillar LN-like inclusions. To determine if the α-syn aggregates were detergent-insoluble, PBS and α-syn-hWT pff-treated neurons were extracted with buffer containing 1% Triton X-100 (Tx-100) during fixation. Under such conditions, endogenous α-syn within neuronal processes in PBS treated neurons was soluble in Tx-100, but cells incubated with α-syn-hWT pffs showed Tx-100-insoluble aggregates (Figure 1A).
Figure 1. α-syn-hWT pffs recruit endogenous α-syn to form pathologic, insoluble aggregates.

A. Two weeks following pff addition, neurons were fixed with paraformaldehyde alone or paraformaldehyde with 1% Tx-100 to extract soluble proteins. In PBS treated neurons, α-syn localized to the presynaptic terminal and was Tx-100 soluble. Addition of α-syn pffs formed Tx-100 insoluble aggregates which recruited α-syn away from synapses. Scale bar = 20 μm. B. Neurons were treated with α-syn-hWT pffs, and 2 weeks later, were sequentially extracted with 1% Tx-100 followed by 2% SDS. Shown are immunoblots from 2 independent sets of samples. Antibodies that either recognize the C-terminus of α-syn (top) or are specific for mouse α-syn (bottom) showed that in PBS treated neurons, α-syn was soluble in Tx-100. α-Syn-hWT pff treatment reduced soluble α-syn and increased Tx-100-insoluble α-syn. The first lane shows α-syn-hWT pffs alone to demonstrate that the C-terminal antibody recognizes both human and mouse α-syn, and the mouse specific antibody recognizes only mouse pffs. Furthermore, the α-syn-hWT pffs themselves are not phosphorylated. C. Addition of α-syn pffs increased pathologic, p-α-syn. Fixation with paraformaldehyde/Tx-100 demonstrated that the phosphorylated aggregates were insoluble. Scale bar = 50 μm. Insert: Within the somata, aggregates appear as LB-like skein-like filaments and dense inclusions (arrow). PBS- treated neurons and neurons from α-syn −/− mice did not show p-α-syn. D. The phosphorylated aggregates are ubiquitin positive (N=2). Scale bar = 50 μm. See also Supplementary Figure 1.
α-Syn recruited into pathologic inclusions undergoes extensive phosphorylation at Ser129 (pSer129), thus antibodies against pSer129 selectively recognize α-syn pathology (Fujiwara et al., 2002). Furthermore, as this modification is absent in recombinant α-syn pffs (Figure 1B, first lane on left, Luk et al., 2009), the accumulation of phosphorylated α-syn (p-α-syn) reflects an intracellular modification. PBS treated neurons did not show staining with 81A, a mAB specific for pSer129 (Figure 1C, Waxman and Giasson, 2008). However, neurons treated with α-syn-hWT pffs showed intense 81A immunostaining that was Tx-100 insoluble (Figure 1C). Pff-induced aggregates exhibited morphologies ranging from small puncta to LN-like inclusions of variable lengths within neurites (Figures 1C, 1D, 2, 4–7). Within neuronal perikarya, these α-syn accumulations resembled LBs observed in human PD brains (Figure 1C inset). The α-syn-hWT pff-induced aggregates also occurred in primary cultures of cortical and midbrain dopaminergic neurons (Supplementary Figure 1A). Furthermore, neurons generated from other strains of mice as well as rats developed LB- and LN-like inclusions when treated with α-syn-hWT pffs, supporting the hypothesis that induction of α-syn pathology is a general feature of primary rodent neurons (data not shown). P-α-syn-positive aggregates (as detected by 81A) did not form in astrocytes (Supplementary Figure 1B). Moreover, the appearance of α-syn pathology required the presence of endogenous α-syn since α-syn-hWT pffs did not induce any pathology in primary neurons from α-syn −/− mice (Figure 1C). Furthermore, monomeric α-syn did not induce α-syn inclusions (data not shown), demonstrating that α-syn pffs alone seed the aggregates.
Figure 2. Minimal α-syn domain necessary for aggregate formation.
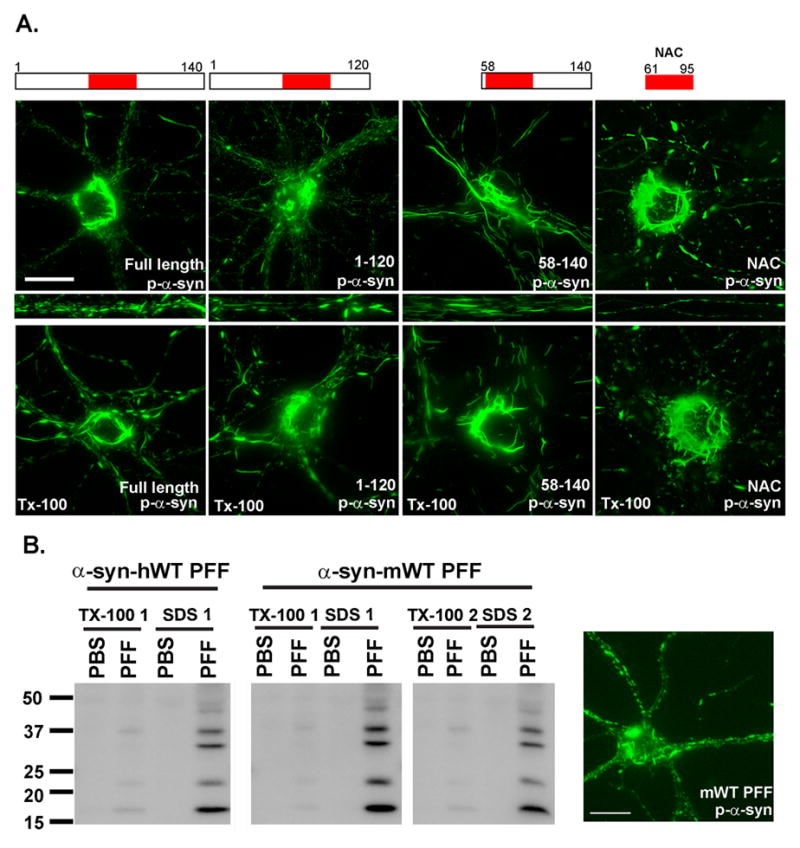
A. Pffs comprised of full length or indicated truncation α-syn mutants were added to DIV5 neurons and fixed 2 weeks later. Immunofluorescence detected p-α-syn, Tx-100 insoluble aggregates after addition of all constructs. Scale bar = 20 μm. B. α-syn-hWT or α-syn-mWT pffs were added to neurons on DIV5 and 2 weeks later were either fixed or extracted with 1% Tx-100 followed by 2% SDS. Immunofluorescence and immunoblots showed that, α-syn-mWT pffs induced the appearance of phosphorylated, Tx-100-insoluble, α-syn. Data represents 2 independent experiments. Scale bar = 20 μm
Figure 4. Time dependence of aggregate formation.

A. Top row: α-syn-hWT pffs were added to DIV5 neurons, and fixed either 4, 7 or 10 days later. Small puncta corresponding to neuritic p-α-syn were visible 4 days after pff addition, and by 7 days, neuritic p-α-syn levels increased, and accumulations were visible in some cell bodies. Ten days following addition of pffs, p-α-syn was seen throughout the neurites as small puncta, longer fibrous structures, and as somal accumulations. Bottom row: α-syn-hWT pffs were added to DIV10 neurons when α-syn expression at the presynaptic terminal is higher. Pathology progresses more quickly and are detectable at 2 days post-pff addition and aggregates in the cell bodies as early as 4 days post-pff addition. Scale bar = 50 μm. B. Immunoblots of DIV5 neurons sequentially extracted with 1% Tx-100 and 2% SDS, 4, 7, 10 and 14 days following PBS or α-syn-hWT pff addition. Over time, soluble α-syn was reduced with a concomitant increase in total and p-α-syn in the Tx-100-insoluble fraction. C, D. Double immunofluorescence for p-α-syn and the axonal marker, mouse tau (T49) (C). or the dendritic marker, MAP2 (D). P-α-syn predominantly colocalized with tau but not MAP2 4 days after pff addition. Two weeks after pff addition, aggregates were found in axons, cell bodies and dendrites where they colocalized with MAP2. Scale bar = 20 μm. See also Supplementary Figure 2.
Figure 7. Effects of aggregate formation on neuronal density and expression of synaptic proteins.

A. Neurons were fixed 2 weeks after treatment with PBS or α-syn-hWT pffs. Formation of α-syn aggregates caused recruitment of α-syn away from the presynaptic terminal such that it no longer colocalized with VAMP2. B. Immunoblots for the indicated synaptic proteins from neurons 2 weeks following treatment with PBS or α-syn-hWT pffs and sequentially extracted with buffer containing 1% Tx-100 followed by 2% SDS. Equal amounts of protein were loaded in each lane. Band intensities were quantified and expressed as average percent change (± SEM) in protein levels from pff treated neurons relative to PBS treated neurons. *indicates p<0.05, ** indicates p<0.01. GAPDH, N=4; βsyn, N=3; CSPα, N= 7; complexin, N=4, Synapsin I, N=3; Synapsin II, N=8; Snap25, N=8; VAMP2, N=6; Syntaxin I, N=7; Synaptophysin, N=7; Dynamin I, N=4; GlurI, N=4; PSD95, N=5. C. Neurons were fixed 4 (N=3), 7 (N=2) or 14 days (N=5) after addition of α-syn-hWT pffs or PBS and immunofluorescence was performed using NeuN to label neuronal nuclei. Numbers of nuclei were counted in cultures from WT neurons and α-syn −/− neurons (N=2). There was an approximately 40% decrease in cell number in α-syn-hWT pff treated, WT, but not α-syn −/− neurons relative to PBS treated controls only following 14 days of pff treatment. See also Supplementary Figure 3.
Immunoblot analyses were conducted on neuron lysates sequentially extracted with 1% Tx-100, followed by 2% SDS (Figure 1B). In contrast to PBS-treated neurons, those treated with α-syn-hWT pffs for 14 days showed >80% reduction of α-syn in the Tx-100-soluble fraction accompanied by a concomitant appearance of α-syn in the SDS-extractable fraction. Immunoblots of the SDS-extractable fraction also showed insoluble p-α-syn. A mouse specific anti-α-syn antibody did not detect α-syn-hWT pffs (Figure 1B, first lane on left), but detected bands in the neuron lysates similar to those labeled by the C-terminus specific α-syn antibody and mAB 81A. In addition, higher molecular weight species of α-syn were detected in the SDS fraction of all α-syn pffs treated cultures, and likely correspond to oligomeric and/or ubiquitinated α-syn (Li et al., 2005; Luk et al., 2009; Sampathu et al., 2003). Sequential extractions of primary hippocampal neurons from α-syn −/− mice 14 days following addition of α-syn-hWT pffs confirmed the absence of pathological α-syn or any other species of immunoreactive α-syn (Supplementary Figure 1C). Thus, these data demonstrate that α-syn pffs induced recruitment of soluble endogenous α-syn into insoluble, hyperphosphorylated α-syn aggregates.
Since α-syn is ubiquitinated in LBs and LNs, we studied α-syn aggregates that formed 14 days after addition of α-syn-hWT pffs and showed they were also ubiquitin positive (Figure 1D), and colocalized with p-α-syn. Because the exogenous α-syn-hWT pffs are not ubiquitinated or phosphorylated (Luk et al., 2009), these post-translational modifications must occur intracellularly as endogenous mouse α-syn accumulates. Thus, these α-syn aggregates share hallmark features of PD-like LNs and LBs allowing us to conclude that misfolded α-syn pffs seed and recruit normal, endogenous α-syn to form pathologic aggregates.
Pffs from the NAC domain of α-syn are sufficient to seed intracellular α-syn aggregates
Previous in vitro studies have shown that recombinant α-syn protein lacking N- or C-terminal residues, or a synthetic peptide containing only the NAC domain (amino acid residues 61–95), assemble into α-syn amyloid fibrils, and nucleate full length α-syn fibrillization (Giasson et al., 2001; Han et al., 1995; Kessler et al., 2003; Luk et al., 2009; Murray et al., 2003; Serpell et al., 2000). Thus, we asked if human α-syn pffs comprised of α-syn-1-120, α-syn-1-89, α-syn-58-140 or α-syn-NAC could seed formation of LBs and LNs in neurons. We observed that α-syn-1-120, and α-syn-1-89 pffs induced robust accumulation of endogenous p-α-syn aggregates that were Tx-100-insoluble (Figure 2A; data not shown), and they are morphologically indistinguishable from those formed by α-syn-hWT pffs. α-Syn-58-140 pffs also seeded formation of endogenous mouse α-syn aggregates that were hyperphosphorylated (Figure 2A). Moreover, pffs comprised of only the central hydrophobic α-syn-NAC domain also resulted in endogenous mouse α-syn fibrillar LB-like aggregates that were Tx-100-insoluble. Overall, our data demonstrate that α-syn pffs containing only the central, hydrophobic portion of α-syn-hWT are sufficient to seed conversion of endogenous α-syn into pathological aggregates.
Mice typically do not develop LBs except in the case of transgenic lines overexpressing mutant human α-syn. We thus asked if the formation of LB-like aggregates required human α-syn or if they can be seeded by α-syn pffs generated from recombinant mouse WT α-syn (α-syn-mWT) (Touchman et al., 2001). Immunoblots demonstrated that 14 days treatment of primary neurons with α-syn-mWT pffs induced appearance of p-α-syn in the Tx-100-insoluble fraction (Figure 2B). Immunofluorescence also showed that α-syn-mWT pffs induced formation of p-α-syn aggregates in neurites and somata. Thus, pathological PD-like α-syn aggregates can be induced by α-syn-mWT pffs and does not require the human protein.
Ultrastructure analysis of α-syn aggregates
Examination of the α-syn aggregates using transmission and immuno-EM demonstrated abundant filaments in neurons treated with either α-syn-hWT or α-syn-1-120 pffs (Figure 3A) for 14 days, but not PBS-treated neurons (data not shown). Remarkably, inclusions comprised of 14–16 nm thick filaments were seen throughout the cytoplasm, visualized by transmission EM. Two different immuno-EM detection systems, horse radish peroxidase (HRP) and immunogold amplification, were used to demonstrate that fibrils comprised of p-α-syn are found throughout the neuron. P-α-syn-positive fibrils were seen in the soma (Figure 3B, 3C), adjacent to the active zone of presynaptic terminals (Figure 3D), the post-synaptic terminal (Figure 3F) and throughout processes (Figure 3E). These data establish that the seeding and recruitment of endogenous mouse α-syn into hyperphosphorylated insoluble, filamentous aggregates recapitulate features of LBs and LNs in PD and other human synucleinopathies.
Figure 3. Ultrastructure of aggregates.

A. Transmission EM of α-syn-hWT pff treated neurons showed filaments in the neuronal soma (see box highlight). B. Immuno-EM of HRP-labeled p-α-syn inclusions were visualized in the neuronal soma near the nucleus. C. Filamentous inclusions in the neuronal soma were labeled with nanogold particles. Insert: higher magnification of labeled filaments. D. Presynaptic nanogold labeled filaments. E. Neuronal process with nanogold labeled α-syn filaments. F. HRP immunoreactivity showed p-α-syn in a postsynaptic ending. (Scale Bars: 1 μm, A, B, C, F; 500 nm, D, E.)
Time and concentration dependence of α-syn aggregate formation
To determine the temporal sequence of α-syn aggregate formation, α-syn-hWT pffs were added to the neurons at DIV5. P-α-syn immunostaining was not detectable until 4 days later when small aggregates began to appear, exclusively in the neurites, albeit at low levels (Figure 4A, upper series). By 7 days after α-syn-hWT pffs addition, there was an increase in α-syn pathology with some cell bodies showing α-syn accumulations. By 10 days post α-syn-hWT pff addition, the overall p-α-syn immunostaining was more intense, and p-α-syn aggregates in the neurites appeared both punctate and fibrillar resembling LNs that were longer than the aggregates observed 4 or 7 days after α-syn-hWT pffs addition.
The sequence of events revealed by immunofluorescence was confirmed by biochemical experiments of sequentially extracted neurons (Figure 4B). Four days after α-syn-hWT pffs addition, the majority of α-syn was found in the Tx-100-soluble fraction and showed levels similar to PBS-treated neurons. In PBS-treated control neurons, there was an increase in α-syn levels by DIV10 as demonstrated previously (Murphy et al., 2000). In contrast, 7–10 days following α-syn-hWT pff treatment, soluble levels of α-syn were reduced, accompanied by a concomitant increase of α-syn into the Tx-100-insoluble fraction. Thus, these data indicate that α-syn-hWT pff-induced recruitment of mouse α-syn into the insoluble fraction with a lag phase of a few days followed by a progressive increase in insoluble p-α-syn.
Since levels of α-syn, and its concentration at the presynaptic terminals increase as primary neurons mature, (Murphy et al., 2000; Figure 4B, day 4 PBS vs. day 10 PBS), we asked if adding pffs to mature neurons would enhance the rate of aggregation. When α-syn-hWT pffs were added to DIV 10 neurons, aggregates were visible in neurites 2 days later (Figure 4A, lower series), in contrast to 4 days required after addition of pffs to DIV5 neurons. By 4 days following α-syn-hWT pff treatment of DIV10 neurons, small punctate aggregates were detected throughout the neurites and some somata also showed accumulations, again unlike 4 days after adding pffs to DIV5 neurons in which α-syn pathology was exclusively in neurites. Seven days after α-syn-hWT pff treatment of DIV10 neurons, the pathology was extensive, similar to 10 days α-syn-hWT pff treatment of DIV5 neurons (Figure 4A). Thus, α-syn aggregates develop faster in mature neurons, consistent with in vitro studies demonstrating that the rate of fibril formation positively correlates with α-syn concentrations (Wood et al., 1999).
We next examined if the amount of α-syn pathology correlated with the amount of fibrils added. We found progressive decreases in the amount of somatic and neuritic pathology correlated with 10-fold serial dilutions of α-syn-hWT pffs added (in ng/mL: 100, 10, 1, 0.1; Supplementary Figure 2). Thus, the rate and extent of pathology depends on the amount of α-syn pffs, and that small quantities of α-syn pffs are sufficient to seed α-syn aggregate formation, consistent with in vitro studies showing that the rate of seeded assembly depends on the initial concentrations of α-syn pffs (Wood et al., 1999).
Initial formation of α-syn pathology within axons
Because α-syn normally localizes to the presynaptic terminal and since α-syn puncta initially appeared in neurites, we hypothesized that α-syn-hWT pffs recruited presynaptic α-syn into insoluble aggregates that then propagate from the axons to the cell bodies. To demonstrate that the pathology initiated in axons, we conducted double labeling immunofluorescence studies using a mAB specific for mouse tau (T49, an axonal marker) and 81A. P-α-syn aggregates colocalized predominately with tau 4 days after pff addition (Figure 4C; upper panel), but not with the dendritic marker, microtubule associated protein 2 (MAP2) (Figure 4D, upper panel), indicating that α-syn accumulations were initiated in axons. However, by 14 days, when more accumulations appeared in the somata, the α-syn aggregates were seen in axons (Figure 4C, lower panel), in cell bodies, and proximal dendrites where they colocalized with MAP2 (Figure 4D, lower panel). Thus, α-syn is recruited away from the presynaptic terminal with subsequent spread via axons to other parts of the polarized neuron.
Enhanced endocytosis of α-syn pffs increases the extent of pathology
To determine if α-syn-hWT pffs can gain access to the cytoplasm to seed recruitment of endogenous α-syn, we performed two-stage immunofluorescence using antibodies that recognize only human α-syn pffs. Live neurons were labeled at 4°C with mAB Syn204 followed by fixation, permeabilization, incubation with the antibody, LB509 (Giasson et al., 2000),. Thus, mAB Syn204 labeled only extracellular hWT pffs whereas LB509 recognized both extracellular and intracellular hWT pffs. Many α-syn-hWT pffs remained outside the neuron and were double-labeled with both mAB Syn204 and LB509 (yellow in the merged image, Figure 5A). However, significant amounts of small puncta labeled exclusively with LB509 (green, arrowheads highlight examples in the merged image), suggesting that α-syn-hWT pffs gain entry inside the neuron, as demonstrated previously for both α-syn and tau amyloid fibrils (Luk et al., 2009; Guo and Lee, 2011). Furthermore, double labeling immunofluorescence in fixed, permeabilized neurons with mAB 81A and mAB Syn204 showed p-α-syn accumulating near seeds of α-syn-hWT pffs (Figure 5B). A 3D view constructed from serial confocal images demonstrated colocalization between α-syn-hWT pffs (Syn204) and p-α-syn (81A) in the XY, XZ and YZ planes (Figure 5C), further confirming that intracellular pffs seed recruitment of endogenous α-syn. Since p-α-syn is exclusively intracellular, our data indicate that pffs enter the cytoplasm where they initiate accumulation of pathologic p-α-syn.
Figure 5. α-syn-hWT pffs are internalized into neurons.

A. Live neurons were incubated with mAB Syn204 (red) to label extracellular α-syn-hWT pffs, followed by fixation, permeabilization and incubation with LB509 (green) to label both intracellular and extracellular α-syn-hWT pffs. Extracellular α-syn-hWT pffs are visualized as yellow in the merged image. Arrowheads highlight examples of internal α-syn-hWT pffs (green). Scale bar = 10 μm. B. Fixed and permeabilized neurons were double labeled with mABs 81A (green) to detect p-α-syn and Syn 204 (red) to detect α-syn-hWT pffs. P-α-syn can be visualized accumulating from seeds of α-syn-hWT pffs. C. α-syn-hWT pffs were added to DIV5 neurons, fixed 14 days later and immunofluorescence was performed to label extracellular α-syn-hWT pffs (red) and p-α-syn (green). A z-stack of confocal images shows that puncta corresponding to α-syn-hWT pffs colocalized with p-α-syn within a neurite, suggesting that pathologic p-a-syn grows from intracellular pffs. D. DIV5 neurons were treated with either α-syn-hWT pffs alone or pffs with 1 μg/mL or 5 μg/mL of WGA. To inhibit WGA endocytosis, neurons were preincubated with 0.1 M GlcNAC followed by incubation with pffs, GlcNAC and 5 μg/mL of WGA. Neurons were fixed 4 days later. Immunoblots and immunofluorescence showed that WGA dose-dependently increased the extent of insoluble p-α-syn.
To begin assessing the mechanism by which pffs gain entry to the cytoplasm, we treated neurons with α-syn-hWT pffs in the presence of wheat germ agglutinin (WGA) which binds N-acetylglucosamine (GlcNAC) and sialic acids at the cell surface and induces adsorptive-mediated endocytosis (Banks et al., 1998; Broadwell et al., 1988; Gonatas and Avrameas, 1973). To determine the effects of WGA on formation of α-syn aggregates, neurons were treated at DIV5 and fixed for immunofluorescence 4 days later. When incubated with α-syn-hWT pffs alone, few p-α-syn puncta were visible in a subset of neurites (Figure 5D). Co-incubation of pffs with WGA dose-dependently increased the extent of p-α-syn pathology. In addition to small puncta, longer, continuous p-α-syn filaments were visible, and α-syn pathology was present in the cell body, particularly with 5 μg/mL of WGA treatment. Furthermore, the addition of 0.1 M GlcNAc, a competitive inhibitor of WGA, reduced the effects of WGA on α-syn pff induced aggregate formation. Immunoblots of sequentially extracted neurons confirm that WGA-mediated endocytosis enhances formation of pathologic α-syn. Four days after treatment with α-syn-hWT pffs alone, the majority of α-syn remained in the Tx-100 extractable fraction, whereas co-incubation of α-syn-hWT pffs with 5 μg/mL of WGA increased the amount of Tx-100 insoluble α-syn. Taken together, our findings indicate that α-syn pffs gain access to the neuronal cytoplasm by adsorptive endocytosis.
Intracellular propagation of pathologic α-syn
To determine if direct addition of α-syn pffs to either neurites or somata leads to propagation of pathologic α-syn aggregates throughout the neuron, we utilized microfluidic culture devices which isolate the neuronal processes from the cell bodies via a series of interconnected microgrooves (Taylor et al., 2005). C-terminally myc-tagged α-syn-1-120 pffs added to the neuritic chamber (Figure 6A) resulted in p-α-syn-positive aggregates within axons and cell bodies (Figure 6B and 6C). Aggregates were morphologically identical to those seen in primary neurons directly exposed to pffs, and they were also insoluble in Tx-100 (Figure 6D). Anti-myc immunostaining suggested that pffs did not enter into the somal compartment (Figure 6C and 6D) or microgrooves. Thus, these data indicate that pathological p-α-syn can form within isolated neurites, and is propagated retrogradely to the cell bodies.
Figure 6. Intracellular propagation of pathologic α-syn aggregates.

A. Hippocampal neurons were grown in microfluidic chambers interconnected by channels accessible only to neuronal processes. α-Syn-1-120-myc pffs were added to the compartment containing exclusively neurites of DIV 5 neurons. B. Neurons stained with SNL-4 (total syn) 7 days following treatment with α-syn pffs showing the distribution of neurites, somata, and exogenous pffs which appear as large puncta in the neuritic compartment. P-α-syn is found within the microchannels as well as cell bodies in the somatic chamber, indicating propagation of α-syn pathology from neurites towards the somata. Inset shows high power images of the somatic (top) and neuritic (bottom) compartments. C, D. α-Syn-1-120-myc pff -treated neurons were double-stained using anti-myc and syn202 (Syn) before (C) and following extraction with Tx-100 (D) demonstrating the presence of insoluble p-α-syn within neurons and processes. myc-positive pffs were confined to the neuritic compartment. E. α-syn-1-120-myc pffs were added to the somal-containing compartment of DIV5 neurons. F. Neurons immunostained for tau and p-α-syn showing pathology extending within axonal processes from the somal compartment into the neuritic compartment. G, H α-Syn-1-120-myc pff -treated neurons double-stained using anti-Myc and Syn202 before (G) and following extraction with Tx-100 (H). α-Syn within the axons is Tx-100-insoluble. α-syn-1-120-myc pffs remained confined to the somal compartment and thus p-α-syn within the neuritic compartment resulted from propagation from the perikarya to the neurites. Scale bar = 50 μm in B, 40 μm in C, D and F-H.
We also exposed neuronal somata that were isolated from neurites in the microfluidic devices to α-syn-1-120-myc pffs and assessed the extent of α-syn pathology in the processes (Figure 6E). As expected, neurons treated with α-syn-1-120-myc pffs formed somatic p-α-syn pathology (Figure 6F). P-α-syn aggregates were also detected in axons that extended through the microgrooves into the neurite chamber, as revealed by co-labeling with tau (Figure 6F). Again, α-syn aggregates throughout the axon were Tx-100-insoluble, and immunofluorescence using the anti-myc antibody demonstrated that α-syn-1-120-myc pffs were confined to the somatic compartment (Figure 6G, 6H). Thus, we conclude that pathologic p-α-syn aggregates also propagate in the anterograde direction.
Formation of aggregates leads to neuron loss and diminished levels of select synaptic proteins
α-syn resides predominantly at the presynaptic terminal and previous reports indicate that it acts as a co-chaperone, in concert with another chaperone, cysteine-string protein α (CSPα), to maintain SNARE complex formation by binding to VAMP2/synaptobrevin 2 (Burre et al., 2010; Chandra et al., 2005; Greten-Harrison et al., 2010). Thus, we examined the consequences of recruitment of α-syn into insoluble aggregates on synaptic protein distribution and expression. In PBS-treated control neurons, α-syn colocalized with VAMP2 at the presynaptic terminal. Addition of α-syn-hWT pffs led to a depletion of α-syn from the presynaptic terminal such that it showed minimal colocalization with presynaptic VAMP2 (Figure 7A). To further investigate the molecular consequences of recruitment of endogenous α-syn into insoluble aggregates, we examined additional synaptic proteins that could be impacted by the pathological sequestration of α-syn into aggregates and away from the presynaptic terminal. Although β-synuclein (β-syn), another member of the same family of neuronal proteins as α-syn, but lacking the NAC domain, colocalized with α-syn at presynaptic terminals in control neurons (Murphy et al., 2000), α-syn-hWT pff addition did not change the presynaptic localization of β-syn (Supplementary Figure 3). Furthermore, Tx-100 extraction showed that, unlike pathological α-syn, which localized to detergent insoluble aggregates, β-syn remained soluble (Supplementary Figure 3). Immunoblot analyses showed that endogenous β-syn was Tx-100 soluble 14 days after adding α-syn pffs (Figure 7B) and protein levels in pff treated neurons were not statistically significantly different from PBS treated neurons. Thus, like LBs in PD brains, the aggregates that developed in primary neurons are comprised of insoluble α-syn, but not β-syn (Spillantini et al., 1998). Importantly, this is consistent with the selective recruitment of α-syn by pffs as opposed to the indiscriminate disruption of adjacent presynaptic components.
Nonetheless, we were able to detect statistically significant reductions in a subpopulation of synaptic proteins two weeks after the addition of α-syn-hWT pffs, including the synaptic vesicle-associated SNARE proteins, Snap25 and VAMP2, as well as soluble proteins that participate in SNARE complex assembly or the exo-endocytic synaptic vesicle cycle such as CSPα, and synapsin II (Figure 7B). Levels of other synaptic proteins showed slight, but not statistically significant reductions. Changes were not observed in GAPDH, the plasma membrane-associated SNARE protein, syntaxin 1, or the transmembrane synaptic protein synaptophysin.
Since loss of synaptic proteins may correlate with neurodegeneration, we asked if the accumulation of α-syn aggregates leads to neuron loss. NeuN-positive neurons were counted in cultures treated with PBS or α-syn-hWT pffs 4, 7 or 14 days after α-syn pff addition. While there was a slight, but not statistically significant decrease in number of neurons 7 days following α-syn-hWT pff treatment, by 14 days post pff treatment, there was a significant 40% decrease in neurons relative to PBS controls (Figure 6C). Cell death did not occur in α-syn-hWT pff treated neurons derived from α-syn −/− mice, demonstrating that intracellular aggregates, rather than the mere addition of exogenous pffs, caused neuron death. Finally, using ethidium homodimer to detect dead cells and Hoechst 33342 to detect total cells, we demonstrated a ~68% increase in cell death 14 days after pff-treatment (56.6%) versus PBS-treated (33.6%) neurons.
Disruption in network activity matches the progression of α-syn pathology
The decreased levels of synaptic proteins suggest impairment in neural network activity following accumulation of α-syn inclusions. Calcium imaging of hippocampal neurons loaded with the calcium-sensitive fluorescent dye, Fluo-4 AM, was performed to investigate the effect of α-syn aggregates on the activity patterns of the in vitro neural network established by these cultured neurons. The spontaneous activity of neurons treated with PBS was characterized by flickering events, intermixed with network-wide bursts when nearly all the neurons were simultaneously firing as reflected by a high synchronization index (Figure 8B). In contrast, neurons treated with α-syn-hWT pffs showed a significant decrease in synchronized activity as early as 4 days after treatment. At this time point, low levels of α-syn aggregates were visualized exclusively in axons by immunofluorescence microscopy, and no pathological α-syn was detected biochemically (Figure 4A, 4B). Yet, this was sufficient to impair coordinated network activity. This reduction in synchronized activity persisted at 7, 10 and 14-days after α-syn-hWT pff treatment (Figure 8B). In contrast, α-syn-hWT pff-treated neurons from α-syn −/− mice showed no impairments in the synchronization index, indicating that these effects are selective for neurons harboring α-syn aggregates and do not result from exogenously added pffs.
Figure 8. Effect of aggregate formation on neural network activity.

Calcium imaging on hippocampal neurons loaded with the calcium-sensitive fluorescent dye, Fluo4-AM was performed. A. PBS-treated WT neurons showed flickering events and simultaneous bursting. The spontaneous activity in α-syn-hWT pff treated WT neurons showed reduced coordination and frequency of oscillations. B. The level of coordinated spontaneous activity was quantified as the synchronization index. α-syn-hWT pff-treated neurons (red) showed a significant decrease in synchronicity by day 4, relative to PBS-treated neurons (blue) and the deficit continued for longer treatment duration. Primary neurons from α-syn −/− mice treated with α-syn-hWT pffs (purple) did not show reductions in the synchronization index relative to PBS-treated neurons (green). C. Excitatory tone in the network was determined by recording spontaneous activity and then by forcing synchronous oscillations via network disinhibition with bicuculline. Incremental concentrations of NBQX were added until coordinated activity stopped and the excitatory tone was reported as [NBQX]/Kd. D. Excitatory tone in PBS (blue) and α-syn-hWT pff (red) treated neurons, showed significant decreases by 10 as well as 14 days post-pff treatment. Addition of α-syn-hWT pffs to α-syn −/− neurons did not affect excitatory tone. E. Functional network connectivity, derived from the rasters in (A), is depicted as nodes (neurons) of varying sizes, where the size of a given node is scaled to reflect the total number of connections to that particular node. F. The average number of connections per neuron was determined from functional connectivity map. Compared to PBS, α-syn-hWT pff treated neurons had fewer numbers of functional connections. Connectivity of α-syn-hWT pff treated α-syn −/− neurons were similar to PBS-treated neurons. PBS-treated: day 4, N=9; day 7, N=11; day 10, N=10; day 14, N=9. PFF-treated: day 4, N=9; day 7, N=12; day 10, N=11; day 14, N=9.
We next determined whether the progressive recruitment of α-syn into pathologic aggregates correlated with changes in the excitatory tone of the network. First, synchronous oscillations were forced using the GABA(A) antagonist, bicuculline, to abolish inhibitory input, followed by increasing doses of the AMPA receptor antagonist, NBQX, until synchronous oscillations stopped (Figure 8C). The final concentration of NBQX required to impair activity within the excitatory network determined the excitatory tone. No significant changes in excitatory tone was detected in cultures 4 or 7 days after α-syn-hWT pff treatment but by 10 and 14 days after treatment, when increasing accumulation of neuritic and perikaryal pathology was observed, there were significant reductions in excitatory tone (Figure 8D), reflecting compromised synaptic activity. Again, neurons from α-syn −/− mice did not show impairments in excitatory tone at 10 and 14 days post-pff treatment, confirming that the effects result from the accumulation of endogenous α-syn aggregates.
Since spatiotemporal patterns of activity are shaped by the underlying connectivity architecture and the relative balance of excitation and inhibition, we used network activity patterns to determine the functional connectivity in PBS and α-syn-hWT pff treated neurons. As neurons matured in vitro, the number of functional connections increased and eventually plateaued (Figure 8F). The timeframe for this correlated well with neurite sprouting and synapse stabilization based on previous studies of developing connections in vitro (Soriano et al., 2008).
However, in α-syn-hWT pff treated WT, but not α-syn −/− neurons, the maturation of functional connections never reached the level achieved in PBS treated cultures, as a significant reduction was observed 10 days after α-syn-hWT pff treatment (Figure 8F). This functional connectivity was severely compromised 14 days post treatment and the network consisted of just a few sparse connections at this time point (Figure 8E-F). In summary, the formation of insoluble aggregates of endogenous α-syn results in early disruption in coordinated network activity. Later, as more α-syn inclusions develop and propagate throughout the neuron, excitatory tone is decreased and functional connectivity is greatly reduced.
Discussion
Here we demonstrate that seeds derived from α-syn amyloid fibrils generated with pure recombinant full length and truncated human WT α-syn, when directly added to mouse primary hippocampal neurons, are internalized and induce the recruitment of endogenous soluble α-syn into insoluble pathologic LB-like and LN-like α-syn aggregates resembling those found in human synucleinopathies. Indeed the verisimilitude of these α-syn aggregates in cultured mouse neurons to LBs and LNs found in PD brains is striking because they are: found in perikarya and extensively in neurites, insoluble, filamentous by EM and immuno-EM, hyperphosphorylated, ubiquitinated, and exclude β-syn (Baba et al., 1998; Duda e al., 2000; Fujiwara et al., 2002; Spillantini et al., 1998). Notably, these aggregates initially formed within axons, sequestering endogenous α-syn away from presynaptic terminals, followed by propagation into the somata. Over time, formation of these α-syn aggregates leads to selective alterations in synaptic proteins, compromises neuronal excitability and connectivity and culminates in neuron death. Thus, we have developed a novel neuronal culture model of PD-like α-syn inclusion formation that allows for the dissection of the mechanisms leading to the formation of LBs and LNs, as well as for studies of the impact of these inclusions on the function and viability of affected neurons. Moreover, since the majority of PD and DLB cases are sporadic and are not caused by mutations or overexpression of α-syn, our neuronal model system provides for the first time a means to study the pathogenesis of α-syn in sporadic PD, as well as other α-synucleinopathies.
We show that α-syn pffs made from pure, recombinant protein are highly potent in the recruitment of the endogenously expressed protein into LB-like and LN-like α-syn pathology, in contrast to previous studies that have relied on experimental manipulations such as protein overexpression of WT and mutant proteins, and/or extrinsic factors to introduce pffs into cells (Clavaguera et al., 2009; Frost et al., 2009; Guo and Lee; Luk et al., 2009). These results thus provide support that α-syn amyloid fibrils alone are sufficient to seed and drive α-syn pathology in healthy neurons. Indeed, our findings can plausibly account for the observation that fetal grafts of embryonic neurons in diseased PD brains develop LBs over time, since this could be caused by the direct uptake of fibrillar α-syn seeds from diseased neurons in the brains of these patients (Kordower et al., 2008a; Kordower et al., 2008b; Li et al., 2008). Furthermore, our data also suggest a pathological mechanism whereby misfolded α-syn species can amplify and propagate in the CNS. Because it is possible that both mature α-syn fibrils and oligomers (Waxman and Giasson, 2009; Winner, et al. 2001) induce α-syn pathology, additional studies are needed to determine the nature of the pathogenic species capable of producing these changes. In addition, although the source of the nidus that initiates α-syn misfolding in PD and related diseases remains enigmatic (e.g. whether it arises from genetic mutations or environmental toxins), we provide provocative evidence that small amounts of misfolded α-syn pffs can trigger the spread of α-syn pathology throughout the entire neuron.
Two-stage immunofluorescence to distinguish extracellular from internal pffs and confocal microscopy to demonstrate colocalization between pffs and p-α-syn suggest that small amounts of α-syn pffs gain access to the neuronal cytoplasm where they can seed α-syn misfolding and accumulation into hyperphosphorylated α-syn inclusions. Co-incubation of α-syn pffs with WGA enhances the extent of pathology, implicating adsorptive-mediated endocytosis as a potential mechanism by which pffs gain entry to the neuron. While the mechanisms by which α-syn pffs are internalized and released into the cytosol require further investigation, it is apparent that they efficiently induce pathology. High concentrations of α-syn are present in presynaptic terminals, where it associates with vesicular membranes and undergoes rapid exchange between bound and unbound states (Fortin et al., 2010). Thus, high local concentrations of presynaptic α-syn, coupled with its dynamic characteristics, may facilitate recruitment of endogenous mouse α-syn by the internalized α-syn pffs to form insoluble fibrils.
We show that formation of α-syn pathology is more efficient in mature neurons with higher levels of α-syn expression at presynaptic terminals. Interestingly, levels of α-syn increase with age (Chu and Kordower, 2007) and α-syn gene duplication and triplication can lead to PD (Singleton et al., 2003). Thus, aging-dependent or gene dosage-dependent increased expression of α-syn may render these neurons more susceptible to α-syn inclusion formation after internalization of α-syn seeds. Future studies will determine if PD-associated mutations in α-syn or overexpression of α-syn caused by gene duplication or triplication enhance the kinetics of α-syn aggregate formation, but it is clear from our data that normal neuronal expression of α-syn is sufficient for seeding of α-syn pathology after exposing these neurons to α-syn pffs.
Our model has allowed us to determine some of the consequences of potential α-syn dysfunction resulting from its recruitment into inclusions, which include reduced levels of synaptic proteins, impaired neuronal function and eventual death of affected neurons. Diminished neuronal synchronization begins early after pff addition when small aggregates are visible only in axons, suggesting that even a minor burden of α-syn pathology can have a major impact on the coordinated activation of neuronal ensembles. By 10 days and 14 days post-pff treatment, when pathology is extensive, neuronal excitability and connectivity is substantially reduced, which may be accounted for by the reductions in presynaptic proteins. Alterations in the expression and localization of these proteins occurs upon ablation of all three synuclein family members or overexpression of α-syn (Burre et al., 2010; Greten-Harrison et al., 2010; Nemani et al., 2010). Sequestration of α-syn away from the presynaptic terminal into insoluble inclusions may impair the homeostasis of presynaptic proteins and consequently, synaptic vesicle exocytosis, as suggested by previous studies demonstrating that α-syn, in cooperation with CSPα, may act as a chaperone to maintain presynaptic SNARE complex assembly (Burre et al., 2010; Chandra et al., 2005). Over time, disruptions in the synaptic vesicle exo-endocytic cycle may contribute to neurodegeneration found in PD and DLB.
Conclusions
For enigmatic reasons, LB pathology in sporadic PD disease progresses in a temporally and topologically sequential manner, and it has been suggested that the pathology is transmitted from neuron-to-neuron, presumably by spreading along axons (Braak and Braak, 1991; Braak et al., 2003). Our findings suggest that LB/LN pathology can be induced by misfolded α-syn and is propagated within neurons. Mounting evidence suggests that propagation of protein aggregates may be a unifying mechanism of disease progression in AD and PD (Clavaguera et al., 2009; Desplats et al., 2009; Frost et al., 2009; Guo and Lee, 2011; Luk et al., 2009). Our findings that small amounts of α-syn pffs directly induce endogenous α-syn to form pathological aggregates that are spread throughout the neuron and accumulate as LB-like and LN-like inclusions support this unifying mechanism of disease progression and therefore have important implications for understanding the onset and progression as well as etiopathogenesis of sporadic PD and other neurodegenerative disorders. Thus, our findings open up new avenues of research into understanding mechanisms underlying the development LB and LN pathology, their impact on neuronal function, and discovering therapies for PD and other α-synucleinopathies.
Experimental Procedures
Primary Neuronal Cultures
Primary neuronal cultures were prepared from E16-E18 C57BL/6 mouse brains (Charles River, Wilmington, MA) and α-syn−/− mice (Abeliovich, et al., 2000). All procedures were performed according to the NIH Guide for the Care and Use of Experimental Animals and were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Dissociated hippocampal neurons were plated onto poly-D-lysine coated coverslips (Carolina Biological Supply, Burlington, NC) or dishes at 20,000–40,000 cells/cm2 or 70–100,000 cells/cm2, respectively. Most experiments were performed at 19 DIV.
Preparation and fibril transduction
Recombinant full length and truncated α-syn with and without a C-terminal myc-tag, were purified as previously described (Giasson et al., 2001). α-Syn pffs were generated by incubating purified α-syn (5 mg/mL in PBS) at 37°C with constant agitation for 5 days, followed by aliquoting and storage at −80 °C. The presence of amyloid was confirmed using Thioflavin T fluorometry. Fibrils of α-syn synthetic NAC peptide (amino acids 61–95) (Biotechnology Resource Center, Yale University) were generated as described (Giasson et al., 2001). Pffs were diluted in PBS at 0.1 mg/mL, sonicated several times and diluted in neuronal media. For a 24 well tray, 1 μg/mL of pffs were added, and 5 μg/mL of α-syn pffs were added to a 60 cm dish. For WGA experiments, α-syn pffs were incubated with 1 μg/mL or 5 μg/mL WGA or preincubated for 1 hour (h) with 0.1 M GlcNAC followed by incubation with media containing α-syn pffs, GlcNAC and WGA.
Indirect Immunofluorescence
Neurons were fixed with 4% paraformaldehyde/4% sucrose in PBS containing followed by permeabilization with 0.1% Triton X-100. To determine if the pathologic α-syn aggregates were detergent-insoluble, neurons were fixed with 4% paraformaldehyde, 4% sucrose and 1% Tx-100 (Luk et al., 2009). Neurons were incubated in primary antibodies (Supplemental Table 1) followed by Alex fluor-conjugated secondary antibodies (Invitrogen; Carlsbad, CA). Two-stage immunofluorescence was performed as described previously (Guo and Lee, 2011) to distinguish between extracellular and intracellular α-syn-hWT pffs using mABs LB509 and Syn204. Unless otherwise stated in the figure legends, all experiments were performed a minimum of 3–10 times.
EM and Immuno-EM
Primary hippocampal neurons were fixed 14 days following addition of pffs. Neurons for transmission EM were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4, post-fixed for 1h in 1% OsO4 and 1.5% potassium ferrocyanide in 0.05 M cacodylate buffer. For immuno-EM, neurons were fixed in periodate-lysine-paraformaldehyde and permeabilized with 0.05% saponin in PBS with 2% fish gelatin (PBS-FG) and 0.05% thimerosal followed by incubation in mAB 81A. Neurons were then incubated in biotinylated horse anti-mouse IgG (Vector Laboratories, Burlingame, CA) followed by Avidin-Biotin Complex Elite (Vector Laboratories, Burlingame, CA) and post-fixed for 15 min in 1.5% glutaraldehyde in 0.1 M cacodylate buffer with 5% sucrose and developed in DAB. For immunogold labeling, neurons were fixed and incubated in mAB 81A as for HRP immuno-EM followed by incubation with goat anti-mouse IgG coupled to nanogold (Nanoprobes, Yaphank, NY). The nanogold labeled neurons were post-fixed, and gold toned with 0.05% gold chloride.
Sequential extraction and Immunoblot analyses
Neurons were scraped into 1% Tx-100 in Tris-buffered saline (TBS) (50 mM Tris, 150 mM NaCl, pH7.4) and protease and phosphatase inhibitor cocktail at 4°C. Lysates were sonicated and centrifuged at 100,000 x g for 30 min. The pellet was washed and suspended in 2% SDS in TBS. Samples were separated by SDS-PAGE and immunoblotting was performed using primary antibodies described in Supplementary Table 1. All immunoblots were performed a minimum of 3–8 times.
Microfluidic Chambers
Microfluidic neuronal culture devices with 2 somal compartments connected by a series of microgrooves were obtained from Xona Microfluidics (Temecula, CA). Glass coverslips (Corning Inc.) were coated with poly-d-lysine and affixed to neuronal devices as per the manufacturer’s instructions. A total of 10,000 dissociated hippocampal neurons were plated. A 50 μL difference in media volume was maintained between the two compartments to regulate the direction of flow. α-Syn 1-120-myc pffs (2 μg) were added to the neuritic compartment (retrograde experiments) or the somal compartment (anterograde experiments) and were fixed 7–12 days later. The retrograde experiments were repeated 4 times and anterograde experiments repeated 3 times, each in triplicates.
Calcium imaging and network analyses
Hippocampal neurons were plated on MatTek dishes at 300,000 cells/dish and treated with PBS or 5 μg/mL α-syn-hWT pffs. Neurons were loaded with Fluo4-AM (1 μM, Invitrogen, Carlsbad, CA). Spontaneous calcium activity from ~200 neurons was recorded for 5 min, at 10Hz acquisition. Synchronous oscillations were forced with bicuculline (100 μM, Tocris) and increasing doses of the AMPAR antagonist, NBQX. When synchronous oscillations stopped, this final concentration of NBQX was used as an indication of excitatory tone (Breskin et al., 2006). Custom-coded MATLAB scripts were used to analyze the images.
Supplementary Material
Acknowledgments
Kurt Brunden, James Soper, Linda Kwong, Eddie Lee and Jing Guo are thanked for reading the manuscript and for helpful discussions, and Patrick O’Brien, Christine Schultheiss, Victoria Kehm, Christina Haas, and Jeffrey Yeh for technical assistance. This work was supported by National Institutes of Health Grants NS053488, the Picower Foundation, the Benaroya Foundation, and the Stein-Bellet Family Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64:783–790. doi: 10.1016/j.neuron.2009.12.016. [DOI] [PubMed] [Google Scholar]
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VMY, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Akerstrom V, Kastin AJ. Adsorptive endocytosis mediates the passage of HIV-1 across the blood-brain barrier: evidence for a post-internalization coreceptor. J Cell Sci. 1998;111(Pt 4):533–540. doi: 10.1242/jcs.111.4.533. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Breskin I, Soriano J, Moses E, Tlusty T. Percolation in living neural networks. Phys Rev Lett. 2006;97:188102. doi: 10.1103/PhysRevLett.97.188102. [DOI] [PubMed] [Google Scholar]
- Broadwell RD, Balin BJ, Salcman M. Transcytotic pathway for blood-borne protein through the blood-brain barrier. Proc Natl Acad Sci U S A. 1988;85:632–636. doi: 10.1073/pnas.85.2.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chu Y, Kordower JH. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol Dis. 2007;25:134–149. doi: 10.1016/j.nbd.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Gur TL, Montine TJ, Robertson D, Biaggioni I, Hurtig HI, Stern MB, Gollomp SM, Grossman M, Lee VMY, Trojanowski JQ. Immunohistochemical and biochemical studies demonstrate a distinct profile of alpha-synuclein permutations in multiple system atrophy. J Neuropathol Exp Neurol. 2000;59:830–841. doi: 10.1093/jnen/59.9.830. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Nemani VM, Nakamura K, Edwards RH. The behavior of alpha-synuclein in neurons. Mov Disord. 2010;25(Suppl 1):S21–26. doi: 10.1002/mds.22722. [DOI] [PubMed] [Google Scholar]
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VMY. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson’s disease. J Neurosci Res. 2000;59:528–533. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Murray IV, Trojanowski JQ, Lee VMY. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- Gonatas NK, Avrameas S. Detection of plasma membrane carbohydrates with lectin peroxidase conjugates. J Cell Biol. 1973;59:436–443. doi: 10.1083/jcb.59.2.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, Chandra SS. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A. 2010;107:19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VMY. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Weinreb PH, Lansbury PT., Jr The core Alzheimer’s peptide NAC forms amyloid fibrils which seed and are seeded by beta-amyloid: is NAC a common trigger or target in neurodegenerative disease? Chem Biol. 1995;2:163–169. doi: 10.1016/1074-5521(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 121:715–725. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler JC, Rochet JC, Lansbury PT., Jr The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry. 2003;42:672–678. doi: 10.1021/bi020429y. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008a;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Olanow CW, Freeman TB. Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord. 2008b;23:2303–2306. doi: 10.1002/mds.22369. [DOI] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Bjorklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jakala P, Hartmann T, Price DL, Lee MK. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci U S A. 2005;102:2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VMY. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VMY. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IV, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, Trojanowski JQ, Lee VMY. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry. 2003;42:8530–8540. doi: 10.1021/bi027363r. [DOI] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampathu DM, Giasson BI, Pawlyk AC, Trojanowski JQ, Lee VMY. Ubiquitination of alpha-synuclein is not required for formation of pathological inclusions in alpha-synucleinopathies. Am J Pathol. 2003;163:91–100. doi: 10.1016/s0002-9440(10)63633-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Soriano J, Rodriguez Martinez M, Tlusty T, Moses E. Development of input connections in neural cultures. Proc Natl Acad Sci U S A. 2008;105:13758–13763. doi: 10.1073/pnas.0707492105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998;251:205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchman JW, Dehejia A, Chiba-Falek O, Cabin DE, Schwartz JR, Orrison BM, Polymeropoulos MH, Nussbaum RL. Human and mouse alpha-synuclein genes: comparative genomic sequence analysis and identification of a novel gene regulatory element. Genome Res. 2001;11:78–86. doi: 10.1101/gr.165801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J Biol Chem. 1999;274:19509–19512. doi: 10.1074/jbc.274.28.19509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.