

Abstract
Protein misfolding and aggregation are considered key features of many neurodegenerative diseases, but biochemical mechanisms underlying protein misfolding and the propagation of protein aggregates are not well understood. Prion disease is a classical neurodegenerative disorder resulting from the misfolding of endogenously expressed normal cellular prion protein (PrPC). Although the exact function of PrPC has not been fully elucidated, studies have suggested that it can function as a metal binding protein. Interestingly, increased brain manganese (Mn) levels have been reported in various prion diseases indicating divalent metals also may play a role in the disease process. Recently, we reported that PrPC protects against Mn-induced cytotoxicity in a neural cell culture model. To further understand the role of Mn in prion diseases, we examined Mn neurotoxicity in an infectious cell culture model of prion disease. Our results show CAD5 scrapie-infected cells were more resistant to Mn neurotoxicity as compared to uninfected cells (EC50 = 428.8 μM for CAD5 infected cells vs. 211.6 μM for uninfected cells). Additionally, treatment with 300 μM Mn in persistently infected CAD5 cells showed a reduction in mitochondrial impairment, caspase-3 activation, and DNA fragmentation when compared to uninfected cells. Scrapie-infected cells also showed significantly reduced Mn uptake as measured by inductively coupled plasma-mass spectrometry (ICP-MS), and altered expression of metal transporting proteins DMT1 and transferrin. Together, our data indicate that conversion of PrP to the pathogenic isoform enhances its ability to regulate Mn homeostasis, and suggest that understanding the interaction of metals with disease-specific proteins may provide further insight to protein aggregation in neurodegenerative diseases.
Keywords: prion, metals, manganese, apoptosis, neurotoxicity
1. Introduction
A conformational isomer of the endogenously expressed prion protein is the putative pathogenic agent in transmissible spongiform encephalopathy (TSE) or prion disease (Prusiner, 1991). Normal cellular prion protein (PrPC) is converted to the pathogenic β-sheet-rich conformation of scrapie prion (PrPSc) through a still unclear mechanism (Bolton et al., 1985; Collinge, 2005; Prusiner and DeArmond, 1990). Prion diseases are fatal neurodegenerative disorders that affect both humans and animals (Prusiner, 1991). The regions of the brain that control motor function, including the basal ganglia, cerebral cortex, thalamus, brain stem, and cerebellum, are severely affected in TSE. The major neurological symptoms of TSE are extrapyramidal motor signs, including tremors, postural instability, ataxia, and myoclonus (Aguzzi and Heikenwalder, 2006; Brandner, 2003; Brown, 2002; Tatzelt and Schatzl, 2007). The neuropathological characterization of prion disease involves massive neuronal degeneration and vacuolization associated with accumulation of PrPSc giving neural tissue the diagnostic spongiform appearance when examined histologically (Collinge, 2001; Owen et al., 1989; Palmer and Collinge, 1992). Increased oxidative stress markers, such as malondialdehyde, 3-nitrotyrosine, 8-hydroxyguanosine, protein carbonyls, and dysregulation of iron homeostasis were observed in the brain tissues of both animal and human prion diseases, suggesting that oxidative damage plays an important role in the pathogenesis of TSE (Freixes et al., 2006; Lee et al., 1999; Petersen et al., 2005; Yun et al., 2006).
Although normal cellular PrPC is abundantly expressed in the central nervous system (CNS), its biological function still remains unclear. PrPC is a glycosylphosphatidylinositol (GPI)-anchored cell surface protein that is believed to function as an antioxidant, a cellular adhesion molecule, a signal transducer, and a metal binding protein (Brown, 2004; Chen et al., 2003; Chiarini et al., 2002; Collinge, 2005; Mange et al., 2002; Prusiner and Kingsbury, 1985; Prusiner et al., 1990; Sakudo et al., 2004). Properly folded prion protein is present on lipid membrane rafts, and is believed to be internalized via clathrin-mediated endocytosis (Nunziante et al., 2003; Peters et al., 2003; Prado et al., 2004). Recent evidence indicates that PrPC is an important metal binding protein for divalent metals, such as copper (Cu), manganese (Mn), and zinc (Zn) (Brazier et al., 2008; Brown, 2009; Choi et al., 2007; Hornshaw et al., 1995; Viles et al., 1999). PrPC contains several octapeptide repeat sequences (PHGGSWGQ) toward the N-terminus, which have binding affinity for divalent metals with preferential binding for Cu (Hornshaw et al., 1995; Viles et al., 1999). Additional higher affinity metal binding sites have been identified at His 95 and 110 (mouse numbering) (Jackson et al., 2001; Jones et al., 2004), but the exact role of these higher affinity metal binding sites remains elusive.
Interestingly, increased Mn content has been observed in the blood and brain of humans infected with Crueztfelt-Jacob Disease (CJD), mice infected with scrapie, and cattle infected with bovine spongiform encephalopathy (BSE) (Hesketh et al., 2008; Hesketh et al., 2007; Thackray et al., 2002; Wong et al., 2001b). Additionally, Mn-bound PrPSc can be isolated from both humans and animals infected with prion disease. Despite these findings, the role of Mn in the pathogenesis of prion disease is currently unknown. Recent studies using recombinant PrP have shown that Mn can irreversibly displace Cu bound to PrP, despite an apparent lower affinity, and this displacement causes conformational changes within the protein (Brazier et al., 2008; Zhu et al., 2008). The biological consequence of Cu replacement by Mn on the prion protein is yet to be established. Recently, we observed that divalent Mn binds to PrP resulting in a reduced neurotoxic response during the early acute phase of the Mn toxicity in an uninfected model of prion disease (Choi et al., 2007). However, prolonged exposure to Mn upregulates PrPC by stabilizing the protein without any change in gene transcription (Choi et al., 2010). In order to determine whether Mn plays a role in the infectious nature of PrP, in the present study we characterized the neurotoxic effect of Mn in a cell culture model of infectious prion disease.
2. Materials and Methods
2.1 Chemicals
Manganese chloride (MnCl2), 3-(4,5-dimethylthiazol-3-yl)-2,5-diphenyltetrazolium bromide (MTT), ethylenediaminetetraacetic acid (EDTA), phenylmethylsulfonyl fluoride (PMSF), sodium chloride (NaCl), Tris HCl, Triton-X, sodium deoxycholate, dithiothreitol (DTT), proteinase K (PK), were purchased from Sigma (St. Louis, MO); Sytox green nucleic dye was purchased from Molecular Probes (Eugene, OR). Cell Death Detection ELISA plus Assay Kit was purchased from Roche Molecular Biochemicals (Indianapolis, IN). Bradford protein assay kit and acrylamide stock solution were purchased from Bio-Rad Laboratories (Hercules, CA). Opti-MEM, fetal bovine serum, penicillin, and streptomycin were purchased from Invitrogen (Carlsbad, CA). 6H4 anti-PrP monoclonal antibody was purchased from Prionics (Schlieren, Switzerland). Anti-4-hydroxynonenal (4-HNE) antibody was purchased from R&D Systems (Minneapolis, MN). Anti-mouse divalent metal transporter (DMT-1) was purchased from Alpha Diagnostic International (San Antonio, TX). Anti-mouse transferrin (Tf) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-β-actin antibody was purchased from Sigma-Aldrich (St. Louis, MO). Alexa Flour 680 conjugated goat anti-mouse IgG was purchased from Invitrogen (Carlsbad, CA). Goat anti-rabbit IgG IR800 Conjugate was purchased from Rockland Immunochemicals (Gilbertsville, PA)
2.2 Cell culture model of prion disease
The CAD-2A2D5 subclone of uninfected and infected Cath.a-differentiated cells (CAD5) were a generous gift from Dr. Charles Weissmann of the Scripps Institute (Jupiter, FL). Lack of suitable cell culture models for prion disease has hindered in vitro mechanistic investigations over the past few decades. Recently, Dr. Weissmann's laboratory has successfully created CAD cells with remarkable susceptibility for infection by the Rocky Mountain Lab (RML) mouse scrapie strain. The development and characterization of CAD5 infectious prion cell culture model is described elsewhere (Mahal et al., 2007). Both uninfected and RML scrapie-infected cells were grown in Opti-MEM media supplemented with 50 units penicillin, 50 μg/ml streptomycin, and 10% qualified fetal bovine serum (Invitrogen) and screened for retention of RML scrapie infection in CAD5 cells over five subsequent passages. Cells were maintained in a humidified atmosphere of 5% CO2 at 37°C.
2.3 Limited proteolysis
Control and RML-infected CAD5 cells were seeded in a T175 flask and allowed to grow to ∼90% confluence. Cells were washed once with ice cold phosphate buffered saline (PBS) and then lysed in the flask by adding 2 ml of lysis buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.4, 5 mM EDTA, 2% Triton X, and 2% sodium deoxycholate) and incubated at 4°C for 30 mins. Protein concentrations were determined by Bradford assay, and 2 mg of protein from each sample were brought to equal volume with addition of excess lysis buffer. Limited proteolysis was begun by addition of PK to each sample at a concentration of 20 μg/ml and incubated at 37°C for 1 h. Reaction was quenched by the addition of 2 mM PMSF (final concentration). After PK digestion, samples were ultracentrifuged at 120,000 × g for 2 h using an Optima Max Ultracentrifuge (Beckman-Coulter, Brea, CA) to pellet the PK-resistant fraction. The supernatant was discarded and 30 μl of PAGE loading buffer and DTT was added directly to the pellet. The samples were then sonicated for 5 mins in a cup horn sonicator, boiled for 10 mins, and separated by SDS-PAGE on a 15% polyacrylamide gel and transferred to nitrocellulose membrane. The membranes were treated with 6H4 anti-PrP monoclonal antibody for 2 h at room temperature. Detection was done with an electrochemiluminescence (ECL) detection kit (GE Healthcare, Piscataway, NJ).
2.4 SDS-PAGE and Western blot
Western blot analysis was performed as described previously (Anantharam et al., 2002; Kitazawa et al., 2005; Latchoumycandane et al., 2005). Following Mn treatment, cells were collected by scraping and washed once with ice cold PBS, and then lysed in RIPA (Radio-Immunoprecipitation Assay) buffer with phosphatase and protease inhibitor cocktail (Thermo Scientific, Rockfort, IL). Protein concentrations of samples were determined using Bradford assay. Samples were then separated by SDS-PAGE using a 12% polyacrylamide gel and transferred onto nitrocellulose membrane. After 1 h in blocking buffer, the membranes were treated with either anti-4-hydroxynonenal antibody, anti-transferrin antibody, or anti-divalent metal transporter 1 at 4°C overnight. Membranes were co-treated with anti-β-actin antibody in blocking buffer to ensure equal protein loading. Membranes were then treated with anti-mouse and anti-rabbit fluorescent secondary antibodies for 1 h at room temperature. Visualization and band quantification was done using an Odyssey scanner (Licor, Lincoln, NE).
2.5 MTT Assay
The MTT cell viability assay was performed as described previously (Choi et al., 2007; Latchoumycandane et al., 2005). 100,000 uninfected and RML scrapie-infected CAD5 cells were seeded on a 96-well microplate, allowed to adhere for 12 h, and then treated for 16 h with manganese chloride (MnCl2) in Opti-MEM in the following concentrations: 0, 10μM, 30μM, 50μM, 100μM, 300μM, 500μM, 1mM, 2mM, 3mM, and 5mM. Following the treatment, the cells were washed with warm PBS and then incubated with 200 μl 0.25% (w/v) MTT in serum free Opti-MEM for 30 mins at 37°C. MTT treatment was removed, cells were washed with warm PBS, and 200 μl of 100% dimethyl sulfoxide (DMSO) was added to each well and pipetted up and down to dissolve the contents of the wells. Yellow MTT is reduced by a mitochondrial dehydrogenase in living cells to dark blue formazan crystals that accumulate in the mitochondria. Absorbance was read at 570 and 630 nm using a SpectroMax microplate reader (model 190; Molecular Devices, Sunnyvale, CA).
2.6 Sytox Staining
One million uninfected and RML scrapie-infected CAD5 cells were plated in a 6-well plate, allowed to adhere to the plate for 12 h, and then were treated with 300 μM and 500 μM MnCl2 in Opti-MEM supplemented with 1 μM Sytox green dye for 24 h. Sytox green is a cell-impermeable nucleic acid dye that intercalates with DNA and produces green fluorescence in dead or dying cells. Pictures were taken with a Nikon inverted fluorescence microscope (model TE-2000U; Nikon, Tokyo, Japan); images were captured with a SPOT digital camera (Diagnostic Instruments, Sterling Heights, MI.)
2.7 Caspase-3 Time Course
Caspase-3 activity assay was performed as described previously (Anantharam et al., 2002; Kitazawa et al., 2005; Latchoumycandane et al., 2005). One million uninfected and RML scrapie-infected CAD5 cells were plated per well in a 6-well plate, allowed to adhere for 12 h, and were then treated with 300 μM MnCl2 in Opti-MEM for 8, 12, 16, 20, and 24 h. Cells were collected with 0.25% trypsin-EDTA, spun down at 300 × g for 5 mins, and the cell pellet was washed with PBS. Cells were resuspended in caspase buffer (50 mM Tris-HCl (pH 7.4), 1 mM EDTA, and 10 mM EGTA) with 10 μM digitonin added and incubated for 30 mins on ice to lyse cells. The lysate was quickly spun down and supernatant was collected. 95 μl of the lysate was incubated with 5 μl of caspase-3 specific substrate Ac-DEVD-AFC for 1 h in a 96-well plate at 37°C. AFC fluorescence was measured with excitation at 400 nm and emission at 505 nm with a SpectraMax fluorescent plate reader (model GeminiXSE; Molecular Devices, Sunnyvale, CA). Protein concentration was determined by Bradford protein assay and data was expressed as fluorescent units per mg of protein per hour.
2.8 DNA fragmentation ELISA on Mn treated CAD5 cells
1 million uninfected and RML scrapie-infected CAD5 cells were plated per well in a 6-well plate, allowed to adhere for 12 h, and were then treated with 300 μM of MnCl2 in Opti-MEM for 24 h. Cells were collected with 0.25% trypsin-EDTA and spun down at 300 × g for 5 mins and the cell pellet was washed with PBS. The amount of histone-associated low molecular weight DNA in the cytoplasm of the cells was determined with the Cell Death Detection ELISA PLUS Kit (Roche, Indianapolis, IN) as described previously (Anantharam et al., 2002; Choi et al., 2007; Kaul et al., 2003). Briefly, cells were lysed in provided lysis buffer for 30 mins at RT. Lysate was centrifuged for 10 mins at 200 × g and 20 μl of supernatant was incubated for 2 h with the mixture of HRP (horseradish peroxidase)-conjugated antibodies that recognize histones and single and double-stranded DNA. After washing away the unbound components, the final reaction product was measured colorimetrically with 2,2′-azino-di-[3-ethylbenzthiazoline sulfonate] as an HRP substrate using a spectrophotometer at 405 nm and 490 nm. The difference in absorbance between 405 and 490 nm was used to determine the amount of DNA fragmentation in each sample. All sample concentrations were normalized to protein concentration using the Bradford protein assay.
2.9 Determination of intracellular manganese levels by IC-PMS
Three million uninfected and RML scrapie-infected CAD5 cells were seeded in a T175, allowed to adhere for 12 h, and then were treated for 16 h with 300 μM MnCl2 in Opti-MEM media. Cells were then collected by scraping and washed twice with PBS. Intracellular Mn concentration was measured at m/z 55 by inductively coupled plasma mass spectrometry (ICP-MS) as described in our previous publications (Afeseh Ngwa et al., 2009; Choi et al., 2007). A magnetic sector instrument (ELEMENT 1, Thermo Finnigan) was operated in medium resolution (m/Δm = 4,000) to resolve the isotope of interest from possible interferences (Shum et al., 1992). Each sample was placed in an acid-washed 5 ml Teflon vial and digested in 200 μl triply distilled high purity nitric acid (TraceMetal Grade, Fisher Scientific, Pittsburg, PA). The digested samples were then diluted to a final volume of 5 ml with 18.2 MΩ deionized water (Elix and Milli-Q Gradient, Millipore, Billerica, MA) resulting in a final acid concentration of 4%. An internal standard method was used for quantification. Gallium (Ga) at m/z 69 was chosen as the internal standard because its m/z ratio is similar to that of Mn, and it has no major spectroscopic interferences. A small spike of Ga standard solution was added to each sample for a final Ga concentration of 10 ppb. A 10 ppb multi-element standard (Mn and Ga) was prepared. The nitric acid blank, the multi-element standard, and each of the samples were introduced into the ICP-MS via a 100 μl/min self-aspirating perfluoroalkoxy (PFA) nebulizer (Elemental Scientific, Inc., Appleton, WI). The nitric acid blank was used to rinse the nebulizer between each sample. The results for each sample were calculated using the integrated average background-subtracted peak intensities from 70 consecutive scans. A normalization factor for Mn was derived from the multi-element standard to account for differences in the ionization efficiency of each element. The Mn concentration was then calculated for each sample.
2.10 Data Analysis
Statistical analysis was performed with Prism 4.0 software (GraphPad Software, San Diego, CA). Statistical significance among treatments was determined by one-way ANOVA analysis with Tukey's multiple comparison testing and is indicated by asterisks with *p<0.05, **p<0.01, and ***p<0.001. Analyses between two data sets were done using Student's t-test. EC50 values were determined by the fitting of a nonlinear regression curve to the data from the MTT assay. Data typically represent three separate experiments and are expressed as mean ± S.E.M.
3. Results
3.1 CAD5 characterization and limited proteolysis
First, we examined the morphological features and prion infection of CAD5 cell model used in our experiments. Fig. 1A details the tendency for the RML scrapie-infected CAD5 cells to grow as aggregate colonies, while the uninfected cells form a more uniform monolayer. Additionally, the RML scrapie-infected cells have an increased doubling time compared to the uninfected cells when cultured over several days (data not shown). We do not see any differences in the growth characteristics such as doubling time, morphology, etc. between infected and uninfected cells because entire treatment is less than 36 hrs. Importantly, the CAD5 strain was able to retain persistent RML scrapie infection over multiple passages as determined by limited proteolytic digestion with PK and subsequent immunoblot using the 6H4 anti-PrP MAb (Fig. 1B). An abundant proteolytically resistant PrPSc was detected in scrapie-infected cells as compared to uninfected cells (Fig. 1B).
Fig. 1.
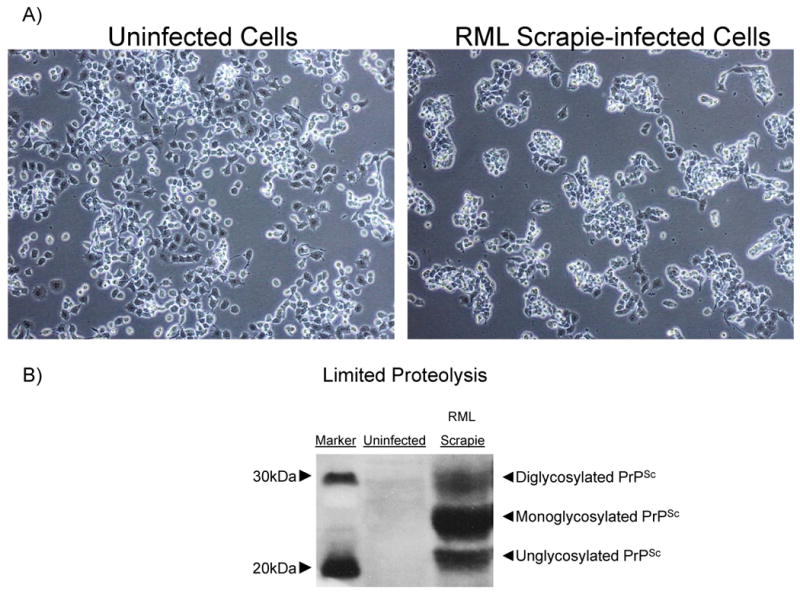
Characterization of CAD5 cells and RML scrapie infection. A) Phase contrast image of uninfected and RML scrapie-infected CAD5 cells. Scrapie-infected cells tend to grow in aggregated colonies and grow more slowly than uninfected cells. B) Lysate from uninfected and infected CAD5 cells was proteolytically digested by addition of 20 μg/ml PK for 1 h at 37°C. Immunoblot with anti-PrP MAb showed complete digestion of PrPC in uninfected cells, and the presence of proteolytically resistant PrPSc in infected cells. The multiple bands represent the three glycoforms of PrP.
3.2 RML scrapie-infected CAD5 cells are more resistant to Mn-induced toxicity
Several lines of evidence from our lab and others have shown that Mn-induced toxicity in neuronal cell models is associated with mitochondrial dysfunction and subsequent generation of reactive oxygen species (ROS) leading to cell death (Aschner et al., 2009; Kitazawa et al., 2005; Latchoumycandane et al., 2005; Roth et al., 2002; Worley et al., 2002). In an effort to establish a cytotoxic dose of Mn for further cell viability studies in the CAD5 cell culture model of prion disease, both uninfected and RML scrapie-infected CAD5 cells were exposed to a full dose range of 10-5000 μM Mn for 16 h and viability was measured by MTT assay. As shown in Fig. 2A, a concentration-dependent decrease in cell viability for both uninfected and RML scrapie-infected cells was observed. A statistically significant difference (p<0.05) was observed between the EC50 values for uninfected and RML-infected cells, as calculated by a three-parameter nonlinear regression of dose response curves. Unexpectedly, the RML scrapie-infected cells showed increased resistance to Mn treatment with a calculated EC50 value of 428.8 μM, greater than double the EC50 value of the uninfected cells, 211.6 μM. The cytotoxicity results were further confirmed by qualitative analysis using Sytox green staining. Uninfected and RML-infected cells were exposed to 300 and 500 μM Mn for 24 h and pictures were taken using a Nikon inverted fluorescent microscope. The difference in the number of Sytox positive cells is evident at both concentrations of Mn, with uninfected CAD5 cells showing a much greater number of Sytox stained cells (Fig. 2B). These studies led us to further examine this disparity in the cytotoxic doses of Mn on the uninfected and RML scrapie-infected CAD5 cell line. Based upon these data, we selected 300 μM Mn as an optimal dose for comparative studies of Mn-induced cytotoxicity.
Fig. 2.
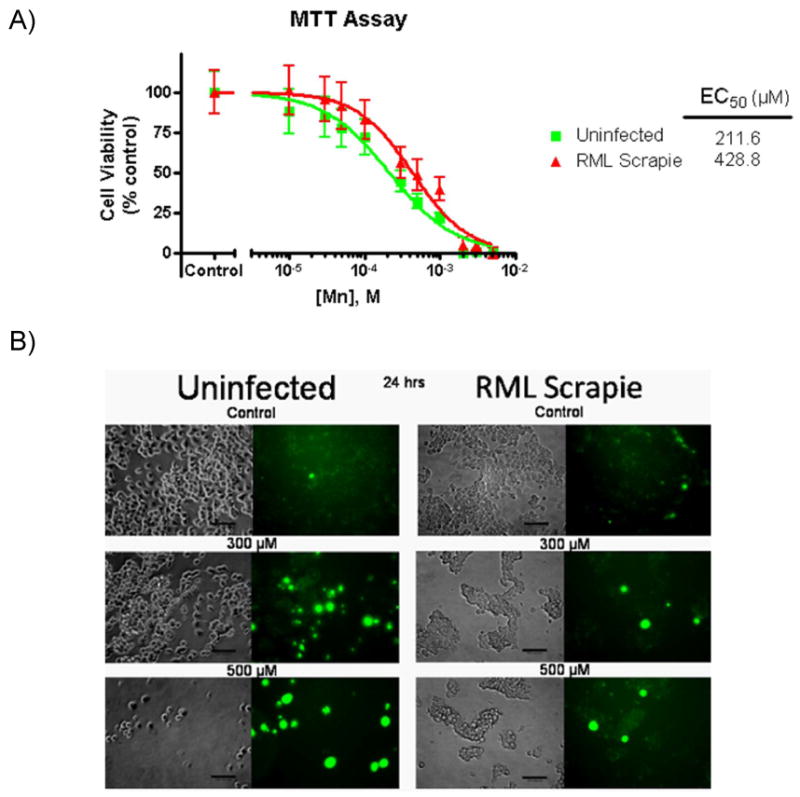
RML scrapie-infected CAD5 cells are more resistant to Mn-induced toxicity. A) Uninfected and RML scrapie-infected CAD5 cells were treated with 10, 30, 50, 100, 300, 500, 1000, and 5000 μM Mn for 16 h and cell viability was measured by MTT assay. The LD50 value of the control cells was determined to be 211.6 μM (95% CI 136.8-304.8 μM, logEC50: -3.675±0.081) while in RML scrapie infected cells the LD50 was calculated to be 428.8 μM (95% CI 290.9-632 μM, logEC50: -3.368±0.086). RML scrapie-infected cells showed reduced susceptibility to Mn-induced toxicity as evidenced by the statistically significant (p<0.05) increase in the EC50 value. Data shown represent two repeats with a total of 16 individual data points per concentration. B) Uninfected and RML scrapie-infected CAD5 cells were treated with 300 μM and 500 μM Mn for 24 h. Qualitative analysis of Mn-induced toxicity was done by Sytox green staining of dead and dying cells. We observed reduced staining by Sytox in RML scrapie-infected CAD5 cells when compared to uninfected cells.
3.3 RML scrapie-infected CAD5 cells are more resistant to Mn-induced apoptotic cell death
Mn-induced neurotoxicity is a known activator of proapoptotic signaling cascades in neuronal cells (Aschner et al., 2009; Kitazawa et al., 2005; Latchoumycandane et al., 2005; Stredrick et al., 2004). In order to explore the activation of proapoptotic caspases by Mn-induced neurotoxicity in uninfected and RML scrapie-infected CAD5 cells, we assayed the activity of the effector caspase, caspase-3, at 8, 12, 16, 20, and 24 h treatments with 300 μM of Mn. At 24 h treatment, a statistically significant increase in the activation of caspase-3 in uninfected cells (p<0.001) was observed, but no such increase was noted in RML-infected cells (Fig. 3A). Furthermore, we quantitatively assessed the level of DNA fragmentation on cells treated for 24 h with 300 μM Mn using a Cell Death Detection ELISA Kit. We found a statistically significant increase in the level of histone-associated low molecular weight DNA in uninfected cells at this time point (p<0.001), while the RML-infected cells showed no significant increase (Fig. 3B). These results indicate that RML scrapie-infected CAD5 cells are more resistant to Mn-induced apoptotic cell death than uninfected cells.
Fig. 3.
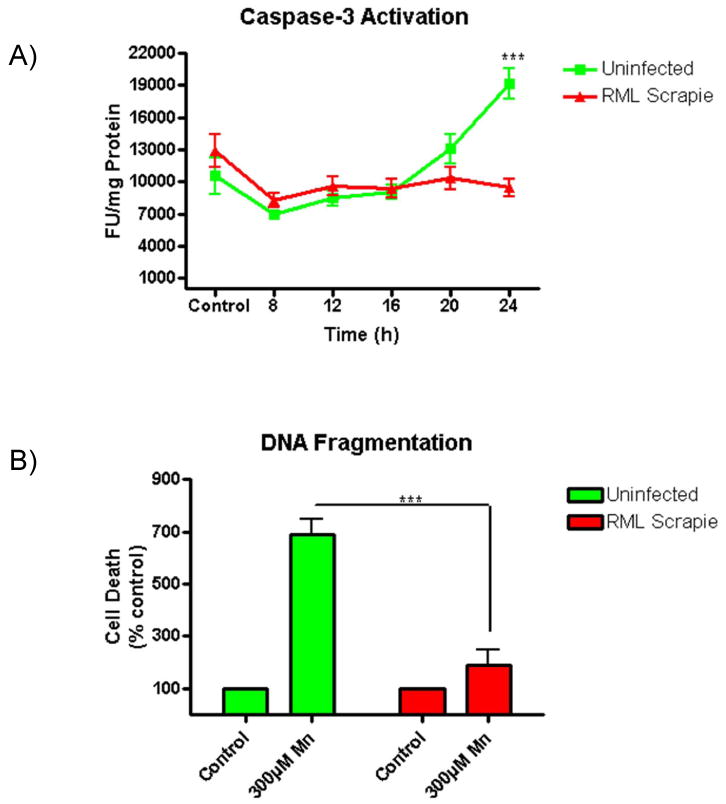
RML scrapie-infected CAD5 cells are more resistant to Mn-induced apoptotic cell death. Uninfected and RML scrapie-infected CAD5 cells were treated with 300 μM Mn and the activity of caspase-3 was determined by caspase-3 specific fluorogenic substrate over time. DNA fragmentation was assayed after 24 h of Mn treatment by ELISA. A) Uninfected CAD5 cells showed a time-dependent increase in caspase-3 activity in response to Mn treatment (p<0.001), while RML scrapie-infected CAD5 cells showed no increase at 24 h. Data shown represents three repeats with a total n=7. B) DNA fragmentation was significantly increased (p<0.001) in uninfected cells treated with Mn when compared to RML scrapie-infected cells.
3.4 RML scrapie infection attenuates Mn-induced oxidative damage
To further probe the decreased susceptibility of RML-infected CAD5 cells to Mn-induced toxicity, we evaluated the incidence of Mn-induced oxidative stress by examining a product of lipid peroxidation, 4-hydroxynonenal (4-HNE), in Mn-treated CAD5 cells (Alikunju et al., 2011; Sango et al., 2008). Multiple studies have shown that neuronal cell lines persistently infected with PrPSc show increased susceptibility to oxidative stress (Fernaeus et al., 2005; Milhavet et al., 2000). As shown in Fig. 4A, uninfected CAD5 cells showed a time-dependent increase in the levels of 4-HNE in response to treatment with 300 μM of Mn. Surprisingly, scrapie-infected CAD5 cells treated with Mn showed reduced levels of 4-HNE when compared to uninfected cells (Fig. 4A). Densitometric analysis revealed a statistically significant (p<0.05) decrease in the 4-HNE band intensity of RML scrapie-infected cells at 24 h (Fig. 4B).
Fig. 4.
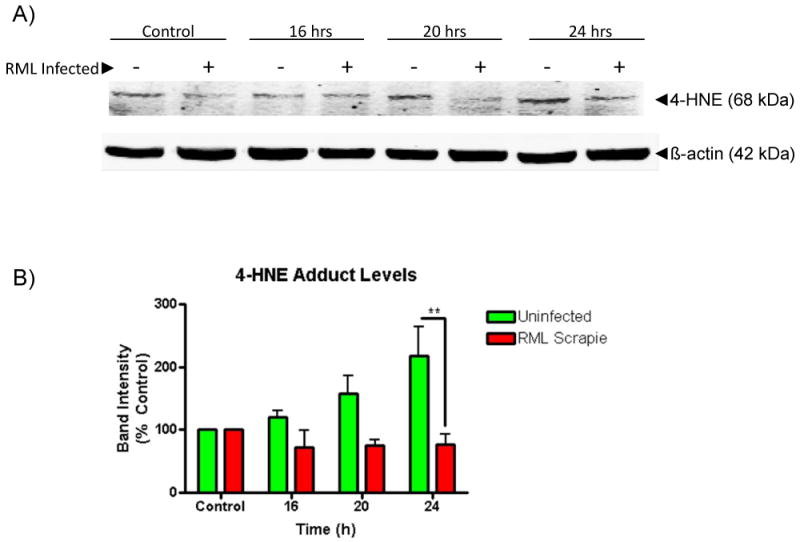
RML scrapie infection attenuates Mn-induced ROS generation. Uninfected and RML scrapie-infected CAD5 cells were treated with 300 μM Mn for 16, 20, and 24 h. A) Generation of Mn-induced ROS was determined by the presence of lipid peroxidation product 4-HNE. Lysate from treated cells was probed after Western blot with anti-4-HNE antibody. To ensure equal protein loading, blots were co-treated with β-actin antibody. B) Uninfected CAD5 cells showed a time-dependent increase in 4-HNE levels as determined by densitometric analysis of three individual repeats. A statistically significantly difference in increases of 4-HNE levels is seen between uninfected and infected cells at 24 h (p<0.01).
3.5 RML Infected CAD5 cells Have Reduced Manganese Accumulation
The binding of divalent cations to PrPC induces clathrin-mediated endocytosis of lipid raft domains and this process is dependent upon the metal binding sites of PrPC (Brown and Harris, 2003; Hooper et al., 2008; Pauly and Harris, 1998; Perera and Hooper, 2001). A recent study from our group showed that knockout of PrPC alters Cu and Mn levels in a neuronal cell culture model (Choi et al., 2007). Also, other studies have shown altered divalent metal content in the brains of PrPC null and TSE-infected animals. These results indicate that PrPC may have a role in divalent metal homeostasis, and that conversion to the disease isoform alters the ability of PrPC to regulate intracellular concentrations. In order to examine the consequence of RML scrapie infection on Mn homeostasis, we treated uninfected and RML scrapie-infected CAD5 cells with 300 μM Mn for 16 h and determined the intracellular Mn concentration by inductively coupled plasma mass spectrometry (ICP-MS). Uninfected and RML cells had similar basal levels of cytoplasmic Mn; however, Mn treatment increased intracellular Mn levels 187-fold in uninfected CAD5 cells and 130-fold in RML infected cells (Fig. 5A). This difference was statistically significant p<0.01.
Fig. 5.
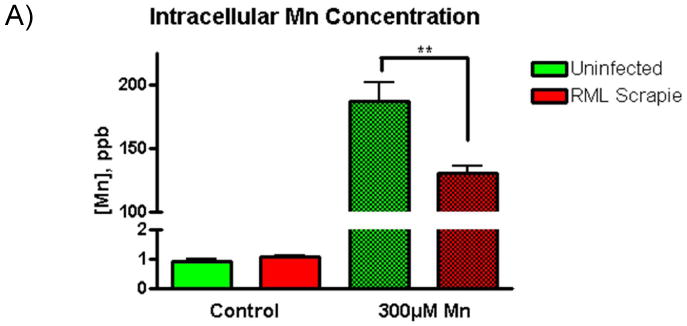
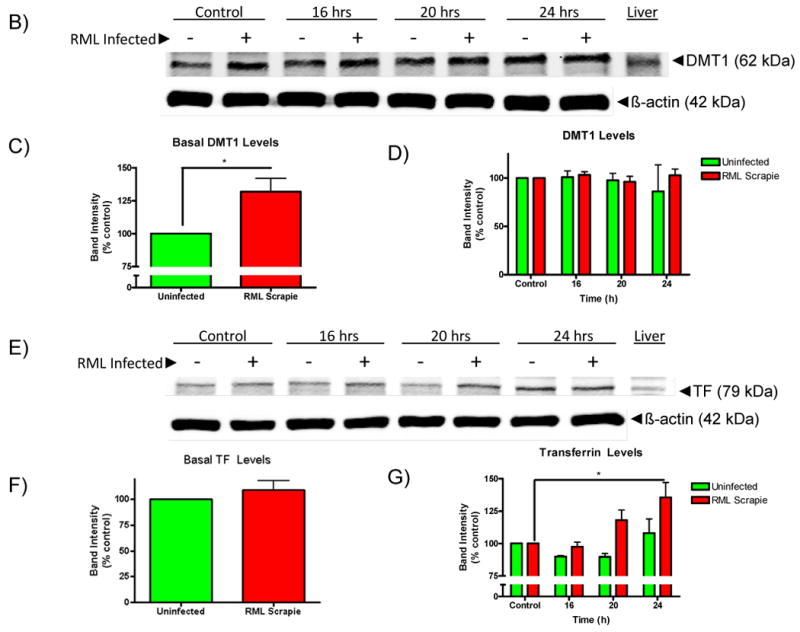
Mn-induced increases in intracellular Mn levels are reduced while the levels of metal uptake proteins are increased in RML-infected CAD5 cells. A) Uninfected and RML scrapie-infected CAD5 cells were treated with 300 μM Mn for 16 h and intracellular Mn levels were determined by ICP-MS. Basal levels of Mn in uninfected and infected CAD5 cells were not significantly different; however, a 187-fold increase was observed in uninfected CAD5 cells while RML infected cells saw a 130-fold increase. This difference was significantly different with p<0.01. B-G) The level of divalent metal regulator proteins, DMT1 and TF were determined by immunoblot. The basal level of DMT1 (C) was significantly increased in RML scrapie-infected cells (p<0.01), while the basal level of TF (E) was unchanged between uninfected and infected cells. After 16, 20, and 24 h of treatment with 300 μM Mn, the level of TF was increased in a time-dependent manner in RML scrapie-infected cells (p<0.01) (G). D) The level of DMT1 was not significantly increased with Mn treatment. These data represent a mean ± S.E.M. from quantification of three individual experiments.
In order to determine a mechanism for the difference in intracellular Mn concentration, we assessed the levels of DMT1 and transferrin in control and Mn-treated CAD5 cells. As seen in Fig. 5B-C, the basal level of DMT1 was increased in RML scrapie-infected CAD5 cells as compared to uninfected cells. However, Mn treatment did not cause any significant change in DMT1 levels in either cell types (Fig. 5B and 5D). Also, the basal level of expression of TF in uninfected and RML scrapie-infected cells is not significantly different (Fig. 5E-F). However, upon Mn treatment, there is a statistically significant, time-dependent increase in the level of TF in RML scrapie-infected cells. There was a slight but not significant increase in the levels of TF in uninfected cells at 24 h (Fig. 5E and 5G). These results show some differences existing in metal transporter protein between uninfected and RML scrapie-infected cells. However, the altered expression of these proteins between the uninfected and infected CAD5 cells are contradictory to the expected intracellular Mn concentrations since both DMT1 and TF are involved in Mn influx yet are increased in the infected cells that have a decreased Mn content. These results indicate that there are other factors influencing the intracellular level of Mn. Altered expression of proteins that are involved in Mn efflux may also play role in Mn-homeostasis. The mechanisms of regulation that are affected by conversion from PrPC to PrPSc remain an interesting target for further study.
4. Discussion
In this study we have shown that RML scrapie-infected catecholaminergic cells are more resistant to Mn-induced neurotoxicity than uninfected cells. Consistent with cytotoxicity, caspase-3 activation, DNA fragmentation, and ROS generation are attenuated in Mn-treated RML scrapie-infected CAD5 cells. Another notable finding of our study was that metal transport proteins DMT and transferrin are altered in scrapie-infected cells as compared to uninfected cells. To our knowledge, this is the first report demonstrating reduced metal neurotoxicity in an infectious model of prion disease.
Our understanding of the role of metals in key neurobiological processes, as well as in the pathogenesis of various neurodegenerative diseases, has greatly expanded over the last two decades. Studies overwhelmingly demonstrate that metal dyshomeostasis, protein aggregation, and oxidative stress are interconnected pathophysiological mechanisms of all neurodegenerative diseases associated with proteinopathies. Significant imbalances in major transition metals such as iron, copper, and zinc are implicated in other major neurodegenerative conditions such as Parkinson's disease (PD), Alzheimer's disease (AD), Amyotrophic lateral sclerosis (ALS) and Huntington Disease (HD) (Bishop et al., 2002; Bossy-Wetzel et al., 2004; Brown, 2009; Bush and Curtain, 2008; Cahill et al., 2009; Gaeta and Hider, 2005; Molina-Holgado et al., 2007; Sayre et al., 1999). Another common pathological feature of these neurodegenerative disorders is the aggregation of β-sheet-rich synaptic proteins associated with each disease (e.g., α-synuclein in PD, a-beta in AD, and huntingtin in HD). Environmental exposure to transition metals is linked to pathological processes of various neurodegenerative conditions since metal exposure is known to augment key degenerative changes including ionic imbalance, oxidative stress, and protein aggregation (Afeseh Ngwa et al., 2009; Anantharam et al., 2002; Aschner et al., 2009; Crossgrove and Zheng, 2004; Kitazawa et al., 2001; Park et al., 2005; Wu et al., 2008).
Emerging evidence indicates that dysregulation of divalent cation homeostasis may play a role in the pathogenesis of prion diseases (Basu et al., 2007; Brown, 2009; Choi et al., 2006; Singh et al., 2009). PrPC contains multiple octapeptide repeat sequences (PHGGSWGQ) toward the N-terminus that have binding affinity for various divalent metals including copper and manganese (Hornshaw et al., 1995; Viles et al., 1999). The number of octapeptide repeats differs from species to species; however, point mutations, deletions or multiple insertions of the octapeptide repeats in the prion protein gene have been linked to inherited prion disease in humans (Krasemann et al., 1995; Mead et al., 2007; Palmer and Collinge, 1993; Yin et al., 2007). Structural studies have suggested that most of the N-terminus of prion protein is rather unstructured, while the C-terminus is highly structured (Di Natale et al., 2005; Jones et al., 2005; Renner et al., 2004; Viles et al., 2001). Binding of divalent cations to PrPC has been speculated to facilitate the folding of the largely unstructured N-terminus, thereby stabilizing the protein conformation (Cereghetti et al., 2003). The antioxidant properties of PrPC have been linked to Cu residency in the octapeptide repeat domain (Brown et al., 2001; Brown et al., 1999; Gaggelli et al., 2008; Treiber et al., 2007). Cu bound by PrPC is able to undergo full and reversible redox chemistry, allowing for the detoxification of superoxide and the reduction of hydroxyl radicals (Nadal et al., 2007). Recently, we showed that prion protein expressing cells are more resistant to oxidative damage as compared to prion knockout cells (Anantharam et al., 2008).
Mn is an important trace elemental metal that is required by most organisms for normal functioning; however, continued exposure to high concentrations of Mn results in adverse neurological deficits commonly referred to manganism. Interestingly, elevated Mn levels have been observed in the brain and blood of humans and animals afflicted with prion diseases and Mn-bound PrPSc has been isolated from TSE-infected neural tissue (Hesketh et al., 2008; Hesketh et al., 2007; Thackray et al., 2002; Wong et al., 2001a). In vitro titration calorimetry studies done using recombinant PrP have shown that Mn can replace Cu residency in the metal binding sites despite a calculated lower affinity (Brazier et al., 2008; Zhu et al., 2008). Mn binding to PrPC induces biophysical properties similar to PrPSc, including increased β-sheet content, proteolytic resistance, and the ability to seed aggregation of soluble oligomers, yet the role of metals in the disease is not very well understood (Abdelraheim et al., 2006; Brazier et al., 2008; Brown et al., 2000; Giese et al., 2004; Kim et al., 2005). Recently, we demonstrated that PrPC effectively attenuates Mn transport into neuronal cells and protects against Mn-induced oxidative stress, mitochondrial dysfunction, cellular antioxidant depletion, and apoptosis (Choi et al., 2007). In addition, we found that Mn treatment upregulates PrPC levels independently of transcription (Choi et al., 2010). Further mechanistic studies revealed that Mn increases stability of prion protein, suggesting that Mn may promote the conversion of PrPC to PrPSc. Our present study shows that the scrapie-infected cells are more resistant to Mn neurotoxicity and the infected cells have altered metal transport as compared to uninfected cells. Taken together, these results suggest that conversion to PrPSc may result in greater affinity for Mn and increased ability for PrPSc to sequester and export excess Mn through the exosomal pathway in neuronal cells (Fevrier et al., 2005). Additionally, the resistance of prion-infected cells to Mn neurotoxicity may aid the propagation of prion protein aggregation from infected cells to healthy cells. Further studies are on-going in our laboratory to address these possibilities. However, the possibility of other factors such as altered expression of Mn efflux proteins or the increase or activation of other survival factors influencing Mn toxicity in infected cells cannot be completely ruled out.
In this study we focused on the CAD cell line, which is derived from catecholaminergic neurons; whereas in humans and animals with prion disease, higher Mn levels in brain tissue may be attributed to an increase in Mn-levels in microglia and astrocytes which constitute at least 90% of the total brain cell population. Consequence of prion infection on glial cells remains an interesting area of study. Unfortunately, the mechanisms involved in the regulation of cellular Mn homeostasis are still not well understood. Influx of Mn across the plasma membrane is believed to be mediated by divalent metal transporter 1 (DMT1) and transferrin (TF), despite dissociation constants in the millimolar range (Aschner et al., 1999; Erikson and Aschner, 2006; Fitsanakis et al., 2007; Garrick et al., 2006; Yokel, 2009). It is possible that ferroportin and ion channels are involved in Mn efflux (Yin et al., 2010). Increased resistance to oxidative stress in RML scrapie-infected cells is not likely to be a contributing factor in reducing the Mn toxicity observed in this work since prion infection has been shown to increase susceptibility of neuronal cells to oxidative stress (Fernaeus and Land, 2005; Fernaeus et al., 2005). Finally, a large body of evidence indicates that presence of dopamine in SH-SY5Y and other dopaminergic cell lines may directly influence the degree of Mn toxicity (For review see (Aschner et al., 2009; Smargiassi and Mutti, 1999.). However, in Cath.a cells, suppression of dopamine production with alpha-methyl-para-tyrosine offered no significant protection from Mn exposure suggesting that dopamine content was not responsible for the enhanced sensitivity to Mn toxicity which rules out the utilization of Mn as a co-factor (Stredrick et al., 2004). Further work is clearly needed to elucidate the interconnectivity of metal dyshomeostasis and protein aggregation in neurodegenerative disorders.
Acknowledgments
This work was supported by National Institutes of Health Grants ES019276 and ES10586. We thank Dr. Charles Weissmann and his colleagues from the Scripps Institute (Jupiter, FL) for providing us the CAD5 cell model for our study. The ICP mass spectrometer was obtained with funds provided by the U. S. Department of Energy, Office of Nuclear Nonproliferation (NA-22), and the Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences through the Ames Laboratory. The Ames Laboratory is operated for the U.S. Department of Energy by Iowa State University under Contract No. DE-AC02-07CH11358. The W. Eugene and Linda Lloyd Endowed Chair for AGK is also acknowledged. We thank Mary Ann deVries for assistance in the preparation of this manuscript.
Footnotes
Conflict of interest: There are no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelraheim SR, Kralovicova S, Brown DR. Hydrogen peroxide cleavage of the prion protein generates a fragment able to initiate polymerisation of full length prion protein. Int J Biochem Cell Biol. 2006;38:1429–40. doi: 10.1016/j.biocel.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Afeseh Ngwa H, Kanthasamy A, Anantharam V, Song C, Witte T, Houk R, et al. Vanadium induces dopaminergic neurotoxicity via protein kinase Cdelta dependent oxidative signaling mechanisms: relevance to etiopathogenesis of Parkinson's disease. Toxicol Appl Pharmacol. 2009;240:273–85. doi: 10.1016/j.taap.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: current status and future outlook. Nat Rev Microbiol. 2006;4:765–75. doi: 10.1038/nrmicro1492. [DOI] [PubMed] [Google Scholar]
- Alikunju S, Abdul Muneer PM, Zhang Y, Szlachetka AM, Haorah J. The inflammatory footprints of alcohol-induced oxidative damage in neurovascular components. Brain Behav Immun. 2011;25 1:S129–36. doi: 10.1016/j.bbi.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam V, Kanthasamy A, Choi CJ, Martin DP, Latchoumycandane C, Richt JA, et al. Opposing roles of prion protein in oxidative stress- and ER stress-induced apoptotic signaling. Free Radic Biol Med. 2008;45:1530–41. doi: 10.1016/j.freeradbiomed.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam V, Kitazawa M, Wagner J, Kaul S, Kanthasamy AG. Caspase-3-dependent proteolytic cleavage of protein kinase Cdelta is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl. J Neurosci. 2002;22:1738–51. doi: 10.1523/JNEUROSCI.22-05-01738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Erikson KM, Herrero Hernandez E, Tjalkens R. Manganese and its role in Parkinson's disease: from transport to neuropathology. Neuromolecular Med. 2009;11:252–66. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Vrana KE, Zheng W. Manganese uptake and distribution in the central nervous system (CNS) Neurotoxicology. 1999;20:173–80. [PubMed] [Google Scholar]
- Basu S, Mohan ML, Luo X, Kundu B, Kong Q, Singh N. Modulation of proteinase K-resistant prion protein in cells and infectious brain homogenate by redox iron: implications for prion replication and disease pathogenesis. Mol Biol Cell. 2007;18:3302–12. doi: 10.1091/mbc.E07-04-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop GM, Robinson SR, Liu Q, Perry G, Atwood CS, Smith MA. Iron: a pathological mediator of Alzheimer disease? Dev Neurosci. 2002;24:184–7. doi: 10.1159/000065696. [DOI] [PubMed] [Google Scholar]
- Bolton DC, Meyer RK, Prusiner SB. Scrapie PrP 27-30 is a sialoglycoprotein. J Virol. 1985;53:596–606. doi: 10.1128/jvi.53.2.596-606.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med. 2004;10(Suppl):S2–9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- Brandner S. CNS pathogenesis of prion diseases. Br Med Bull. 2003;66:131–9. doi: 10.1093/bmb/66.1.131. [DOI] [PubMed] [Google Scholar]
- Brazier MW, Davies P, Player E, Marken F, Viles JH, Brown DR. Manganese binding to the prion protein. J Biol Chem. 2008;283:12831–9. doi: 10.1074/jbc.M709820200. [DOI] [PubMed] [Google Scholar]
- Brown DR. Mayhem of the multiple mechanisms: modelling neurodegeneration in prion disease. J Neurochem. 2002;82:209–15. doi: 10.1046/j.1471-4159.2002.01004.x. [DOI] [PubMed] [Google Scholar]
- Brown DR. Metallic prions. Biochem Soc Symp. 2004:193–202. doi: 10.1042/bss0710193. [DOI] [PubMed] [Google Scholar]
- Brown DR. Brain proteins that mind metals: a neurodegenerative perspective. Dalton Trans. 2009:4069–76. doi: 10.1039/b822135a. [DOI] [PubMed] [Google Scholar]
- Brown DR, Clive C, Haswell SJ. Antioxidant activity related to copper binding of native prion protein. J Neurochem. 2001;76:69–76. doi: 10.1046/j.1471-4159.2001.00009.x. [DOI] [PubMed] [Google Scholar]
- Brown DR, Hafiz F, Glasssmith LL, Wong BS, Jones IM, Clive C, et al. Consequences of manganese replacement of copper for prion protein function and proteinase resistance. Embo J. 2000;19:1180–6. doi: 10.1093/emboj/19.6.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR, Wong BS, Hafiz F, Clive C, Haswell SJ, Jones IM. Normal prion protein has an activity like that of superoxide dismutase. Biochem J. 1999;344(Pt 1):1–5. [PMC free article] [PubMed] [Google Scholar]
- Brown LR, Harris DA. Copper and zinc cause delivery of the prion protein from the plasma membrane to a subset of early endosomes and the Golgi. J Neurochem. 2003;87:353–63. doi: 10.1046/j.1471-4159.2003.01996.x. [DOI] [PubMed] [Google Scholar]
- Bush AI, Curtain CC. Twenty years of metallo-neurobiology: where to now? Eur Biophys J. 2008;37:241–5. doi: 10.1007/s00249-007-0228-1. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Lahiri DK, Huang X, Rogers JT. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim Biophys Acta. 2009;1790:615–28. doi: 10.1016/j.bbagen.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereghetti GM, Schweiger A, Glockshuber R, Van Doorslaer S. Stability and Cu(II) binding of prion protein variants related to inherited human prion diseases. Biophys J. 2003;84:1985–97. doi: 10.1016/S0006-3495(03)75007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Mange A, Dong L, Lehmann S, Schachner M. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol Cell Neurosci. 2003;22:227–33. doi: 10.1016/s1044-7431(02)00014-3. [DOI] [PubMed] [Google Scholar]
- Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. Embo J. 2002;21:3317–26. doi: 10.1093/emboj/cdf324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CJ, Anantharam V, Martin DP, Nicholson EM, Richt JA, Kanthasamy A, et al. Manganese upregulates cellular prion protein and contributes to altered stabilization and proteolysis: relevance to role of metals in pathogenesis of prion disease. Toxicol Sci. 2010;115:535–46. doi: 10.1093/toxsci/kfq049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CJ, Anantharam V, Saetveit NJ, Houk RS, Kanthasamy A, Kanthasamy AG. Normal Cellular Prion Protein Protects against Manganese-Induced Oxidative Stress and Apoptotic Cell Death. Toxicol Sci. 2007;98:495–509. doi: 10.1093/toxsci/kfm099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CJ, Kanthasamy A, Anantharam V, Kanthasamy AG. Interaction of metals with prion protein: possible role of divalent cations in the pathogenesis of prion diseases. Neurotoxicology. 2006;27:777–87. doi: 10.1016/j.neuro.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–50. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- Collinge J. Molecular neurology of prion disease. J Neurol Neurosurg Psychiatry. 2005;76:906–19. doi: 10.1136/jnnp.2004.048660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossgrove J, Zheng W. Manganese toxicity upon overexposure. NMR Biomed. 2004;17:544–53. doi: 10.1002/nbm.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Natale G, Grasso G, Impellizzeri G, La Mendola D, Micera G, Mihala N, et al. Copper(II) interaction with unstructured prion domain outside the octarepeat region: speciation, stability, and binding details of copper(II) complexes with PrP106-126 peptides. Inorg Chem. 2005;44:7214–25. doi: 10.1021/ic050754k. [DOI] [PubMed] [Google Scholar]
- Erikson KM, Aschner M. Increased manganese uptake by primary astrocyte cultures with altered iron status is mediated primarily by divalent metal transporter. Neurotoxicology. 2006;27:125–30. doi: 10.1016/j.neuro.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Fernaeus S, Land T. Increased iron-induced oxidative stress and toxicity in scrapie-infected neuroblastoma cells. Neurosci Lett. 2005;382:217–20. doi: 10.1016/j.neulet.2005.03.069. [DOI] [PubMed] [Google Scholar]
- Fernaeus S, Reis K, Bedecs K, Land T. Increased susceptibility to oxidative stress in scrapie-infected neuroblastoma cells is associated with intracellular iron status. Neurosci Lett. 2005;389:133–6. doi: 10.1016/j.neulet.2005.07.032. [DOI] [PubMed] [Google Scholar]
- Fevrier B, Vilette D, Laude H, Raposo G. Exosomes: a bubble ride for prions? Traffic. 2005;6:10–7. doi: 10.1111/j.1600-0854.2004.00247.x. [DOI] [PubMed] [Google Scholar]
- Fitsanakis VA, Piccola G, Marreilha dos Santos AP, Aschner JL, Aschner M. Putative proteins involved in manganese transport across the blood-brain barrier. Hum Exp Toxicol. 2007;26:295–302. doi: 10.1177/0960327107070496. [DOI] [PubMed] [Google Scholar]
- Freixes M, Rodriguez A, Dalfo E, Ferrer I. Oxidation, glycoxidation, lipoxidation, nitration, and responses to oxidative stress in the cerebral cortex in Creutzfeldt-Jakob disease. Neurobiol Aging. 2006;27:1807–15. doi: 10.1016/j.neurobiolaging.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Gaeta A, Hider RC. The crucial role of metal ions in neurodegeneration: the basis for a promising therapeutic strategy. Br J Pharmacol. 2005;146:1041–59. doi: 10.1038/sj.bjp.0706416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaggelli E, Jankowska E, Kozlowski H, Marcinkowska A, Migliorini C, Stanczak P, et al. Structural characterization of the intra- and inter-repeat copper binding modes within the N-terminal region of “prion related protein” (PrP-rel-2) of zebrafish. J Phys Chem B. 2008;112:15140–50. doi: 10.1021/jp804759q. [DOI] [PubMed] [Google Scholar]
- Garrick MD, Kuo HC, Vargas F, Singleton S, Zhao L, Smith JJ, et al. Comparison of mammalian cell lines expressing distinct isoforms of divalent metal transporter 1 in a tetracycline-regulated fashion. Biochem J. 2006;398:539–46. doi: 10.1042/BJ20051987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese A, Levin J, Bertsch U, Kretzschmar H. Effect of metal ions on de novo aggregation of full-length prion protein. Biochem Biophys Res Commun. 2004;320:1240–6. doi: 10.1016/j.bbrc.2004.06.075. [DOI] [PubMed] [Google Scholar]
- Hesketh S, Sassoon J, Knight R, Brown DR. Elevated manganese levels in blood and CNS in human prion disease. Mol Cell Neurosci. 2008;37:590–8. doi: 10.1016/j.mcn.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Hesketh S, Sassoon J, Knight R, Hopkins J, Brown DR. Elevated manganese levels in blood and central nervous system occur before onset of clinical signs in scrapie and bovine spongiform encephalopathy. J Anim Sci. 2007;85:1596–609. doi: 10.2527/jas.2006-714. [DOI] [PubMed] [Google Scholar]
- Hooper NM, Taylor DR, Watt NT. Mechanism of the metal-mediated endocytosis of the prion protein. Biochem Soc Trans. 2008;36:1272–6. doi: 10.1042/BST0361272. [DOI] [PubMed] [Google Scholar]
- Hornshaw MP, McDermott JR, Candy JM. Copper binding to the N-terminal tandem repeat regions of mammalian and avian prion protein. Biochem Biophys Res Commun. 1995;207:621–9. doi: 10.1006/bbrc.1995.1233. [DOI] [PubMed] [Google Scholar]
- Jackson GS, Murray I, Hosszu LL, Gibbs N, Waltho JP, Clarke AR, et al. Location and properties of metal-binding sites on the human prion protein. Proc Natl Acad Sci U S A. 2001;98:8531–5. doi: 10.1073/pnas.151038498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CE, Abdelraheim SR, Brown DR, Viles JH. Preferential Cu2+ coordination by His96 and His111 induces beta-sheet formation in the unstructured amyloidogenic region of the prion protein. J Biol Chem. 2004;279:32018–27. doi: 10.1074/jbc.M403467200. [DOI] [PubMed] [Google Scholar]
- Jones CE, Klewpatinond M, Abdelraheim SR, Brown DR, Viles JH. Probing copper2+ binding to the prion protein using diamagnetic nickel2+ and 1H NMR: the unstructured N terminus facilitates the coordination of six copper2+ ions at physiological concentrations. J Mol Biol. 2005;346:1393–407. doi: 10.1016/j.jmb.2004.12.043. [DOI] [PubMed] [Google Scholar]
- Kaul S, Kanthasamy A, Kitazawa M, Anantharam V, Kanthasamy AG. Caspase-3 dependent proteolytic activation of protein kinase C delta mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: relevance to oxidative stress in dopaminergic degeneration. Eur J Neurosci. 2003;18:1387–401. doi: 10.1046/j.1460-9568.2003.02864.x. [DOI] [PubMed] [Google Scholar]
- Kim NH, Choi JK, Jeong BH, Kim JI, Kwon MS, Carp RI, et al. Effect of transition metals (Mn, Cu, Fe) and deoxycholic acid (DA) on the conversion of PrPC to PrPres. Faseb J. 2005;19:783–5. doi: 10.1096/fj.04-2117fje. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Anantharam V, Kanthasamy AG. Activation of oxidative stress-dependent cell signaling pathways in methylcyclopentadienyl manganese tricarbonyl (MMT)-induced apoptosis: Downstream events and regulatory mechanisms. 40th annual meeting of Society of Toxicology; San Francisco, CA. 2001. [Google Scholar]
- Kitazawa M, Anantharam V, Yang Y, Hirata Y, Kanthasamy A, Kanthasamy AG. Activation of protein kinase C delta by proteolytic cleavage contributes to manganese-induced apoptosis in dopaminergic cells: protective role of Bcl-2. Biochem Pharmacol. 2005;69:133–46. doi: 10.1016/j.bcp.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Zerr I, Weber T, Poser S, Kretzschmar H, Hunsmann G, et al. Prion disease associated with a novel nine octapeptide repeat insertion in the PRNP gene. Brain Res Mol Brain Res. 1995;34:173–6. doi: 10.1016/0169-328x(95)00175-r. [DOI] [PubMed] [Google Scholar]
- Latchoumycandane C, Anantharam V, Kitazawa M, Yang Y, Kanthasamy A, Kanthasamy AG. Protein kinase Cdelta is a key downstream mediator of manganese-induced apoptosis in dopaminergic neuronal cells. J Pharmacol Exp Ther. 2005;313:46–55. doi: 10.1124/jpet.104.078469. [DOI] [PubMed] [Google Scholar]
- Lee DW, Sohn HO, Lim HB, Lee YG, Kim YS, Carp RI, et al. Alteration of free radical metabolism in the brain of mice infected with scrapie agent. Free Radic Res. 1999;30:499–507. doi: 10.1080/10715769900300541. [DOI] [PubMed] [Google Scholar]
- Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci U S A. 2007;104:20908–13. doi: 10.1073/pnas.0710054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mange A, Milhavet O, Umlauf D, Harris D, Lehmann S. PrP-dependent cell adhesion in N2a neuroblastoma cells. FEBS Lett. 2002;514:159–62. doi: 10.1016/s0014-5793(02)02338-4. [DOI] [PubMed] [Google Scholar]
- Mead S, Webb TE, Campbell TA, Beck J, Linehan JM, Rutherfoord S, et al. Inherited prion disease with 5-OPRI: phenotype modification by repeat length and codon 129. Neurology. 2007;69:730–8. doi: 10.1212/01.wnl.0000267642.41594.9d. [DOI] [PubMed] [Google Scholar]
- Milhavet O, McMahon HE, Rachidi W, Nishida N, Katamine S, Mange A, et al. Prion infection impairs the cellular response to oxidative stress. Proc Natl Acad Sci U S A. 2000;97:13937–42. doi: 10.1073/pnas.250289197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Holgado F, Hider RC, Gaeta A, Williams R, Francis P. Metals ions and neurodegeneration. Biometals. 2007;20:639–54. doi: 10.1007/s10534-006-9033-z. [DOI] [PubMed] [Google Scholar]
- Nadal RC, Abdelraheim SR, Brazier MW, Rigby SE, Brown DR, Viles JH. Prion protein does not redox-silence Cu2+, but is a sacrificial quencher of hydroxyl radicals. Free Radic Biol Med. 2007;42:79–89. doi: 10.1016/j.freeradbiomed.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Nunziante M, Gilch S, Schatzl HM. Essential role of the prion protein N terminus in subcellular trafficking and half-life of cellular prion protein. J Biol Chem. 2003;278:3726–34. doi: 10.1074/jbc.M206313200. [DOI] [PubMed] [Google Scholar]
- Owen F, Poulter M, Lofthouse R, Collinge J, Crow TJ, Risby D, et al. Insertion in prion protein gene in familial Creutzfeldt-Jakob disease. Lancet. 1989;1:51–2. doi: 10.1016/s0140-6736(89)91713-3. [DOI] [PubMed] [Google Scholar]
- Palmer MS, Collinge J. Human prion diseases. Curr Opin Neurol Neurosurg. 1992;5:895–901. [PubMed] [Google Scholar]
- Palmer MS, Collinge J. Mutations and polymorphisms in the prion protein gene. Hum Mutat. 1993;2:168–73. doi: 10.1002/humu.1380020303. [DOI] [PubMed] [Google Scholar]
- Park RM, Schulte PA, Bowman JD, Walker JT, Bondy SC, Yost MG, et al. Potential occupational risks for neurodegenerative diseases. Am J Ind Med. 2005;48:63–77. doi: 10.1002/ajim.20178. [DOI] [PubMed] [Google Scholar]
- Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem. 1998;273:33107–10. doi: 10.1074/jbc.273.50.33107. [DOI] [PubMed] [Google Scholar]
- Perera WS, Hooper NM. Ablation of the metal ion-induced endocytosis of the prion protein by disease-associated mutation of the octarepeat region. Curr Biol. 2001;11:519–23. doi: 10.1016/s0960-9822(01)00147-6. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Mironov A, Jr, Peretz D, van Donselaar E, Leclerc E, Erpel S, et al. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J Cell Biol. 2003;162:703–17. doi: 10.1083/jcb.200304140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RB, Siedlak SL, Lee HG, Kim YS, Nunomura A, Tagliavini F, et al. Redox metals and oxidative abnormalities in human prion diseases. Acta Neuropathol. 2005;110:232–8. doi: 10.1007/s00401-005-1034-4. [DOI] [PubMed] [Google Scholar]
- Prado MA, Alves-Silva J, Magalhaes AC, Prado VF, Linden R, Martins VR, et al. PrPc on the road: trafficking of the cellular prion protein. J Neurochem. 2004;88:769–81. doi: 10.1046/j.1471-4159.2003.02199.x. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–22. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, DeArmond SJ. Prion diseases of the central nervous system. Monogr Pathol. 1990:86–122. [PubMed] [Google Scholar]
- Prusiner SB, Kingsbury DT. Prions--infectious pathogens causing the spongiform encephalopathies. CRC Crit Rev Clin Neurobiol. 1985;1:181–200. [PubMed] [Google Scholar]
- Prusiner SB, Scott M, Foster D, Pan KM, Groth D, Mirenda C, et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell. 1990;63:673–86. doi: 10.1016/0092-8674(90)90134-z. [DOI] [PubMed] [Google Scholar]
- Renner C, Fiori S, Fiorino F, Landgraf D, Deluca D, Mentler M, et al. Micellar environments induce structuring of the N-terminal tail of the prion protein. Biopolymers. 2004;73:421–33. doi: 10.1002/bip.20015. [DOI] [PubMed] [Google Scholar]
- Roth JA, Horbinski C, Higgins D, Lein P, Garrick MD. Mechanisms of manganese-induced rat pheochromocytoma (PC12) cell death and cell differentiation. Neurotoxicology. 2002;23:147–57. doi: 10.1016/s0161-813x(01)00077-8. [DOI] [PubMed] [Google Scholar]
- Sakudo A, Lee DC, Yoshimura E, Nagasaka S, Nitta K, Saeki K, et al. Prion protein suppresses perturbation of cellular copper homeostasis under oxidative conditions. Biochem Biophys Res Commun. 2004;313:850–5. doi: 10.1016/j.bbrc.2003.12.020. [DOI] [PubMed] [Google Scholar]
- Sango K, Yanagisawa H, Kato K, Kato N, Hirooka H, Watabe K. Differential Effects of High Glucose and Methylglyoxal on Viability and Polyol Metabolism in Immortalized Adult Mouse Schwann Cells. The Open Diabetes Journal. 2008:1–11. [Google Scholar]
- Sayre LM, Perry G, Smith MA. Redox metals and neurodegenerative disease. Curr Opin Chem Biol. 1999;3:220–5. doi: 10.1016/S1367-5931(99)80035-0. [DOI] [PubMed] [Google Scholar]
- Shum SC, Neddersen R, Houk RS. Elemental speciation by liquid chromatography-inductively coupled plasma mass spectrometry with direct injection nebulization. Analyst. 1992;117:577–82. doi: 10.1039/an9921700577. [DOI] [PubMed] [Google Scholar]
- Singh A, Isaac AO, Luo X, Mohan ML, Cohen ML, Chen F, et al. Abnormal brain iron homeostasis in human and animal prion disorders. PLoS Pathog. 2009;5:e1000336. doi: 10.1371/journal.ppat.1000336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smargiassi A, Mutti A. Peripheral biomarkers and exposure to manganese. Neurotoxicology. 1999;20:401–6. [PubMed] [Google Scholar]
- Stredrick DL, Stokes AH, Worst TJ, Freeman WM, Johnson EA, Lash LH, et al. Manganese-induced cytotoxicity in dopamine-producing cells. Neurotoxicology. 2004;25:543–53. doi: 10.1016/j.neuro.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Tatzelt J, Schatzl HM. Molecular basis of cerebral neurodegeneration in prion diseases. Febs J. 2007;274:606–11. doi: 10.1111/j.1742-4658.2007.05633.x. [DOI] [PubMed] [Google Scholar]
- Thackray AM, Knight R, Haswell SJ, Bujdoso R, Brown DR. Metal imbalance and compromised antioxidant function are early changes in prion disease. Biochem J. 2002;362:253–8. doi: 10.1042/0264-6021:3620253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiber C, Pipkorn R, Weise C, Holland G, Multhaup G. Copper is required for prion protein-associated superoxide dismutase-I activity in Pichia pastoris. Febs J. 2007;274:1304–11. doi: 10.1111/j.1742-4658.2007.05678.x. [DOI] [PubMed] [Google Scholar]
- Viles JH, Cohen FE, Prusiner SB, Goodin DB, Wright PE, Dyson HJ. Copper binding to the prion protein: structural implications of four identical cooperative binding sites. Proc Natl Acad Sci U S A. 1999;96:2042–7. doi: 10.1073/pnas.96.5.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viles JH, Donne D, Kroon G, Prusiner SB, Cohen FE, Dyson HJ, et al. Local structural plasticity of the prion protein. Analysis of NMR relaxation dynamics. Biochemistry. 2001;40:2743–53. doi: 10.1021/bi002898a. [DOI] [PubMed] [Google Scholar]
- Wong BS, Brown DR, Pan T, Whiteman M, Liu T, Bu X, et al. Oxidative impairment in scrapie-infected mice is associated with brain metals perturbations and altered antioxidant activities. J Neurochem. 2001a;79:689–98. doi: 10.1046/j.1471-4159.2001.00625.x. [DOI] [PubMed] [Google Scholar]
- Wong BS, Chen SG, Colucci M, Xie Z, Pan T, Liu T, et al. Aberrant metal binding by prion protein in human prion disease. J Neurochem. 2001b;78:1400–8. doi: 10.1046/j.1471-4159.2001.00522.x. [DOI] [PubMed] [Google Scholar]
- Worley CG, Bombick D, Allen JW, Suber RL, Aschner M. Effects of manganese on oxidative stress in CATH. a cells Neurotoxicology. 2002;23:159–64. doi: 10.1016/s0161-813x(02)00028-1. [DOI] [PubMed] [Google Scholar]
- Wu J, Basha MR, Zawia NH. The environment, epigenetics and amyloidogenesis. J Mol Neurosci. 2008;34:1–7. doi: 10.1007/s12031-007-0009-4. [DOI] [PubMed] [Google Scholar]
- Yin S, Pham N, Yu S, Li C, Wong P, Chang B, et al. Human prion proteins with pathogenic mutations share common conformational changes resulting in enhanced binding to glycosaminoglycans. Proc Natl Acad Sci U S A. 2007;104:7546–51. doi: 10.1073/pnas.0610827104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Jiang H, Lee ES, Ni M, Erikson KM, Milatovic D, et al. Ferroportin is a manganese-responsive protein that decreases manganese cytotoxicity and accumulation. J Neurochem. 2010;112:1190–8. doi: 10.1111/j.1471-4159.2009.06534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokel RA. Manganese flux across the blood-brain barrier. Neuromolecular Med. 2009;11:297–310. doi: 10.1007/s12017-009-8101-2. [DOI] [PubMed] [Google Scholar]
- Yun SW, Gerlach M, Riederer P, Klein MA. Oxidative stress in the brain at early preclinical stages of mouse scrapie. Exp Neurol. 2006;201:90–8. doi: 10.1016/j.expneurol.2006.03.025. [DOI] [PubMed] [Google Scholar]
- Zhu F, Davies P, Thompsett AR, Kelly SM, Tranter GE, Hecht L, et al. Raman optical activity and circular dichroism reveal dramatic differences in the influence of divalent copper and manganese ions on prion protein folding. Biochemistry. 2008;47:2510–7. doi: 10.1021/bi7022893. [DOI] [PubMed] [Google Scholar]