

Abstract
Sequencing DNA in a synthetic solid-state nanopore is potentially a low-cost and high-throughput method. Essential to the nanopore-based DNA sequencing method is the ability to control the motion of a single-stranded DNA (ssDNA) molecule at single-base resolution. Experimental studies showed that the average translocation speed of DNA driven by a biasing electric field can be affected by ionic concentration, solvent viscosity, or temperature. Even though it is possible to slow down the average translocation speed, instantaneous motion of DNA is too diffusive to allow each DNA base to stay in front of a sensor site for its measurement. Using extensive all-atom molecular dynamics simulations, we study the diffusion constant, friction coefficient, electrophoretic mobility, and effective charge of ssDNA in a solid-state nanopore. Simulation results show that the spatial fluctuation of ssDNA in 1 ns is comparable to the spacing between neighboring nucleotides in ssDNA, which makes the sensing of a DNA base very difficult. We demonstrate that the recently proposed DNA transistor could potentially solve this problem by electrically trapping ssDNA inside the DNA transistor and ratcheting ssDNA base-by-base in a biasing electric field. When increasing the biasing electric field, we observed that the translocation of ssDNA changes from ratcheting to steady-sliding. The simulated translocation of ssDNA in the DNA transistor was theoretically characterized using Fokker-Planck analysis.
Introduction
In the last decade, a significant amount of work has been focused on developing low-cost and high-throughput methods for sequencing DNA. Besides methods of sequencing DNA by synthesis, such as fluorescent in situ sequencing (1) and pyrosequencing (2), nanopore-based DNA sequencing methods are very promising and have attracted extensive interest (3–5). Protein pores (6–9), especially engineered ones (10,11), have been used for sensing DNA bases by reading ionic current through a pore (3). When a nucleotide is in the constriction site of a protein pore, the ionic current is modified from an open-pore current and is very sensitive to the type of a nucleotide. Generally, a protein pore can be used as a sensor for a polymeric molecule (12). Inspired by protein pores, a synthetic solid-state nanopore was proposed to sequence DNA (3,13–15). Current fabrication techniques allow thousands of pores drilled in a silicon-based chip (16), yielding a potentially cheap and scalable method. Another advantage of using a solid-state nanopore is that nanoelectrodes can be integrated to sense a DNA base via a transversal tunneling current (17–19).
To achieve a single-base resolution, the motion of single-stranded DNA (ssDNA) should be slow and steady enough to allow an accurate sensing of a DNA base. One important way to sense a DNA base is to measure a tunneling current through the base. Only an average of repeated independent measurements of tunneling currents can yield a base-sensitive electric signal (10,18,19). Therefore, a fast DNA translocation, despite being desirable as a high-throughput method, could cause large uncertainties in measured electric signals, limiting the accuracy of DNA sequencing.
Many methods have been suggested to slow down the motion of DNA in a solid-state nanopore. Increasing the friction coefficient is one way to reduce the translocation velocity of DNA. It has been experimentally demonstrated that a viscous electrolyte can dramatically slow down the motion of DNA (20). Decreasing temperature (20,21) and increasing ion concentration (20,22) can also increase the friction coefficient of DNA in an electrolyte. Surrounding DNA, an electroosmotic flow driven by ionic motion in an electric field can impose extra friction on DNA (22–25). Such a hydrodynamic friction force on DNA depends on properties of a pore surface that serves as a boundary for the electroosmotic flow (23) inside a pore. After DNA enters a solid-state nanopore, reversing the biasing electric field can exert a resistant force on DNA (26). Additionally, the translocation velocity of DNA can be reduced using a nanopore smaller than the double-helix (27). Although the mean translocation velocity can be greatly reduced using these methods, diffusive motion may cause difficulties in sensing DNA at single-base resolution.
The motion of DNA can also be slowed down by a mechanical force externally applied by a magnetic (28) or an optical (29,30) tweezer. These marvelous single-molecule techniques can be used to pull DNA at an arbitrarily slow velocity, but they may not be cheap or scalable sequencing methods. Instead of using a mechanical force, alternatively, an electric trapping force can be applied on DNA in a nanopore with embedded nanoelectrodes (a device we called the “DNA transistor”) (31). It has been shown in simulation that ssDNA can be base-by-base ratcheted through the DNA transistor (32). In this controlled motion, ssDNA alternatively stops (a trapped state) and moves forward by one-nucleotide spacing. If a sensor is integrated in the DNA transistor, during each trapped state, a base paused in front of the sensor could be measured, permitting single-base resolution.
Extensive experimental work has been focused on double-stranded DNA in a solid-state nanopore (14). Recently, it was demonstrated in experiment that ssDNA with or without a secondary structure can also be electrically driven through a solid-state nanopore (33,34), yielding a scaling relation, between translocation times and biasing voltages, that is different from the one for double-stranded DNA (34). Using extensive (∼3.5 μs) molecular dynamics simulations, we studied the ratcheting motion of ssDNA in different electrolytes in the presence of electric trapping fields provided by the DNA transistor. To characterize the driven motion of ssDNA in the DNA transistor beyond timescales affordable by simulation (hundreds of nanoseconds per run), we developed a theoretical approach (based on the Fokker-Planck equation) that builds up from previous theoretical analyses of polymer translocation through a narrow pore (35). The simulated driven motion of ssDNA in the DNA transistor was quantitatively compared with theoretical results, using independently simulated physical parameters such as diffusion and friction coefficients, electrophoretic mobility, and effective charge of ssDNA.
Methods
Simulation system
A crystalline SiO2 solid was melted and quenched in silico to obtain an amorphous SiO2 solid. The force field used for simulating melting and quenching processes of an SiO2 solid was developed by van Beest et al. (36). During the quenching process, atoms inside the 2-nm-radius cylinder whose symmetry axis coincides with the z axis were driven out, forming a cylindrical channel. The quenched solid measures 65 × 65 × 148 Å3. A linear ssDNA fragment comprising 20 adenine nucleotides was placed on the symmetry axis of the channel. The ssDNA fragment is covalently linked to itself through periodic boundary of the system. Because DNA could be electrically (37) and hydrodynamically (23,38) stretched during the translocation through a solid-state nanopore, the ssDNA in simulation was kept in a stretched form because of the periodic boundary condition.
The average spacing d between neighboring phosphate groups in the backbone of ssDNA is 7.4 Å (which is attained at several hundred picoNewtons of tension in ssDNA), in contrast to a curly conformation (d ∼ 2.5 Å) of ssDNA in the α-hemolysin nanopore (39). ssDNA was solvated with a 0 M, 0.1 M, or 1 M NaCl electrolyte. The channel pressure is ∼1 bar. The total charge of −20e (where e denotes the electron charge) was uniformly distributed to SiO2 atoms on the channel surface, which was suggested to enhance the capture rate of ssDNA (40). The whole system is electrically neutral. The system (shown later in Fig. 3 a) was minimized for 10 ps, followed by 5-ns equilibration in the NVT ensemble at 300 K. In a previous study (41), we showed that a hydrophobic coating of a self-assembled monolayer can prevent ssDNA from adhering to the SiO2 solid surface and can confine ssDNA near the symmetry axis of the pore. This confinement was imposed in our simulations by constraining the center of mass of all phosphorus atoms with a harmonic spring (1.44 kcal/(Å2 ⋅ mol)) to the symmetry axis of the channel.
Figure 3.
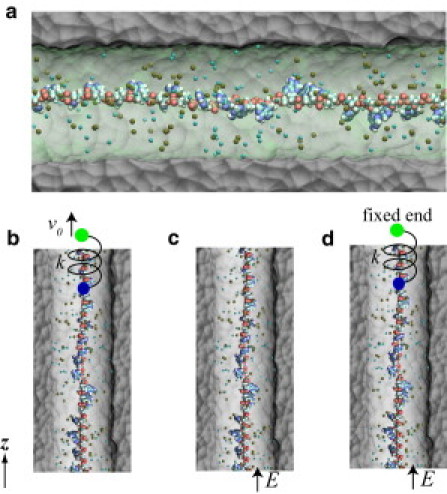
Simulation of ssDNA in a solid-state channel. (a) A cross-section view of the simulated system. (Gray molecular surface) SiO2 solid. ssDNA is solvated in a 0.1 M NaCl electrolyte, containing water molecules (green and transparent), sodium (tan), and chloride (cyan) ions. (b) The setup of simulation when DNA is pulled by a harmonic spring. One end (green) of the spring moves at a constant velocity v0 and the other end (blue) is attached to the center of mass of all phosphorus atoms in ssDNA. (c) The setup of simulation when DNA is driven by a biasing electric field E across the channel. (d) The setup of simulation when ssDNA is under simultaneous actions of a harmonic pulling and an electric driving force. The end (green) of spring, not attached to ssDNA, is fixed. In panels b–d, water is not shown.
Simulation of ssDNA in the DNA transistor
The DNA transistor (Fig. 1 a) contains a sandwichlike, multilayered structure in which metal and dielectric layers are alternatively stacked on top of each other (31). To model the DNA transistor, we assume that the thicknesses of each metal and each dielectric layer are 2d and 2.5d (where d denotes the spacing between neighboring phosphate groups in ssDNA), respectively. As a typical voltage setup in the DNA transistor, two outer metal-layers are grounded and the voltage on the middle metal-layer is 2 V. To mimic electric fields in the DNA transistor, ±Et(Et = 108 mV/Å) are applied (using a grid-force approach (42)) to all atoms in the whole layers aligned with corresponding dielectric regions, as illustrated in Fig. 1 b. Previous simulations of ssDNA in these electric fields showed that ssDNA can be trapped in the DNA transistor (43). The experimental procedure for fabrication of the device will be published elsewhere (H. Peng, unpublished).
Figure 1.
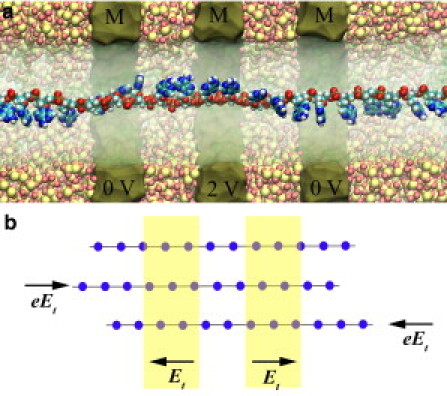
Simulation of ssDNA in the DNA transistor. (a) A cross-section view of the simulation setup. SiO2 atoms are shown as van der Waals spheres. DNA is solvated in a NaCl electrolyte (not shown). The metal layers (tan) are labeled with M. The voltage on the middle metal-layer is 2 V and the two outer metal-layers are grounded. (b) Schematic illustration of trapping of ssDNA in the DNA transistor. (Lines) Backbone of ssDNA. (Dots) Negatively charged nucleotides.
MD methods
All MD simulations were carried out using the program NAMD (45). The AMBER (parm-bsc0) force field (46) was used for ssDNA; the TIP3P model (47) was used for water molecules; the standard force field was used for ions (48); and the silica force field (49) was used for a SiO2 solid interacting with water. Periodic boundary conditions are applied in all three dimensions. Long-range Coulomb interactions are computed using particle-mesh Ewald full electrostatics over a 64 × 64 × 136 grid. The temperature was kept at 300 K by applying the Langevin thermostat (50) to all SiO2 atoms (see the Supporting Material) that are harmonically constrained to their respective original positions. The simulation time-step was 1 fs and the standard multiple-time-stepping approach (51) was used. The van der Waals interaction between atoms were calculated using a smooth (10–12 Å) cutoff.
Results
Ratcheting of ssDNA in the DNA transistor
To sequence ssDNA at single-base resolution, it would be convenient if each nucleotide could be stopped in front of a sensor for a sufficiently long time before the ssDNA molecule moves forward by one-nucleotide spacing. The DNA transistor can potentially provide such a functionality. When appropriate voltages are applied on the three metal electrodes (indicated by M in Fig. 1 a) in the DNA transistor, electric fields inside the nanopore (Fig. 1 b) are applied on the charged phosphate groups in ssDNA. When ssDNA moves a little (<0.5d) toward the right (left) side, the number of phosphate groups in the right (left) dielectric region will be one more than that in the left (right) dielectric region. Therefore, a force of eE (assuming a point charge of each phosphate group) opposite the direction of ssDNA motion is exerted on ssDNA. In a previous theoretical study (43) of ssDNA in the DNA transistor solvated by a 0.1 M NaCl electrolyte, ssDNA was pulled by a harmonic spring (100 pN/Å) through the DNA transistor (without a biasing electric field). To maintain a constant velocity, pulling forces by the spring periodically increase and decrease (43), indicating that the ssDNA molecule is subject to a periodic trapping potential due to the electric fields provided by the DNA transistor.
Fig. 2 shows the electrically driven motion of ssDNA in the DNA transistor when the trapping voltage on the middle electrode is 2 V and the ion concentration is 0.1 M. The electric driving force on the bare ssDNA is QE, where Q is the charge of bare ssDNA and E the biasing electric field. When the biasing electric field is weak (QE < 75 pN), ssDNA moves in a ratchetlike manner, i.e., ssDNA alternatively stays in a trapped state for some time and moves quickly by one-nucleotide spacing. Ideally, a sensor could identify the base located in its proximity during each trapped state of ssDNA. The ratchet motion of ssDNA is more pronounced in a weaker biasing electric field. However, it is also possible for ssDNA to move backward when the biasing electric field is too weak (such as the case when QE = 20 pN), as shown in Fig. 2. When the biasing electric field is big (QE > 100 pN), ssDNA is barely trapped and moves forward steadily. In an intermediate field strength (QE = 75 or 100 pN), both ratchetlike and steady motion of ssDNA are present (Fig. 2).
Figure 2.
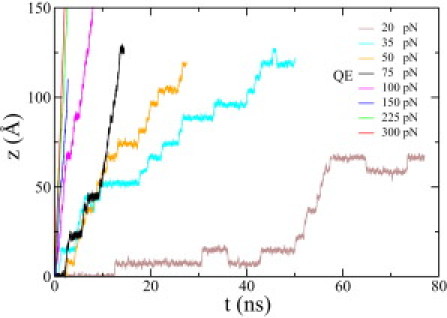
Electrically driven motion of ssDNA in a 0.1 M NaCl electrolyte in the DNA transistor. The biasing electric field varies from 6.25 to 93.75 mV/nm, corresponding to an electrical driving force QE changing from 20 to 300 pN.
Similar ratcheting motion of ssDNA is observed in the same channel filled with a 1 M NaCl electrolyte (see Fig. S1 in the Supporting Material). Overall, we found that the driven motion of ssDNA is slower in the 1 M electrolyte than in the 0.1 M electrolyte. Experimentally, it was also found that the translocation velocity of DNA could be reduced by increasing the ion concentration (20,22). In a trapped state, the mean trapping time is longer for ssDNA in the 1 M electrolyte, in accord with the result that the friction coefficient of ssDNA is bigger in the 1 M electrolyte (see below).
Theory of DNA translocation in the DNA transistor
When ssDNA is in the trapping potential provided by the DNA transistor, the motion of ssDNA is overdamped (where viscous effects dominate inertial effects) because ∼ 5(> 1). Here m is the mass of ssDNA; kmax is the maximum stiffness of the trapping potential F, i.e., kmax = |d2F/dz2|max; and ξ is the friction coefficient (see below). Thus, the probability P(z,t) of finding the center of mass of ssDNA at position z and at time t satisfies the Smoluchowski equation,
| (1) |
where J is the probability current that is defined as
| (2) |
In Eq. 2, T is the absolute temperature and kB is the Boltzmann constant. The value f(z) is the position-dependent force applied to the center of mass of a ssDNA molecule, which derives from the potential
| (3) |
where F(z) is the periodic trapping potential in the DNA transistor, with periodicity d (d is the interphosphate distance), and f0 is the effective driving force exerted on the DNA due to the biasing electric field E. Here f0 = qeffE, where qeff is the effective charge of ssDNA.
We assume that the system is in the stationary state. In this case, the left-hand side of Eq. 1 is zero, and, therefore, the probability current is constant—that is, J(z,t) = J0.
To find the average translocation velocity v of ssDNA as a function of trapping and biasing fields, we need to solve for J0, because
| (4) |
We rewrite Eq. 2 as
| (5) |
Multiplying both terms of Eq. 5 with and integrating between −d/2 and d/2, and using the fact that the center of mass has a periodic probability density function with periodicity d (i.e., P(−d/2) = P(d/2) ≡ P0), we obtain that
| (6) |
where FM ≡ F(−d/2) = F(d/2), the latter equality justified by the periodicity of F(z). The equation for J0 is determined except for P0, which can be computed from the constraint that the P(z) has to normalize to 1. Again, using Eq. 5, multiplying both terms with and integrating between −d/2 and z, we obtain that
| (7) |
from where P0 can be found as
| (8) |
Before we can compare predictions of the theory and the simulation results shown in Fig. 2, physical parameters, such as friction coefficient, electrophoretic mobility, effective ssDNA charge, and the electric trapping energy for ssDNA in the DNA transistor, are required. In the following, we use MD simulations to obtain these parameters.
Physical parameters for DNA motion in the channel
Diffusion and friction coefficients
In the simulation setup shown in Fig. 3 a, ssDNA was allowed to diffuse freely along the z axis in different electrolytes. Fig. 4, a, c, and e, shows time-dependent displacements of ssDNA in 0 M, 0.1 M, and 1 M NaCl electrolytes confined in the channel, respectively. During tens of nanoseconds of simulation of ssDNA in each electrolyte, ssDNA moved back and forth randomly and the variation in ssDNA positions shown in Fig. 4 is ∼3 nm, about the length of four nucleotides in simulated ssDNA. Thus, the diffusive motion of ssDNA is a major obstacle for sequencing ssDNA at single-base resolution.
Figure 4.
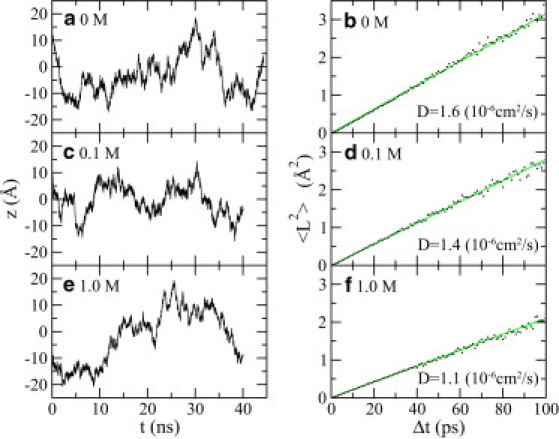
One-dimensional diffusive motion of ssDNA in the channel. (a, c, and e) Time-dependent ssDNA positions when ssDNA is in a 0 (a), 0.1 M (c), or 1 M (e) NaCl electrolyte. (b, d, and f) Mean-square displacement 〈L2〉 versus time interval Δt when ssDNA is in a 0 (b), 0.1 M (d), or 1 M (f) NaCl electrolyte. The slope of each fitting line is equal to twice the diffusion constant D of ssDNA.
From the trajectory of ssDNA, the mean-square displacements 〈L2〉 of ssDNA can be computed at each time interval Δt. This is shown in Fig. 4, b, d, and f, for different ion concentrations. For a one-dimensional random motion,
| (9) |
where D is the diffusion coefficient of ssDNA. Therefore, the diffusion coefficient D = 1.6 × 10−6cm2/s, 1.4 × 10−6cm2/s, and 1.1 × 10−6cm2/s for ssDNA in 0, 0.1, and 1 M electrolytes, respectively, i.e., the diffusion coefficient of ssDNA decreases with an increase of the ion concentration. Note that these numerically computed diffusion coefficients are of the same order of magnitude as the experimentally measured diffusion coefficient of a 20-base ssDNA fragment in free solution (52).
In our simulations, ssDNA is in a linear conformation along its moving direction. As a consequence, the relaxation time of ssDNA conformation is negligible. Therefore, the friction coefficient ξ of ssDNA can be obtained from the Einstein relation,
| (10) |
At 300 K, the calculated friction coefficients of ssDNA in 0, 0.1, and 1 M electrolytes are 25.6, 29.3, and 37.3 pN ⋅ ns/nm, respectively. These values are consistent with directly measured friction coefficients using the steered molecular dynamics (53) method to pull ssDNA along the z axis (Fig. 3 b and see the Supporting Material).
Electrophoretic mobility of ssDNA
Fig. 3 c shows that ssDNA can be driven through the channel by a biasing electric field E. The electric driving force on DNA can be written as qeffE, where qeff is the effective charge of ssDNA. In an electric field pointing in the +z direction, negatively charged ssDNA moves opposite the field direction. When the electric driving force on ssDNA is balanced by the hydrodynamic friction force, ssDNA moves forward with a time-independent average velocity v. Fig. 5 shows that, upon increasing the biasing electric field, the translocation velocity v of ssDNA in the 0.1 M electrolyte also increases. Note that the lowest electric field (3.13 mV/nm) is very close to the biasing electric field typically used in experiments (6,54). The inset in Fig. 5 shows that the translocation velocity of ssDNA increases linearly with the biasing electric field. Thus,
| (11) |
where μ is the electrophoretic mobility of ssDNA. A linear fit of data yields that μ = 0.09 nm2/(ns ⋅ mV), very close to the experimentally measured mobility of ssDNA (52) in a bulk electrolyte, and much bigger than the mobility of ssDNA in the α-hemolysin pore where the interaction between ssDNA and the protein pore is important (6).
Figure 5.
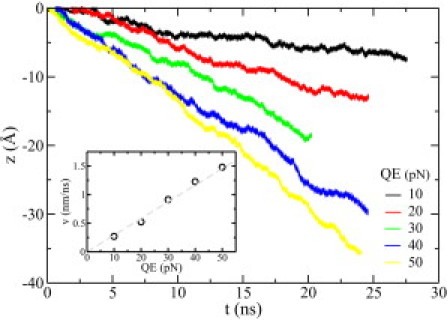
Electrophoretic motion of ssDNA in the channel, driven by a biasing electric field E. E = 3.13 (black), 6.25 (red), 9.38 (green), 12.5 (blue), and 15.63 (yellow) mV/nm, corresponding to electric driving forces (on bare ssDNA with charge Q) of 10, 20, 30, 40, and 50 pN, respectively. (Inset) Absolute value of the mean translocation velocity v versus the electric driving force. The electrophoretic mobility can be obtained from the slope of the fitting line (dashed).
Effective charge of ssDNA
The effective charge of DNA in a solid-state nanopore has been investigated experimentally, using an optical tweezer to hold one end of DNA and simultaneously applying a biasing electric field to drive DNA through the pore (25,29). The effective charge of DNA qeff is defined as the mean force 〈f〉 exerted by the optical tweezer divided by the electric field strength E, i.e., qeff = 〈f〉/E. Fig. 3 d shows the setup of a simulation designed to mimic the optical-tweezer experiment. Following the method described in Luan (23), one end of a harmonic spring (1 pN/Å) is fixed while the other end is attached to the center of mass of all phosphorus atoms in ssDNA. After applying a biasing electric field, ssDNA moves opposite the field direction. Meanwhile, the spring stretches and the force applied on ssDNA increases. Eventually, the force in the spring balances the effective electric driving force, and ssDNA stalls. Fig. 6 a shows time-dependent forces in the spring in an electric field. After ∼30 ns, the spring force saturates and fluctuates around a mean value 〈f〉 that increases with the applied field strength E.
Figure 6.
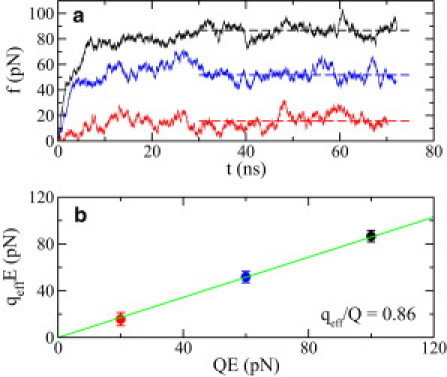
The effective charge qeff of ssDNA in a 0.1 M NaCl electrolyte. (a) Time-dependent spring forces f that balance the effective electric driving force qeffE on ssDNA. The biasing electric field E = 6.25 (red), 15.63 (blue), and 28.13 (black) mV/nm. (Dashed lines) Mean spring forces. (b) Effective electric driving force versus electric driving force on bare ssDNA. The slope of the fitting line is the ratio between the effective charge qeff of ssDNA and the charge Q of bare ssDNA. (Error bars) Standard deviations.
Fig. 6 b shows that the mean force in the spring (that is also qeffE) increases linearly with the electric force applied on bare ssDNA whose charge is Q. The slope of a linear fit yields the ratio between qeff and Q. Here the effective charge of ssDNA is ∼86% of the charge of bare ssDNA. Theoretical studies (22,23) showed that the effective charge of DNA cannot be explained by the counterion condensation (55) but depends on the electroosmotic flow between charged surfaces of DNA and the pore. Thus, the effective charge of ssDNA depends on the ion concentration, as well as on the radius, surface charge density, and surface roughness of the pore (22,23). However, a simple relation can be used to account for both electric and hydrodynamic screening effects (23), i.e.,
| (12) |
From the independently obtained friction coefficient and electrophoretic mobility of ssDNA, the effective charge of ssDNA predicted from the Eq. 12 is 17.8 e. Because Q = 20e, the effective charge is ∼89% of the charge of bare ssDNA. Thus, the effective charge from Eq. 12 is very close to the one directly measured from simulation.
When the ion concentration is 1 M, Fig. S4 shows that effective driving forces depend linearly on electric driving forces (QE) on bare ssDNA. From this relation, the effective charge of ssDNA can be derived to be 6e (i.e., ∼30% of the charge of bare ssDNA).
Note that the effective charge of ssDNA results from two effects: the binding of counterions on ssDNA (electric screening) and the motion of counterions against the ssDNA translocation (electroosmotic screening). These effects were also found for a charged PEG molecule in the α-hemolysin pore (12). In that case, the ratio between viscous and electric forces (FV/FE = qeff/Q) for PEG in a 4 M electrolyte was ∼0.72. Additionally, the effective charge of ssDNA, poly(dC)30 in the α-hemolysin pore is ∼0.07Q (56). The variation in measured effective charges results from different ionic strengths and interaction of ssDNA with a pore. The reduction of charge in ssDNA will affect the translocation velocity of ssDNA in a nanopore (23,25,57,58) and the trapping of ssDNA in the DNA transistor (31,32).
Trapping energy of ssDNA in the DNA transistor
To obtain the profile of the trapping potential for ssDNA in the DNA transistor, we performed six independent steered molecular dynamics simulations to pull ssDNA in 0.1 M NaCl electrolyte through the DNA transistor. The pulling velocity v was 1 Å/ns and a stiff spring (1000 pN/Å) was used. The time-dependent pulling forces f are obtained from the extension of the pulling spring. The work can be calculated as and the mean work was averaged over six sets of data. Using the Jarzynski relation (59) and cumulant expansion (60), the potential of mean force (PMF) F is shown in Fig. 7. As ssDNA moves forward by a distance of d, the PMF in the end of the pulling process returns back to the initial value, showing a periodic trapping potential.
Figure 7.
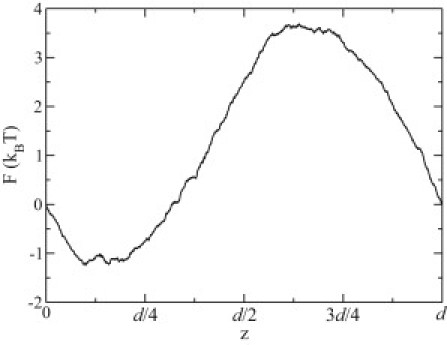
The potential of mean force for ssDNA solvated in 0.1 M NaCl electrolyte in the DNA transistor. The label d is the spacing between two neighboring phosphate groups in the backbone of ssDNA.
Estimated from the profile of the PMF, the energy barrier or the trapping energy is ∼4.8 kBT for ssDNA in a 0.1 M NaCl electrolyte. The trapping energy increases linearly with the applied trapping voltage. From Fig. S3, pulling forces on ssDNA moving through the DNA transistor are quite similar for different ion concentrations, indicating that the trapping energy is independent of the ion concentration.
Comparison between simulation and theory
For the implementation of Eqs. 4–8, we assume that the periodic trapping potential for ssDNA in the DNA transistor,
| (13) |
according to the profile of PMF (Fig. 7) and we take ET to be 4.8 kBT (T = 300 K) as measured from the potential of mean force of ssDNA in the DNA transistor.
Combining Eqs. 4–8 and 13 and using diffusion coefficients measured from MD simulations, the force-dependent translocation velocity v of ssDNA in the DNA transistor can be theoretically calculated. From MD simulation, the translocation velocities v of ssDNA inside the DNA transistor and in 0.1 M and 1 M NaCl electrolytes can be obtained from motion of ssDNA, which is shown in Fig. 2 (and see also Fig. S1). Fig. 8 shows that theoretical predictions (without any fitting parameters) agree well with simulation data.
Figure 8.
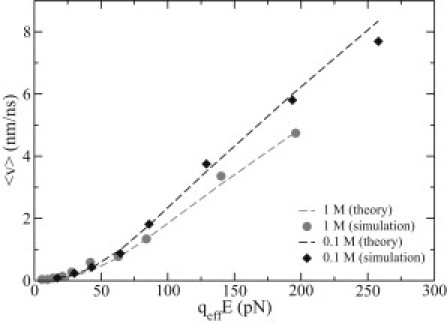
Dependence of the translocation velocity of DNA in the DNA transistor on the effective electric driving force. Ion concentrations are 0.1 M (shaded) and 1 M (black). (Dashed lines) Results from the theoretical calculation, without fitting parameters.
Having a Fokker-Planck parameterization of the dynamics of ssDNA is useful to estimate trapping times that depend on the trapping energy ET and the diffusivity D of ssDNA. This is particularly useful when the trapping time is approximately a microsecond or longer, as it is difficult to extend MD simulations to such timescales.
The current J is a balance between a forward current JF going in the direction of the external force f0, and a backward JB current going in the opposite direction, i.e.,
| (14) |
Assuming that the forward and backward currents decrease exponentially with the heights of the forward and backward energy barriers, respectively, we find that
| (15) |
and noticing that the average time of trapping before a jump forward or backward are, respectively, and , we obtain
| (16) |
| (17) |
Fig. 9 shows the dependence of residence times on the different parameters of the system. Fig. 9 a shows the forward and backward residence times as a function of the effective electric driving force qeffE. When E = 0, the forward and backward residence times are same. As the biasing electric field E increases, the forward residence time becomes much smaller than the backward one, and the dynamics is dominated by motion in the forward direction. Fig. 9 b shows the dependence of the residence time as a function of the trapping energy at zero biasing force (i.e., τF = τB). When increasing the trapping energy from 4.8 kBT to 10 kBT by raising the voltage on the middle electrode in the DNA transistor, the residence time increases to the timescale of a few microseconds. Decreasing the diffusivity of ssDNA can also increase the residence time of ssDNA, as shown in Fig. 9 c. If decreasing the diffusivity by two orders of magnitude (e.g., adding glycerol molecules to the solution) and raising the trapping energy to 10 kBT, the residence time can approach the millisecond range. A trapping (or residence) time of ssDNA of approximately milliseconds may be necessary if we want to increase the signal/noise ratio in the electric measurement of a single DNA base (10,18,19).
Figure 9.
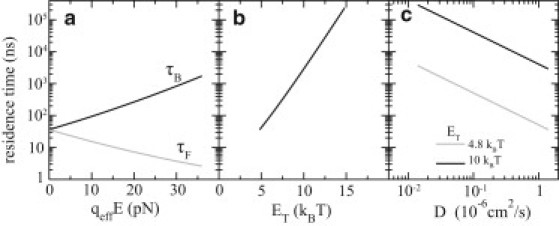
Dependencies of the residence time of ssDNA in the DNA transistor on the effective electric driving force (a), on the trapping energy (b), and on the diffusivity of ssDNA (c). In panels a and b, ssDNA is assumed to be solvated in 0.1 M NaCl electrolyte and the diffusion coefficient of ssDNA is 1.4 × 106 cm2/s. In panels b and c, the biasing electric fields are off.
Conclusion
Using both theory and simulation, we have investigated the hydrodynamics and electrokinetics of ssDNA translocation through a solid-state nanochannel, when ssDNA is in a stretched conformation that is ideal for sensing each base in ssDNA. The friction coefficient of ssDNA in an electrolyte increases with the ion concentration, whereas the diffusion coefficient of ssDNA decreases with increasing ion concentrations. The electrophoretic mobility of ssDNA was obtained in simulation of ssDNA electrophoresis in the nanochannel and is comparable to experimentally measured ones. Under simultaneous actions of a mechanic and an electric driving force on ssDNA, we found the effective charges of ssDNA in 0.1 and 1 M NaCl electrolytes. In a 1 M NaCl electrolyte, the effective charge of ssDNA is approximately three times less than that of ssDNA in a 0.1 M electrolyte. These physical parameters for ssDNA confined in a nanopore are useful for understanding and analyzing experimental results.
Typically, when the electric driving force is bigger than the trapping force on ssDNA, ssDNA moves forward nearly at a constant velocity. When the biasing electric field is weak enough, ssDNA can stay in a potential well for some time and thereafter is thermally activated to jump into the next potential well, i.e., the ratcheting process. This imposes a criterion for the biasing electric field to achieve the ratcheting motion of ssDNA in experiment. In a very weak biasing electric field, energy barriers for forward and backward motion are similar. Thus, it is possible for ssDNA to ratchet backward. Increasing the biasing electric field can prevent ssDNA from ratcheting backward but also reduce the energy barrier for the forward motion. To have a reasonable amount of trapping time for ssDNA in a trapped state, the friction coefficient of ssDNA should be increased. Thus, it is desirable to use a viscous solution (such as glycerol (20)). We plan to study the translocation of ssDNA in the DNA transistor filled with a glycerol electrolyte in the near future.
Our simplifying simulation assumptions did not take into account effects such as an ionic flux from a reservoir and ionic screening of metal electrodes. The trapping and ratcheting of ssDNA in a 1 M electrolyte might be difficult to achieve experimentally. Therefore, experiments should be carried out in an electrolyte with a low (e.g., 10 mM) ionic strength.
To achieve single-base resolution in ssDNA sequencing, a well-controlled motion of ssDNA in a solid-state nanopore is required. To our best knowledge, this fine translocation control has not yet been demonstrated in experiment. The ratcheting motion of ssDNA observed in our simulations could be an important component to achieve high-resolution ssDNA sequencing. We have carried out extensive experimental work ((61–63) and H. Peng, unpublished) toward the implementation of the DNA transistor. We hope that the DNA transistor would provide a platform not only for future nanopore-based DNA sensing technology but also for other single molecule nanotechnology applications.
Acknowledgments
Discussions with the members in the IBM DNA transistor team are gratefully acknowledged. We profited from the suggestions of three anonymous reviewers.
This work is supported by a grant from the National Institutes of Health (No. R01-HG05110-01).
Contributor Information
Binquan Luan, Email: bluan@us.ibm.com.
Gustavo Stolovitzky, Email: gustavo@us.ibm.com.
Supporting Material
References
- 1.Mitra R.D., Shendure J., Church G.M. Fluorescent in situ sequencing on polymerase colonies. Anal. Biochem. 2003;320:55–65. doi: 10.1016/s0003-2697(03)00291-4. [DOI] [PubMed] [Google Scholar]
- 2.Ronaghi M. Pyrosequencing sheds light on DNA sequencing. Genome Res. 2001;11:3–11. doi: 10.1101/gr.11.1.3. [DOI] [PubMed] [Google Scholar]
- 3.Branton D., Deamer D.W., Schloss J.A. The potential and challenges of nanopore sequencing. Nat. Biotechnol. 2008;26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kasianowicz J.J., Robertson J.W., Stanford V.M. Nanoscopic porous sensors. Annu. Rev. Anal. Chem. 2008;1:737–766. doi: 10.1146/annurev.anchem.1.031207.112818. [DOI] [PubMed] [Google Scholar]
- 5.Aksimentiev A. Deciphering ionic current signatures of DNA transport through a nanopore. Nanoscale. 2010;2:468–483. doi: 10.1039/b9nr00275h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kasianowicz J.J., Brandin E., Deamer D.W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. USA. 1996;93:13770–13773. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Purnell R.F., Schmidt J.J. Discrimination of single base substitutions in a DNA strand immobilized in a biological nanopore. ACS Nano. 2009;3:2533–2538. doi: 10.1021/nn900441x. [DOI] [PubMed] [Google Scholar]
- 8.Wong C.T., Muthukumar M. Polymer translocation through α-hemolysin pore with tunable polymer-pore electrostatic interaction. J. Chem. Phys. 2010;133:045101. doi: 10.1063/1.3464333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noskov S.Y., Im W., Roux B. Ion permeation through the α-hemolysin channel: theoretical studies based on Brownian dynamics and Poisson-Nernst-Plank electrodiffusion theory. Biophys. J. 2004;87:2299–2309. doi: 10.1529/biophysj.104.044008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke J., Wu H.C., Bayley H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009;4:265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- 11.Derrington I.M., Butler T.Z., Gundlach J.H. Nanopore DNA sequencing with MspA. Proc. Natl. Acad. Sci. USA. 2010;107:16060–16065. doi: 10.1073/pnas.1001831107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reiner J.E., Kasianowicz J.J., Robertson J.W. Theory for polymer analysis using nanopore-based single-molecule mass spectrometry. Proc. Natl. Acad. Sci. USA. 2010;107:12080–12085. doi: 10.1073/pnas.1002194107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J., Gershow M., Golovchenko J.A. DNA molecules and configurations in a solid-state nanopore microscope. Nat. Mater. 2003;2:611–615. doi: 10.1038/nmat965. [DOI] [PubMed] [Google Scholar]
- 14.Dekker C. Solid-state nanopores. Nat. Nanotechnol. 2007;2:209–215. doi: 10.1038/nnano.2007.27. [DOI] [PubMed] [Google Scholar]
- 15.Aksimentiev A., Heng J.B., Schulten K. Microscopic kinetics of DNA translocation through synthetic nanopores. Biophys. J. 2004;87:2086–2097. doi: 10.1529/biophysj.104.042960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim M., Wanunu M., Meller A. Rapid fabrication of uniformly sized nanopores and nanopore arrays for parallel DNA analysis. Adv. Mater. 2006;18:3149–3153. [Google Scholar]
- 17.Zwolak M., Di Ventra M. Electronic signature of DNA nucleotides via transverse transport. Nano Lett. 2005;5:421–424. doi: 10.1021/nl048289w. [DOI] [PubMed] [Google Scholar]
- 18.Huang S., He J., Lindsay S. Identifying single bases in a DNA oligomer with electron tunneling. Nat. Nanotechnol. 2010;5:868–873. doi: 10.1038/nnano.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsutsui M., Taniguchi M., Kawai T. Identifying single nucleotides by tunneling current. Nat. Nanotechnol. 2010;5:286–290. doi: 10.1038/nnano.2010.42. [DOI] [PubMed] [Google Scholar]
- 20.Fologea D., Uplinger J., Li J. Slowing DNA translocation in a solid-state nanopore. Nano Lett. 2005;5:1734–1737. doi: 10.1021/nl051063o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meller A., Nivon L., Branton D. Rapid nanopore discrimination between single polynucleotide molecules. Proc. Natl. Acad. Sci. USA. 2000;97:1079–1084. doi: 10.1073/pnas.97.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luan B., Aksimentiev A. Control and reversal of the electrophoretic force on DNA in a charged nanopore. J. Phys. Condens. Matter. 2010;22:454123. doi: 10.1088/0953-8984/22/45/454123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luan B.Q., Aksimentiev A. Electro-osmotic screening of the DNA charge in a nanopore. Phys. Rev. E. 2008;78:021912. doi: 10.1103/PhysRevE.78.021912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosal S. Electrokinetic-flow-induced viscous drag on a tethered DNA inside a nanopore. Phys. Rev. E. 2007;76:061916. doi: 10.1103/PhysRevE.76.061916. [DOI] [PubMed] [Google Scholar]
- 25.van Dorp S., Keyser U., Lemay S. Origin of the electrophoretic force on DNA in solid-state nanopores. Nat. Phys. 2009;5:347–351. [Google Scholar]
- 26.Sigalov G., Comer J., Aksimentiev A. Detection of DNA sequences using an alternating electric field in a nanopore capacitor. Nano Lett. 2008;8:56–63. doi: 10.1021/nl071890k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mirsaidov U., Comer J., Timp G. Slowing the translocation of double-stranded DNA using a nanopore smaller than the double helix. Nanotechnology. 2010;21:395501. doi: 10.1088/0957-4484/21/39/395501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng H., Ling X.S. Reverse DNA translocation through a solid-state nanopore by magnetic tweezers. Nanotechnology. 2009;20:185101–185108. doi: 10.1088/0957-4484/20/18/185101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keyser U., Koeleman B., Dekker C. Direct force measurements on DNA in a solid-state nanopore. Nat. Phys. 2006;2:473–477. [Google Scholar]
- 30.Trepagnier E.H., Radenovic A., Liphardt J. Controlling DNA capture and propagation through artificial nanopores. Nano Lett. 2007;7:2824–2830. doi: 10.1021/nl0714334. [DOI] [PubMed] [Google Scholar]
- 31.Polonsky S., Rossnagel S., Stolovitzky G. Nanopore in metal-dielectric sandwich for DNA position control. Appl. Phys. Lett. 2007;91:153103. [Google Scholar]
- 32.Luan B., Aksimentiev A. Electric and electrophoretic inversion of the DNA charge in multivalent electrolytes. Soft Matter. 2010;6:243–246. doi: 10.1039/b917973a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fologea D., Gershow M., Li J. Detecting single stranded DNA with a solid state nanopore. Nano Lett. 2005;5:1905–1909. doi: 10.1021/nl051199m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kowalczyk S.W., Tuijtel M.W., Dekker C. Unraveling single-stranded DNA in a solid-state nanopore. Nano Lett. 2010;10:1414–1420. doi: 10.1021/nl100271c. [DOI] [PubMed] [Google Scholar]
- 35.Lubensky D.K., Nelson D.R. Driven polymer translocation through a narrow pore. Biophys. J. 1999;77:1824–1838. doi: 10.1016/S0006-3495(99)77027-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Beest B.W., Kramer G.J., van Santen R.A. Force fields for silicas and aluminophosphates based on ab initio calculations. Phys. Rev. Lett. 1990;64:1955–1958. doi: 10.1103/PhysRevLett.64.1955. [DOI] [PubMed] [Google Scholar]
- 37.Heng J.B., Aksimentiev A., Timp G. Stretching DNA using the electric field in a synthetic nanopore. Nano Lett. 2005;5:1883–1888. doi: 10.1021/nl0510816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schroeder C.M., Babcock H.P., Chu S. Observation of polymer conformation hysteresis in extensional flow. Science. 2003;301:1515–1519. doi: 10.1126/science.1086070. [DOI] [PubMed] [Google Scholar]
- 39.Henrickson S.E., DiMarzio E.A., Kasianowicz J.J. Probing single nanometer-scale pores with polymeric molecular rulers. J. Chem. Phys. 2010;132:135101. doi: 10.1063/1.3328875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong C.T.A., Muthukumar M. Polymer capture by electro-osmotic flow of oppositely charged nanopores. J. Chem. Phys. 2007;126:164903. doi: 10.1063/1.2723088. [DOI] [PubMed] [Google Scholar]
- 41.Luan B., Afzali A., Martyna G. Tribological effects on DNA translocation in a nanochannel coated with a self-assembled monolayer. J. Phys. Chem. B. 2010;114:17172–17176. doi: 10.1021/jp108865q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wells D.B., Abramkina V., Aksimentiev A. Exploring transmembrane transport through α-hemolysin with grid-steered molecular dynamics. J. Chem. Phys. 2007;127:125101. doi: 10.1063/1.2770738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luan B., Peng H., Martyna G. Base-by-base ratcheting of single stranded DNA through a solid-state nanopore. Phys. Rev. Lett. 2010;104:238103. doi: 10.1103/PhysRevLett.104.238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reference deleted in proof.
- 45.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pérez A., Marchán I., Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 48.Beglov D., Roux B. Finite representation of an infinite bulk system: solvent boundary potential for computer simulations. J. Chem. Phys. 1994;100:9050–9063. [Google Scholar]
- 49.Cruz-Chu E.R., Aksimentiev A., Schulten K. Water-silica force field for simulating nanodevices. J. Phys. Chem. B. 2006;110:21497–21508. doi: 10.1021/jp063896o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brünger A.T. The Howard Hughes Medical Institute and Department of Molecular Biophysics and Biochemistry, Yale University; New Haven, CT: 1992. X-PLOR, Version 3.1: A System for X-Ray Crystallography and NMR. [Google Scholar]
- 51.Batcho P.F., Case D.A., Schlick T. Optimized particle-mesh Ewald/multiple-time step integration for molecular dynamics simulations. J. Chem. Phys. 2001;115:4003–4018. [Google Scholar]
- 52.Stellwagen E., Stellwagen N.C. Determining the electrophoretic mobility and translational diffusion coefficients of DNA molecules in free solution. Electrophoresis. 2002;23:2794–2803. doi: 10.1002/1522-2683(200208)23:16<2794::AID-ELPS2794>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 53.Isralewitz B., Gao M., Schulten K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001;11:224–230. doi: 10.1016/s0959-440x(00)00194-9. [DOI] [PubMed] [Google Scholar]
- 54.Meller A., Nivon L., Branton D. Voltage-driven DNA translocations through a nanopore. Phys. Rev. Lett. 2001;86:3435–3438. doi: 10.1103/PhysRevLett.86.3435. [DOI] [PubMed] [Google Scholar]
- 55.Manning G.S. Limiting laws and counterion condensation in polyelectrolyte solutions. I. Colligative properties. J. Chem. Phys. 1969;51:924. [Google Scholar]
- 56.Henrickson S.E., Misakian M., Kasianowicz J.J. Driven DNA transport into an asymmetric nanometer-scale pore. Phys. Rev. Lett. 2000;85:3057–3060. doi: 10.1103/PhysRevLett.85.3057. [DOI] [PubMed] [Google Scholar]
- 57.Ambjörnsson T., Apell S.P., Kasianowicz J.J. Charged polymer membrane translocation. J. Chem. Phys. 2002;117:4063–4073. [Google Scholar]
- 58.Ghosal S. Effect of salt concentration on the electrophoretic speed of a polyelectrolyte through a nanopore. Phys. Rev. Lett. 2007;98:238104. doi: 10.1103/PhysRevLett.98.238104. [DOI] [PubMed] [Google Scholar]
- 59.Jarzynski C. Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 1997;78:2690–2693. [Google Scholar]
- 60.Park S., Schulten K. Calculating potentials of mean force from steered molecular dynamics simulations. J. Chem. Phys. 2004;120:5946–5961. doi: 10.1063/1.1651473. [DOI] [PubMed] [Google Scholar]
- 61.Harrer S., Ahmed S., Stolovitzky G.A. Electrochemical characterization of thin film electrodes toward developing a DNA transistor. Langmuir. 2010;26:19191–19198. doi: 10.1021/la102671g. [DOI] [PubMed] [Google Scholar]
- 62.Harrer S., Waggoner P.S., Stolovitzky G.A. Electrochemical protection of thin film electrodes in solid state nanopores. Nanotechnology. 2011;22:275–304. doi: 10.1088/0957-4484/22/27/275304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Waggoner P., Kuan A., Rossnagel S. Increasing the speed of solid-state nanopores. J. Vac. Sci. Technol. B, Microelectron. Nanometer Struct. 2011;29 032206–032206. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.