

Abstract
Identification of deamidated sites in proteins is commonly used for assignment of N-glycosylation sites. It is also important for assessing the role of deamidation in vivo. However, nonenzymatic deamidation occurs easily in peptides under conditions commonly used in treatment with trypsin and PNGase F. The impact on proteomic sample preparation has not yet been evaluated systematically. In addition, the 13C peaks of amidated peptides can be misassigned as monoisotopic peaks of the corresponding deamidated ones in database searches. The 19.34 mDa mass difference between them is proposed as a means for eliminating the resulting false positive identifications in large-scale proteomic analysis. We evaluated five groups of proteomic data, obtained mainly through an electrostatic repulsion-hydrophilic interaction chromatography (ERLIC)-reverse phase (RP) chromatography sequence, and ascertained that nonenzymatic asparagine deamidation occurred to some extent on 4–9% of the peptides, resulting in the false positive identification of many N-glycosylation sites. A comprehensive investigation indicated that the chief causative factors were the mildly alkaline pH and prolonged incubations at 37 °C during proteomic sample preparation. An improved protocol is proposed featuring tryptic digestion at pH 6 and deglycosylation at pH 5, resulting in a significant decrease in nonenzymatic deamidation while conserving adequate digestion efficiency. The number of identified deamidation sites was improved significantly by increasing the sample loading amount in liquid chromatography-tandem MS. This permitted the identification of a significant number of glutamine deamidation sites, which featured sequence motifs largely different from those for asparagine deamidation: -Q-V-, -Q-L- and -Q-G- and, to a lesser extent, -Q-A- and -Q-E-.
Accurate identification of protein deamidation sites is important to understanding the role of deamidation in human diseases and aging and in the correct location of N-glycosylation using the PNGase F deglycosylation method. However, determination of protein deamidation with liquid chromatography-tandem MS (LC-MS/MS)1 is compromised by current methods of proteomics sample preparation and database searching. Nonenzymatic deamidation of Asn and Gln occurs spontaneously on proteins and peptides both in vivo and in vitro. In vivo, deamidation is reported to be related to Alzheimer's disease and cataracts (1–3). It has also been proposed as a molecular clock in studies of aging (4, 5). In vitro, deamidation leads to the degradation of therapeutic proteins during production and storage (6, 7). It takes place relatively slowly in intact proteins with a half-life of 1–500 days for Asn and 100–500 days for Gln (8). However, the deamidation rate of peptides increases significantly under conditions typical of tryptic digestion, i.e. incubation in digestion buffers at pH 8 for 8–16 h at 37 °C (9). Under mildly alkaline conditions, Asn deamidation happens mainly through the formation of a succinimide ring intermediate that is quickly hydrolyzed to d,l-Asp and d,l-isoAsp with isoAsp predominating (10). Deamidation of Gln (Gln-deamidation) is as much as ten times slower because it is thermodynamically less favorable to form a six-member glutarimide ring (11).
Most of the published papers about deamidation have focused on its mechanism and how to detect it in specific proteins (12–14). Recently, several papers have been published about distinguishing deamidation in vivo from that happening during sample preparation (15, 16). Interest is also increasing in distinguishing l-isoAsp from l-Asp in synthesized peptides or biological samples (17, 18). The deamidation of peptides with -Asn-Gly- sequences has been evaluated during proteomic sample preparation (19) but the general impact of nonenzymatic deamidation has not been evaluated comprehensively, although it is known to be facilitated by the conditions of tryptic digestion, the workhorse digestion method of proteomics.
Because nonenzymatic deamidation could potentially occur on every Asn and Gln residue at various rates (12, 20), all peptides containing either residue could exhibit deamidated counterparts after trypsin digestion, which would reduce the intensities of native peptides and complicate peptide identification. It is especially harmful when the peptides containing either residue are of low abundance. In addition, for large-scale proteomic analysis, the convention for identification of N-linked glycosylation sites generally depends on the detection of Asn deamidation in the consensus sequence N-X-S/T (with X not proline) in peptides after PNGase F treatment. Because nonenzymatic deamidation can also occur on the Asn in the consensus sequence if it is not occupied by a glycan prior to PNGase F treatment (which is also customarily performed at pH 8), this results in some false-positive identification of N-glycosylation sites. H218O has been used in differentiating native deamidation sites from those occurring during sample preparation (15, 16), but this approach cannot distinguish the nonenzymatic deamidation that occurs in the consensus sequence during the PNGase F treatment from the real glycosylation sites. Our preliminary inspection of a large data set (see Table II) revealed that the incidence of such false positive identifications was not negligible. There is a pressing need, then, to reduce this artifact in proteomic sample preparation.
Table II. Estimation of nonenzymatic deamidated peptides and N-glycopeptides in five groups of proteomic data.
| ERLIC1-RPc | ERLIC2-RPc | ERLIC3-RPd | ERLIC4-RPd | In-Gel-RP | |
|---|---|---|---|---|---|
| Unique peptides | 17003 | 16518 | 10396 | 11939 | 5267 |
| Unique glycopeptidesa | 96 | 108 | 107 | 781 | 35 |
| Unique Asn-deamidated peptides | 815 | 845 | 821 | 1020 | 288 |
| Ratio of Asn-deamidationb | 4.79% | 5.12% | 7.90% | 8.54% | 5.45% |
| Unique Gln-deamidated peptides | 320 | 366 | 307 | 418 | 89 |
| Ratio of Gln-deamidationb | 1.88% | 2.22% | 2.95% | 3.50% | 1.69% |
a It should be noted that all N-glycopeptides identified in ERLIC1-RP, ERLIC2-RP, ERLIC3-RP, and In-Gel-RP are false positive ones because these samples were not treated with PNGase F to remove N-glycans.
b Ratio of deamidation is calculated as the number of unique deamidated peptides divided by that of all identified unique peptides in the run.
c ERLIC1-RP and ERLIC2-RP were two replicates for whole proteome analysis.
d ERLIC3-RP and ERLIC4-RP were used for the concurrent analysis of proteome, phosphoproteome and glycoproteome. The only difference between them was that ERLIC4-RP was deglycosylated with PNGase F, but ERLIC3-RP was not.
The pH of digestion buffers, the temperature, and the digestion time are three major factors that control the deamidation rate during proteolytic digestion (9). To minimize digestion-induced deamidation, two improved digestion protocols have been proposed. Endoproteinase Lys-C digestion at pH 6.0 and 4 °C for 24–120 h was used to reduce nonenzymatic deamidation for several model proteins (21). The protocol is effective but is difficult to apply to routine proteomic sample preparation because of the prolonged digestion time and the impact of the 4 °C temperature on the digestion efficiency for complex samples. Another protocol minimized nonenzymatic deamidation by reducing the trypsin digestion time to 0.5h with detergents for denaturing proteins omitted (9). However, this also has not been widely used in proteomic sample preparation, due perhaps to the same consideration of digestion efficiency for complex samples. Because the temperature and incubation time are essential for efficient digestion, we proposed to solve the problem by digesting samples at lower pH in this study.
During the identification of deamidated peptides, database searches usually return false-positive identifications because of the wrong assignment of the 13C peaks of amidated peptides as the corresponding deamidated peptides (22, 23). A possible reason is that the 13C or 13C2 peaks of amidated peptides are selected for matching by peak-picking software, and the MS/MS is not accurate enough to differentiate them from corresponding deamidated ones unambiguously. This is still a serious problem for data acquired with a high resolution mass spectrometer. This issue further compromises the LC-MS/MS method for accurate characterization of deamidation of Asn. Here, the resulting false-positive identifications were eliminated based on the accurate mass difference between the 13C peaks of amidated peptides and the corresponding deamidated ones. We have also evaluated several factors that might affect the deamidation rate and proposed an improved protocol for sample preparation.
MATERIALS AND METHODS
Sample Preparation
Male Sprague-Dawley rats were handled in accordance with the guidelines of Nangyang Technological University Institutional Animal Care and Use Committee (NTU-IACUC), NTU, Singapore. Rat livers were snap-frozen in liquid nitrogen immediately after collection and kept at −80 °C until use. The tissue was cut into small pieces and ground into fine powder in liquid nitrogen with a pestle. The powder was then suspended in 4% SDS. The suspension was sonicated for 10 s thrice on ice and centrifuged at 20,000 × g at room temperature (RT) for 30 min. The protein concentration of the supernatant was then determined by the bicinchoninic acid assay. Rat kidneys were treated in the same way as rat livers.
Traditional In-Gel Tryptic Digestion and PNGase F Treatment
Rat liver samples were run on a SDS-PAGE gel. Protein bands were cut out and transferred to Eppendorf tubes. They were washed with Milli-Q water, mixed with 50% acetonitrile (ACN)/50% 25 mm NH4HCO3 via vigorous vortexing for 30 min and dehydrated with 100% ACN until the gel particles became white. They were then reduced with 10 mm dithiothreitol at 56 °C for 1 h and alkylated with 55 mm iodoacetamide for 45 min in the dark followed by successive washes with 25 mm NH4HCO3 and 50% ACN/50% 25 mm NH4HCO3. Finally, they were dehydrated with 100% ACN and dried in vacuum. Trypsin (V5111, Promega, Madison, WI) was added in the approximate weight ratio of 1:30. V5111 is side-chain protected sequencing grade porcine trypsin. All of the trypsin used in this study is from Promega unless otherwise specified. After the trypsin solution was completely absorbed by gel particles, 25 mm NH4HCO3 was added to completely cover the particles. They were then incubated at 37 °C overnight.
Peptides were extracted from gel particles with 50% ACN containing 0.1% trifluoroacetic acid under sonication for 20 min twice. The combined extracts were dried in vacuum and redissolved in 25 mm NH4HCO3. To the solution, PNGase F (P0705L, New England Biolabs Inc.) was added as per the manufacturer's instructions and incubated at 37 °C for 6h for complete deglycosylation.
Improved In-Gel Tryptic Digestion and PNGase F Treatment
The improved In-Gel tryptic digestion and PNGase F treatment were conducted using the same procedures as the traditional one except that 50 mm CH3COONH4, pH 6 and 50 mm CH3COONH4, pH 5 were used instead of 25 mm NH4HCO3, respectively.
LC-MS/MS
LC-MS/MS was performed as previously described (24). Briefly, peptides were separated and analyzed on a Shimadzu UFLC system coupled to a linear quadrupole ion trap-Fourier transform (LTQ-FT) Ultra (Thermo Electron, Bremen, Germany). Mobile phase A (0.1% formic acid (FA) in H2O) and mobile phase B (0.1% FA in ACN) were used to establish the 60 min gradient comprised of 45 min of 8–35% B, 8 min of 35–50% B and 2 min of 80% B followed by re-equilibration at 5% B for 5 min. Peptides were then analyzed on LTQ-FT with an ADVANCE™ CaptiveSpray™ Source (Michrom BioResources) at an electrospray potential of 1.5 kV. A gas flow of 2, ion transfer tube temperature of 180 °C, and collision gas pressure of 0.85 mTorr were used. The LTQ-FT was set to perform data acquisition in the positive ion mode as previously described (25). Briefly, a full MS scan (350–1600 m/z range) was acquired in the FT-ICR cell at a resolution of 100,000 and a maximum ion accumulation time of 1000 msec. The AGC target for FT was set at 1e+06 and precursor ion charge state screening was activated. The linear ion trap was used to collect peptides and to measure peptide fragments generated by collision-activated dissociation. The default AGC setting was used (full MS target at 3.0e+04, MSn1e+04) in linear ion trap. The 10 most intense ions above a 500 counts threshold were selected for fragmentation in collision-activated dissociation (MS2), which was performed concurrently with a maximum ion accumulation time of 200 msec. Dynamic exclusion was activated for the process, with a repeat count of 1 and exclusion duration of 20 s. Single charged ions were excluded from MS/MS. Isolation width was 2 Da, and default charge state was 5. For collision-activated dissociation, normalized collision energy was set to 35%, activation Q was set to 0.25, and activation time 30 ms. Spectra were acquired in centroid format in raw data files with XCalibur (version 2.0 SR2).
Data Analysis
The raw data were first converted into the dta format using the extract_msn (version 4.0) in Bioworks Browser (version 3.3, Thermo Fisher Scientific, Inc.), and then the dta files were converted into Mascot generic file format using an in-house program. Intensity values and fragment ion m/z ratios were not manipulated. The International Protein Index rat protein database (version 3.40, 40381 sequences, 20547209 residues) and its reversed complement were combined and used for database searches. The database search was performed using an in-house Mascot server (version 2.2.04, Matrix Science, Boston, MA, USA) with MS tolerance of 5.1 ppm, #13C of 2 and MS/MS tolerance of 0.5 Da. Two missed cleavage sites of trypsin were allowed. Carbamidomethylation (C) was set as a fixed modification, and oxidation (M), phosphorylation (S, T, and Y) and deamidation (N and Q) were set as variable modifications. The obtained peptide and protein list for each fraction was exported to Microsoft Excel or processed using an in-house script for further analysis. The dta files of peptides for which the Mascot score was over 20 in each fraction were combined and converted into Mascot generic file format using an in-house program. It was then searched again using Mascot to generate the protein and peptide list.
For high confidence peptide identification, only peptides with an E-value of less than 0.05 were used for statistical calculation. The false discovery rate (FDR) of peptide identification was estimated to be less than 1% (FDR = 2.0 × decoy_hits/total_hits). The FDR rate is based on the assigned spectra. Peptides identified with a consensus N-X-S/T (with X not proline) and a modification of deamidation at the asparagine were regarded as N-linked glycopeptides, and those identified with a modification of deamidation at glutamine or at asparagine but not in the consensus sequence were regarded as nonenzymatically deamidated peptides.
Evaluation of the Ratio of Deamidation in Five Groups of Proteomic Data
Electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) has recently been shown to be both effective and convenient for fractionation of complex tryptic digests in a sequence with RP (24). The ratio of deamidation was evaluated in five groups of proteomic data obtained from rat kidney tissue, i.e. ERLIC1-RP, ERLIC2-RP, ERLIC3-RP, ERLIC4-RP, and In-Gel-RP. For ERLIC-RP, tryptic peptides were obtained as described (24). Each of 2 mg peptides were fractionated using a PolyWAX LP anion-exchange column (4.6 × 200 mm, 5 μm, 300 Å, PolyLC, Columbia, MD) using the ERLIC gradients in Table I, and one third of the collected fractions were used for LC-MS/MS. For In-Gel-RP, 300 μg proteins were run on a SDS-PAGE gel, which was then cut into 21 slices. In-Gel tryptic digestion was performed as described above using the traditional protocol.
Table I. Detailed information about the four ERLIC fractionation methods used.
| ERLIC1 | ERLIC2 | ERLIC3 | ERLIC4 | |
|---|---|---|---|---|
| Mobile phase A | 90% ACN/0.1% acetic acid | 80% ACN/0.1% FA | ||
| Mobile phase B | 30% ACN/0.1% FA | 10% ACN/2% FA | ||
| Gradient | Forty-six fractions were collected with a 140 min gradient of 100% Mobile phase A for 10 min, 0–8% Mobile phase B for 20 min, 8–27% B for 30 min, 27–45% B for 10 min, 45–81% B for 20 min, and 81–100% B for 20 min followed by 30 min at 100% B at a flow rate of 0.5 ml/min | |||
| PNGase F treatment of each fraction | No | No | No | Yes |
Evaluation of Factors Potentially Effecting Deamidation during Sample Preparation
Six hundred micrograms of proteins from rat liver tissue were run on a SDS-PAGE gel, and then about one third of the gel slice was cut from the middle part and digested with trypsin in 25 mm NH4HCO3 at 37 °C for 8 h. Peptides were extracted and divided into 18 aliquots. To evaluate the effect of trypsin digestion time on deamidation, 2 aliquots were incubated at 37 °C an additional 4 h, and another 2 aliquots were incubated at 37 °C an additional 8 h. Because both In-Gel and in-solution digested samples have to be dried in vacuum after peptide extraction or desalting, several commonly used vacuum drying conditions were evaluated. Two milliliters of 70% ACN/0.1% trifluoroacetic acid (TFA) or 50% ACN/2% FA were added to the peptide aliquot, which was then put in a SpeedVac (Thermo Electron, Waltham, MA) at RT for 12 h or at 60 °C for 6 h. In order to keep peptides in the solution for the long duration, the buffers were topped up before drying. Because PNGase F was used for deglycosylation during the determination of N-glycosylation sites, we also investigated its effect on the deamidation ratio. Two aliquots of peptides were incubated in 25 mm NH4HCO3 at 37 °C for 6 h with PNGase F added for deglycosylation, and another two similarly treated as the control without addition of PNGase F. LC-MS/MS was performed in replicate on each aliquot.
Comparison of the Digestion Efficiency of Trypsin at pH6 and pH8 Using Bovine Serum Albumin (BSA) as the Substrate
BSA was dissolved in 8 m urea and 1% SDS at a concentration of 8 mg/ml. The protein was reduced with 10 mm dithiothreitol at 56 °C for 1 h and alkylated with 55 mm iodoacetamide for 45 min in the dark. The concentration of urea was diluted to 1 m with 25 mm NH4HCO3, pH 8, or 50 mm CH3COONH4, pH 6, before trypsin was added in a weight ratio of 1:30. Similarly, the concentration of SDS was diluted to 0.1% before trypsin was added. It was then incubated at 37 °C for 2 h, 4 h, 8 h, and 24 h, respectively. Each portion of 5 μg protein digest was run on a SDS-PAGE gel in order to compare the difference in trypsin digestion efficiency at different time points. For comparison, unmodified trypsin (T8802) from Sigma-Aldrich (St. Louis, MO) was also tested in the same way.
Comparison of the Deamidation Rate at pH6 and pH8 Using Synthetic Peptides
The peptide DGNGYISAAELR was synthesized at the Peptide Synthesis Core Facility, Nangyang Technological University. It was incubated in 25 mm NH4HCO3, pH 8, or 50 mm CH3COONH4, pH 6, for 2 h, 4 h, 8 h, and 24 h at 37 °C. The mass spectra of the untreated and treated peptide were acquired with an ABI 4800 matrix-assisted laser desorption ionization/time of flight (MALDI-TOF)/TOF (Applied Biosystems). Both the untreated and treated peptide was analyzed using a PolySULFOETHYL A column (4.6 × 200 mm, 5 μm, 200 Å, PolyLC, Columbia, MD) on a Shimadzu Prominence UFLC system. Buffer A was 0.1% FA in 80% ACN and buffer B was 30 mm CH3COONH4 in 30% ACN. Twenty fractions were collected during a 30 min gradient: 5–50% buffer B for 5 min, 50–100% buffer B for 20 min, and 100% B for 5 min at a flow rate of 1 ml/min. The absorbance was monitored at 280 nm. The identifications of the peptide and two deamidated products were verified with MALDI-TOF/TOF. The reporter ion of yn - 46 is used to distinguish between the two deamidated products as shown in supplemental Fig. S1 (26).
Comparison of the Traditional and Improved Sample Preparation Protocol
One hundred micrograms of protein from rat liver tissue were run on each lane of a SDS-PAGE gel, and about one tenth of the gel was cut from the middle part and transferred to Eppendorf tubes. To evaluate the effect of pH 6 on tryptic digestion efficiency and the deamidation ratios, gel particles were digested in 50 mm CH3COONH4, pH 6, and 25 mm NH4HCO3, pH 8, for both 8 h and 16 h time courses. To evaluate whether the deglycosylation at pH 5 reduces the deamidation ratio significantly compared with that at pH 8, tryptic peptides were treated with PNGase F for 6 h in 50 mm CH3COONH4, pH 5, or 25 mm NH4HCO3, pH 8. Each test was performed in replicate, and LC-MS/MS was performed in replicate on each test sample.
Enhanced Detection of Nonenzymatic Deamidation from Complex Samples
Two milligrams of protein from rat kidney tissue were run on a SDS-PAGE gel, which was then cut into 20 slices. In-Gel tryptic digestion and PNGase F treatment were performed as described above using the traditional protocol. LC-MS/MS was performed as described above; the loading amount was about 100 μg per injection. This approach is hereafter referred to as In-Gel-RP-En.
RESULTS AND DISCUSSION
The Reliable Identification of Deamidated Peptides in Database Searches
LC-MS/MS is the method of choice for determination of protein deamidation. However, database searches can return some false-positive identification of deamidated peptides because the 13C peaks of amidated peptides can be wrongly assigned as the monoisotopic peaks of the corresponding deamidated peptides. This problem is still severe even when deamidated peptides are identified from complex proteomic samples using mass spectrometers with high resolution. Thus, we need to find a way to differentiate the real deamidated peptides from such false positive ones. In this study, LC-MS/MS was performed on a LTQ-FT Ultra with sufficiently good mass accuracy to solve the problem based on the mass difference between the 13C peaks of amidated peptides and the corresponding deamidated ones. As shown in Fig. 1A, the mass difference between the first isotopic peak M and the 13C peak M' of an amidated peptide is 1.00335 Da; the mass difference between peak M and the first isotopic peak N of the corresponding deamidated peptide is 0.98401 Da; the mass difference between peak M′ and N is 19.34 mDa. In order to differentiate peak N from peak M′ in database searches, a mass error of 5.1 ppm was used here. Thus, deamidated peptides with mass of less than 3868 Da (19.34 mDa/5.1 ppm) could be identified unambiguously. The lower limit of mass error that can be used depends on the mass accuracy of the mass spectrometer in use. The 19.34 mDa mass difference has been used in differentiating the amidated and deamidated forms of synthetic peptides or intact proteins using FTMS (22, 27). In this study we have extended its application to automated database searches in large-scale proteomic analysis. Jung et al. has also developed several programs to aid the accurate assignment of monoisotopic precursor masses to MS/MS data (28).
Fig. 1.

The reliable identification of deamidated peptides. A, Schematic mass spectrum of an amidated peptide and its corresponding deamidated peptide. Peak M is the first isotopic peak of the amidated peptide; M′ and M″ are its 13C and 13C2 peaks, respectively. Peak N is the first isotopic peak of the corresponding deamidated peptide. The mass difference between M′ and N is 19.34 mDa. B, An example of the wrong assignment of the 13C peak as the Asn-deamidation product in Mascot database searches; C, An example of correctly assigned Asn-deamidation in Mascot database searches; D, The extracted ion chromatograms of the peptides identified in Fig. 1B and 1C, in the form of a chromatogram obtained via reversed-phase.
Because nonenzymatic deamidation usually does not run to completion with peptides, the experimental mass difference between the amidated peptides and the corresponding deamidated ones can also be used in determining the false positive identification of deamidated peptides. An example of wrong assignment of the 13C peak of the amidated peptide as deamidated peptides is shown in Fig. 1B. It was determined based on the mass difference between the experimental mass of the assumed deamidated peptide and the amidated one, i.e. 3118.4132–3117.4159 = 0.9973. This is nearer to 1.00335 than 0.98401, so the putative deamidated peptide is actually the 13C peak of the amidated peptide. The mass difference can also be used in determining false positive identifications of deamidated peptides over 3868 Da. An example of a correctly assigned deamidated peptide is shown in Fig. 1C. The mass difference between the experimental masses of the assumed deamidated peptide and the amidated one was 0.9831 Da, which is nearer to 0.98401 than 1.00335. In addition, the identification of deamidated peptides can also be confirmed based on the retention time. As shown in Fig. 1D, an amidated peptide and its 13C peak have the same retention time in RP chromatography, but a deamidated peptide has a different retention time than that of the corresponding amidated peptide. This has been used in the reliable identification of deamidated peptides (19, 29). However, the method that we propose here is decidedly more convenient because it can be achieved easily by merely changing the parameters in database searches.
Evaluation of the Ratio of Deamidation in Five Groups of Proteomic Data
To investigate the effect of nonenzymatic deamidation on proteomic sample preparation, the extent of deamidation was evaluated in five groups of proteomic data using the above-mentioned data analysis method. As shown in Table II, Asn-deamidation occurs to some extent with 4–9% of all peptides, evidence of a significant effect of proteomic sample preparation conditions. Gln-deamidation occurs at a lower ratio: 1–4%. Because Gln-deamidation happens via a mechanism similar to Asn-deamidation (formation of a glutarimide ring intermediate) but at a slower rate, we focus mainly on Asn-deamidation here. It is worthy of notice that all N-glycopeptides identified in ERLIC1-RP, ERLIC2-RP, ERLIC3-RP, and In-Gel-RP are false positive ones because these samples were not treated with PNGase F to remove N-glycans. As the only difference between ERLIC3-RP and ERLIC4-RP was that each fraction from ERLIC was deglycosylated with PNGase F in ERLIC4-RP, the number of their false positive N-glycopeptides should be nearly identical. Thus, about 13.7% (107/781) of the N-glycopeptides in ERLIC4-RP are false positive identifications because of nonenzymatic deamidation, confirming the desirability of reducing the occurrence of this artifact in the identification of N-glycopeptides. To the best of our knowledge, this is the first report about the large-scale evaluation of nonenzymatically deamidated peptides in complex proteomic samples. The database search results for the five groups of data were stored in supplemental data 1–5. The information listed in Table II can be obtained from them using an in-house PERL program.
It may be noted that the modifications to the mobile phase that distinguish ERLIC3 and ERLIC4 from ERLIC1 and ERLIC2 conditions were designed to improve the isolation and identification of peptides with post-translational modifications at the expense of resolution of unmodified peptides. Thus, in Table II, appreciably more glycopeptides were identified using ERLIC4 and significantly less unmodified peptides.
Evaluation of Factors Potentially Affecting Deamidation during Sample Preparation
Because nonenzymatic deamidation occurs at a high ratio in proteomics samples and results in the false positive identification of many N-glycopeptides, it is instructive to determine how it happens and how to prevent it. Deamidation proceeds readily under typical trypsin digestion conditions, i.e. incubation of proteins in digestion buffers of pH 8 at 37 °C for 8–16 h. We first evaluated the effect of trypsin digestion time on deamidation with gel-separated rat liver tissue proteins. As shown in Fig. 2A, the deamidation ratio increases significantly during trypsin digestion for 16 h compared with that for 8 h, indicating that many deamidation sites were newly induced or became detectable during trypsin digestion. Thus, deamidation can be reduced by shortening trypsin digestion time or improving the digestion conditions. It should be noted that the number of detectable deamidated peptides is closely related to the sensitivity of the mass spectrometer used. Because this may change slightly with time, it is preferable to do the LC-MS/MS on samples consecutively for comparing the deamidation ratio.
Fig. 2.

Effect of trypsin digestion time (A), vaccum drying condition (B), and PNGase F treatment (C) on Asn deamidation. BLK = Blank; the control corresponding to treatment using PNGase F conditions but without the PNGase F, as described in Materials and Methods. Proteins extracted from rat liver tissue were used in the test.
Tryptic peptides were also subjected to some other treatments before LC-MS/MS. Generally, 50% ACN, 2% FA was used in the extraction of peptides from gel particles, and 70% ACN, 0.1% TFA was used in the elution of desalted peptides. Both steps were followed by vacuum drying. Thus, we tested whether the vacuum drying with these solutions significantly affected the deamidation ratio. As shown in Fig. 2B, compared with untreated samples, the deamidation ratio did not significantly increase in the four tested vacuum drying conditions, i.e. with 70% ACN/0.1% TFA at RT for 12h, 50% ACN/2% FA at RT for 12h, 70% ACN/0.1% TFA at 60 °C for 6h and 50% ACN/2% FA at 60 °C for 6h. This indicates that nonenzymatic deamidation proceeds slowly under acidic conditions even at 60 °C.
The effect of PNGase F treatment on the deamidation ratio is shown in Fig. 2C. Compared with untreated samples, the deamidation ratio increased significantly in both PNGase F treated samples and the control, the two being comparable. This indicates that the incubation of tryptic peptides with 25 mm NH4HCO3 is responsible for the increase of deamidation, not the PNGase F treatment itself, and suggests that the incubation conditions could be improved in order to reduce the occurrence of deamidation. The database search results of the tests listed in Fig. 2 are shown in supplemental data 6.
Improved Protocol for N-Glycosylation Site Determination from Complex Samples
Based on our results, the standard trypsin digestion and deglycosylation protocol induces a significant degree of nonenzymatic deamidation because of the mildly alkaline pH of the reaction buffers. As shown by the schematic in Fig. 3, we propose to solve this problem via an improved protocol that involves conducting protein reduction, alkylation, and tryptic digestion in 50 mm CH3COONH4, pH 6 and performing the deglycosylation in 50 mm CH3COONH4, pH 5. The deamidation rate is minimal at pH 5 (15), but tryptic digestion cannot be performed at so low a pH according to the instructions from the manufacturer. Accordingly, we evaluated tryptic digestion at pH 6. For the deglycosylation of N-glycopeptides, the suggested reaction buffer for PNGase F is 50 mm Na2HPO4, pH 7.5, or 25 mm NH4HCO3, but the enzyme activity was found not to be compromised at a pH as low as 5 (30). Accordingly, we proposed to do the deglycosylation at pH 5 in this study.
Fig. 3.
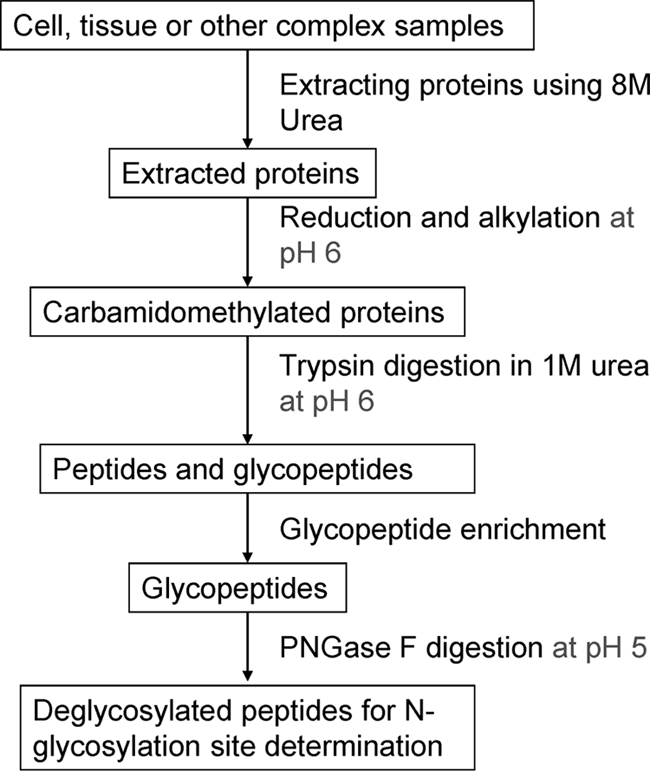
Proposed improved tryptic digestion and PNGase F treatment protocol for N-glycosylation site determination.
Comparison of the Digestion Efficiency of Trypsin at pH6 and pH8 Using Bovine Serum Albumin (BSA) as the Substrate
The digestion efficiency of trypsin at pH 6 was evaluated with BSA before it was applied to complex samples. As shown in Fig. 4A and 4C, when urea is used as the denaturant, five μg BSA disappeared completely after being digested with either unmodified or side-chain protected trypsin for 2–24 h, indicating that even 2 h digestion at pH 6 at 37 °C was enough for complete digestion. However, when SDS was used as the denaturant, the band of BSA remained unchanged even after 24 h digestion at pH 6 at 37 °C if unmodified trypsin was used (Fig. 4B), indicating that it was completely deactivated at pH 6. BSA was digested into large fragments at pH 8 if SDS and unmodified trypsin were used. In contrast, side-chain protected trypsin performed much better than unmodified when SDS was used as the denaturant. As shown in Fig. 4D, although it only digested BSA into large fragments after 24 h at pH 6 at 37 °C, digestion was complete at pH 8 in 2 h. Thus, it was preferable to use urea as the denaturant when the trypsin digestion was done at pH 6, and the resulting activity of trypsin was good enough for proteomic analysis. The difference in the performance of V5111 and T8802 trypsin is possibly because V5111 trypsin is modified by reductive methylation, rendering it more resistant to SDS denaturation. Because most researchers are using side-chain protected trypsin to prevent autolysis, its high activity at pH 6 seems to be an additional benefit in reducing digestion-induced deamidation.
Fig. 4.

SDS-PAGE images of trypsin digested BSA at pH 6 and pH 8 for different time periods using urea or SDS as the denaturant. A, SDS-PAGE image of unmodified trypsin-digested BSA at pH 6 and pH 8 in urea buffer; B, SDS-PAGE image of unmodified trypsin digested BSA at pH 6 and pH 8 in SDS buffer; C, SDS-PAGE image of side-chain protected trypsin digested BSA at pH 6 and pH 8 in urea buffer; D, SDS-PAGE image of side-chain protected trypsin digested BSA at pH 6 and pH 8 in SDS buffer. Lane 1, MW mark; 2, BSA without adding trypsin; 3, digestion at pH 6 for 2 h; 4, digestion at pH 8 for 2 h; 5, digestion at pH 6 for 4 h; 6, digestion at pH 8 for 4 h; 7, digestion at pH 6 for 8 h; 8, digestion at pH 8 for 8 h; 9, digestion at pH 6 for 24 h; 10, digestion at pH 8 for 24 h.
Comparison of the Deamidation Rate at pH6 and pH8 Using Synthetic Peptides
The deamidation rate at pH 6 and pH 8 at 37 °C was determined using the synthetic peptide DGNGYISAAELR. As shown in Fig. 5, the strong cation exchange (SCX) chromatograms showed the gradual transformation of the undeamidated peptide into two deamidation products containing -βDG- and -DG- at pH 8. The identification of the peptide and two deamidated products was verified with MALDI-TOF/TOF. The deamidation ratio was calculated as the area of the two deamidation products divided by the sum of the area of both deamidated and undeamidated peptides. It was estimated to be 5.7%, 11.4%, 25.8%, and 64.8% after 2 h, 4 h, 8 h, and 24 h incubation at pH 8 at 37 °C. However, the chromatograms remained unchanged when the synthetic peptide was incubated at pH 6 at 37 °C for 2 h, 4 h, and 8 h [data not shown]. Even after 24 h incubation at pH 6 at 37 °C, the deamidation ratio was estimated to be only 0.52%. The same trend was also semiquantitatively reflected in the MALDI-TOF mass spectra of the solution at different time points shown in the insets of Fig. 5. This confirms that deamidation happens much more slowly at pH 6 than that at pH 8. Krokhin et al. reported that the half-life of the deamidation of peptides with -NG- was ∼8h in 100 mm NH4HCO3 at 37 °C (19). This is two times faster than the deamidation rate in 25 mm NH4HCO3 at 37 °C detected in this study. The difference might be because of some difference in the incubation buffer and/or the peptide sequence.
Fig. 5.

Monitoring the deamidation of the synthetic peptide DGNGYISAAELR at pH 6 and pH 8 by HPLC and MALDI-TOF. Solutions of the synthetic peptide in 50 mm CH3COONH4, pH 6, or 25 mm NH4HCO3 were incubated at 37 °C for 2 h, 4 h, 8 h and 24 h, respectively. They were resolved with a SCX column with UV detection at 280 nm. Each unfractionated mixture was also analyzed semiquantitatively with MALDI-TOF with the mass spectra shown in the insets.
Comparison of the Traditional and Improved Sample Preparation Protocol Using Complex Samples
Gel-separated rat liver tissue proteins were used in testing the improved sample preparation protocol. The digestion efficiency of trypsin was compared at pH 6 and 8. Although the kinetics of tryptic digestion were slower at pH 6, the number of peptides actually identified in the four test groups was comparable (Fig. 6A). This suggests that a pH of 6 can be used without sacrificing the extent of proteomic identification. However, this comparison should be performed anew if different samples are used because their properties may affect the digestion efficiency. In addition, we tried using Glu-C from two companies (Sigma-Aldrich and Roche Diagnostics (Basel, Switzerland)) in digesting proteins at pH 4–6. A significant decrease in the deamidation ratio was observed, but the digestion efficiency in both cases was much lower than that of trypsin [data not shown].
Fig. 6.
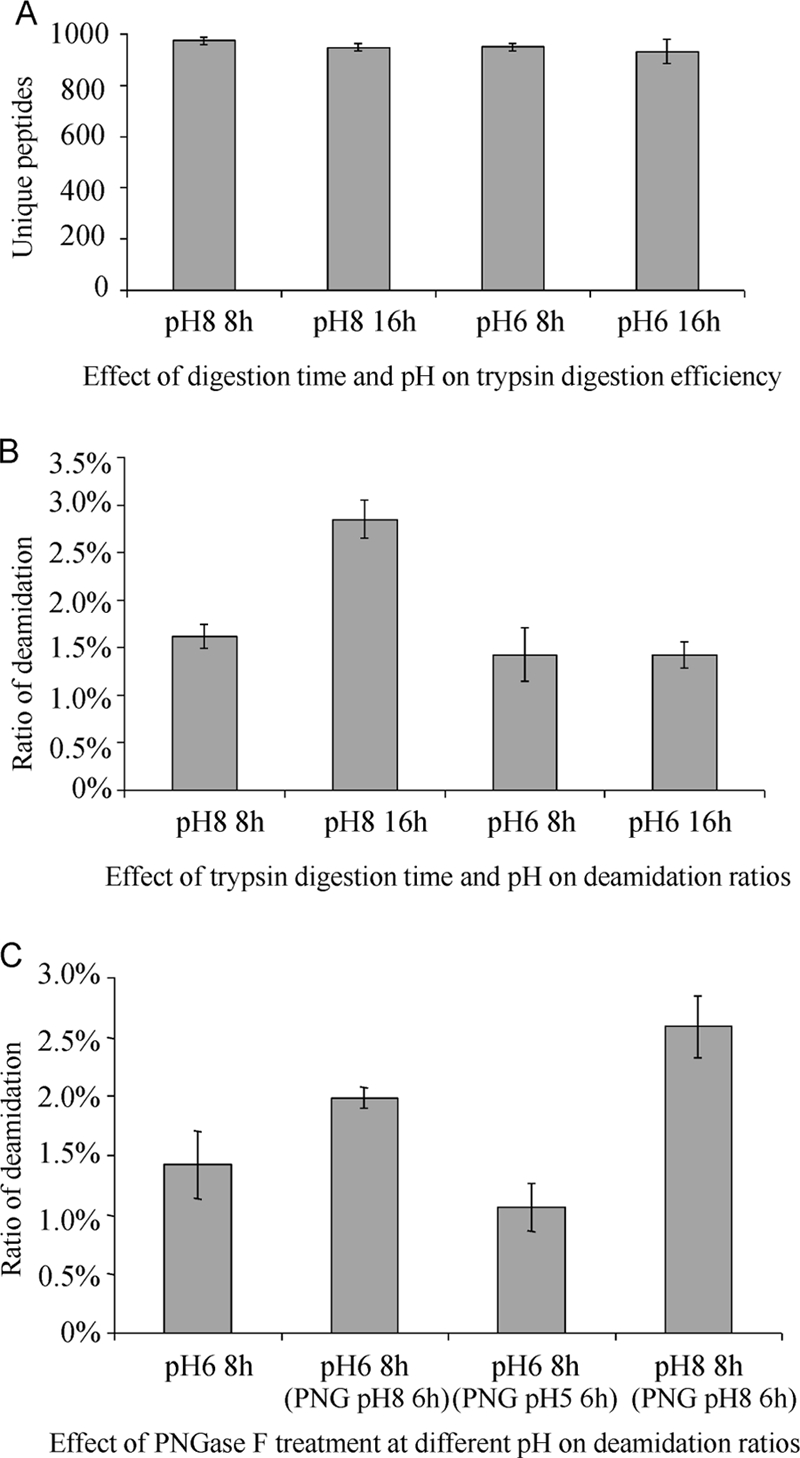
Comparison of the traditional and improved tryptic digestion and PNGase F treatment protocol. A, Effect of digestion time and pH on trypsin digestion efficiency; B, Effect of trypsin digestion time and pH on deamidation ratios; C, Effect of PNGase F treatment at different pH on deamidation ratios. Proteins extracted from rat liver tissue were used in the test.
The deamidation ratio was also compared among the four tested trypsin digestion conditions. Trypsin digestion at pH 6 for 8 h and 16 h produced comparable deamidation ratios, which were slightly less than that at pH 8 for 8h (Fig. 6B). This indicates that some deamidations exist in vivo in the native proteins or occur easily in the first 8 h of tryptic digestion, but incubation of tryptic peptides with the reaction buffer of pH 6 did not lead to a significant increase in deamidation after the initial 8 h digestion time. Apparently, prolonged trypsin digestion at pH 6 can be used safely with complex samples in order to ensure thorough digestion. However, trypsin digestion at pH 8 for 16 h produced a significantly higher deamidation ratio than in the other groups (Fig. 6B), further confirming the liability to deamidation from prolonged incubation with reaction buffers of pH 8. Thus, decreasing trypsin digestion time is an alternative solution for reducing deamidation while maintaining digestion efficiency.
The effect of PNGase F treatment on deamidation was evaluated at various pH values as well. As shown in Fig. 6C, when tryptic peptides obtained from 8 h digestion at pH 6 were treated with PNGase F in 25 mm NH4HCO3, pH 8, at 37 °C for 6 h, the deamidation ratio increased 40.1%. However, it showed no significant increase when they were treated with PNGase F in 50 mm CH3COONH4, pH 5, at 37 °C for 6 h. The number of N-glycopeptides identified from these two different treatments was comparable. It appears that the PNGase F treatment at pH 5 is much better than that at pH 8 in reducing nonenzymatic deamidation while maintaining adequate enzyme activity. The deamidation ratio was 1.07% ± 0.20% when samples were prepared using the improved sample preparation protocol, i.e. trypsin digestion at pH 6 for 8 h and PNGase F treatment at pH 5 for 6 h, a 58.7% decrease compared with the results from the traditional sample preparation protocol, i.e. trypsin digestion at pH 8 for 8 h and PNGase F treatment at pH 8 for 6 h. The database search results of the tests listed in Fig. 6 are shown in supplemental data 7. Actually, many labs routinely perform tryptic digestion and PNGase F treatment overnight (8 h–16 h) in order to ensure complete digestion, in which case the deamidation ratio could be even higher.
Comparison of the Motifs of Asn-deamidation and Gln-deamidation
Many papers have been published on various aspects of the deamidation of peptides and proteins, most of them about Asn-deamidation. Asn-deamidation is particularly prone to occurring with the sequences -N-G- and -N-S-. As best we know, no large-scale analysis of Gln-deamidated peptides in complex samples has been done yet, and the relevant motifs of susceptible sequences remain undiscovered. The rate of Gln-deamidation is ten times slower than that of Asn-deamidation, making it more difficult to analyze. In this study, we occasionally found that an increase in the amount of sample loaded in LC-MS/MS was helpful for the detection of deamidated peptides. When the loading amount per LC-MS/MS injection increases from about 7.5 μg (In-Gel-RP) to about 100 μg (In-Gel-RP-En), the figure for the ratio of Asn-deamidation increases from 5.45% to 10.10%, and the figure for the ratio of Gln-deamidation increases from 1.69% to 10.41%. Obviously there has been a dramatic increase in the ability to identify Gln-deamidated peptides of low abundance, presumably because of enhancement of mass signals, as well as a less dramatic increase in identification of Asn-deamidated peptides. However, the increase in the loading amount does not have a significant effect on the identification of unmodified peptides. With 794 Gln-deamidated peptides identified in In-Gel-RP-En (supplemental Table 1), motifs for Gln-deamidation susceptibility could be compiled and compared with those for Asn-deamidation in ERLIC4-RP, in which 1020 Asn-deamidated peptides were identified (supplemental Table 2). As shown in Fig. 7, Asn-deamidation tends to occur at -N-G-, -N-S-, and -N-D- sequences, consistent with previous studies. Gln-deamidation tends to occur at -Q-V-, -Q-L- and -Q-G- sequences, and to a lesser extent at Q-A- and Q-E-. This contrasts with Asn-deamidation motifs. Gln-deamidation proceeds through a mechanism similar to that of Asn-deamidation except that a six-member glutarimide ring is formed. Presumably the effect of steric hindrance on the formation of a glutarimide ring is not as strong as that on the formation of a five-member ring so that the kinetics of Gln-deamidation are comparatively rapid with -Q-V- and -Q-L- sequences. Interesting, -Q-V-, -Q-L-, -Q-A-, and -Q-I- have relatively high deamidation rate. We speculate that the electron-donating property of alkyl- groups (of V, L, A, and I residues) promotes the nucleophilic attack that results in the intermediate glutarimide ring; the inductive effect makes it easier for the attacking nitrogen to push its unshared pair of electrons onto the electropositive carbonyl that gets attacked to form the ring. The database search results of In-Gel-RP-En are shown in supplemental data 8.
Fig. 7.

Comparison of the incidence of Asn-deamidation and Gln-deamidation in proteomic samples. A, Experimental distribution of Asn-deamidation and Gln-deamidation; B, Distribution of Asn-Xxx/Gln-Xxx in IPI rat 3.40 database; C, Normalized Distribution of Asn/Gln deamidation. The x axis lists the residue on the C-terminal side of the deamidated N- or Q-residue.
CONCLUSIONS
Nonenzymatic asparagine deamidation was found to occur to some extent in 4–9% of all peptides during workup of samples for proteomic analysis and resulted in the false positive identification of numerous N-glycopeptides. An improved protocol of trypsin digestion at pH 6 and PNGase F treatment at pH 5 was proposed and validated in order to solve this problem. The minimization of nonenzymatic deamidation will not only improve the reliability of N-glycopeptide identification but also be helpful for the identification of low-abundance peptides from complex samples. It should be noted that nonenzymatic deamidation during sample preparation cannot be avoided completely even with the improved protocol (unless the minimal deamidation in question was present in vivo). In the future, H218O could also be used during PNGase F treatment in the improved protocol to reduce further the false positive identification of N-glycopeptides.
Footnotes
* This work is supported by grants from the Ministry of Education (ARC: T206B3211) and the Agency for Science, Technology and Research (BMRC: 08/1/22/19/575) of Singapore.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- LC-MS/MS
- liquid chromatography coupled to tandem mass spectrometry
- ERLIC
- electrostatic repulsion-hydrophilic interaction chromatography
- LTQ
- linear quadrupole ion trap
- FA
- formic acid
- FDR
- false discovery rate
- RT
- room temperature
- ACN
- acetonitrile
- TFA
- trifluoroacetic acid
- BSA
- bovine serum albumin.
REFERENCES
- 1. Roher A. E., Lowenson J. D., Clarke S., Wolkow C., Wang R., Cotter R. J., Reardon I. M., Zürcher-Neely H. A., Heinrikson R. L., Ball M. J. (1993) Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer's disease. J. Biol. Chem. 268, 3072–3083 [PubMed] [Google Scholar]
- 2. Takata T., Oxford J. T., Demeler B., Lampi K. J. (2008) Deamidation destabilizes and triggers aggregation of a lens protein, betaA3-crystallin. Protein Sci. 17, 1565–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wilmarth P. A., Tanner S., Dasari S., Nagalla S. R., Riviere M. A., Bafna V., Pevzner P. A., David L. L. (2006) Age-related changes in human crystallins determined from comparative analysis of post-translational modifications in young and aged lens: does deamidation contribute to crystallin insolubility? J. Proteome Res. 5, 2554–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robinson N. E., Robinson A. B. (2001) Molecular clocks. Proc. Natl. Acad. Sci. U.S.A. 98, 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Robinson N. E., Robinson A. B. (2004) Amide molecular clocks in drosophila proteins: potential regulators of aging and other processes. Mech. Ageing Dev. 125, 259–267 [DOI] [PubMed] [Google Scholar]
- 6. Solstad T., Flatmark T. (2000) Microheterogeneity of recombinant human phenylalanine hydroxylase as a result of nonenzymatic deamidations of labile amide containing amino acids. Effects on catalytic and stability properties. Eur. J. Biochem. 267, 6302–6310 [DOI] [PubMed] [Google Scholar]
- 7. Huang L., Lu J., Wroblewski V. J., Beals J. M., Riggin R. M. (2005) In vivo deamidation characterization of monoclonal antibody by LC/MS/MS. Anal. Chem. 77, 1432–1439 [DOI] [PubMed] [Google Scholar]
- 8. Robinson N. E., Robinson A. B. (2001) Deamidation of human proteins. Proc. Natl. Acad. Sci. U.S.A. 98, 12409–12413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ren D., Pipes G. D., Liu D., Shih L. Y., Nichols A. C., Treuheit M. J., Brems D. N., Bondarenko P. V. (2009) An improved trypsin digestion method minimizes digestion-induced modifications on proteins. Anal. Biochem. 392, 12–21 [DOI] [PubMed] [Google Scholar]
- 10. Geiger T., Clarke S. (1987) Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 262, 785–794 [PubMed] [Google Scholar]
- 11. Li X., Lin C., O'Connor P. B. (2010) Glutamine deamidation: differentiation of glutamic acid and gamma-glutamic acid in peptides by electron capture dissociation. Anal. Chem. 82, 3606–3615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Robinson N. E., Robinson A. B. (2004) Prediction of primary structure deamidation rates of asparaginyl and glutaminyl peptides through steric and catalytic effects. J. Pept. Res. 63, 437–448 [DOI] [PubMed] [Google Scholar]
- 13. Catak S., Monard G., Aviyente V., Ruiz-López M. F. (2006) Reaction mechanism of deamidation of asparaginyl residues in peptides: effect of solvent molecules. J. Phys. Chem. A 110, 8354–8365 [DOI] [PubMed] [Google Scholar]
- 14. Zabrouskov V., Han X., Welker E., Zhai H., Lin C., van Wijk K. J., Scheraga H. A., McLafferty F. W. (2006) Stepwise deamidation of ribonuclease A at five sites determined by top down mass spectrometry. Biochemistry 45, 987–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li X., Cournoyer J. J., Lin C., O'Connor P. B. (2008) Use of 18O labels to monitor deamidation during protein and peptide sample processing. J. Am. Soc. Mass. Spectrom. 19, 855–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gaza-Bulseco G., Li B., Bulseco A., Liu H. C. (2008) Method to differentiate asn deamidation that occurred prior to and during sample preparation of a monoclonal antibody. Anal. Chem. 80, 9491–9498 [DOI] [PubMed] [Google Scholar]
- 17. Cournoyer J. J., Pittman J. L., Ivleva V. B., Fallows E., Waskell L., Costello C. E., O'Connor P. B. (2005) Deamidation: Differentiation of aspartyl from isoaspartyl products in peptides by electron capture dissociation. Protein Sci. 14, 452–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang H., Fung E. Y., Zubarev A. R., Zubarev R. A. (2009) Toward proteome-scale identification and quantification of isoaspartyl residues in biological samples. J. Proteome Res. 8, 4615–4621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krokhin O. V., Antonovici M., Ens W., Wilkins J. A., Standing K. G. (2006) Deamidation of -Asn-Gly- sequences during sample preparation for proteomics: Consequences for MALDI and HPLC-MALDI analysis. Anal. Chem. 78, 6645–6650 [DOI] [PubMed] [Google Scholar]
- 20. Robinson N. E., Robinson Z. W., Robinson B. R., Robinson A. L., Robinson J. A., Robinson M. L., Robinson A. B. (2004) Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J. Pept. Res. 63, 426–436 [DOI] [PubMed] [Google Scholar]
- 21. Stroop S. D. (2007) A modified peptide mapping strategy for quantifying site-specific deamidation by electrospray time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 21, 830–836 [DOI] [PubMed] [Google Scholar]
- 22. Robinson N. E., Lampi K. J., McIver R. T., Williams R. H., Muster W. C., Kruppa G., Robinson A. B. (2005) Quantitative measurement of deamidation in lens betaB2-crystallin and peptides by direct electrospray injection and fragmentation in a Fourier transform mass spectrometer. Mol. Vis. 11, 1211–1219 [PMC free article] [PubMed] [Google Scholar]
- 23. Segu Z. M., Hussein A., Novotny M. V., Mechref Y. (2010) Assigning N-glycosylation sites of glycoproteins using LC/MSMS in conjunction with endo-M/exoglycosidase mixture. J. Proteome Res. 9, 3598–3607 [DOI] [PubMed] [Google Scholar]
- 24. Hao P., Guo T., Li X., Adav S. S., Yang J., Wei M., Sze S. K. (2010) Novel application of electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) in shotgun proteomics: comprehensive profiling of rat kidney proteome. J. Proteome Res. 9, 3520–3526 [DOI] [PubMed] [Google Scholar]
- 25. Gan C. S., Guo T., Zhang H., Lim S. K., Sze S. K. (2008) A comparative study of electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) versus SCX-IMAC-based methods for phosphopeptide isolation/enrichment. J. Proteome Res. 7, 4869–4877 [DOI] [PubMed] [Google Scholar]
- 26. González L. J., Shimizu T., Satomi Y., Betancourt L., Besada V., Padrón G., Orlando R., Shirasawa T., Shimonishi Y., Takao T. (2000) Differentiating alpha- and beta-aspartic acids by electrospray ionization and low-energy tandem mass spectrometry. Rapid Commun. Mass Spectrom. 14, 2092–2102 [DOI] [PubMed] [Google Scholar]
- 27. Robinson N. E., Zabrouskov V., Zhang J., Lampi K. J., Robinson A. B. (2006) Measurement of deamidation of intact proteins by isotopic envelope and mass defect with ion cyclotron resonance Fourier transform mass spectrometry. Rapid Commun. Mass Spectrom. 20, 3535–3541 [DOI] [PubMed] [Google Scholar]
- 28. Jung H. J., Purvine S. O., Kim H., Petyuk V. A., Hyung S. W., Monroe M. E., Mun D. G., Kim K. C., Park J. M., Kim S. J., Tolic N., Slysz G. W., Moore R. J., Zhao R., Adkins J. N., Anderson G. A., Lee H., Camp D. G., 2nd, Yu M. H., Smith R. D., Lee S. W. (2010) Integrated post-experiment monoisotopic mass refinement: an integrated approach to accurately assign monoisotopic precursor masses to tandem mass spectrometric data. Anal. Chem. 82, 8510–8518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dasari S., Wilmarth P. A., Rustvold D. L., Riviere M. A., Nagalla S. R., David L. L. (2007) Reliable detection of deamidated peptides from lens crystallin proteins using changes in reversed-phase elution times and parent ion masses. J. Proteome Res. 6, 3819–3826 [DOI] [PubMed] [Google Scholar]
- 30. Liu Y., Salas-Solano O., Gennaro L. A. (2009) Investigation of sample preparation artifacts formed during the enzymatic release of N-linked glycans prior to analysis by capillary electrophoresis. Anal. Chem. 81, 6823–6829 [DOI] [PubMed] [Google Scholar]