

Abstract
A synthesis of representative monohydroxy derivatives of valinomycin (VLM) was achieved under mild conditions by direct hydroxylation at the side-chains of the macrocyclic substrate using dioxiranes. Results demonstrate that the powerful methyl(trifluoromethyl)dioxirane 1b should be the reagent of choice to carry out these key transformations. Thus, a mixture of compounds derived from the direct dioxirane attack at the β-(CH3)2C-H alkyl chain of one Hyi residue (compound 3a) or of one Val moiety (compounds 3b and 3c) could be obtained. Following convenient mixture separation, each of the new oxyfunctionalized macrocycles became completely characterized.
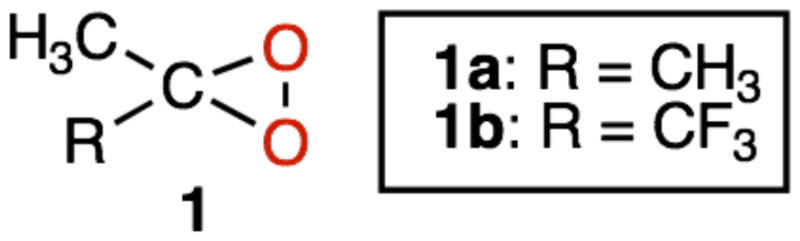
Chemical modification of natural peptides through selective backbone and/or side-chain functionalization constitutes a powerful approach to novel drug design and to the development of peptide-based nanomaterials.1 To achieve this, it is desirable to employ efficient reagents which operate selectively under mild conditions that are compatible with natural biomolecules. In this context, the dioxiranes (1)2,3 represent a useful new entry. In fact, this class of powerful oxidants proved capable of performing selectively under remarkably mild conditions, i.e. at subambient temperatures and at a pH close to neutrality.2–4
In recent papers,5 we have shown that direct and selective hydroxylation of N-protected alkyl amino acids, as well as of di- and tripeptides bearing alkyl side chains, are successfully achieved with no loss of configuration at the chiral centers using dimethyldioxirane (1a)2 or its trifluoro analogue 1b.3
We observed that the powerful methyl(trifluoromethyl)dioxirane (1b) is the reagent of choice to carry out these trasformations because of its higher reactivity, which is accompained by remarkable selectivity. These reactions can be conveniently tuned employing a suitable peptide N-protecting group. For instance, in the selective hydroxylation of N-protected di- and tripeptides, dioxirane attack at the γ-CH of leucine or at the β-CH of valine residues could be selectively directed at the alkyl side chain using amide-type protecting groups, such as acetyl (Ac), trifluoroacetyl (Tfa) or pivaloyl (Piv).5a,b
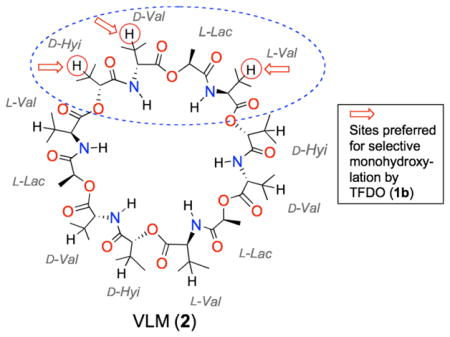
In our view, these selective oxidative transformations of peptides using dioxiranes are promising; in fact, they pave the road to access hydroxy derivatives of attractive target peptides as well as of macrocyclic peptides. These constitute a major class of natural and unnatural molecules encompassing a wide range of structures and functional groups, which makes them versatile compounds in diverse areas such as, ion transport, biological recognition, and catalysis.6 Among the various naturally occurring cyclopeptides, valinomycin (VLM, 2) (Fig. 1) has attracted much attention as an ion carrier deriving its antibiotic, antitumoral, and antiviral properties from specific binding to potassium ion.7 This dodecadepsipeptide presents a threefold repeated pattern, i.e. (D-Hyi-D-Val-L-Lac-L-Val)3. Unfortunately, the renowned VLM cytotoxicity appears has so far prevented its clinical use.8
Figure 1.
Common Dioxiranes in the isolated form.
To our knowledge, VLM analogues which present hydroxy groups at specific sites in the backbone and/or in the side-chains have not been reported to date. In view of the high versatility of the hydroxy functionalities in further leading to a variety of functional groups, we considered that hydroxylated VLM derivatives would be valuable for designing new analogues of this macrocycle. In fact, they might allow access to new complex structures with modified properties. We report herein our preliminary findings concerning the direct dioxirane oxidation of VLM establishing this as an efficient method to access VLM hydroxy derivatives under remarkably mild conditions.
Our initial screening experiments pointed at the powerful methyl(trifluoromethyl)dioxirane 1b (TFDO) as the reagent of choice to carry out efficient VLM hydroxylations. In view of the volatility and high reactivity of TFDO, oxidation experiments were performed most conveniently at sub-ambient temperature.
Using a suitable excess of dioxirane 1b (TFDO) standardized solution,9 oxidations of VLM substrate were carried out on the 50–100 mg scale. The simple procedure merely involved addition of an aliquot (0.5–1 mL) of a cold (0 °C) dioxirane solution to the macrocyclic peptide dissolved in acetone (2–3 mL) at 0 °C. The reaction was kept at 0 °C and its progress monitored by HPLC. Product isolation simply entailed solvent removal.
Initially, we verified that the poly-oxyfunctionalization of the macrocyclic peptide takes place using a substantial excess of the TFDO oxidant. In fact, when the oxidation reaction is carried out with a 30-fold molar excess of TFDO, oxidation products resulting from sequential O-insertions are obtained.
The MALDI mass spectrum of the reaction mixture resulting from the oxidation of VLM with 30 equiv of TFDO in acetone at 0 °C (18 h reaction time) suggested the formation of mono-, di-, tri-, tetra-, and even of penta-hydroxy derivatives, which accompany some residual unreacted VLM, [M + K]+= 1149.39 (Fig. 3).
Figure 3.

MALDI-ToF spectrum of the reaction mixture resulting from oxidation of VLM (2) with 30 equiv of TFDO (1b) in the presence of KCl (DHB matrix).
Having established the efficiency of TFDO as an oxidant, we sought reaction conditions that allow for the selective introduction of a single hydroxyl in conjunction with optimal substrate conversion. We found that, running the oxidation reaction at 0° C in acetone solvent using 10 equiv. of the oxidant, substrate conversion during 6 h was 45% (based on the recovered unreacted substrate). In particular, monooxidation products 3a–c (cf. Figure 4) were formed exclusively, as indicated by HPLC and MALDI-ToF analysis of the crude reaction mixture.
Figure 4.
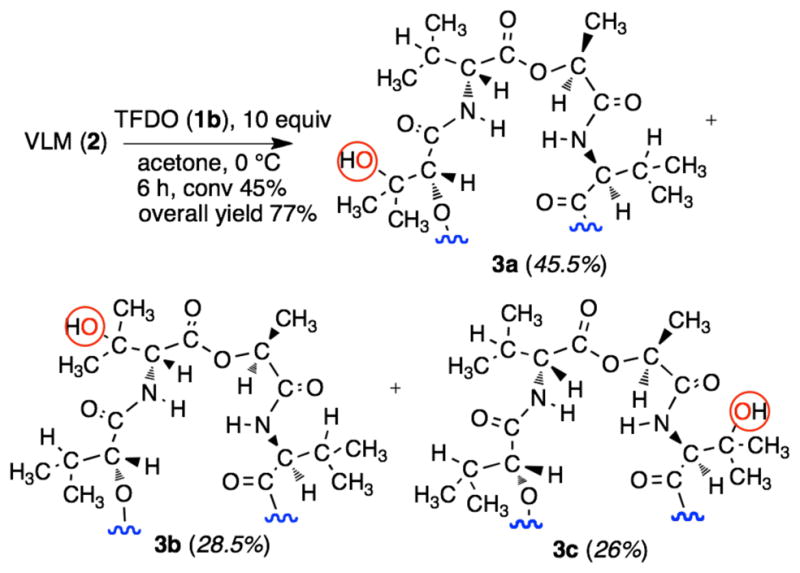
Valinomycin oxidation by methyl(trifluoromethyl)-dioxirane. Products distribution shown in parentheses.
Isolation of products 3a–c simply entailed a column chromatography separation of the residual starting material from the product mixture. The reaction products were subsequently individually separated and purified by preparative HPLC. Each of the compounds 3a–c (Figure 4) was obtained in adequate yield with high purity (>95%, HPLC). None of the amorphous solid products isolated were found amenable to direct structure determination using X-rays crystallography. However, all three mono hydroxylated products (β-OH-D-Hyi)1-VLM (3a), (β-OH-D-Val)1-VLM (3b), and (β-OH-L-Val)1-VLM (3c) could be fully characterized by using a combination of spectroscopic techniques. The MALDI mass spectra of 3a, 3b, and 3c are consistent with the incorporation of a single oxygen atom. The actual site of hydroxylation could be assessed by careful inspection of the corresponding 1D NMR (1H, 13C, DEPT-135) and 2D NMR (COSY, NOESY, HMQC, HMBC) spectra. To this end, our strategy relied on sequence-specific assignments based on the residual NHi+1-αHi correlations provided by the corresponding 1H-1H NOESY NMR spectra.10 This approach was especially useful in discriminating compound 3b from 3c, since their structural divergence derives almost entirely from the different residues that are proximal to the hydroxylated valine moiety. Details are given in the Supporting Information section.
For example, the NOESY NMR spectrum of 3b relative to the NH resonance region (Fig. 5) shows that the hydroxylated amino acid residue is sequential to a D-Hyi residue, since the NH proton signal (7.60 ppm) of the former shows a cross signal with the α-CH proton resonance (5.04 ppm) of the latter (blue dashed line, Fig. 3).
Figure 5.

NOESY NMR spectrum of compound 3b (acetone-d6, 400 MHz, tmix= 0.35 sec).
Conversely, in the NOESY spectrum of 3c the amidic proton of the hydroxylated residue shows an interresidual correlation with the α-CH proton of an L-Lac residue, which is consistent with the structure envisaged. Furthermore, the 13C NMR spectrum of 3b presents a signal at 72.59 ppm that can be assigned to the resonance of the tertiary alcohol carbon (C-OH), since this signal disappears in the DEPT-135 spectrum. Additiony, in the HMBC spectrum (cf., Supporting Info), this signal displays cross-peaks with the OH proton at 4.57 ppm, as well as with the doublet at 4.45 ppm (α-CH) and with the two singlets at 1.35, 1.31 ppm (γ,γ′-CH3).
A comment is in order concerning the high chemoselectivity observed in the oxidation reactions presented in Figure 4. As already mentioned, we previously reported that the TFDO oxidation of noncyclic, N-acyl protected peptides bearing alkyl side chains leads to selective hydroxylation at the side chain of leucine and valine residues, while the N-H bonds remain unaffected.5a,b This seems to apply also to the oxidation of the cyclic peptide valinomycin (2) with TFDO (1b); in fact, we observe that the D-, and L-Val residues experience selective hydroxylation at the β-CH bonds.
The remarkable reactivity of methyl(trifluoromethyl)-dioxirane 1b (TFDO) in the selective hydroxylation of “unactivated” hydrocarbon C—H bonds, was previously demonstrated. In a decidedly stereoselective process.4a high tertiary vs. secondary selectivities (Rst from 15 to > 250) can be routinely achieved.9 This was attributed to a high sensitivity to steric shielding, due to the stringent stereoelectonic alignement demands posed by the “oxenoid” process of dioxirane O-insertion into C—H bonds. Actually, concerning the application of dioxiranes to the oxyfunctionalization of complex targets, the remarkable site-selectivity observed herein in the mono-hydroxylation at VLM side chains finds an instructive precedent in the reported selective hydroxylation at the tertiary carbon (CH3)2C—H in the side-chain moiety of Vitamin D3 and of Vitamin D2.11
On the other hand, on the basis of electronic factors it is problematic to rationalize the different regioisomeric ratio observed in the distribution of hydroxylation products 3a–c (i.e., 1.75 : 1.1 : 1). In fact, TFDO hydroxylation at the β-CH of a D-Hyi residue is seen to prevail over oxyfunctionalization at the β-CH of D- and L-Val residues by a factor of ca. 2; and this, despite the stronger deactivating effect exercised of the proximal ester functionality in the D-Hyi residue with respect to the amidic group nearby the D- and L-Val fragments.
Complementing previous NMR investigations,12 recent Raman Optical Activity spectroscopy and multiscale molecular modeling investigations have addressed the item of valinomycin preferred conformations.13
These pointed out that in solution free (uncomplexed) valinomycin adopts variable conformations depending on solvent polarity; while a symmetric “bracelet” form dominates in non-polar solvents an asymmetric “bracelet” or a “propeller” conformation (akin to that shown in Fig. 6) was suggested for solvents of medium polarity.12,13 The latter are rather flexible and it was reported that dynamic conformational equilibria of valinomycin in solution occur with relaxation times shorter than 10−6 s.13,14 Such being the case, in acetone (the reaction solvent employed in our experiments), the VLM macrocycle should consist of a mixture of various interconverting conformers. It is likely that, under the reaction conditions applied herein, important details of the favored macrocycle conformations play a major role in dictating the optimal steric exposure to dioxirane attack at the various isopropyl residues. Hence the preferential hydoxylation sites ensue.
Figure 6.

One representative VLM “propeller” conformation in solvents of medium polarity (ref. 12,13). For clarity, most H atoms have been omitted.
The results reported herein offer easy access to VLM hydroxy derivatives using dioxiranes under mild conditions. Through further transformation of the hydroxyl functionalities introduced, access to novel valinomycin analogues exhibiting enhanced bioactivities is possible.15 Alternatively, such hydroxy derivatives might themselves display distinctive activity. In fact, β-hydroxy valine derivatives occur frequently in natural cyclopeptides that display special bioactivity, including novel antibiotic and antiproliferative character.16
Supplementary Material
Figure 2.
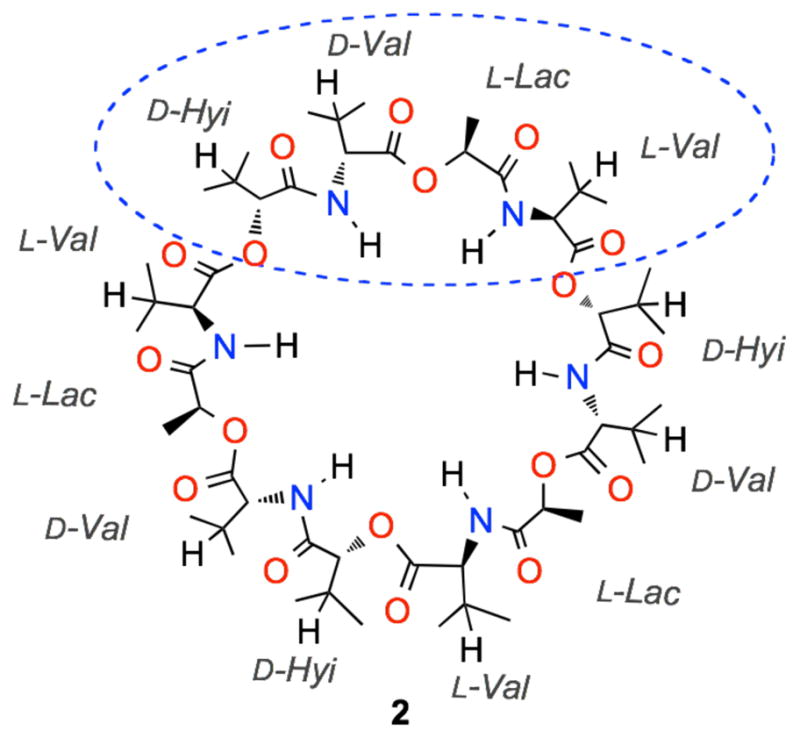
Valinomycin structure (D-Hyi = D-α-Hydroxyiso-valeric acid; D-Val = D-Valine; L-Lac = L-Lactic acid; L-Val = L -Valine).
Acknowledgments
Thanks are due to the Ministry of Education of Italy (MIUR, grant PRIN 2008), to the National Research Council (CNR, Rome, Italy), the NIH (GM-35982) and the NSF (CHE-0718275) for financial support.
Footnotes
Supporting Information Available General experimental details, NMR and MALDI mass spectra of products 3a–c. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Chatterjee J, Gilon C, Hoffmann A, Kessler H. Acc Chem Res. 2008;41:1331. doi: 10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]; (b) Ross SA, Gulve EA, Wang M. Chem Rev. 2004;104:1255. doi: 10.1021/cr0204653. [DOI] [PubMed] [Google Scholar]
- 2.(a) Cassidei L, Fiorentino M, Mello R, Sciacovelli O, Curci R. J Org Chem. 1987;52:699. [Google Scholar]; (b) Murray RW, Jeyaraman R. J Org Chem. 1985;50:2847. [Google Scholar]
- 3.Mello R, Fiorentino M, Sciacovelli O, Curci R. J Org Chem. 1988;53:3890. [Google Scholar]
- 4.For recent dioxirane reviews, see: Curci R, D’Accolti L, Fusco C. Acc Chem Res. 2006;39:1. doi: 10.1021/ar050163y.Adam W, Zhao C-G, Kavitha J. Organic Reactions. Vol. 69. Wiley; Hoboken, NJ: 2007. pp. 1–346. See also references cited therein.
- 5.(a) Annese C, D’Accolti L, De Zotti M, Fusco C, Toniolo C, Williard PG, Curci R. J Org Chem. 2010;75:4812. doi: 10.1021/jo100855h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rella MR, Williard PG. J Org Chem. 2007;72:525. doi: 10.1021/jo061910n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Detomaso A, Curci R. Tetrahedron Lett. 2001;42:755. [Google Scholar]
- 6.Gibson SE, Lecci C. Angew Chem Int Ed. 2006;45:1364. doi: 10.1002/anie.200503428. [DOI] [PubMed] [Google Scholar]
- 7.For instance, see: Park, Nam C, Lee JM, Lee D, Kim BS. J Microbiol Biotechnol. 2008;18:880.Watanabe H, Azuma M, Igarashi K, Ooshima H. J Antibiot. 2005;58:753. doi: 10.1038/ja.2005.102.
- 8.Paananen A, Jarvinen K, Sareneva T, Salkinoja-Salonen MS, Timonen T, Holtta Toxicology. 2005;212:37. doi: 10.1016/j.tox.2005.04.003. See references cited therein. [DOI] [PubMed] [Google Scholar]
- 9.Mello R, Fiorentino M, Fusco C, Curci R. J Am Chem Soc. 1989;111:6749. [Google Scholar]
- 10.For instance, see: Cavanagh J, Fairbrother WJ, Palmer AG, III, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. Vol. 8 Academic Press; 1996.
- 11.(a) Curci R, Detomaso A, Lattanzio ME, Carpenter GB. J Am Chem Soc. 1996;118:11089. [Google Scholar]; (b) Bovicelli P, Lupattelli P, Mincione E, Prencipe T, Curci R. J Org Chem. 1992;57:5052. [Google Scholar]
- 12.Bystrov VF, Gavrilov YD, Ivanov VT, Ovchinnikov YA. Eur J Biochem. 1977;78:63. doi: 10.1111/j.1432-1033.1977.tb11714.x. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto S, Watarai H, Bouř P. ChemPhysChem. 2011;12:1509. doi: 10.1002/cphc.201000917. See also references cited therein. [DOI] [PubMed] [Google Scholar]
- 14.Grell E, Funck T. J Supramol Struct. 1973;1:307. doi: 10.1002/jss.400010408. [DOI] [PubMed] [Google Scholar]
- 15.Cheng YQ. ChemBioChem. 2006;7:471. doi: 10.1002/cbic.200500425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Yun BS, George NP, Wendt-Pienkowski E, Galm U, Oh TJ, Coughlin JM, Zhang G, Tao M, Shen B. J Nat Prod. 2007;70:402. doi: 10.1021/np060592k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.