

Abstract
Heat shock protein 70 (Hsp70) has been shown to have an anti-apoptotic function, but its mechanism is not clear in heart. In this study, we examined the effect of Hsp70 deletion on AIF-induced apoptosis during ischemia/reperfusion (I/R) in vivo. Although Hsp70 KO and WT mice demonstrated similar amounts of AIF released from mitochondria after I/R surgery, Hsp70 KO mice showed a significantly greater increase in apoptosis, larger infarct size, and decreased cardiac output. There was also a significant fourfold increase in the nuclear accumulation of AIF in Hsp70 KO mice compared with WT mice. Treatment with 4-AN (4-amino-1,8-napthalimide, 3 mg/kg), a potent inhibitor of PARP-1, which is a critical regulator of AIF-induced apoptosis, significantly blocked the release of AIF from mitochondria and the translocation of AIF into the nuclei after I/R in both WT and Hsp70 KO mice. In addition, 4-AN treatment resulted in a significant inhibition of apoptosis, a reduction of infarct size, and attenuated cardiac dysfunction in both WT and Hsp70 KO mice after I/R. The anti-apoptotic function of Hsp70 occurs through the inhibition of AIF-induced apoptosis by blocking the mitochondria to nucleus translocation of AIF. PARP-1 inhibition improves cardiac function by blocking AIF-induced apoptosis.
Keywords: Apoptosis, Caspase, PARP-1, Cardiac function, Infarct size
Introduction
Apoptosis has been shown to play an important role in various cardiovascular diseases [19]. Mitochondria have a central role in the induction of apoptosis by releasing various apoptotic factors into the cytosol. One such factor is cytochrome c, which is critical in initiating cardiac apoptosis through the activation of caspases [18, 24]. Yet, caspase inhibition alone does not completely inhibit apoptosis in heart [1, 30]. Apoptosis inducing factor (AIF) is also released from the mitochondria and mediates apoptosis independent of the caspases [40]. AIF is a 57 kDa mitochondrial protein that under normal circumstances acts as an oxidoreductase [40, 43]. However, after apoptotic stimulation, AIF is released from mitochondria followed by nuclear translocation [33, 45]. The mitochondria to nucleus translocation of AIF has been observed in many models of apoptosis, and the subcellular localization of AIF, analogous to the role of cytochrome c, appears to be particularly important for its dual functions as a survival and an apoptotic factor [8, 10, 26].
Although, there are currently no known specific inhibitors of AIF, stress-inducible heat shock protein 70 (Hsp70) has been shown to delay or prevent AIF-mediated toxicity [13, 36]. Overexpression of Hsp70 provides protection from apoptosis induced by serum withdrawal in cultured cells and cardiac ischemia/reperfusion in animals [36, 41]. Hsp70 is known to regulate apoptotic cell death by directly interfering with the function of several proteins that induce apoptotic cell death [5, 36], as well as by indirectly increasing levels of the anti-death protein Bcl-2 [39]. In addition, Hsp70 has been shown to act as a molecular chaperone and is involved in protein folding [11, 14]. However, the protective mechanisms of Hsp70 against AIF-mediated apoptosis in heart remain to be defined. In this study, we investigate the mechanism of Hsp70 in AIF-induced apoptosis after ischemia/reperfusion (I/R) in heart in vivo, and examine whether PARP-1 inhibition is effective in blocking AIF-induced apoptosis.
Methods
Materials
Antibodies were obtained from the following sources: AIF and cytochrome c (BD Pharmingen, Franklin Lakes, NJ, USA), Hsp70 (Calbiochem, Gibbstown, NJ, USA), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Sigma-Aldrich, St. Louis, MO, USA), connexin 43 (Cell Signaling Technology, Danvers, MA, USA), cytochrome c oxidase IV (Molecular Probes, Eugene, OR, USA), and anti-mouse HRP and anti-rabbit HRP (Jackson Immuno Research Laboratory, West Grove, PA, USA). 4-amino-1,8-napthalimide (4-AN) was obtained from Biomol International (Plymouth Meeting, PA, USA).
Animal models
Experiments were conducted using 10-week old male Hsp70 knockout (KO) mice (The Jackson Laboratory, Bar Harbor, ME, USA) and their littermates in a C57BL/6 background. The mice were housed at the Animal Research Facility at Beth Israel Deaconess Medical Center under pathogen-free conditions with a reverse daily 12:12 h light:dark cycle. The animal care standards were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and all experimental procedures were approved by the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center.
Animal surgeries
I/R surgery was performed at 10 weeks of age as described previously [28]. Briefly, mice were anaesthetized, intubated and placed on a rodent ventilator. After thoracotomy, the left anterior descending artery (LAD) artery was identified and tied around a specialized 30G-catheter with a 7-0 silk suture. The animal remained under anesthesia and ventilated for a specified duration of ischemia. Reperfusion was achieved by cutting the suture and reestablishing arterial perfusion. Sham operated mice underwent the same procedure without LAD occlusion/reperfusion.
Assays for caspase-3 and PARP-1 activities
Caspase-3 activities were measured using synthetic caspase substrate AcDEVD-pNa. Release of pNa was measured at a wavelength of 405 nm by spectrometer, and adjusted to the background. PARP activity was measured from cell extracts with an ELISA based, PARP Universal Colorimetric Assay Kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instruction. Details of the assays for caspase-3 and PARP-1 activities are provided in the Supplemental information.
Subcellular fractionation, immunoblots and immunoprecipitation
Subcellular fractionations were obtained according to the previously published protocol [2, 38]. Immunoblotting was performed as described previously [9, 38]. Protein concentration of whole heart tissue lysate was determined by the Bradford method, and GAPDH was used as a loading control. Immunoprecipitation was performed using Catch and Release v2.0 according to the manufacturer’s instructions (Millipore, Billerica, MA, USA). Details of the methods are provided in the Supplemental information.
Measurement of the outer membrane integrity of mitochondria
The integrity of the outer membrane was assessed by measuring cytochrome c oxidase activity using a cytochrome c Oxidase Assay Kit in accordance with the manufacturer’s instructions (Sigma, St. Louis, MO, USA). Details of the method are provided in the Supplemental information.
Infarct size determination
Infarct size was measured 24 h after I/R surgery. At the time of LAD occlusion, 50 μL of fluorescent microspheres (Molecular Probe, Carlsbad, CA, USA) was injected directly into the left ventricle (LV) via the apex using a 25 gauge needle. Since microspheres are not deposited in areas supplied by the occluded LAD, the fluorescent microsphere negative area accurately demarcates the total area at risk (AAR). At the time of killing 48 h after I/R surgery, the heart was excised and cut into 1 mm perpendicular sections. Sections were stained with 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma-Aldrich, St. Louis, MO, USA) solution to visualize the infarcted myocardium (TTC negative area). Total LV area, AAR, and area of infarction were determined for each slice using computer planimetry and NIH Image software, and the standardized size of the infarct calculated in each slice as a proportion of the AAR (TTC negative area/AAR).
Cardiac functional analysis
Cardiac functional analysis was performed 2 weeks after I/R surgery using LV pressure–volume loop measurements. Pressure–volume parameters were measured under isoflurane (2%) inhalant anesthesia using a 1.4 Fr. micro-tip pressure–volume catheter (Scisense Inc, Ontario, Canada) inserted into the right common carotid artery [32]. The catheter was then gently advanced into the LV. Initially, LV pressure and volume were measured at the steady state to obtain LV hemodynamic parameters, such as heart rate, stroke volume and cardiac output, as well as dp/dtmax and dp/dtmin. After the steady-state measurements, we occluded the inferior vena cava (IVC) by gently lifting the tie around the IVC while briefly suspending mechanical ventilation. We obtained LV end-systolic pressure–volume relationship (ESPVR) during this maneuver. Data were recorded using a Powerlab system (ADInstruments, Colorado Springs, CO, USA), and analyzed using CardioSoft Pro software (CardioSoft, Houston, TX, USA).
Immunohistochemistry
Immunofluorescent staining was performed as described on frozen sections of the heart in the ischemic area 24 h after I/R surgery [25, 38]. Apoptosis was quantified using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining. To distinguish cardiomyocytes from other cells, we stained for cardiac α-actinin. We used 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes, Eugene, OR, USA) to stain nuclei. The amount of apoptosis was expressed as the number of TUNEL positive cardiomyocyte nuclei (TUNEL+) as a proportion of the total number of all cardiomyocyte nuclei (N). At least 10 high power fields (~2,000 nuclei/field) were analyzed.
Statistical analyses
Statistical analyses were conducted using one-way ANOVA and Tukey’s tests for post hoc differences between group means. Statistical significance was accepted at P < 0.05. Data is given as the mean ± standard error (SE).
Results
I/R induces AIF and caspase activation in WT and Hsp70 KO mice hearts in vivo
We first sought to define an I/R protocol that yields maximal activation of AIF in WT mice in vivo. We examined three different durations of ischemia by occluding the coronary artery for 30, 45, or 60 min followed by reperfusion for various lengths of time from 0 to 24 h. For all three durations of ischemia, we observed a significant increase in cytosolic AIF that peaked at 9 h of reperfusion (Fig. 1a, b). In contrast, cytochrome c release from the mitochondria, an indication of the activation of caspase-dependent apoptosis, peaked earlier at 6 h of reperfusion. There was a very modest increase in cytosolic AIF and cytochrome c after sham operation, which most likely is due to the effect of anesthesia (Supplemental Figure 1).
Fig. 1.
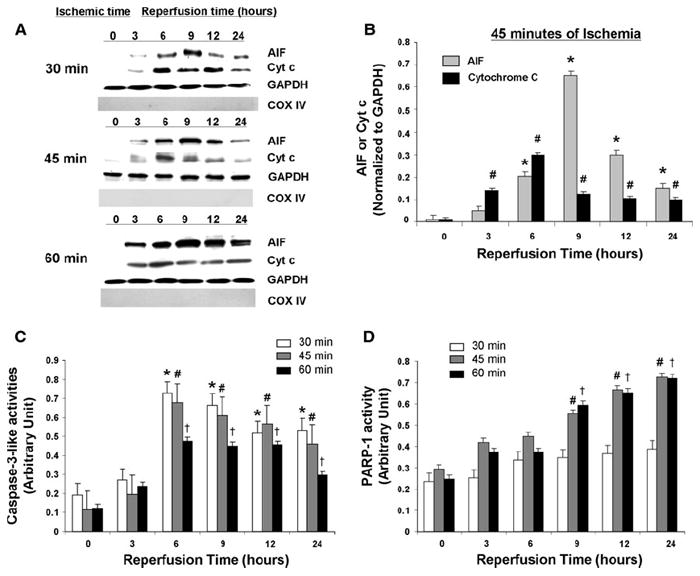
Effect of different I/R protocols on WT mice hearts. a Western blot analysis of cytosolic AIF and cytochrome c in WT mice after different I/R conditions. GAPDH is internal loading control for cytosolic fractions. COX IV is to confirm the absence of significant mitochondrial contamination in cytosolic fractions. b Quantitative analysis of cytosolic AIF and cytochrome c release after 45 min of ischemia and reperfusion. Cytosolic AIF and cytochrome c levels were normalized to GAPDH levels. N = 4. *P < 0.05 versus AIF 0 h reperfusion, #P < 0.05 versus cytochrome c 0 h reperfusion. c Quantitative analysis of caspase-3-like activities after different I/R conditions. N = 6. *P < 0.05 versus 30 min of ischemia and 0 h reperfusion, #P < 0.05 versus 45 min of ischemia and 0 h reperfusion, †P < 0.05 versus 60 min of ischemia and 0 h reperfusion. d Quantitative analysis of PARP activities after different I/R conditions. N = 6. #P < 0.05 versus 45 min of ischemia and 0 h reperfusion, †P < 0.05 versus 60 min of ischemia and 0 h reperfusion
The caspases and PARP-1 are important regulators of caspase-dependent and caspase-independent apoptosis, respectively [1, 3, 7, 29]. To examine the temporal relationship between PARP-1 and caspase activation, we measured PARP-1 and caspase-3 activities in response to I/R. We found a significant increase in caspase-3 activity in all three ischemic protocols. It was particularly prominent after 30 and 45 min of ischemia and peaked at 6 h of reperfusion (Fig. 1c). In comparison, PARP-1 activity was significantly increased after 9 h reperfusion with 45 and 60 min of ischemia, but not with 30 min of ischemia (Fig. 1d). PARP-1 activity continued to increase up to 24 h, which was the longest duration of reperfusion examined. These findings suggest that a more intense apoptotic insult results in a greater propensity to promote caspase-independent apoptosis in heart, and that the activation of AIF and PARP-1, factors involved in caspase-independent apoptosis, is delayed relative to the activation of cytochrome c and caspase-3, which are involved in caspase-dependent apoptosis. We chose 45 min of ischemia for subsequent experiments since this I/R protocol showed a significant increase in AIF and PARP activation.
To investigate whether Hsp70 deletion affects I/R-induced AIF release in vivo, we examined the effect of 45 min of ischemia with reperfusion in Hsp70 KO mice. We observed similar patterns of AIF and cytochrome c release in Hsp70 KO and WT mice after I/R surgery. AIF release peaked at 9 h after I/R, whereas cytochrome c release peaked at 3–6 h after I/R (Fig. 2a, b). To determine the effects of I/R on changes in Hsp70, we measured the expression of Hsp70 mRNA and protein. The endogenous expression of both Hsp70 mRNA (Fig. 2c, e) and protein (Fig. 2d, f) were significantly increased after I/R in WT mice. As expected, however, there was no detectable mRNA or protein expression of Hsp70 in Hsp70 KO mice either at baseline or after I/R.
Fig. 2.
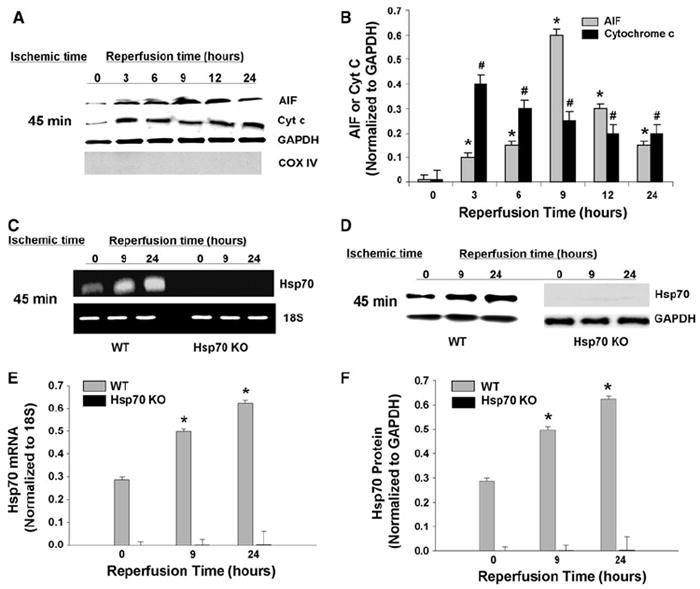
Effect of I/R on Hsp70 KO mice. a Western blot analysis of cytosolic AIF and cytochrome c in Hsp70 KO mice hearts after 45 min of ischemia and reperfusion. GAPDH is internal loading control for cytosolic fractions. COX IV is to confirm the absence of significant mitochondrial contamination in cytosolic fractions. b Quantitative analysis of cytosolic AIF and cytochrome c after 45 min of ischemia and reperfusion. Cytosolic AIF and cytochrome c levels were normalized to GAPDH levels. N = 4. *P < 0.05 versus AIF 0 h reperfusion, #P < 0.05 versus cytochrome c 0 h reperfusion. c–d Hsp70 mRNA (c) and protein (d) levels after 45 min of ischemia followed by 9 and 24 h of reperfusion in WT and Hsp70 KO mice. 18S was used as an internal control for mRNA expression, and GAPDH was used as a loading control for western blots. e–f Quantitative analysis of mRNA (e) and protein (f) levels after 45 min of ischemia followed by 9 and 24 h of reperfusion in WT and Hsp70 KO mice. Hsp70 mRNA expression was normalized to 18S expression, and Hsp70 protein expression was normalized to GAPDH expression. N = 4. *P < 0.05 versus WT 0 h reperfusion
PARP-1 inhibition blocks AIF activation by blocking AIF release and nuclear translocation after I/R
Since we observed a significant increase in PARP-1 activity after I/R in both WT and Hsp70 KO mice, we next examined whether PARP-1 might regulate the release of AIF. WT and Hsp70 KO mice were pretreated before I/R surgery with 3 mg/kg of 4-AN, a potent PARP-1 inhibitor that has been shown to inhibit PARP-1 activation during cerebral I/R [16, 50]. Pretreatment with 4-AN blocked both PARP-1 activation (Fig. 3a) and decreased the amount of AIF released from the mitochondria in both WT and Hsp70 KO mice (Fig. 3b, c). These data suggest that I/R induced release of AIF is regulated by PARP-1. To determine if there is a direct interaction between AIF and Hsp70, we immunoprecipitated cytosolic AIF from WT and Hsp70 KO mice heart lysates after I/R (both 30 and 45 min of ischemia and 9 h of reperfusion) or sham operation using anti-AIF antibodies, and then performed western blotting with anti-Hsp70 antibodies. We found that cytosolic AIF from WT, but not Hsp70 KO mice hearts, showed an interaction with Hsp70 (Fig. 3d). In addition, we confirmed that the mitochondrial fractions we used contained connexin 43, which indicates that they are subsarcolemmal and not interfibrillar mitochondria (Fig. 3e) [6].
Fig. 3.
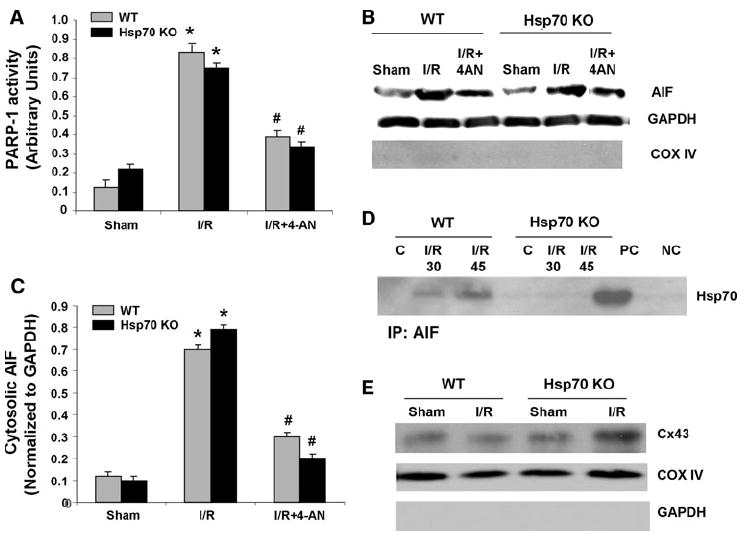
Effect of PARP inhibition on AIF release and PARP-1 activity after I/R. a PARP activity assay in WT and Hsp70 KO mice after 45 min of ischemia and 9 h of reperfusion with or without 4-AN. N = 4. *P < 0.05 versus Sham, #P < 0.05 versus I/R. b Representative immunoblots of cytosolic AIF in WT and Hsp70 KO mice heart after 45 min of ischemia and 9 h of reperfusion with or without 4-AN. GAPDH is an internal loading control for cytosolic fractions. COX IV is used to confirm the absence of significant mitochondrial contamination in cytosolic fractions. c Quantitative analysis of cytosolic AIF after 45 min of ischemia and 9 h of reperfusion with or without 4-AN. AIF level was normalized to GAPDH. N = 4. * P < 0.05 versus Sham, # P < 0.05 versus I/R. d Interaction between cytosolic AIF and Hsp70. Cytosolic AIF was immunoprecipitated (IP) with anti-AIF antibodies from cytosolic fractions in WT and Hsp70 KO mice hearts after 30 or 45 min of ischemia and 9 h of reperfusion or sham operation. Heart lysate without AIF IP and normal rabbit serum were used as a positive control (PC) and a negative control (NC), respectively. Western blot analysis was performed using anti-Hsp70 antibodies. e Representative immunoblot of connexin 43 in the mitochondrial fractions to distinguish the subsarcolemmal and the interfibrillar mitochondria
Our data did not show significant differences in the amount of AIF released from mitochondria in Hsp70 KO compared to WT mice. Since it has been suggested that Hsp70 inhibits AIF nuclear translocation, we used immunofluorescent staining to assess whether I/R is associated with increased nuclear translocation of AIF in Hsp70 KO mice hearts. We observed a significant fourfold increased accumulation of AIF in the nuclei in Hsp70 KO as compared to WT mice after I/R (Fig. 4a, b). Pretreatment with 4-AN, however, significantly inhibited AIF nuclear translocation. To address the possibility of a pleiotropic effect of PARP-1 inhibition extending beyond its effect on Hsp70, we examined the mitochondrial outer membrane. We found that PARP-1 inhibition significantly attenuated the integrity of the mitochondrial outer membrane (Fig. 4c). We also found that the level of Bcl-2, which was significantly downregulated by I/R, did not change significantly with 4-AN treatment (Supplemental Figure 2). These findings suggest that PARP-1 inhibition results in anti-apoptotic effects via modulating the nuclear translocation of AIF via Hsp70, and maintaining mitochondrial membrane integrity in heart.
Fig. 4.
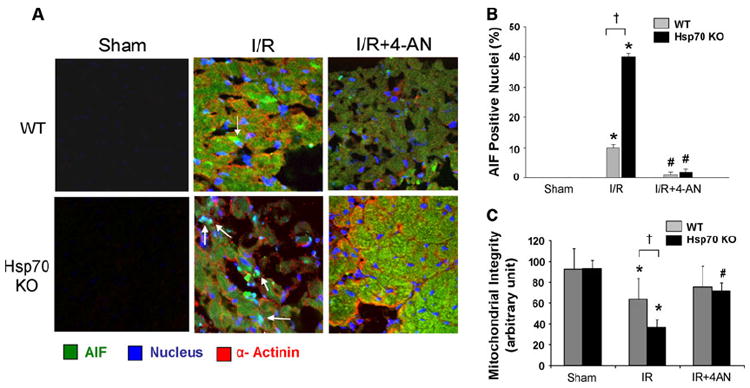
Nuclear translocation of AIF after I/R in hearts. a Representative triple stained images of AIF nuclear translocation in WT and Hsp70 KO mouse heart after 45 min of ischemia and 9 h of reperfusion with or without 4-AN. AIF green, nuclei blue, α-actinin red, Magnification 40x. b Quantification of nuclear AIF in WT and Hsp70 KO heart after 45 min of ischemia and 9 h of reperfusion with or without 4-AN. N = 6. *P < 0.05 versus Sham, #P < 0.05 versus I/R. c Effect of a PARP-1 inhibitor on mitochondrial outer membrane integrity. Cytochrome c oxidase assay in WT and Hsp70 Ko mice after 45 min of ischemia and 9 h of reperfusion with or without 4-AN. N = 4. *P < 0.05 versus Sham, #P < 0.05 versus I/R, †P < 0.05 WT I/R
Effect of Hsp70 deletion on infarct size and cardiac apoptosis after I/R
We then evaluated the influence of Hsp70 deletion on infarct size 24 h after I/R using fluorescent microspheres and TTC. We observed significant increases in infarct size in both WT and Hsp70 KO mice; the increase in infarct size, however, was 26% greater in Hsp70 KO mice (Fig. 5a, b). Pre-treatment with 4-AN resulted in a significant reduction of infarct size to similar levels in both WT and Hsp70 KO mice.
Fig. 5.
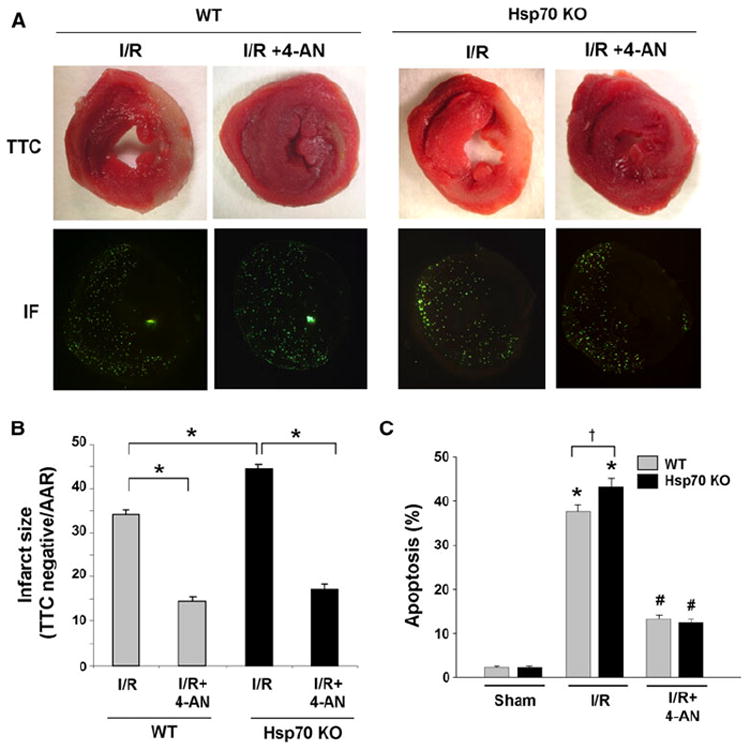
The effect of PARP-1 inhibition on infarct size and cardiac apoptosis. a Representative heart sections from WT and Hsp70 mice after 45 min of ischemia and 24 h of reperfusion with or without 4-AN. Infarct sizes were determined as infarct area (IA)/AAR using TTC and fluorescent microspheres. Infarct areas were determined using TTC staining (the infarct is the pale area) (upper panels), and areas at risk were measured using fluorescent microspheres (florescent negative area) (lower panels). b Quantitative analysis of infarct size (infarct area/AAR) in WT and Hsp70 mice after 45 min of ischemia and 24 h of reperfusion with or without 4-AN. N = 8, *P < 0.05. c Quantitative analysis of TUNEL staining in WT and Hsp70 mice after 45 min of ischemia and 24 h of reperfusion with or without 4-AN. N = 8 *P < 0.05 versus Sham, †P < 0.05 WT I/R
To examine whether the increase in infarct size after I/R is mediated by increased cardiac apoptosis, we measured the rate of apoptosis 24 h after I/R using TUNEL staining. We found that I/R induced significantly more TUNEL-positive nuclei in Hsp70 KO heart (43%) than WT (38%) (Fig. 5c). Furthermore, pretreatment with 4-AN resulted in a significant reduction of cardiac apoptosis in both WT and Hsp70 KO. We also determined apoptosis rate in the non-infarcted and non-ischemia area of the myocardium after I/R. We found that there was no significant increase in apoptosis in the remote myocardium between sham and I/R groups (Supplemental Figure 3). These data suggest that the deletion of Hsp70 results in an exaggerated and deleterious response in heart to I/R by inducing cardiac apoptosis and increasing myocardial damage; PARP-1 inhibition results in significant protection in both WT and Hsp70 KO mice.
Effect of I/R and Hsp70 deletion on cardiac function
To determine whether the reduction in infarct size and cardiac apoptosis with PARP-1 inhibition is associated with improved cardiac function, we performed pathological and functional analyses 2 weeks after I/R. Pathological analysis showed that Hsp70 KO mice developed a significant increase in heart weight (HW) and body weight (BW) ratio (HW/BW) and lung weight (LuW) and BW ratio (LuW/BW) when compared with WT mice after I/R (Table 1). Pretreatment with 4-AN significantly attenuated HW/BW and LuW/BW ratios in both WT and Hsp70 KO mice.
Table 1.
Morphological data 7 days after I/R
| Sham
|
I/R
|
I/R + 4-AN
|
||||
|---|---|---|---|---|---|---|
| WT | Hsp70 KO | WT | Hsp70 KO | WT | Hsp70 KO | |
| Body weight (g) | 20.1 ± 0.2 | 17.9 ± 0.3 | 19.8 ± 0.5 | 16.7 ± 0.5 | 19.5 ± 0.2 | 17.3 ± 1.4 |
| Heart weight (mg) | 96.5 ± 1.5 | 76.5 ± 1.3 | 118.8 ± 1.5* | 125.3 ± 1.2* | 99.45 ± 1.7# | 93.42 ± 1.8# |
| Lung weight (mg) | 90.5 ± 2.1 | 94.9 ± 2.1 | 114.8 ± 3.1* | 118.6 ± 1.2* | 89.7 ± 1.2# | 86.5 ± 1.6# |
| HW/BW (mg/g) | 4.8 ± 0.02 | 4.2 ± 0.03 | 6.0 ± 0.1* | 7.5 ± 0.1*, † | 5.1 ± 0.9# | 5.4 ± 0.2# |
| LW/BW (mg/g) | 4.5 ± 0.1 | 5.3 ± 0.1 | 5.8 ± 0.3* | 7.1 ± 0.1*, † | 4.6 ± 0.2# | 5.0 ± 0.1# |
HW/BW heart weight to body weight ratio, LW/BW lung weight to body weight ratio.
P < 0.05 compared to the corresponding WT or Hsp70 KO sham.
P < 0.05 WT I/R.
P < 0.05 compared to the corresponding WT or Hsp70 KO I/R. N = 6–9
We also measured cardiac function using pressure–volume loop analysis. Although, there were no significant differences in the basal hemodynamic parameters of WT and Hsp70 KO mice (Fig. 6a–d), various cardiac functional parameters decreased significantly in both groups 2 weeks after I/R. There were 2.3-, 1.9-, 2.3- and 0.12-fold decreases in stroke volume, stroke work, cardiac output, and +dp/dt in Hsp70 KO mice as compared to WT mice (Fig. 6a–d). PARP-1 inhibition, however, resulted in significant improvement of these parameters to near baseline levels in both WT and Hsp70 KO mice. These findings suggest that Hsp70 KO mice develop significantly greater cardiac dysfunction than WT mice after I/R, and these parameters are significantly improved after PARP-1 inhibition.
Fig. 6.
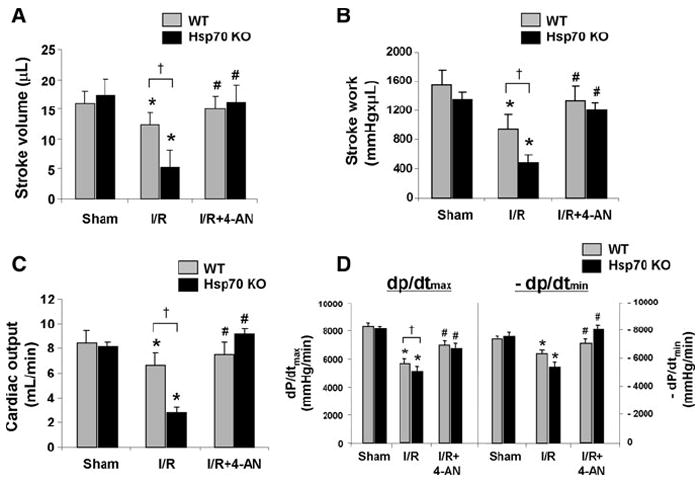
The effect of PARP-1 inhibition on cardiac function. Stroke volume (a), stroke work (b), cardiac output (c), +dp/dt and −dp/dt (d) in WT and HSP 70 KO mice after 45 min of ischemia and 2 weeks of reperfusion with and without 4AN. N = 6. *P < 0.05 versus Sham, #P < 0.05 versus I/R, †P < 0.05 WT I/R
Discussion
The main finding in this study is that deletion of Hsp70 exaggerates I/R-induced cardiac dysfunction, which is associated with increased cardiac apoptosis mediated by AIF activation. The mechanism of increased AIF-induced apoptosis involves an increase in the nuclear translocation of AIF in Hsp70 KO mouse heart. We also found that PARP-1 inhibition by 4-AN effectively inhibited apoptosis by inhibiting AIF release from mitochondria and AIF nuclear translocation.
AIF-mediated cell death is a two-step process that involves the initial release of AIF from the mitochondria into the cytosol, which is followed by nuclear translocation and accumulation [40, 45, 49]. Recent data based on the crystallographic structure of AIF suggest that AIF translocates into the nucleus, where it interacts with DNA and induces chromatin condensation to exert its apoptotic function [48]. We and other investigators have shown that blocking AIF release from the mitochondria into the cytosol can block apoptosis in various models [8, 9, 45]. However, the significance of AIF nuclear translocation in heart has not been well understood, and the effectiveness of blocking AIF nuclear translocation is not clear. In this study, we demonstrate that Hsp70 is involved in the nuclear translocation of AIF, and that the inhibition of AIF nuclear translocation can inhibit apoptosis in heart.
Opening of mitochondrial permeability transition pore has been implicated in I/R-injuries in heart [4, 15]. We observed that the mitochondrial release of cytochrome c and caspase-3 activation were most intense after 30 min of ischemia, and peaked with 3–6 h of reperfusion. However, the mitochondrial release of AIF and PARP-1 activation were most intense after 45 or 60 min of ischemia, and the maximal activation occurred after 9 h or more of reperfusion. These findings demonstrate the differences in the time course of factors involved in caspase-dependent apoptosis (e.g. cytochrome c and caspase-3) and caspase-independent apoptosis (e.g. AIF and PARP-1). They suggest that the caspase-independent pathway is activated in a setting of more prolonged or intense apoptotic stimulation. It has been suggested that apoptosis has dual phases related to pore size. Pores that are formed initially are smaller and allow only smaller molecules, such as cytochrome c, to be released, but over time, the size of the pores increases, allowing larger molecules, such as AIF and Endo G, to be released [27, 42]. We have previously shown that release of AIF is preferentially initiated after prolonged apoptotic stimulation, or in a setting of caspase inhibition, such as treatment with pan-caspase inhibitor, zVAD.fmk [9, 18]. Other mechanisms of mitochondrial apoptotic factor release may involve loss of mitochondrial membrane potential, mitochondrial swelling and ultimate outer membrane rupture [44].
The anti-apoptotic function of Hsp70 involves interactions with several components of the apoptotic machinery. It has been shown that Hsp70 can also inhibit caspase-independent apoptosis by directly interacting with AIF and thereby neutralizing AIF-induced nuclear modifications [13], and that the down-regulation of Hsp70 by an anti-sense construct increases the sensitivity of cells to AIF-mediated killing [36]. Previous studies suggested that the deletion of Hsp70 is associated with cardiac dysfunction and impaired stress response to I/R ex vivo [20]. In contrast, Hsp70 has been suggested to play a cardioprotection role early after myocardial infarction [31]. In this study, we showed the cardioprotective effect of Hsp70 against I/R injury, and the regulation of caspase-independent apoptosis by Hsp70 in heart in vivo. Since an upregulation of Hsp70 after I/R is evident as early as 9 h of reperfusion, the increase in Hsp70 likely contributes to the cardioprotection. However, other mechanisms of Hsp70’s cardioprotective effect, in addition to the blocking of AIF nuclear translocation, cannot be ruled out. In addition, we cannot rule out that Hsp70 overexpression during I/R may be AIF-mediated. However, Hsp70 is overexpressed in conditions that are not known to cause apoptosis, such as heat stress. Thus, the effect of AIF-mediated Hsp70 overexpression, if it occurs, will likely be insignificant.
PARP-1 is a highly conserved, 116-kDa nuclear enzyme involved in DNA repair [49] and has been shown to facilitate both the release of AIF from mitochondria and AIF nuclear translocation [10, 34]. We also observed increased PARP-1 activity with increasing cytosolic AIF release in response to I/R in both WT and Hsp70 KO mice. PARP-1 inhibition effectively blocked the release of AIF from mitochondria and the nuclear translocation of AIF in Hsp70 KO mice. Supporting this finding, PARP-1 inhibition using either isoindolinone-based PARP-1 inhibitor (INO-1001) or PARP-1 genetic deficient mice has been found to reduce the mitochondrial-to-nuclear translocation of AIF in a mouse model of heart failure [46]. The mechanism responsible for PARP-1-dependent release of AIF from mitochondria remains to be clarified.
In our study, the protective effect of Hsp70 was relatively modest as compared to the robust protection achieved by PARP-1. Since PARP-1 regulates a multitude of factors, Hsp70 regulation of PARP-1 may represent only a partial effect on myocardial protection. In fact, overexpression of human Hsp70 does not appear to affect normal protein synthesis or the stress response in transgenic mice when compared with nontransgenic mice [35] suggesting that Hsp70 may not be the primary targeted molecule. Nevertheless, PARP-1 activity appears to be important for AIF to translocate to the nucleus after apoptotic stimulation, a process most likely mediated by small PAR fragments signaling into the mitochondria [49]. As such, AIF is currently believed to play an important role in PARP-1-dependent cell death. Another potential mechanism may involve a PARP-1-dependent activation of c-Jun N-terminal kinase via a pathway using members of the tumor necrosis factor signaling cascade [47].
In this study, we focused on AIF-mediated apoptosis after I/R. However, other mitochondria-mediated mechanisms [12, 21] or other types of cell death, such as necrosis and autophagy, may also be involved in this model. Necrosis, which has often been viewed as an accidental and uncontrolled cell death process, may also be a regulated type of programmed cell death [37, 51]. Autophagy has features resembling apoptosis, including a possible association with the caspases and Bcl-2 [22, 23]. Studies show that different types of cell death can occur simultaneously in the same tissue [17]. In fact, the various types of cell death may represent a spectrum of cell death processes, and they may have triggering events in common. On the other hand, the intensity and duration of insult and as well as the tissue type may contribute to which cell death mechanisms predominate. We did not specifically take account of other cell death processes in this paper, but we nevertheless recognize that they have the potential to make a contribution to overall cell death in our model. More work is needed to address these issues.
Supplementary Material
Acknowledgments
This study was supported by the grants from National Institutes of Health RO1 HL091998 (PMK) and the World Class University program (R31-20029) from Ministry of Education, Science and Technology, South Korea (PMK).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00395-011-0164-1) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no conflict of interest.
Contributor Information
Sangita Choudhury, Cardiovascular Institute, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, USA.
Soochan Bae, Cardiovascular Institute, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, USA.
Qingen Ke, Cardiovascular Institute, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, USA.
Ji Yoo Lee, Cardiovascular Institute, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, USA.
Jacob Kim, Cardiovascular Institute, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, USA.
Peter M. Kang, Cardiovascular Institute, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, USA, Department of BIN Fusion Technology, Chonbuk National University, Jeonju, South Korea, Cardiovascular Institute, Beth Israel Deaconess Medical Center, 3 Blackfan Circle, CLS-910, Boston, MA 02215, USA, pkang@bidmc.harvard.edu
References
- 1.Abraham MC, Shaham S. Death without caspases, caspases without death. Trends Cell Biol. 2004;14:184–193. doi: 10.1016/j.tcb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Bae S, Siu PM, Choudhury S, Ke Q, Choi JH, Koh YY, Kang PM. Delayed activation of caspase-independent apoptosis during heart failure in transgenic mice overexpressing caspase inhibitor CrmA. Am J Physiol Heart Circ Physiol. 2010;299:H1374–H1381. doi: 10.1152/ajpheart.00168.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bae S, Yalamarti B, Kang PM. Role of caspase-independent apoptosis in cardiovascular diseases. Front Biosci. 2008;13:2495–2503. doi: 10.2741/2861. [DOI] [PubMed] [Google Scholar]
- 4.Baines CP. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res Cardiol. 2009;104:181–188. doi: 10.1007/s00395-009-0004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 6.Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz-Meana M, Garcia-Dorado D, Heusch G, Schulz R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol. 2009;104:141–147. doi: 10.1007/s00395-009-0007-5. [DOI] [PubMed] [Google Scholar]
- 7.Chen M, Zsengeller Z, Xiao CY, Szabo C. Mitochondrial-to-nuclear translocation of apoptosis-inducing factor in cardiac myocytes during oxidant stress: potential role of poly(ADP-ribose) polymerase-1. Cardiovasc Res. 2004;63:682–688. doi: 10.1016/j.cardiores.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 8.Cheung EC, Joza N, Steenaart NA, McClellan KA, Neuspiel M, McNamara S, MacLaurin JG, Rippstein P, Park DS, Shore GC, McBride HM, Penninger JM, Slack RS. Dissociating the dual roles of apoptosis-inducing factor in maintaining mitochondrial structure and apoptosis. EMBO J. 2006;25:4061–4073. doi: 10.1038/sj.emboj.7601276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choudhury S, Bae S, Kumar SR, Ke Q, Yalamarti B, Choi JH, Kirshenbaum LA, Kang PM. Role of AIF in cardiac apoptosis in hypertrophic cardiomyocytes from Dahl salt-sensitive rats. Cardiovasc Res. 2010;85:28–37. doi: 10.1093/cvr/cvp261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 12.Di Lisa F, Kaludercic N, Carpi A, Menabo R, Giorgio M. Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66(Shc) and monoamine oxidase. Basic Res Cardiol. 2009;104:131–139. doi: 10.1007/s00395-009-0008-4. [DOI] [PubMed] [Google Scholar]
- 13.Gurbuxani S, Schmitt E, Cande C, Parcellier A, Hammann A, Daugas E, Kouranti I, Spahr C, Pance A, Kroemer G, Garrido C. Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene. 2003;22:6669–6678. doi: 10.1038/sj.onc.1206794. [DOI] [PubMed] [Google Scholar]
- 14.Hartl FU, Martin J, Neupert W. Protein folding in the cell: the role of molecular chaperones Hsp70 and Hsp60. Annu Rev Biophys Biomol Struct. 1992;21:293–322. doi: 10.1146/annurev.biophys.21.1.293. [DOI] [PubMed] [Google Scholar]
- 15.Heusch G, Boengler K, Schulz R. Inhibition of mitochondrial permeability transition pore opening: the Holy Grail of cardioprotection. Basic Res Cardiol. 2010;105:151–154. doi: 10.1146/annurev.biophys.21.1.293. [DOI] [PubMed] [Google Scholar]
- 16.Kabra DG, Thiyagarajan M, Kaul CL, Sharma SS. Neuroprotective effect of 4-amino-1, 8-napthalimide, a poly(ADP ribose) polymerase inhibitor in middle cerebral artery occlusion-induced focal cerebral ischemia in rat. Brain Res Bull. 2004;62:425–433. doi: 10.1016/j.brainresbull.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Kajstura J, Cheng W, Reiss K, Clark WA, Sonnenblick EH, Krajewski S, Reed JC, Olivetti G, Anversa P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- 18.Kang PM, Haunstetter A, Aoki H, Usheva A, Izumo S. Morphological and molecular characterization of adult cardiomyocyte apoptosis during hypoxia and reoxygenation. Circ Res. 2000;87:118–125. doi: 10.1161/01.res.87.2.118. [DOI] [PubMed] [Google Scholar]
- 19.Kang PM, Izumo S. Apoptosis in heart: basic mechanisms and implications in cardiovascular diseases. Trends Mol Med. 2003;9:177–182. doi: 10.1016/S1471-4914(03)00025-X. [DOI] [PubMed] [Google Scholar]
- 20.Kim YK, Suarez J, Hu Y, McDonough PM, Boer C, Dix DJ, Dillmann WH. Deletion of the inducible 70-kDa heat shock protein genes in mice impairs cardiac contractile function and calcium handling associated with hypertrophy. Circulation. 2006;113:2589–2597. doi: 10.1161/CIRCULATIONAHA.105.598409. [DOI] [PubMed] [Google Scholar]
- 21.Lacerda L, McCarthy J, Mungly SF, Lynn EG, Sack MN, Opie LH, Lecour S. TNF alpha protects cardiac mitochondria independently of its cell surface receptors. Basic Res Cardiol. 2010;105:751–762. doi: 10.1007/s00395-010-0113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee CY, Baehrecke EH. Steroid regulation of autophagic programmed cell death during development. Development. 2001;128:1443–1455. doi: 10.1242/dev.128.8.1443. [DOI] [PubMed] [Google Scholar]
- 23.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/S0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 25.Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330–335. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
- 26.Modjtahedi N, Giordanetto F, Madeo F, Kroemer G. Apoptosis-inducing factor: vital and lethal. Trends Cell Biol. 2006;16:264–272. doi: 10.1016/j.tcb.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 27.Munoz-Pinedo C, Guio-Carrion A, Goldstein JC, Fitzgerald P, Newmeyer DD, Green DR. Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc Natl Acad Sci USA. 2006;103:11573–11578. doi: 10.1073/pnas.0603007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, Hemmings BA, Kass DA, Champion HC, Rosenzweig A. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest. 2005;115:2128–2138. doi: 10.1172/JCI23073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/S0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 30.Okamura T, Miura T, Takemura G, Fujiwara H, Iwamoto H, Kawamura S, Kimura M, Ikeda Y, Iwatate M, Matsuzaki M. Effect of caspase inhibitors on myocardial infarct size and myocyte DNA fragmentation in the ischemia-reperfused rat heart. Cardiovasc Res. 2000;45:642–650. doi: 10.1016/S0008-6363(99)00271-0. [DOI] [PubMed] [Google Scholar]
- 31.Pantos C, Mourouzis I, Dimopoulos A, Markakis K, Panagiotou M, Xinaris C, Tzeis S, Kokkinos AD, Cokkinos DV. Enhanced tolerance of the rat myocardium to ischemia and reperfusion injury early after acute myocardial infarction. Basic Res Cardiol. 2007;102:327–333. doi: 10.1007/s00395-007-0645-4. [DOI] [PubMed] [Google Scholar]
- 32.Paolocci N, Tavazzi B, Biondi R, Gluzband YA, Amorini AM, Tocchetti CG, Hejazi M, Caturegli PM, Kajstura J, Lazzarino G, Kass DA. Metalloproteinase inhibitor counters high-energy phosphate depletion and AMP deaminase activity enhancing ventricular diastolic compliance in subacute heart failure. J Pharmacol Exp Ther. 2006;317:506–513. doi: 10.1124/jpet.105.099168. [DOI] [PubMed] [Google Scholar]
- 33.Penninger JM, Kroemer G. Mitochondria, AIF and caspases—rivaling for cell death execution. Nat Cell Biol. 2003;5:97–99. doi: 10.1038/ncb0203-97. [DOI] [PubMed] [Google Scholar]
- 34.Pieper AA, Brat DJ, Krug DK, Watkins CC, Gupta A, Blackshaw S, Verma A, Wang ZQ, Snyder SH. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci USA. 1999;96:3059–3064. doi: 10.1073/pnas.96.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plumier JC, Ross BM, Currie RW, Angelidis CE, Kazlaris H, Kollias G, Pagoulatos GN. Transgenic mice expressing the human heat shock protein 70 have improved post-ischemic myocardial recovery. J Clin Invest. 1995;95:1854–1860. doi: 10.1172/JCI117865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, Mak T, Jaattela M, Penninger JM, Garrido C, Kroemer G. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3:839–843. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu S, Eguchi Y, Kamiike W, Waguri S, Uchiyama Y, Matsuda H, Tsujimoto Y. Retardation of chemical hypoxia-induced necrotic cell death by Bcl-2 and ICE inhibitors: possible involvement of common mediators in apoptotic and necrotic signal transductions. Oncogene. 1996;12:2045–2050. [PubMed] [Google Scholar]
- 38.Siu PM, Bae S, Bodyak N, Rigor DL, Kang PM. Response of caspase-independent apoptotic factors to high salt diet-induced heart failure. J Mol Cell Cardiol. 2007;42:678–686. doi: 10.1016/j.yjmcc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stankiewicz AR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem. 2005;280:38729–38739. doi: 10.1074/jbc.M509497200. [DOI] [PubMed] [Google Scholar]
- 40.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki K, Sawa Y, Kagisaki K, Taketani S, Ichikawa H, Kaneda Y, Matsuda H. Reduction in myocardial apoptosis associated with overexpression of heat shock protein 70. Basic Res Cardiol. 2000;95:397–403. doi: 10.1007/s003950070039. [DOI] [PubMed] [Google Scholar]
- 42.Tait SW, Green DR. Caspase-independent cell death: leaving the set without the final cut. Oncogene. 2008;27:6452–6461. doi: 10.1038/onc.2008.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schagger H, Rustin P, Kroemer G. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/S0092-8674(00)80450-X. [DOI] [PubMed] [Google Scholar]
- 45.Wang X, Yang C, Chai J, Shi Y, Xue D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science. 2002;298:1587–1592. doi: 10.1126/science.1076194. [DOI] [PubMed] [Google Scholar]
- 46.Xiao CY, Chen M, Zsengeller Z, Li H, Kiss L, Kollai M, Szabo C. Poly(ADP-Ribose) polymerase promotes cardiac remodeling, contractile failure, and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. J Pharmacol Exp Ther. 2005;312:891–898. doi: 10.1124/jpet.104.077164. [DOI] [PubMed] [Google Scholar]
- 47.Xu Y, Huang S, Liu ZG, Han J. Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J Biol Chem. 2006;281:8788–8795. doi: 10.1074/jbc.M508135200. [DOI] [PubMed] [Google Scholar]
- 48.Ye H, Cande C, Stephanou NC, Jiang S, Gurbuxani S, Larochette N, Daugas E, Garrido C, Kroemer G, Wu H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat Struct Biol. 2002;9:680–684. doi: 10.1038/nsb836. [DOI] [PubMed] [Google Scholar]
- 49.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, Zhang X, Park TS, Gidday JM. Cerebral endothelial cell apoptosis after ischemia-reperfusion: role of PARP activation and AIF translocation. J Cereb Blood Flow Metab. 2005;25:868–877. doi: 10.1038/sj.jcbfm.9600081. [DOI] [PubMed] [Google Scholar]
- 51.Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.