

Background: Cold-adapted enzymes remain catalytically active at low temperatures.
Results: Mutants of a cold-adapted α-amylase stabilized by engineered weak interactions and a disulfide bond have lost the kinetic optimization to low temperatures.
Conclusion: The disappearance of stabilizing interactions in psychrophilic enzymes increases the dynamics of active site residues at low temperature, leading to a higher activity.
Significance: An experimental support to the activity-stability relationships.
Keywords: Enzyme Mutation, Glycosidases, Microbiology, Mutagenesis Site-specific, Protein Engineering, Protein Folding, Protein Stability, Cold-active Enzyme, Extremophiles, Psychrophiles
Abstract
The mutants Mut5 and Mut5CC from a psychrophilic α-amylase bear representative stabilizing interactions found in the heat-stable porcine pancreatic α-amylase but lacking in the cold-active enzyme from an Antarctic bacterium. From an evolutionary perspective, these mutants can be regarded as structural intermediates between the psychrophilic and the mesophilic enzymes. We found that these engineered interactions improve all the investigated parameters related to protein stability as follows: compactness; kinetically driven stability; thermodynamic stability; resistance toward chemical denaturation, and the kinetics of unfolding/refolding. Concomitantly to this improved stability, both mutants have lost the kinetic optimization to low temperature activity displayed by the parent psychrophilic enzyme. These results provide strong experimental support to the hypothesis assuming that the disappearance of stabilizing interactions in psychrophilic enzymes increases the amplitude of concerted motions required by catalysis and the dynamics of active site residues at low temperature, leading to a higher activity.
Introduction
One frequently overlooks that over 80% of the Earth's biosphere is permanently cold (1) but also that these cold biotopes have been successfully colonized by diverse organisms. Psychrophiles are such cold-adapted organisms thriving permanently at temperatures close to 0 °C and are found, for instance, in Antarctica, the Arctic regions, deep sea water, and sediments, or in the permafrost. They include a large range of representatives from all three domains (Bacteria, Archaea, and Eukarya) and are the most abundant extremophiles in terms of biomass, diversity, and distribution (2, 3). The observation of metabolically active bacteria at −20 °C in the brine veins between sea ice crystals illustrates the remarkable adaptations of psychrophiles (4).
Among the numerous adverse effects of low temperature on the cell (1, 5, 6), the major constraint is exerted on enzyme activity, which is exponentially reduced by temperature decreases. In psychrophiles, this constraint is alleviated by the synthesis of cold-active enzymes to maintain metabolic fluxes compatible with life. The prevailing hypothesis assumes that cold-adapted enzymes have acquired a high catalytic activity at low temperature by improving their conformational flexibility at the expense of stability (7–10). It has been shown that the crystal structure of psychrophilic enzymes is characterized by the disappearance of various noncovalent stabilizing interactions, resulting in both an improved dynamics of the enzyme conformation and in a weak stability (11–17). There is indeed a clear decrease in the number and strength of all known weak interactions and structural factors involved in protein stability, from thermophiles, mesophiles to psychrophiles (17–19).
The heat-labile α-amylase (AHA)3 from the Antarctic bacterium Pseudoalteromonas haloplanktis (20) is the best characterized psychrophilic enzyme. It displays striking sequence and structure similarities with its mesophilic homologue from pig pancreas (PPA) (11, 21–23). For instance, all 24 residues forming the catalytic cleft (supplemental Fig. S1) and involved in substrate binding are strictly conserved in both the psychrophilic and the mesophilic homologues (11, 24). This outstanding example of active site identity demonstrates that cold activity is reached without any amino acid substitution in the reaction center. As a consequence, changes occurring elsewhere in the molecule are responsible for the optimization of the catalytic parameters at low temperature, by adjustments of the active site residue dynamics. To probe the activity-flexibility-stability hypothesis, numerous mutants of AHA have been constructed, each bearing additional weak interactions mediated by the replacement of a specific residue, as well as a disulfide bond as found in PPA. It has been shown that single amino acid side chain substitutions significantly modify the stability parameters, the cooperativity and reversibility of unfolding, the thermal inactivation rate constant, and the kinetic parameters kcat and Km (25–27). Here, we report the activity and mainly the stability properties of two stabilized multiple mutants (Fig. 1 and supplemental Fig. S1) of the heat-labile α-amylase AHA as follows: Mut5, bearing five mutations and Mut5CC, also bearing an extra disulfide bond specific to α-amylases from warm-blooded animals (28). The five mutations shared by both multiple mutants have been selected on the basis of the DSC thermograms of single mutants (25) that showed a global stabilization of the protein (higher melting point Tm and calorimetric enthalpy ΔHcal), without evidence for local destabilization (identical or higher temperature of transition start). The lack of interferences between individual mutations, modeled in the crystal structure of AHA, was also checked. These multiple mutants provide clear insights into the structure-function relationships allowing psychrophilic enzymes to fulfill their biochemical functions at low temperatures.
FIGURE 1.

Mutations engineered in the psychrophilic α-amylase AHA (blue) based on the structure of the mesophilic homologue PPA (red). The mutation N150D introduces a salt bridge with the corresponding Lys side chain. V196F restores a triple face-to-edge aromatic interaction. K300R provides a bidentate coordination of the chloride ion via short H-bonds as demonstrated by the crystal structure of the single mutant (43). T232V and Q164I increase the apolarity of hydrophobic core clusters (only AHA side chains are labeled for clarity), and the double mutation Q58C/A99C creates a disulfide bond. Protein Data Bank coordinate for AHA is 1AQH and for PPA is 1PPI.
EXPERIMENTAL PROCEDURES
Mutagenesis and Protein Purification
The multiple mutants Mut5 and Mut5CC were constructed by combining restriction fragments of the single mutants and by reverse PCR as described previously (25). The recombinant AHA, Mut5, and Mut5CC were expressed in Escherichia coli at 18 °C and purified by DEAE-agarose, Sephadex G-100, and Ultrogel AcA54 column chromatography, as described previously (25). The following parameters were used for calculation: AHA (49,343.1 Da), Mut5 (49,403.2 Da), and Mut5CC (49,410.3 Da) on 453 aa.
Enzyme Assays
α-Amylase activity was recorded using 3.5 mm 4-nitrophenyl-α-d-maltoheptaoside-4,6-O-ethylidene as substrate and by the dinitrosalicylic acid method using 1% soluble starch as substrate (25). Microcalorimetric determination of activity toward various polysaccharides and malto-oligosaccharides was performed using an isothermal titration calorimeter as described previously (29).
Differential Scanning Calorimetry
Measurements were performed using a MicroCal VP-DSC instrument at a scan rate of 60 K h−1 and under ∼25 p.s.i. positive cell pressure. Samples (∼2 mg/ml) were dialyzed overnight against 30 mm MOPS, 50 mm NaCl, 1 mm CaCl2, pH 7.2, and, when required, were brought to 1 m 3-(1-pyridinio)-1-propanesulfonate (i.e. a nondetergent sulfobetaine) as detailed previously (30). Thermograms of enzyme-acarbose complexes were recorded in the presence of 1 mm acarbose (Bayer). Thermograms were analyzed according to a non-two-state model in which the melting point Tm, the calorimetric enthalpy ΔHcal, and the van't Hoff enthalpy ΔHeff of individual transitions are fitted independently using the MicroCal Origin software (version 7). The magnitude and source of the errors in the Tm and enthalpy values have been discussed elsewhere (31).
Kinetically driven unfolding was recorded without nondetergent sulfobetaine addition, and the rate constant ku,i was calculated from the relation (32) shown in Equation 1,
where v is the scan rate (K s−1); Cp is the excess heat capacity at a temperature T; Δcal is the total heat of the process, and Δ(T) is the heat evolved at a given temperature T.
Unfolding Recorded by Intrinsic Fluorescence
Heat-induced unfolding was recorded using an SML-AMINCO model 8100 spectrofluorometer (Spectronic Instruments) at an excitation wavelength of 280 nm and at an emission wavelength of 350 nm (22). GdmCl-induced unfolding was monitored at 20 °C after overnight incubation of the samples at this temperature in 30 mm MOPS, 50 mm NaCl, 1 mm CaCl2, pH 7.2, on an LS50B spectrofluorometer (PerkinElmer Life Sciences) (22). The equilibrium condition was ascertained by recording unfolding as a function of time. Least squares analysis of ΔG0 values as a function of GdmCl concentrations allowed estimating the conformational stability in the absence of denaturant, ΔGH2O0, according to Equation 2,
Dynamic Quenching of Fluorescence
The acrylamide-dependent quenching of intrinsic protein fluorescence was monitored as described previously (22). The Stern-Volmer quenching constants KSV were calculated according to Equation 3,
where F and F0 are the fluorescence intensity in the presence and absence of molar concentration of the quencher Q, respectively (33).
Kinetics of Unfolding and Refolding
All kinetic experiments were performed using a Bio-Logic (Claix, France) SFM-3 stopped-flow, coupled with a MOS-200 spectrophotometer and a MPS-51 power supply. Temperature was maintained at 15 °C by a Julabo F30-C thermostated bath. Fluorescence measurements were performed using a 1.5-mm path length cell (FC-15), and the dead time of the apparatus was found to be ∼10 ms under all experimental settings. This value was estimated by monitoring the fluorescence of the reduction of dichlorophenolindophenol by ascorbic acid, as described by the manufacturer. All experiments were performed in 30 mm MOPS, 50 mm NaCl, 1 mm CaCl2, pH 7.2, using a protein final concentration of 0.1 mg ml−1 (∼2 μm). To initiate refolding reactions, unfolded α-amylase (1 mg ml−1) in 3 m GdmCl was diluted 10-fold with aqueous buffer or with GdmCl solutions of varying concentrations to give the desired final concentrations of GdmCl. Conversely, unfolding reactions were initiated by an 10-fold dilution of native α-amylase with the same buffer containing various amounts of GdmCl, to yield final concentrations ranging from 0.3 to 0.9, 0.3 to 1.1, and 0.3 to 1.25 m, for AHA, Mut5, and Mut5CC, respectively. Unfolding and refolding kinetics were followed by intrinsic fluorescence with an excitation wavelength of 280 nm, and total emission above 320 nm was monitored using a high pass filter. For each experiment, 7000 data points were sampled over the entire time course.
Kinetic traces resulting from the accumulation of five identical experiments were analyzed according to the sum of two exponential terms (Equation 4) in the case of refolding and to a simple exponential term (Equation 5) in the case of unfolding.
where yt is the intensity of fluorescence at time t; y∞ is the signal value for an infinite time; A is the amplitude of the signal associated with rate constant k. The data sets were averaged to obtain the rate constant, and errors were calculated as standard deviations.
The dependence of unfolding and refolding rate constants on denaturant concentration was analyzed according to the following linear relationship (Equation 6) (34, 35),
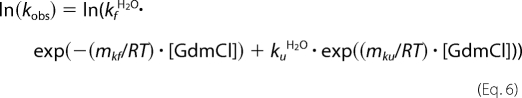 |
where kobs is the rate of unfolding or refolding measured at various GdmCl concentrations, kfH2O and kuH2Oare the values for folding and unfolding rates, respectively, in the absence of denaturant, and mkf/RT and mku/RT are proportionality constants, which describe the denaturant dependence. The program Bio-Kine 32 version 4.51 was used for nonlinear least squares analysis of the data.
RESULTS AND DISCUSSION
Activity of the Mutants Mut5 and Mut5CC
The effects of the single, double, and combined mutations on activity toward a chromogenic substrate were described previously (25–27) and are summarized in Fig. 2. The general trend of the mutations combined to create both multiple mutants is to decrease kcat and Km concomitantly. Furthermore, a mesophilic like activity was engineered in the multiple mutants Mut5 and Mut5CC as both kinetic parameters are similar to those recorded for the mesophilic homologue PPA.
FIGURE 2.

Correlation of kinetic parameters kcat and Km in the psychrophilic AHA, the mesophilic PPA (open symbols), and in single and multiple mutants of AHA. Activity toward the chromogenic substrate Et-G7-NP at 25 °C. Data are from Refs. 25–27.
The specificity of both mutants for various polysaccharides was further investigated by microcalorimetry. Table 1 reports the relative activities of the α-amylases, taking soluble starch, the natural substrate, as a reference. Interestingly, the specificity profile of Mut5 and Mut5CC closely follows that of the parent AHA, indicating that the mode of interaction with these polysaccharides has not been modified. By contrast, comparison of the absolute catalytic constants kcat shows that the activity of the mutants is similar to that of the mesophilic homologue, revealing a less efficient active site.
TABLE 1.
Relative activity and catalytic constant of Mut5 and Mut5CC on various polysaccharides recorded by microcalorimetry at 25 °C
| Substrate | Relative activity |
kcat |
||||||
|---|---|---|---|---|---|---|---|---|
| AHAa | Mut5 | Mut5CC | PPAa | AHAa | Mut5 | Mut5CC | PPAa | |
| % | s−1 | |||||||
| Starch | 100 | 100 | 100 | 100 | 663 | 352 | 314 | 327 |
| Amylopectin | 96 | 94 | 90 | 68 | 636 | 331 | 283 | 222 |
| Dextrin | 108 | 111 | 112 | 95 | 716 | 391 | 352 | 311 |
| Glycogen | 74 | 73 | 71 | 59 | 491 | 257 | 223 | 193 |
| Maltopentaose | 69 | 50 | 48 | 145 | 457 | 176 | 151 | 474 |
| Et-G7-NP | 105 | 97 | 97 | 101 | 642 | 341 | 304 | 330 |
a Data are from Ref. 29.
Fig. 3b illustrates the increased stability of both multiple mutants as probed by their heat-induced unfolding recorded by intrinsic fluorescence. Despite their higher thermal stability, there was no significant differences in the apparent optimal temperature for activity, which is recorded near 35 °C (Fig. 3a). However, Mut5CC displayed a marked protection against heat inactivation at the upper temperatures. This indicates that the disulfide bond bridging the domains forming the active site cleft contributes to thermal stability of activity in the mesophilic homologue PPA. It should be noted that thermal inactivation above 35 °C (Fig. 3a) is recorded before any detectable structural change (Fig. 3b). As a result, the active site remains the most heat-labile structural element in both mutants.
FIGURE 3.

Temperature dependence of activity (a) and heat-induced unfolding recorded by fluorescence (b). Data are shown for the psychrophilic AHA (○), the multiple mutants Mut5 (●) and Mut5CC (■), and the mesophilic PPA (▴) (data from Refs. 22, 44). Activity toward starch as substrate. All experiments were performed at similar protein concentrations (5–40 μg/ml).
Dynamic Fluorescence Quenching
Determination of molecular flexibility requires the definition of the types and amplitudes of atomic motions as well as a time scale for these motions. In this respect, dynamic fluorescence quenching usefully averages most of these parameters into a single signal (33). The structural permeability of the enzymes was investigated by dynamic fluorescence quenching of aromatic residues (dominated by 12 tryptophan residues in AHA and its mutants) by acrylamide. The individual Stern-Volmer plots for AHA and its multiple mutants at 4 and 30 °C are provided in supplemental Fig. S2, and the difference between the slopes of the Stern-Volmer plots at 30 and 4 °C were used to illustrate the difference in structural permeability of the enzymes with an increase in temperature (Fig. 4). The lower slope for Mut5 and Mut5CC indicates a reduced accessibility of aromatic residues relative to AHA, arising from a lower permeability of the mutant structures to the small quencher molecule. This reveals a more compact conformation undergoing reduced micro-unfolding events of the native state and shorter native state fluctuations for both mutants. The flexibility of these proteins and of the mesophilic homologue also correlates with their difference in stability (i.e. AHA < Mut5 < Mut5CC < PPA).
FIGURE 4.

Variation of fluorescence quenching by acrylamide between 4 and 30 °C. Graph was constructed by subtracting the regression lines of Stern-Volmer plots at individual temperatures, as shown in supplemental Fig. S2. Data for the mesophilic PPA (22) are shown at similar temperature intervals.
Irreversible Unfolding of Stabilized Mutants
Fig. 5 illustrates the correlation between stability of the activity and unfolding reversibility in single and multiple mutants. With the noticeable exception of the disulfide-containing mutant (26), the general trend of the mutations was to protect against heat inactivation but to decrease concomitantly the unfolding reversibility (25). In this respect, both multiple mutants Mut5 and Mut5CC also display mesophilic like properties.
FIGURE 5.

Correlation of stability parameters in the psychrophilic AHA, the mesophilic PPA (open symbols), and in single and multiple mutants of AHA. kinact is the first order rate constant for activity decay at 45 °C, and unfolding reversibility was calculated from two consecutive DSC scans.
According to the pronounced unfolding irreversibility of both Mut5 and Mut5CC mutants shown in Fig. 5, the kinetically driven stability was analyzed by DSC to determine the rate constant for heat-induced irreversible unfolding, ku,i (32). Such kinetic analysis is not possible for AHA (fully reversible unfolding), and data were obtained from its mutant N12R. The latter displays the same microcalorimetric properties as AHA but only 30% denaturation reversibility (25), ensuring that unfolding is kinetically driven. The temperature dependence of ku,i is shown as an Arrhenius plot in Fig. 6, and the corresponding activation parameters are provided in Table 2.
FIGURE 6.

Arrhenius plot of the rate constant for heat-induced irreversible unfolding, ku,i. Data were derived from irreversible DSC endotherms according to Ref. 32 and Equation 1.
TABLE 2.
Thermodynamic parameters for heat-induced irreversible unfolding of AHA and its stabilized mutants at 316 K (43 °C)
| Protein | ku,i | ΔG* | ΔH* | TΔS* |
|---|---|---|---|---|
| s−1 | kcal mol−1 | kcal mol−1 | kcal mol−1 | |
| AHAN12R | 1.8·10−1 | 19.6 | 112 | 92 |
| Mut5 | 4.6·10−4 | 23.3 | 61 | 38 |
| Mut5CC | 3.1·10−5 | 25.0 | 92 | 67 |
At an identical temperature of 316 K (43 °C), the rate constants for irreversible unfolding differ by several orders of magnitude, which correspond to higher activation energy barriers ΔG* in the stabilized mutants (Table 2). Both mutants also display reduced activation enthalpy ΔH* as calculated from the slope of the plot in Fig. 6. This mainly reflects a lower temperature dependence of irreversible unfolding and therefore a larger resistance toward unfolding in a given temperature interval, as compared with the parent protein AHA. The lower activation enthalpy value of Mut5 (Table 2) arises from its broader DSC endotherm (see Fig. 7). The weaker entropic contribution in both mutants can be tentatively explained by a less disordered transition state. In other words, the stabilized mutants would resist against unfolding (before irreversible denaturation) to a larger extent as compared with AHA. As also shown in Fig. 6, PPA unfolds at higher temperatures, which preclude direct comparison. Nevertheless, it is worth noting that Mut5CC tends toward the behavior of PPA in terms of temperature intervals for irreversible unfolding and ΔH* (slope of the plot).
FIGURE 7.

Normalized reversible endotherms of AHA and of its mutants recorded by DSC in the presence of a nondetergent sulfobetaine. Thermograms were recorded in 30 mm MOPS, 50 mm NaCl, 1 mm CaCl2, pH 7.2, and 1 m 3-(1-pyridinio)-1-propanesulfonate in the case of Mut5 and Mut5CC. Raw data were base line-subtracted and normalized for protein concentration. Inset, same graph including the irreversible thermogram of PPA (44).
Thermodynamic Stability Recorded by DSC
Although heat-induced unfolding of both mutants was irreversible under standard microcalorimetric conditions, we found that addition of the nondetergent sulfobetaine 3-(1-pyridinio)-1-propanesulfonate promotes full unfolding reversibility as illustrated in the supplemental Fig. S3. This compound has been reported to protect unfolded proteins from nonspecific interactions by both charge screening and hydrophobic screening effects (30, 36, 37). Such reversibility of the heat-induced unfolding reaction allowed us to re-investigate the thermodynamic stability (reversible reaction characterized by an equilibrium constant) of Mut5 and Mut5CC, assuming that nondetergent sulfobetaine does not alter the unfolding parameters, as already demonstrated for the parent enzyme AHA (36). Fig. 7 illustrates the normalized DSC thermograms. The thermodynamic parameters of unfolding derived from reversible DSC endotherms are close to those previously obtained under nonreversible conditions (27) and are therefore reported in the supplemental material. As shown in supplemental Table 1, the improved stability of both mutants is characterized by increased melting points (Tm) and calorimetric enthalpies (ΔHcal). This indicates a significant contribution of the engineered interactions in the enthalpic stability of the protein. Furthermore, the stability curves (supplemental Fig. S4) obtained by plotting the Gibbs free energy of unfolding, i.e. the work required to disrupt the native state at any temperature (38), clearly illustrate the stability increases brought by the mutations, with, in the case of Mut5CC, values approaching those of the mesophilic homologue PPA.
Stability of α-Amylase-Acarbose Complexes
Acarbose is a pseudosaccharide inhibitor containing an N-glycosidic bond and acting as a transition state analogue (39). It has been shown that stabilization induced by acarbose binding closely follows the activation parameters of the amylolytic reaction (22) between the ground state (free enzyme) and the transition state intermediate (enzyme-acarbose complex). The thermograms of AHA and of its mutants were therefore recorded by DSC in the presence of acarbose (supplemental Fig. S5). Table 3 provides the relevant parameters, and the full data set is presented in supplemental Table S2.
TABLE 3.
Microcalorimetric parameters of thermal unfolding for α-amylases in complex with the pseudosaccharide inhibitor acarbose
The values refer to the differences in Tmax (top of the transition) and in ΔHcal (area of the transition) with respect to the free enzyme. Activation parameters of the amylolytic reaction at 15 °C are also indicated (27).
Upon acarbose binding, AHA is strongly stabilized, as indicated by the large increase of Tmax and of the calorimetric enthalpy ΔHcal. It has been argued (22) that this reflects the weak number of interactions to be broken to reach the activated state in the cold-active enzyme (low ΔH# in Table 3), which in turn implies large structural motions of the loose free enzyme upon substrate binding (high ΔS# in Table 3). It is worth mentioning that Mut5 and Mut5CC are less stabilized by acarbose and display intermediate values with PPA (Table 3). Concomitantly, their activation parameters come close to those of PPA. This is a strong indication that the stabilizing interactions engineered in the mutants directly impair the cold activity optimization of the parent psychrophilic α-amylase.
Equilibrium Unfolding in GdmCl
The resistance toward chemical denaturation using GdmCl was probed by equilibrium unfolding recorded by fluorescence (Fig. 8 and Table 4). Both mutants unfold reversibly at higher GdmCl concentrations (C½ values) with respect to AHA and in accordance with their increased thermal stability. The cooperativity of unfolding remains similar to that of AHA, as indicated by the m value. Estimation of the conformational stability in the absence of denaturant (ΔGH2O0) at 20 °C using Equation 2 provides a ratio of 1:1.5:1.8:2 for AHA, Mut5, Mut5CC, and PPA, respectively, in reasonable agreement with the values derived from the DSC stability curves (supplemental Fig. S4).
FIGURE 8.

Equilibrium unfolding in GdmCl of AHA and of its mutants as recorded by fluorescence emission. From left to right: AHA (open circles), Mut5 (closed circles), Mut5CC (closed squares), and PPA (closed triangles, data from (22)) are shown.
TABLE 4.
Equilibrium unfolding parameters in GdmCl of AHA and of its mutants
Kinetics of Unfolding and Refolding
To address the kinetic origin of the gain in stability of the mutants, the folding kinetics in GdmCl were recorded. Indeed, the equilibrium constant KN-U between the native state N and the unfolded state U (KN-U = [U]/[N]) is also expressed by the ratio kunfold/kfold of the kinetic constants for unfolding and folding, respectively. As shown in Fig. 9, kinetics of unfolding and refolding were determined between 0.3 and 2 m GdmCl. Under all conditions, unfolding was found to be a monophasic process, although refolding was a biphasic process comprising a fast phase (∼80% amplitude) and a slow phase (∼20% amplitude). In the context of our analysis, the corresponding parameters (Table 5) were restricted to the rate constants in the absence of denaturant (kfH2O and kuH2O) obtained by linear regression (dashed lines in Fig. 9), the corresponding time constants (τ = 1/k), their dependences on the denaturant concentration (mkf and mku) and, when relevant, the transition midpoint C½. Interestingly, the folding rate constants kfH2O for both phases converge toward very similar values in the three enzymes. By contrast, the unfolding rate constants kuH2O of both stabilized mutants are reduced as follows: Mut5 unfolds 3.5 times slower and Mut5CC unfolds 13.4 times slower with respect to AHA. It can be concluded that the decreased unfolding rate is the main kinetic determinant of the improved stability in both mutants.
FIGURE 9.

Chevron plots of the GdmCl concentration dependence of the rate constants for unfolding and refolding. Data for AHA (open circles), Mut5 (closed circles), and Mut5CC (closed squares) are shown. Unfolding rate constants (right arms of the plot) and refolding rate constants (left arms, slow phase) were fitted on Equation 6 (solid lines). Dashed lines are extrapolations to 0 m GdmCl allowing the determination of kfH2O and kuH2O, the folding and unfolding rate constants in the absence of denaturant. Upper data set indicates fast refolding phases.
TABLE 5.
Kinetic parameters of unfolding and refolding in GdmCl
| lnkuH2O | ln kfH2O | kuH2O | kfH2O | τu | τ f | mku | mkf | C½ | |
|---|---|---|---|---|---|---|---|---|---|
| s−1 | s−1 | s | s | kcal m−1mol−1 | kcal m−1mol−1 | m | |||
| Slow phases | |||||||||
| AHA | −7.15 | −0.62 | 7.8e-4 | 0.53 | 1282 | 1.88 | 1.15 | −3.51 | 0.81 |
| Mut5 | −8.41 | −0.58 | 2.2e-4 | 0.55 | 4545 | 1.81 | 1.67 | −2.22 | 1.14 |
| Mut5CC | −9.74 | −0.61 | 5.8e-5 | 0.54 | 17241 | 1.85 | 1.97 | −2.11 | 1.26 |
| Fast refolding phase | |||||||||
| AHA | 1.44 | 4.22 | 0.24 | −2.01 | |||||
| Mut5 | 1.31 | 3.69 | 0.27 | −2.55 | |||||
| Mut5CC | 1.26 | 4.58 | 0.22 | −1.92 | |||||
Conclusions
The mutants Mut5 and Mut5CC bear representative stabilizing interactions found in the heat-stable PPA but lacking in the psychrophilic AHA, i.e. a salt bridge (N150D), weakly polar interactions (V196F), short H-bonds (K300R), better hydrophobic effect in core clusters (T232V and Q194I), and a covalent disulfide bond (Q58C/A99C) specific to warm-blooded animals (Fig. 1). From an evolutionary perspective, these mutants can be regarded as structural intermediates between the psychrophilic and the mesophilic enzymes. We have shown here that these engineered interactions improve all the investigated parameters related to protein stability as follows: the compactness (as probed by fluorescence quenching), the kinetically driven stability (irreversible heat-induced unfolding), the thermodynamic stability (from DSC endotherms), the resistance toward chemical denaturation (equilibrium unfolding in GdmCl), and the kinetics of unfolding/refolding. Concomitantly to this improved stability, both mutants have lost the kinetic optimization to low temperature activity displayed by the parent enzyme AHA, as demonstrated by their kinetic parameters toward a model substrate (Fig. 2), by their activity toward various polysaccharides and oligosaccharides (Table 1), and by their stability in complex with the transition state analog acarbose (Table 3). These results provide strong experimental support to the prevailing hypothesis (7, 27), i.e. the disappearance of stabilizing interactions in psychrophilic enzymes increases the amplitude of concerted motions required by catalysis and the dynamics of active site residues at low temperature, leading to a higher activity but also to a lower substrate binding strength (Fig. 2). We have argued elsewhere that losing stability is apparently the easiest strategy to improve molecular dynamics in cold environments where the selective pressure for stable proteins is lacking (8, 40).
Two aspects of the present stability studies are also worth comments. (i) The role of disulfide bonds has been frequently debated in the general context of protein stability (26). In the case of Mut5CC, there is an unquestionable contribution of the disulfide to all recorded stability parameters. Furthermore, a fascinating aspect, which deserve further investigations, is the observation that the disulfide has a higher contribution to stability when added to Mut5 than to its effect on AHA alone (26), as far as Tm and ΔHcal variations are concerned. This reflects a very strong cooperativity of stabilizing interactions, rather than a simple additivity, in the maintenance of the native folded state of proteins. (ii) The kinetics of unfolding/refolding of Mut5 and Mut5CC demonstrates that the gain in stability is governed by a slow unfolding rate, although the folding rate remains unchanged (Fig. 9 and Table 5). This perfectly fits with similar experiments performed on hyperthermophilic proteins and aimed at explaining their unusual stability (41, 42). Altogether, these results suggest the occurrence of a transition state on the unfolding pathway, the free energy level of which modulates the stability of proteins adapted to extreme biological temperatures. This aspect should stimulate further investigations, using appropriate extremophilic protein models.
Supplementary Material
Acknowledgments
We thank H. Bichoff (Bayer AG, Germany) for the kind gift of acarbose. The support of the Institut Polaire Français was also appreciated.
This work was supported by the Fonds de la Recherche Scientifique-Fonds, National de la Recherche Scientifique, Belgium Fonds de la Recherche Fondamentale et Collective Contracts 2.4535.08 (to G. F.) and 2.4530.09 (to A. M.) and the Belgian Program of Interuniversity Attraction Poles initiated by the Federal Office for Scientific, Technical and Cultural Affaires P6/19.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5 and Tables S1 and S2.
- AHA
- heat-labile α-amylase from P. haloplanktis
- PPA
- α-amylase from pig pancreas
- DSC
- differential scanning calorimetry
- GdmCl
- guanidinium chloride.
REFERENCES
- 1. Rodrigues D. F., Tiedje J. M. (2008) Appl. Environ. Microbiol. 74, 1677–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerday C., Glansdorff N. (2007) Physiology and Biochemistry of Extremophiles, American Society for Microbiology, Washington, D. C [Google Scholar]
- 3. Margesin R., Schinner F., Marx J. C., Gerday C. (2008) Psychrophiles, from Biodiversity to Biotechnology, Springer-Verlag, Berlin [Google Scholar]
- 4. Deming J. W. (2002) Curr. Opin. Microbiol. 5, 301–309 [DOI] [PubMed] [Google Scholar]
- 5. D'Amico S., Collins T., Marx J. C., Feller G., Gerday C. (2006) EMBO Rep. 7, 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Casanueva A., Tuffin M., Cary C., Cowan D. A. (2010) Trends Microbiol. 18, 374–381 [DOI] [PubMed] [Google Scholar]
- 7. Fields P. A., Somero G. N. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 11476–11481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feller G., Gerday C. (2003) Nat. Rev. Microbiol. 1, 200–208 [DOI] [PubMed] [Google Scholar]
- 9. Feller G. (2010) J. Phys.: Condens Mat. 22, 323101 doi 10.1088/0953–8984/1022/1032/323101 [DOI] [PubMed] [Google Scholar]
- 10. Siddiqui K. S., Cavicchioli R. (2006) Annu. Rev. Biochem. 75, 403–433 [DOI] [PubMed] [Google Scholar]
- 11. Aghajari N., Feller G., Gerday C., Haser R. (1998) Structure 6, 1503–1516 [DOI] [PubMed] [Google Scholar]
- 12. Russell R. J., Gerike U., Danson M. J., Hough D. W., Taylor G. L. (1998) Structure 6, 351–361 [DOI] [PubMed] [Google Scholar]
- 13. Smalås A. O., Leiros H. K., Os V., Willassen N. P. (2000) Biotechnol. Annu. Rev. 6, 1–57 [DOI] [PubMed] [Google Scholar]
- 14. Bell G. S., Russell R. J., Connaris H., Hough D. W., Danson M. J., Taylor G. L. (2002) Eur. J. Biochem. 269, 6250–6260 [DOI] [PubMed] [Google Scholar]
- 15. Tehei M., Franzetti B., Madern D., Ginzburg M., Ginzburg B. Z., Giudici-Orticoni M. T., Bruschi M., Zaccai G. (2004) EMBO Rep. 5, 66–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bae E., Phillips G. N., Jr. (2004) J. Biol. Chem. 279, 28202–28208 [DOI] [PubMed] [Google Scholar]
- 17. Coquelle N., Fioravanti E., Weik M., Vellieux F., Madern D. (2007) J. Mol. Biol. 374, 547–562 [DOI] [PubMed] [Google Scholar]
- 18. Gianese G., Bossa F., Pascarella S. (2002) Proteins 47, 236–249 [DOI] [PubMed] [Google Scholar]
- 19. Tronelli D., Maugini E., Bossa F., Pascarella S. (2007) FEBS J. 274, 4595–4608 [DOI] [PubMed] [Google Scholar]
- 20. Médigue C., Krin E., Pascal G., Barbe V., Bernsel A., Bertin P. N., Cheung F., Cruveiller S., D'Amico S., Duilio A., Fang G., Feller G., Ho C., Mangenot S., Marino G., Nilsson J., Parrilli E., Rocha E. P., Rouy Z., Sekowska A., Tutino M. L., Vallenet D., von Heijne G., Danchin A. (2005) Genome Res. 15, 1325–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feller G., Payan F., Theys F., Qian M., Haser R., Gerday C. (1994) Eur. J. Biochem. 222, 441–447 [DOI] [PubMed] [Google Scholar]
- 22. D'Amico S., Marx J. C., Gerday C., Feller G. (2003) J. Biol. Chem. 278, 7891–7896 [DOI] [PubMed] [Google Scholar]
- 23. Da Lage J. L., Feller G., Janecek S. (2004) Cell. Mol. Life Sci. 61, 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aghajari N., Feller G., Gerday C., Haser R. (1998) Protein Sci. 7, 564–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. D'Amico S., Gerday C., Feller G. (2001) J. Biol. Chem. 276, 25791–25796 [DOI] [PubMed] [Google Scholar]
- 26. D'Amico S., Gerday C., Feller G. (2002) J. Biol. Chem. 277, 46110–46115 [DOI] [PubMed] [Google Scholar]
- 27. D'Amico S., Gerday C., Feller G. (2003) J. Mol. Biol. 332, 981–988 [DOI] [PubMed] [Google Scholar]
- 28. D'Amico S., Gerday C., Feller G. (2000) Gene 253, 95–105 [DOI] [PubMed] [Google Scholar]
- 29. D'Amico S., Sohier J. S., Feller G. (2006) J. Mol. Biol. 358, 1296–1304 [DOI] [PubMed] [Google Scholar]
- 30. D'Amico S., Feller G. (2009) Anal. Biochem. 385, 389–391 [DOI] [PubMed] [Google Scholar]
- 31. Matouschek A., Matthews J. M., Johnson C. M., Fersht A. R. (1994) Protein Eng. 7, 1089–1095 [DOI] [PubMed] [Google Scholar]
- 32. Sánchez-Ruiz J. M., López-Lacomba J. L., Cortijo M., Mateo P. L. (1988) Biochemistry 27, 1648–1652 [DOI] [PubMed] [Google Scholar]
- 33. Lakowicz J. (1983) Principles of Fluorescence Spectroscopy, Plenum Press, New York [Google Scholar]
- 34. Khorasanizadeh S., Peters I. D., Roder H. (1996) Nat. Struct. Biol. 3, 193–205 [DOI] [PubMed] [Google Scholar]
- 35. Fersht A. R. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, W. H. Freeman & Co., New York [Google Scholar]
- 36. Collins T., D'Amico S., Georlette D., Marx J. C., Huston A. L., Feller G. (2006) Anal. Biochem. 352, 299–301 [DOI] [PubMed] [Google Scholar]
- 37. Vuillard L., Braun-Breton C., Rabilloud T. (1995) Biochem. J. 305, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Privalov P. (1992) in Protein Folding (Creighton T. ed) pp. 83–126, W.H. Freeman & Co., New York [Google Scholar]
- 39. Qian M., Haser R., Buisson G., Duée E., Payan F. (1994) Biochemistry 33, 6284–6294 [DOI] [PubMed] [Google Scholar]
- 40. Roulling F., Piette F., Cipolla A., Struvay C., Feller G. (2011) in Extremophiles Handbook (Horikoshi K. ed) pp. 891–916, Springer Verlag, Tokyo [Google Scholar]
- 41. Luke K. A., Higgins C. L., Wittung-Stafshede P. (2007) FEBS J. 274, 4023–4033 [DOI] [PubMed] [Google Scholar]
- 42. Perl D., Welker C., Schindler T., Schröder K., Marahiel M. A., Jaenicke R., Schmid F. X. (1998) Nat. Struct. Biol. 5, 229–235 [DOI] [PubMed] [Google Scholar]
- 43. Aghajari N., Feller G., Gerday C., Haser R. (2002) Protein Sci. 11, 1435–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Feller G., d'Amico D., Gerday C. (1999) Biochemistry 38, 4613–4619 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.