

Abstract
DNA mismatch recognition and repair is vital for preserving the fidelity of the genome. Conserved across prokaryotes and eukaryotes, MutS is the primary protein that is responsible for recognizing a variety of DNA mismatches. From molecular dynamics simulations of the Escherichia coli MutS-DNA complex, we describe significant conformational dynamics in the DNA surrounding a G·T mismatch that involves weakening of the basepair hydrogen bonding in the basepair adjacent to the mismatch and, in one simulation, complete base opening via the major groove. The energetics of base flipping was further examined with Hamiltonian replica exchange free energy calculations revealing a stable flipped-out state with an initial barrier of ∼2 kcal/mol. Furthermore, we observe changes in the local DNA structure as well as in the MutS structure that appear to be correlated with base flipping. Our results suggest a role of base flipping as part of the repair initiation mechanism most likely leading to sliding-clamp formation.
Introduction
The integrity of the genome is safeguarded from replication errors by an evolutionarily conserved DNA mismatch repair (MMR) pathway. MMR in Escherichia coli begins with the mismatch recognition protein, MutS, scanning the DNA for base-base mismatches and small insertion/deletion loops (1). Upon mismatch recognition, MutL binds to MutS followed by further downstream repair events to ultimately restore the parental genotype (2–9). Defects in the MMR pathway lead to replication and recombination errors and have been linked to hereditary nonpolyposis colorectal cancer in humans (10) and are likely to play a role in other types of cancer as well (11).
Crystal structures of prokaryotic MutS and one of its human homologs, MSH2-MSH6, bound to various DNA mismatches, have provided mechanistic insight into the mismatch recognition process (12–21). Heteroduplex DNA bound to MutS (Fig. 1 A) is bent by ∼45–60° toward the major groove at the site of the mismatch. Mismatch specific contacts are made by a conserved F36-X-E38 motif (Fig. 1 B). The F36, first identified in cross-linking studies (22), forms an aromatic ring stack on the 3′ side of the mismatched base. Mutation of F36 abolishes mismatch binding in vitro and is associated with defective MMR in vivo (23–25).
Figure 1.
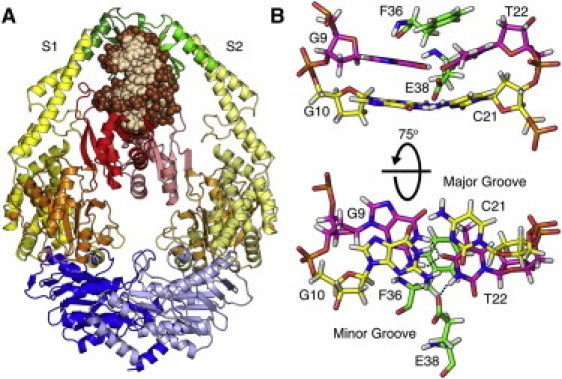
X-ray crystal structure of E. coli MutS (12). (A) MutS is colored with respect to its DNA binding domains (red/pink), connector domains (orange or pale orange), core domains (yellow or pale yellow), clamp domains (green or pale green), and ATPase domains (blue or pale blue). DNA (beige) bases and (brown) backbone. Bound nucleotides are omitted for clarity. (B) A conserved Phe36-Xaa-Glu38 motif interacts with the G·T mismatch through the DNA minor groove. (Green) Protein; (pink) the mismatch; and (yellow) G/C(−1) basepair with the 5′ adjacent base C21. (Black dotted lines) Bifurcated basepair hydrogen bond in the G·T mismatch and hydrogen bonding between Glu38 and T22.
The intrusion of a Phe residue into the duplex stack resembles intercalating residues commonly found in other DNA repair systems such as DNA glycosylases, T4 endonuclease V, and DNA demethylases, all of which involve a base-flipping mechanism (26,27). A similar base-flipping mechanism has also been proposed for MutS (19,28–31) but direct evidence has been lacking to date.
A recent FRET study has indicated that the MutS-DNA complex may involve transient intermediate states and exhibit more dynamics than suggested by the crystal structures (32). More detailed insight into the dynamics of the MutS-DNA complex during mismatch recognition is difficult to obtain with experiments but can be gained from computer simulations. Previous computational studies of MutS and homologs include normal mode analysis (33) and limited molecular dynamics (MD) simulations (34–36). Here, we present results from submicrosecond MD simulations of the MutS-DNA complex to focus on the details of the postmismatch recognition process. In particular, we describe the observation of spontaneous base flipping of the base adjacent to the mismatch site when bound to MutS. Quantitative aspects of the base-opening transition were additionally analyzed with the Hamiltonian replica exchange method (HREM) (37). Our results suggest that flipping of the base adjacent to the mismatch is energetically likely in the MutS-DNA complex. Furthermore, it appears that base flipping may be coupled to conformational changes in the protein, suggesting a mechanistic role during repair initiation by MutS.
Materials and Methods
Simulated systems and molecular dynamics protocol
Molecular dynamics (MD) simulations of E. coli MutS in complex with DNA containing a G·T mismatch were carried out with explicit solvent. The starting conformation of the MutS-DNA complex was taken from the crystal structure 1W7A (18). Missing residues 660–667 in the S1 (mismatch binding) monomer were completed using the loop modeling (38) function in MODELER, version 9 (39). Visual comparison of the model with a recent crystal structure of MutS where the disordered loop was resolved (20) showed no appreciable differences. Missing residues in the homodimeric S2 subunit were modeled after the S1 chain. Histidine ionization states were predicted using PROPKA3.1 (40) and confirmed visually based on the local protein environment. Nine simulations were carried out with all possible combinations of bound ATP, bound ADP, or no nucleotide at either the S1 or S2 ATPase domain. Positioning of the nucleotides was based on resolved nucleotides in the 1E3M (12) and 1W7A (18) crystal structures. The “X:X” notation is used here to denote which nucleotides are bound to the S1 and S2 subunits, respectively (e.g., ATP:ADP means that ATP is bound to S1 and ADP is bound to S2 while NONE:NONE is free of nucleotides). In addition to the wild-type system, simulations of an S1-F36A mutant with four different nucleotide combinations (ADP:NONE, ADP:ADP, ADP:ATP, NONE:NONE) were also carried out (see Supporting Material for more details).
Each structure was solvated using the TIP3P water model (41) and electrically neutralized with sodium ions. The total dimension of each system was ∼155 Å × 117 Å × 94 Å and contained >165,000 atoms. The particle-mesh Ewald method (42) was employed to account for electrostatic interactions. The direct electrostatic sum and Lennard-Jones interactions were truncated at 10 Å with a switching function becoming effective at 8.5 Å and a nonbonded list cutoff at 12.5 Å. The all-atom CHARMM27/CMAP force field was used for all calculations (43–45) and chosen because it has been extensively validated in many other simulations of protein-nucleic acid simulations (46–48) including simulations describing base flipping (49). Each system was independently minimized and equilibrated followed by over 200 ns of production simulation (see in the Supporting Material).
Hamiltonian replica exchange simulations and free energy calculations
To investigate the energetics of base flipping, umbrella sampling simulations were carried out. A harmonic biasing potential was applied to enhance base flipping and to obtain sufficient statistical sampling for estimating the free energy profile associated with base flipping. The reaction coordinate used for the biasing potential is a pseudodihedral angle introduced earlier (50). The pseudodihedral is based on the following four heavy atom sites: 1, center of mass of the G9, T22, C11, and G20 bases (flanking the base of interest, i.e., the cytosine base next to the mismatch site, C21); 2), T22 phosphate; 3), C21 phosphate; and 4), center of mass of the C21 base (see Fig. S4 A). Although other reaction coordinates have been utilized in the past to study base flipping (49,51–53), this pseudodihedral angle definition provides an improvement over previous methods (50) and has been shown to produce results that are in good agreement with experiment (54). The biasing potential was applied using the miscellaneous mean-field potential module (55) of CHARMM and has the form
| (1) |
where ki is the force constant set to 100 kcal/mol/rad2, θ is the pseudodihedral angle, and θi is the target value for the ith window. A total range of 0–162.5° was covered in 2.5° increments to result in 66 windows. Instead of conventional umbrella sampling, we used HREM (37) with 66 replicas corresponding to the umbrella windows to enhance sampling efficiency further. These simulations involved the entire E. coli MutS-DNA complex in explicit solvent. They were carried out using CHARMM (56) in conjunction with the MMTSB Tool Set (57). Starting structures for different replicas were taken from one of the unbiased simulations where base flipping was observed spontaneously. Each starting structure was initially subjected to 200 ps of equilibration with the biasing potential of a given replica. Each replica was then simulated for 10.5 ns (for a total simulation time of 693 ns for all 66 replicas). Exchanges between neighboring replicas were attempted every 1 ps. A quantity of 23–37% of the exchanges was successful.
Analysis
Most of the analysis was carried out with the MMTSB Tool Set and CHARMM, version c35a1, based on the 200-ns production time for the unbiased simulations. Protein root mean-square deviation (RMSD) values were calculated using Cα atoms. The DNA RMSD was calculated using all heavy atoms omitting the ultimate and penultimate bases. One-dimensional potentials of mean force (PMFs) were generated from the replica exchange simulation using the weighted histogram analysis method (58) after discarding the first 5 ns as equilibration. Two-dimensional PMFs along additional degrees of freedom were estimated from the HREM simulations (also with the first 5 ns removed as equilibration) using the standard weighted histogram analysis method under the assumption that all other degrees of freedom orthogonal to the pseudodihedral angle are thoroughly sampled (59). All structural figures were generated using PyMOL (60).
Results and Discussion
A series of nine 200-ns MD simulations of MutS in complex with a G·T mismatch containing DNA were analyzed with a primary focus on MutS-DNA interactions and the dynamics of mismatch DNA when bound to MutS. The simulations differed in the nucleotide(s) bound in the ATPase sites because the simulations were initially set up to study the effect of different nucleotides on the MutS structure. During the course of the simulations reported here, we did not see significant structural perturbations that could be correlated with the type of nucleotide bound to the ATPase domain. In fact, we found that the MutS-DNA complex sampled similar conformations in all nine simulations. The Cα RMSDs were all within 3–4 Å relative to the x-ray structure (see Fig. S1 A). Furthermore, clustering analysis shows that structures from all simulations fall into closely related conformations, with overlapping sampling of conformations belonging to the four largest clusters (see Fig. S1 B). This suggests that different nucleotides bound in the ATPase domain do not dramatically affect the overall MutS structure on the submicrosecond timescales covered here. Consequently, the simulations discussed here are treated as nine independent simulations of essentially the same system, providing a total of 1.8 μs of sampling of the MutS-DNA complex.
Dynamics of DNA and base flipping in the MutS-DNA complex
Overall, the DNA bound to MutS maintained its bent structure in all simulations as indicated by a heavy-atom RMSD of 1–4 Å (see Fig. S1 A). However, a more detailed analysis of basepair hydrogen bonding revealed significant base dynamics near the mismatch. More specifically, the G/C(−1) basepair adjacent to the mismatch site on the 5′-side of the thymine of the G·T basepair lost Watson-Crick hydrogen bonding in most of the simulations (Fig. 2 A). The X/Y(±N) notation is used here to denote the X/Y basepair relative to the thymine of the G·T mismatch (see Table S1 in the Supporting Material). The G·T mismatch remained stable in all but one of the simulations. In that simulation (NONE:ADP), a new N3-O6 hydrogen bond was formed within the same basepair due to shearing of the G·T basepair. The next-neighbor A/T(+1) and C/G(−2) basepairs stably maintained standard hydrogen bonding in all simulations (Fig. 2 A).
Figure 2.
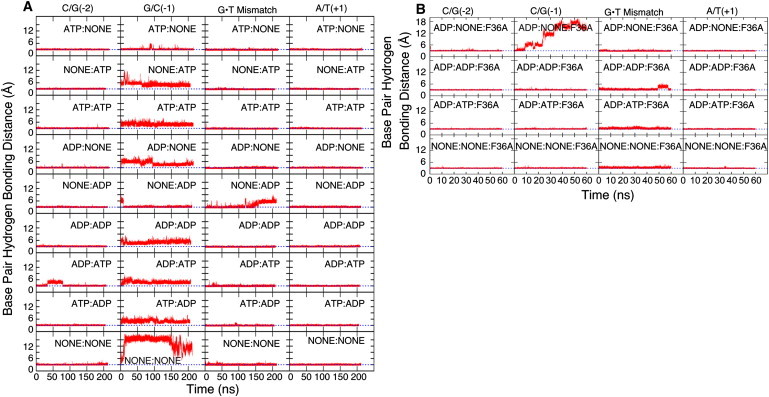
DNA basepair hydrogen bonding for C/G(−2), G/C(−1), G·T mismatch, and A/T(+1) basepairs from N3-N1 (C/G basepairs), N1-N3 (A/T basepairs), and N1-O4 (G·T mismatch) distance time series in each simulation are described here. (Blue dotted lines) Typical hydrogen-bond distances of 3 Å. (A) Wild-type simulations with different nucleotide combinations. (B) F36A mutant simulations.
The instability of the G/C(−1) basepair was unexpected and involved the loss of N1–N3 hydrogen bonding and at least partial opening of the C21 base into the major groove. In one of the simulations (NONE:NONE) all of the G/C(−1) hydrogen bonds were lost within the first 10 ns and the base subsequently flipped out into the major groove, where it remained for the rest of the simulation. This observation appears to be in conflict with previous nuclear magnetic resonance and molecular dynamics studies where significant instability of G·T pairs over canonical basepairs has been established (61,62). However, these studies were not conducted in the presence of MutS and therefore do not account for the severe bend in the DNA caused by interactions with MutS (12,13,28). The bending leads to significant distortions of the grooves near the mismatch site. In particular, the major groove width is reduced to only 13 Å at the G·T mismatch but increased to 18 Å at the G/C(−1) basepair (see Fig. S2) compared to the major groove width of canonical B-DNA at ∼17 Å (63). The narrow major groove at the G·T pair effectively prevents base opening whereas the wider major groove at the G/C(−1) basepair is more favorable for base opening.
To test a possible role of F36 in stabilizing mismatch basepairing and promoting G/C(−1) base flipping, we ran four additional 60-ns simulations of a S1-F36A mutant. We find that mismatch basepairing is stably maintained without F36 (see Fig. 2 B), although the T22 base reorients with different glycosyl rotation angles (see Fig. S3, C and D). Interestingly, we again observed spontaneous base opening of the G/C(−1) basepair in one of the simulations (ADP:NONE:F36A) in a very similar manner as in the NONE:NONE simulation (see Fig. 2 B). These results suggest that F36 does not play a significant role in either stabilizing mismatch basepairing or promoting base opening of the C/G(−1) basepair.
Progression of the base-flipping process was quantified with the help of a pseudodihedral angle, θ (see Materials and Methods), with negative values as the base opens into the major groove (see Fig. 3 A). Fig. 3 K shows snapshots of key time points during the base-opening process. Initially, G/C(−1) was perfectly basepaired (θ ≈ 0). The base then rapidly lost basepair hydrogen bonds and stacking interactions to reach a semiopen state (θ = −40°) that was stable for a few nanoseconds. Further opening led to another intermediate state that was stabilized by hydrogen-bonding interactions to the DNA backbone (θ = −81°). This state also persisted for a few nanoseconds. Eventually, the C21 base opened entirely at ∼10 ns from the beginning of the production phase of the simulation. The base was briefly fully exposed to the solvent environment (θ = −130°) but then began to interact with the DNA backbone of the opposing strand (θ = −120°). This conformation persisted from t ≈ 20 ns to t ≈ 120 ns. During the remainder of the simulation, C21 moved back toward various semiopen states but without re-forming a fully stacked configuration. C21 base flipping was associated with a change in the C21 backbone ζ-torsion angle from ∼−150° to 150° (see Figs. 3 B and 4 B, and see Fig. S3 A) as generally expected for DNA base flipping (64). Otherwise, the DNA structure remained largely unaffected by the opening of the C21 base on the timescale of our simulations.
Figure 3.
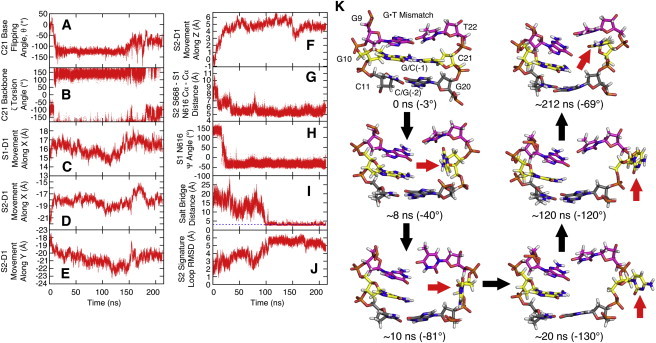
Correlation of C21 base flipping in NONE:NONE simulation with various structural quantities (see Materials and Methods for definitions): (A) Pseudodihedral angle. (B) C21 backbone ζ-torsion angle. (C) Movement of the S1 DNA binding domain (S1-D1) along X. (D) Movement of S2-D1 along X. (E) Movement of S2-D1 along Y. (F) Movement of S2-D1 along Z. (G) S2 Ser668 to S1 Asn616 Cα-Cα distance. (H) S1 Asn616 Ψ backbone torsion angle. (I) Salt bridge distance between S2 Arg667 and S1 Glu594 measured between heavy atoms. (Blue line) A distance of 3 Å corresponding to hydrogen bonding. (J) Cα-RMSD of the S2 signature loop. (K) Snapshots of base-flipping progress viewed from the major groove. Protein, water, and additional DNA are omitted for clarity. (Pink) G·T. (Yellow) G/C(−1). (Gray) C/G(−2). (Red arrow) C21.
Figure 4.
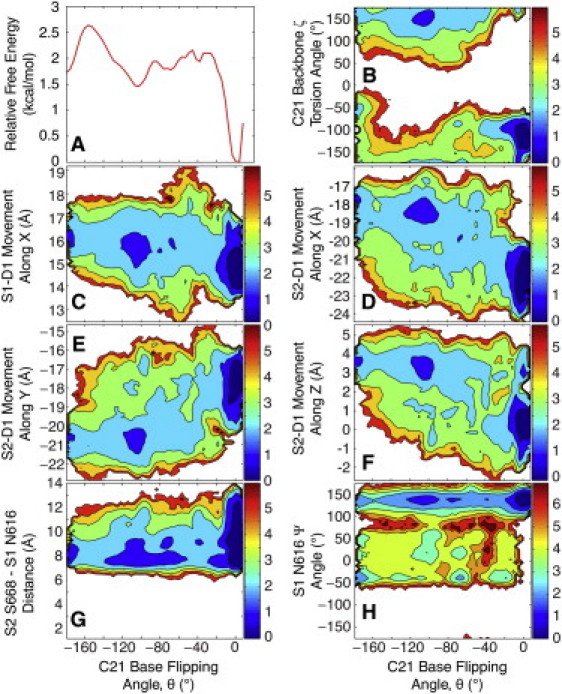
Free energy profiles from the HREM simulation: (A) Free energy of base flipping (10.5 ns/replica). (B) C21 backbone ζ-torsion angle versus base flipping. (C) Movement of S1-D1 along X versus base flipping. (D) Movement of S2-D1 along X versus base flipping. (E) Movement of S2-D1 along Y versus base flipping. (F) Movement of S2-D1 along Z versus base flipping. (G) S2 Ser668 to S1 Arg616 Cα-Cα distance versus base flipping. (H) S1 Arg616 Ψ backbone torsion angle versus base flipping.
Our observation of spontaneous base flipping in DNA complexed to MutS provides new molecular-level evidence for the previously proposed idea that base flipping may play a role in mismatch recognition (19,28–31). To gain more quantitative insight we also carried out an HREM simulation of the NONE:NONE MutS-DNA complex where sampling along the base-flipping reaction coordinate was enhanced with a total 10.5 ns of simulation time for each replica. The main result is a PMF free energy profile along the base-flipping reaction coordinate (see Fig. 4 A). The PMF has a prominent minimum near 0° for the fully basepaired state and a second minimum at ∼−105° corresponding to the flipped-out state. The two states are estimated to be separated by a 2 kcal/mol energy barrier. To examine the convergence of the PMF we compared it to PMFs with shorter simulation lengths (7.5 ns/replica and 9 ns/replica) and found negligible change between the 9.5 ns/replica and 10.5 ns/replica PMFs (see Fig. S4 C). Based on the variation of the PMF over time, we roughly estimate the uncertainty to be between 0.1 and 0.5 kcal/mol. Thus, the HREM simulation confirms the existence of a favorable, flipped-out state. Based on the PMF we calculate that the G/C(−1) basepair is intact (θ ≥ −20°) for 69% of the time, but the C21 is flipped-out to varying degrees during the remaining 31% with an estimated uncertainty of 5–10% based on the uncertainty of the PMF.
The observed 2 kcal/mol barrier suggests conformational transitions on nanosecond timescales. This is in apparent contradiction with the rarity of full base-opening/-closing events in the unbiased simulations. In the replica exchange simulations, complete base-opening/-closing was also never observed for any individual replica, although significant sampling overlap from many replicas at each pseudodihedral value (see Fig. S4 B) suggests that the PMF presented in Fig. 4 A is realistic. This suggests the presence of significantly higher kinetic barriers in orthogonal degrees of freedom not captured by the projection onto the C21 pseudodihedral angle. One source for such barriers is likely the torsional dynamics of the ζ-backbone dihedral with a barrier height estimated to be >5 kcal/mol in previous simulations of base opening (65). Another source for slow base-opening/-closing kinetics appears to be the presence of long-lived water molecules at the constrained protein-DNA interface (see Fig. S5, A and B). An analysis of residence times of water molecules located within 4 Å of the G10 base from the NONE:NONE simulation found that waters located on the major groove side and within the cavity left by the flipped out C21 base had residence times up to 500 ps (compared to ∼50 ps of surface-bound waters, see Fig. S5, C and D), whereas waters that managed to enter the cramped minor groove side essentially become trapped near the N2, N3, and N9 atoms of the G10 base with even longer residence times in the nanosecond range (see Fig. S5, E and F). The presence of these long-lived water molecules likely hinders base closing, which cannot be accomplished unless these waters are displaced. This would explain why C21 never fully restacked in the NONE:NONE simulation despite the ζ-torsion reverting to the −150° range near the end of the simulation.
Our results suggest that base opening may occur on submicrosecond timescales because it was observed spontaneously in two of our simulations. Most likely, base-opening kinetics is dominated by the kinetic barrier for ζ-backbone dihedral transitions. Base closing, on the other hand, appears to involve much longer timescales due to obstruction by long-lived water molecules. This would imply that the flipped-out state may be kinetically stabilized for a long time despite being thermodynamically slightly less favorable than the fully stacked state according to our analysis.
Opening of the 5′ adjacent base next to the mismatch is in agreement with experiment
Direct structural evidence for DNA base flipping in the MutS-DNA complex is lacking, but there is indirect experimental evidence for at least partial opening of the 5′ adjacent base next to the mismatch: before the discovery of the MutS structure, chemical footprinting was used to uncover the interactions between Thermus aquaticus MutS and the DNA minor groove (66). MutS-bound DNA with a G·T mismatch was found to be protected on the 3′ side of the lesion but not on the 5′ side of the mismatched thymine where the −4, −2, and −1 positions were hyperreactive to oxidative attack. This was attributed to widening of the minor groove when the crystal structure became available (13). To further understand this data, we analyzed the effect of MutS on solvent-accessibility of H1′ (the hydrogen attached to the C1′ attack site located in the minor groove) from our simulations. We found that without base flipping (in ATP:NONE), access to the −4 base is fully maintained, access to the −2 base is partially hindered, but the −1 base is largely occluded (see Fig. S5 G). Base flipping (in NONE:NONE), on the other hand, fully exposes the −1 base so that all three bases become vulnerable to oxidative attack as indicated by experiment.
In a more recent study, 2-aminopurine, a fluorescent adenine analog often used to probe DNA base flipping, was incorporated into various positions next to a G·T mismatch (67). It was found that the mean fluorescence lifetime increased when the mismatch was bound by MutS. Furthermore, the level of increase in the observed mean fluorescence lifetime was significantly higher when the probe was placed on the 5′ side of the mismatch as compared to other positions. This increase was attributed to an increased amplitude of the longest lifetime component, which could be explained by an increased fraction of extrahelical states. Interestingly, the relative population with the increased fluorescence lifetime, calculated by summing up the fractional amplitude of the two longest lifetimes, was estimated to be ∼31% (67), the same percentage as the fraction of flipped-out conformations in our HREM simulation. Although we believe that a qualitative comparison with the experiment is meaningful, the surprisingly good quantitative agreement is likely fortuitous because of uncertainties in both the experimental and computational results as well as differences between experiment and simulation. In the experimental results, each reported lifetime results from numerous conformers with comparable quenching rates. Furthermore, the experimental study describes opening of an A/T (or 2-aminopurine/T) basepair that is known to have different base-opening rates than G/C basepairs (62), as in the MutS-DNA complex studied here.
Changes in MutS as a result of base flipping
Experiments suggest that initial mismatch recognition is followed by three changes in MutS: First, biochemical data indicate altered activity of the ATPase domain as a result of mismatch recognition where ADP is exchanged for ATP and hydrolysis is stalled (21,68–71). Second, the affinity for MutS-MutL complex formation is increased. Third, a transition to a sliding clamp formation has been suggested to allow MutS to leave the mismatch site after initial recognition so that DNA repair can take place (7,8,71,72). Thus, if base flipping plays a role in postmismatch recognition, sliding-clamp formation, and/or initiation of repair, one would expect that there are correlated changes in the MutS structure in the DNA binding domains, the core and connector domains where MutL is proposed to bind (73), or in the ATPase domains. In our simulations we identified changes in the DNA binding domains of both chains and local rearrangements in the ATPase domains, but no significant changes in the core and connector domains of MutS correlated with base flipping.
DNA binding domain motions were characterized by a local coordinate system: X corresponds to motion along the DNA helical axis, Y to motion perpendicular to the helical axis toward the tips of the clamp domains, and Z to motion across the S1-S2 dimer interface (see Fig. S6 A). The S1 DNA binding domain (S1-D1), which interacts specifically with the DNA mismatch, showed a significant shift by, on average, 1–1.5 Å along X in the simulation where the base is flipped out (Fig. 3 C) compared to all of the other simulations where the base did not flip out (see Fig. S6, C and E). Motion of S1 along Y and Z was not correlated with base flipping (see Fig. S7, A and B). Due to the bent shape of the DNA, this motion effectively moves the domain away from the DNA (see Fig. S6 B). The S2 DNA binding domain shows a significant shift along Z, laterally away from the DNA toward the S1 core and a moderate shift along X and Y (see Fig. S6, F–H). As a result of the motion of the S2 domain, MutS-DNA interactions are also reduced (see Fig. S6 B) and these motions appear to be closely coupled to base flipping (Fig. 3, D–F). Additional analysis based on data from the HREM simulations confirms a strong correlation between base flipping and the motion of S2 along X, Y, and Z (see Fig. 4, D–F) and to a lesser extent for S1 along X (see Fig. 4 C) but not along Y or Z (see Fig. S7, C and D). An apparent correlation between motion of the DNA binding domain and base flipping, points at a possible connection with sliding-clamp formation that is assumed to involve reduced MutS-DNA interactions. Recently, it was demonstrated that MutS undergoes an ATP-induced conformational change that involves interactions between the DNA binding domain and the connector domain (74). These observations may be related to the domain motions described by our simulations.
Functional coupling between mismatch recognition and ATPase activity requires allosteric signaling over 90 Å from the DNA binding domains to the ATPase domains (see Fig. 1 A). Based on the MutS structure, it appears that such communication would involve the core and connector domains that provide the structural connection. In fact, there is a string of highly conserved residues from the DNA-binding to the ATPase domains (see Fig. S8 A). In our simulations we did not observe motions along this pathway that could be uniquely attributed to base flipping, but we did identify changes in the ATPase domain itself in the vicinity of the S1 nucleotide binding pocket that appear to be correlated to base flipping.
Ser668, a conserved residue in MutS homologs, was previously implicated in ATP hydrolysis (18,20,75). Crystal structures suggest that Ser668 in the S2 monomer may move closer to the opposing S1 nucleotide binding site by ∼5 Å to take part in ATP hydrolysis (18,20) (see Fig. S8 B). Although the exact mechanism is unclear, it has been postulated that S2 Ser668, located at the end of an α-helix, could convey the positive charge generated from the helix dipole to the γ-phosphate to assist with catalysis of the hydrolysis reaction (18,20). For this structural rearrangement to occur, Asn616, situated in the P-loop (see Fig. S8 B), has to move away from the dimer interface to allow Ser668 to complete the active site (18). Significant reduction in ATPase activity after mutation of either Asn616 or Ser668 supports such a mechanism (18,75).
In our simulations, we found that the S2 Ser668-to-S1 Asn616 distance, the backbone conformation of Asn616 in the S1 monomer, and the ability to form a salt bridge between Glu594 of the S1 monomer and Arg667 of the S2 monomer, are all correlated with base flipping. Changes in the backbone of Asn616 were measured by the Ψ (N-Cα-C-N) torsion angle. Fig. 3 G shows a significant decrease in the Ser668-to-Asn616 Cα-Cα distance from ∼9 Å to 5 Å upon base flipping that may promote nucleotide hydrolysis as indicated by the biochemical data. As shown in Fig. 3 H, the Asn616 Ψ angle changes from 125° to −30° at the same time as the base flips out and, as a result, allows Ser668 to approach the S1 active site. A correlation with base flipping is confirmed from the HREM data where base opening appears to limit the Cα-Cα distance between Ser668 and Asn616 to 6–8 Å instead of 6–12 Å when the base is fully stacked (Fig. 4 G). Similarly, base flipping seems to broaden the conformational sampling of the Asn616 Ψ value to the full range from −50 to 180° whereas only extended conformations are observed for fully base-stacked DNA (see Fig. 4 H).
Repositioning of S1 Asn616 also allows the S2 Arg667 side chain to relocate and form a salt bridge with S1 Glu594 (Fig. 3 I and see Fig. S8 C). The interaction between these two residues further stabilizes the S2 signature loop in which Ser668 resides (see Fig. 3 J) and Arg667 is also positioned to hydrogen-bond with the ribose of adenosine (see Fig. S8 C). Based on these results, we speculate that base flipping may promote (or restore) the ability to hydrolyze ATP in the S1 binding site. However, there remains uncertainty about the exact mechanism of how variations in MutS-DNA interactions are communicated to the distant ATPase domain.
Known crystal structures of MutS are very similar with either ATP (18) or ADP bound in the ATPase domains (12). This suggests that they more likely represent the postmismatch recognition state where ATP hydrolysis is stalled and MutS is poised for sliding-clamp formation. The simulation results suggest that this structure may promote base flipping in DNA, which in turn seems to initiate sliding-clamp formation. The correlated changes in the ATPase domain suggest a connection to ATPase activity. The coincidence of apparent changes in the ATPase domain and DNA binding domain as a result of base flipping would be consistent with a previously suggested role of ATP hydrolysis during sliding-clamp formation (8,76). However, this idea is inconsistent with a hydrolysis-independent model for sliding-clamp formation that is supported by other studies (7,71). A recent study appears to reconcile these two models by demonstrating that two nonequivalent MutS dimers bind to the mismatched DNA and, in the presence of ATP, one MutS dimer remains bound to (or near the site of) the mismatch, and at the same time, the second dimer is free to move along the DNA while retaining its interactions with the first dimer (77). We speculate that the MutS-DNA conformation observed in our simulations may correspond to the dimer that can slide along the DNA.
Conclusions
Submicrosecond computer simulations were used to report direct structural evidence for DNA base flipping in the large MutS-DNA system. The instability in DNA basepairing was found to be specific for the 5′ adjacent basepair instead of the mismatch. This is in contrast to previous hypotheses but appears to be in good agreement with experimental data. Energetic analysis of the base-flipping process confirmed the existence of a stable flipped-out state in the presence of MutS and revealed an estimated 2–2.5 kcal/mol activation energy barrier for base flipping. Kinetic rates for base flipping were estimated to be in the microsecond range due to slow DNA backbone and water rearrangements. Further analysis of changes in the MutS structure suggests that base flipping leads to motions of the DNA-binding domains away from the DNA and more subtle changes in the ATPase domain.
Taken together, our simulations suggest that base flipping may be the key step that allows MutS to transition from the postmismatch recognition complex to the sliding-clamp formation. We hope that our results will motivate further computational and experimental studies to better understand the mechanistic details of DNA mismatch repair initiation by MutS.
Acknowledgments
We thank Dr. Shayantani Mukherjee, Dr. Katarzyna Maksimiak, Dr. Yi-Ming Cheng, Dr. Richard Venable, and Hugh Crosmun for discussions and Dr. Chresten Søndergaard for help with the determination of protein ionization states in the presence of DNA. We also acknowledge access to computational resources from the High Performance Computing Center at Michigan State University.
This work was supported by National Science Foundation MCB-0447799, National Institutes of Health GM 092949, TeraGrid TG-MCB090003, and the Center for Biological Modeling at Michigan State University (to S.M.L.).
Supporting Material
References
- 1.Su S.S., Modrich P. Escherichia coli MutS-encoded protein binds to mismatched DNA base pairs. Proc. Natl. Acad. Sci. USA. 1986;83:5057–5061. doi: 10.1073/pnas.83.14.5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lahue R.S., Au K.G., Modrich P. DNA mismatch correction in a defined system. Science. 1989;245:160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- 3.Grilley M., Griffith J., Modrich P. Bidirectional excision in methyl-directed mismatch repair. J. Biol. Chem. 1993;268:11830–11837. [PubMed] [Google Scholar]
- 4.Cooper D.L., Lahue R.S., Modrich P. Methyl-directed mismatch repair is bidirectional. J. Biol. Chem. 1993;268:11823–11829. [PubMed] [Google Scholar]
- 5.Au K.G., Welsh K., Modrich P. Initiation of methyl-directed mismatch repair. J. Biol. Chem. 1992;267:12142–12148. [PubMed] [Google Scholar]
- 6.Grilley M., Welsh K.M., Modrich P. Isolation and characterization of the Escherichia coli MutL gene product. J. Biol. Chem. 1989;264:1000–1004. [PubMed] [Google Scholar]
- 7.Gradia S., Subramanian D., Fishel R. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol. Cell. 1999;3:255–261. doi: 10.1016/s1097-2765(00)80316-0. [DOI] [PubMed] [Google Scholar]
- 8.Allen D.J., Makhov A., Griffith J.D. MutS mediates heteroduplex loop formation by a translocation mechanism. EMBO J. 1997;16:4467–4476. doi: 10.1093/emboj/16.14.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hargreaves V.V., Shell S.S., Kolodner R.D. Interaction between the Msh2 and Msh6 nucleotide-binding sites in the Saccharomyces cerevisiae Msh2-Msh6 complex. J. Biol. Chem. 2010;285:9301–9310. doi: 10.1074/jbc.M109.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolodner R.D., Hall N.R., Fishel R. Human mismatch repair genes and their association with hereditary non-polyposis colon cancer. Cold Spring Harb. Symp. Quant. Biol. 1994;59:331–338. doi: 10.1101/sqb.1994.059.01.037. [DOI] [PubMed] [Google Scholar]
- 11.Li G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 12.Lamers M.H., Perrakis A., Sixma T.K. The crystal structure of DNA mismatch repair protein MutS binding to a G × T mismatch. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 13.Obmolova G., Ban C., Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 14.Junop M.S., Obmolova G., Yang W. Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol. Cell. 2001;7:1–12. doi: 10.1016/s1097-2765(01)00149-6. [DOI] [PubMed] [Google Scholar]
- 15.Alani E., Lee J.Y., Yang W. Crystal structure and biochemical analysis of the MutS·ADP·beryllium fluoride complex suggests a conserved mechanism for ATP interactions in mismatch repair. J. Biol. Chem. 2003;278:16088–16094. doi: 10.1074/jbc.M213193200. [DOI] [PubMed] [Google Scholar]
- 16.Lamers M.H., Winterwerp H.H., Sixma T.K. The alternating ATPase domains of MutS control DNA mismatch repair. EMBO J. 2003;22:746–756. doi: 10.1093/emboj/cdg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Natrajan G., Lamers M.H., Sixma T.K. Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Res. 2003;31:4814–4821. doi: 10.1093/nar/gkg677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamers M.H., Georgijevic D., Sixma T.K. ATP increases the affinity between MutS ATPase domains. Implications for ATP hydrolysis and conformational changes. J. Biol. Chem. 2004;279:43879–43885. doi: 10.1074/jbc.M406380200. [DOI] [PubMed] [Google Scholar]
- 19.Warren J.J., Pohlhaus T.J., Beese L.S. Structure of the human MutSα DNA lesion recognition complex. Mol. Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 20.Lebbink J.H.G., Fish A., Sixma T.K. Magnesium coordination controls the molecular switch function of DNA mismatch repair protein MutS. J. Biol. Chem. 2010;285:13131–13141. doi: 10.1074/jbc.M109.066001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lebbink J.H.G., Georgijevic D., de Wind N. Dual role of MutS glutamate 38 in DNA mismatch discrimination and in the authorization of repair. EMBO J. 2006;25:409–419. doi: 10.1038/sj.emboj.7600936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malkov V.A., Biswas I., Hsieh P. Photocross-linking of the NH2-terminal region of Taq MutS protein to the major groove of a heteroduplex DNA. J. Biol. Chem. 1997;272:23811–23817. doi: 10.1074/jbc.272.38.23811. [DOI] [PubMed] [Google Scholar]
- 23.Bowers J., Sokolsky T., Alani E. A mutation in the MSH6 subunit of the Saccharomyces cerevisiae MSH2-MSH6 complex disrupts mismatch recognition. J. Biol. Chem. 1999;274:16115–16125. doi: 10.1074/jbc.274.23.16115. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto A., Schofield M.J., Hsieh P. Requirement for Phe36 for DNA binding and mismatch repair by Escherichia coli MutS protein. Nucleic Acids Res. 2000;28:3564–3569. doi: 10.1093/nar/28.18.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drotschmann K., Yang W., Kunkel T.A. Asymmetric recognition of DNA local distortion. Structure-based functional studies of eukaryotic Msh2-Msh6. J. Biol. Chem. 2001;276:46225–46229. doi: 10.1074/jbc.C100450200. [DOI] [PubMed] [Google Scholar]
- 26.Yang W. Structure and mechanism for DNA lesion recognition. Cell Res. 2008;18:184–197. doi: 10.1038/cr.2007.116. [DOI] [PubMed] [Google Scholar]
- 27.Yang C.G., Yi C., He C. Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature. 2008;452:961–965. doi: 10.1038/nature06889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H., Yang Y., Erie D.A. DNA bending and unbending by MutS govern mismatch recognition and specificity. Proc. Natl. Acad. Sci. USA. 2003;100:14822–14827. doi: 10.1073/pnas.2433654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kunkel T.A., Erie D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 30.Holmes S.F., Scarpinato K.D., Kunkel T.A. Specialized mismatch repair function of Glu339 in the Phe-X-Glu motif of yeast Msh6. DNA Repair (Amst.) 2007;6:293–303. doi: 10.1016/j.dnarep.2006.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tessmer I., Yang Y., Erie D.A. Mechanism of MutS searching for DNA mismatches and signaling repair. J. Biol. Chem. 2008;283:36646–36654. doi: 10.1074/jbc.M805712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sass L.E., Lanyi C., Erie D.A. Single-molecule FRET TACKLE reveals highly dynamic mismatched DNA-MutS complexes. Biochemistry. 2010;49:3174–3190. doi: 10.1021/bi901871u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukherjee S., Law S.M., Feig M. Deciphering the mismatch recognition cycle in MutS and MSH2-MSH6 using normal-mode analysis. Biophys. J. 2009;96:1707–1720. doi: 10.1016/j.bpj.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salsbury F.R., Jr. Effects of cisplatin binding to DNA on the dynamics of the E. coli MutS dimer. Protein Pept. Lett. 2010;17:744–750. doi: 10.2174/092986610791190318. [DOI] [PubMed] [Google Scholar]
- 35.Salsbury F.R., Jr., Clodfelter J.E., Scarpinato K.D. The molecular mechanism of DNA damage recognition by MutS homologs and its consequences for cell death response. Nucleic Acids Res. 2006;34:2173–2185. doi: 10.1093/nar/gkl238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mukherjee S., Feig M. Conformational change in MSH2-MSH6 upon binding DNA coupled to ATPase activity. Biophys. J. 2009;96:L63–L65. doi: 10.1016/j.bpj.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fukunishi H., Watanabe O., Takada S. On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: application to protein structure prediction. J. Chem. Phys. 2002;116:9058–9067. [Google Scholar]
- 38.Fiser A., Feig M., Sali A. Evolution and physics in comparative protein structure modeling. Acc. Chem. Res. 2002;35:413–421. doi: 10.1021/ar010061h. [DOI] [PubMed] [Google Scholar]
- 39.Sali A., Blundell T.L. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 40.Bas D.C., Rogers D.M., Jensen J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 41.Jorgensen W.L. Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers—application to liquid water. J. Am. Chem. Soc. 1981;103:335–340. [Google Scholar]
- 42.Darden T., York D., Pedersen L. Particle mesh Ewald—an Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 43.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 44.MacKerell A.D., Jr., Feig M., Brooks C.L., 3rd Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 2004;126:698–699. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- 45.Foloppe N., MacKerell A.D. All-atom empirical force field for nucleic acids. I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000;21:86–104. [Google Scholar]
- 46.Golosov A.A., Karplus M. Analysis of the translocation step in DNA replication by DNA Polymerase I with computer simulations. Biophys. J. 2007:225a. doi: 10.1016/j.str.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woo H.J., Liu Y., Sousa R. Molecular dynamics studies of the energetics of translocation in model T7 RNA polymerase elongation complexes. Proteins. 2008;73:1021–1036. doi: 10.1002/prot.22134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villa E., Balaeff A., Schulten K. Structural dynamics of the Lac repressor-DNA complex revealed by a multiscale simulation. Proc. Natl. Acad. Sci. USA. 2005;102:6783–6788. doi: 10.1073/pnas.0409387102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang N., Banavali N.K., MacKerell A.D., Jr. Protein-facilitated base flipping in DNA by cytosine-5-methyltransferase. Proc. Natl. Acad. Sci. USA. 2003;100:68–73. doi: 10.1073/pnas.0135427100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song K., Cambell A.J., Simmerling C. An improved reaction coordinate for nucleic acid base flipping studies. J. Chem. Theory Comput. 2009;5:3105–3113. doi: 10.1021/ct9001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banavali N.K., MacKerell A.D., Jr. Free energy and structural pathways of base flipping in a DNA GCGC containing sequence. J. Mol. Biol. 2002;319:141–160. doi: 10.1016/S0022-2836(02)00194-8. [DOI] [PubMed] [Google Scholar]
- 52.Várnai P., Lavery R. Base flipping in DNA: pathways and energetics studied with molecular dynamic simulations. J. Am. Chem. Soc. 2002;124:7272–7273. doi: 10.1021/ja025980x. [DOI] [PubMed] [Google Scholar]
- 53.Hagan M.F., Dinner A.R., Chakraborty A.K. Atomistic understanding of kinetic pathways for single base-pair binding and unbinding in DNA. Proc. Natl. Acad. Sci. USA. 2003;100:13922–13927. doi: 10.1073/pnas.2036378100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nikolova E.N., Kim E., Al-Hashimi H.M. Transient Hoogsteen base pairs in canonical duplex DNA. Nature. 2011;470:498–502. doi: 10.1038/nature09775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beglov D., Roux B. An integral equation to describe the solvation of polar molecules in liquid water. J. Phys. Chem. B. 1997;101:7821–7826. [Google Scholar]
- 56.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feig M., Karanicolas J., Brooks C.L., 3rd MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. Model. 2004;22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 58.Kumar S., Bouzida D., Rosenberg J.M. The weighted histogram analysis method for free-energy calculations of biomolecules. I. The method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 59.Banavali N.K., Roux B. Free energy landscape of A-DNA to B-DNA conversion in aqueous solution. J. Am. Chem. Soc. 2005;127:6866–6876. doi: 10.1021/ja050482k. [DOI] [PubMed] [Google Scholar]
- 60.Schrödinger L. Schrödinger, LLC, New York; New York: 2010. The PyMOL Molecular Graphics System, Version 1.3. [Google Scholar]
- 61.Várnai P., Canalia M., Leroy J.L. Opening mechanism of G·T/U pairs in DNA and RNA duplexes: a combined study of imino proton exchange and molecular dynamics simulation. J. Am. Chem. Soc. 2004;126:14659–14667. doi: 10.1021/ja0470721. [DOI] [PubMed] [Google Scholar]
- 62.Moe J.G., Russu I.M. Kinetics and energetics of base-pair opening in 5′-d(CGCGAATTCGCG)-3′ and a substituted dodecamer containing G·T mismatches. Biochemistry. 1992;31:8421–8428. doi: 10.1021/bi00151a005. [DOI] [PubMed] [Google Scholar]
- 63.Drew H.R., Wing R.M., Dickerson R.E. Structure of a B-DNA dodecamer—conformation and dynamics. 1. Proc. Natl. Acad. Sci. USA. 1981;78:2179–2183. doi: 10.1073/pnas.78.4.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Y.Z., Mohan V., Griffey R.H. Effect of backbone ζ-torsion angle on low energy single base opening in B-DNA crystal structures. Chem. Phys. Lett. 1998;287:570–574. [Google Scholar]
- 65.Feig M., Zacharias M., Pettitt B.M. Conformations of an adenine bulge in a DNA octamer and its influence on DNA structure from molecular dynamics simulations. Biophys. J. 2001;81:352–370. doi: 10.1016/S0006-3495(01)75705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Biswas I., Hsieh P. Interaction of MutS protein with the major and minor grooves of a heteroduplex DNA. J. Biol. Chem. 1997;272:13355–13364. doi: 10.1074/jbc.272.20.13355. [DOI] [PubMed] [Google Scholar]
- 67.Nag N., Rao B.J., Krishnamoorthy G. Altered dynamics of DNA bases adjacent to a mismatch: a cue for mismatch recognition by MutS. J. Mol. Biol. 2007;374:39–53. doi: 10.1016/j.jmb.2007.08.065. [DOI] [PubMed] [Google Scholar]
- 68.Bjornson K.P., Allen D.J., Modrich P. Modulation of MutS ATP hydrolysis by DNA cofactors. Biochemistry. 2000;39:3176–3183. doi: 10.1021/bi992286u. [DOI] [PubMed] [Google Scholar]
- 69.Acharya S., Foster P.L., Fishel R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol. Cell. 2003;12:233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- 70.Antony E., Hingorani M.M. Mismatch recognition-coupled stabilization of Msh2-Msh6 in an ATP-bound state at the initiation of DNA repair. Biochemistry. 2003;42:7682–7693. doi: 10.1021/bi034602h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mazur D.J., Mendillo M.L., Kolodner R.D. Inhibition of Msh6 ATPase activity by mispaired DNA induces a Msh2(ATP)-Msh6(ATP) state capable of hydrolysis-independent movement along DNA. Mol. Cell. 2006;22:39–49. doi: 10.1016/j.molcel.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 72.Pluciennik A., Modrich P. Protein roadblocks and helix discontinuities are barriers to the initiation of mismatch repair. Proc. Natl. Acad. Sci. USA. 2007;104:12709–12713. doi: 10.1073/pnas.0705129104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mendillo M.L., Hargreaves V.V., Kolodner R.D. A conserved MutS homolog connector domain interface interacts with MutL homologs. Proc. Natl. Acad. Sci. USA. 2009;106:22223–22228. doi: 10.1073/pnas.0912250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Winkler I., Marx A.D., Friedhoff P. Chemical trapping of the dynamic MutS-MutL complex formed in DNA mismatch repair in Escherichia coli. J. Biol. Chem. 2011;286:17326–17337. doi: 10.1074/jbc.M110.187641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Acharya S., Patterson K. Mutations in the conserved glycine and serine of the MutS ABC signature motif affect nucleotide exchange, kinetics of sliding clamp release of mismatch and mismatch repair. Mutat. Res. 2010;684:56–65. doi: 10.1016/j.mrfmmm.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 76.Blackwell L.J., Bjornson K.P., Modrich P. Distinct MutS DNA-binding modes that are differentially modulated by ATP binding and hydrolysis. J. Biol. Chem. 2001;276:34339–34347. doi: 10.1074/jbc.M104256200. [DOI] [PubMed] [Google Scholar]
- 77.Jiang Y., Marszalek P.E. Atomic force microscopy captures MutS tetramers initiating DNA mismatch repair. EMBO J. 2011;30:2881–2893. doi: 10.1038/emboj.2011.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.