

Background: GlgE is a maltosyltransferase involved in bacterial α-glucan biosynthesis and is a genetically validated anti-tuberculosis target.
Results: We have determined the catalytic properties of Streptomyces coelicolor GlgE and solved its structure.
Conclusion: The enzyme has the same catalytic properties as Mycobacterium tuberculosis GlgE and the structure reveals how GlgE functions.
Significance: The structure will help guide the development of inhibitors with therapeutic potential.
Keywords: Enzyme Inhibitors, Enzyme Kinetics, Enzyme Mechanisms, Enzyme Structure, Glycoside Hydrolases, Glycosyltransferases, Phosphorylation Enzymes, GlgE, Maltosyltranferase, Tuberculosis Target
Abstract
GlgE is a recently identified (1→4)-α-d-glucan:phosphate α-d-maltosyltransferase involved in α-glucan biosynthesis in bacteria and is a genetically validated anti-tuberculosis drug target. It is a member of the GH13_3 CAZy subfamily for which no structures were previously known. We have solved the structure of GlgE isoform I from Streptomyces coelicolor and shown that this enzyme has the same catalytic and very similar kinetic properties to GlgE from Mycobacterium tuberculosis. The S. coelicolor enzyme forms a homodimer with each subunit comprising five domains, including a core catalytic α-amylase-type domain A with a (β/α)8 fold. This domain is elaborated with domain B and two inserts that are specifically configured to define a well conserved donor pocket capable of binding maltose. Domain A, together with domain N from the neighboring subunit, forms a hydrophobic patch that is close to the maltose-binding site and capable of binding cyclodextrins. Cyclodextrins competitively inhibit the binding of maltooligosaccharides to the S. coelicolor enzyme, showing that the hydrophobic patch overlaps with the acceptor binding site. This patch is incompletely conserved in the M. tuberculosis enzyme such that cyclodextrins do not inhibit this enzyme, despite acceptor length specificity being conserved. The crystal structure reveals two further domains, C and S, the latter being a helix bundle not previously reported in GH13 members. The structure provides a framework for understanding how GlgE functions and will help guide the development of inhibitors with therapeutic potential.
Introduction
The crucial need to develop new drugs against tuberculosis (1), one of the world's most pervasive and lethal infectious diseases (2), drives much research into the causative agent Mycobacterium tuberculosis. In this context, we recently identified a new α-glucan pathway in this bacterium (Fig. 1) (3). Its defining enzyme, GlgE, is a (1→4)-α-d-glucan:phosphate α-d-maltosyltransferase and member of the glycoside hydrolase family subfamily GH13_3 (4). It is capable of transferring maltosyl units not only from maltose 1-phosphate to maltooligosaccharides but also between maltooligosaccharides. We have genetically validated GlgE to be a potential new drug target (3) that has some attractive features as discussed at length elsewhere (5). The bactericidal mechanism of the blockage of GlgE is novel because rather than preventing the formation of an essential metabolic product, it is the auto-amplified build up of GlgE's donor substrate, maltose 1-phosphate, that leads to pleiotropic effects, toxicity and cell death.
FIGURE 1.

α-Glucan pathways of actinomycetes. The classical GlgA cytosolic glycogen pathway and the newly identified GlgE pathway (highlighted in red (3)) are common to both S. coelicolor and M. tuberculosis. The Rv3032 pathway for methylglucose lipopolysaccharide biosynthesis is present in M. tuberculosis. Which pathway is responsible for capsular glucan biosynthesis in M. tuberculosis is not yet clear, and there may be redundancy between the pathways.
The GlgE pathway generates a branched α-glucan from trehalose (Fig. 1) (3). M. tuberculosis is known to produce three α-glucans as follows: cytosolic glycogen, capsular α-glucan, and methylglucose lipopolysaccharide (6). These are either involved or implicated in the storage of carbon (7), evasion of the immune system (8–11), and chaperoning/regulating fatty acid biosynthesis (12), respectively. It is not yet known how much the GlgE pathway contributes to the biosynthesis of each of the three α-glucans. Nevertheless, synthetic lethality has been observed between the GlgE and methylglucose lipopolysaccharide pathways, implying the essentiality of at least one type of α-glucan and the role of GlgE in its biosynthesis (3).
The GlgE pathway is present in many other actinomycetes. For example, it is involved in carbon management in Streptomyces coelicolor (13–15). The genes of this pathway are duplicated and separately and developmentally regulated in this organism, such that each is respectively associated with transient glycogen deposition at the initiation of aerial growth (phase I) and during the first stages of sporulation (phase II). The pathway is not restricted to actinomycetes and is remarkably widespread (6). Fourteen percent of sequenced microbial genomes contain all of the GlgE pathway genes, which are usually clustered, making the pathway half as common as the more well known glycogen pathway involving GlgA and GlgC.
Structures have not previously been reported for GlgE or any other GH13_3 subfamily member. In parallel studies of the mycobacterial and Streptomyces GlgE enzymes, we have found that S. coelicolor GlgE isoform I is particularly amenable to structural analysis. This enzyme comprises domains in common with other members of the GH13 α-amylase family of enzymes together with a helix bundle domain that is novel in this structural context. The location of the donor-binding site has been defined together with a site capable of binding cyclodextrins that overlaps with the acceptor-binding site. The structure is consistent with evidence that maltooligosaccharide acceptors are extended at their nonreducing ends. The S. coelicolor and M. tuberculosis GlgE enzymes have the same catalytic and very similar kinetic properties, with well conserved donor-binding sites. This allows the structure of the former to be used to guide inhibitor development for the latter in the search for new therapies against tuberculosis.
EXPERIMENTAL PROCEDURES
Chemical Synthesis
α- and β-maltose 1-phosphate, 1a and 1b, were synthesized from 2,3,6,2′,3′,4′,6′-hepta-O-acetyl-d-maltose (2) that was readily prepared from d-maltose using known procedures (see supplemental “Experimental Procedures for details of preparation of 2) (16). α-Maltosyl fluoride (17) was prepared via 2,3,6,2′,3′,4′,6′-hepta-O-acetyl-α-d-maltosyl fluoride using published procedures (18). TLC was performed on pre-coated silica plates (Merck 60 F254, 0.25 mm) containing a fluorescence indicator. Compounds were visualized under UV light (254 nm) and/or by heating after dipping in a solution of 5% H2SO4 in ethanol. Flash column chromatography was performed on silica gel columns (Biotage KP-SilTM Silica, 60 Å, 32–63 μm) fitted to a Biotage SP1® automated purification system (Uppsala, Sweden). High resolution MS was carried out using a Thermo Fisher Scientific (Waltham, MA) LTQ Orbitrap XL. Low resolution mass spectra were recorded with a Thermo Fisher Scientific Finnigan LCQ Deca XP Plus ion trap mass spectrometer. 1H and 13C NMR spectra were recorded at 300 K on a Bruker Avance II 600 MHz spectrometer with Bruker TCI cryoprobe (Bruker Biospin Ltd.). Water peaks were suppressed with presaturation, and data were analyzed with Topspin 2.1 software (Bruker Biospin Ltd.). Chemical shifts are reported in parts/million relative to tetramethylsilane (δH 0.0) or, for samples in D2O, residual water (δH 4.70). Full assignment of 1H and 13C spectra was achieved with the aid of COSY, DEPT, HMBC, HSQC, and HSQC-TOCSY experiments. 31P spectra were obtained at 161 MHz on a Bruker Avance III 400 MHz spectrometer with and without decoupling and were referenced with external D3PO4 (δP 0.0). J values are in Hz.
Dibenzyl 2,3,6,2′,3′,4′,6′-Hepta-O-acetyl-α-d-maltosyl Phosphate (3a) and Dibenzyl 2,3,6,2′,3′,4′,6′-Hepta-O-acetyl-β-d-maltosyl Phosphate (3b)
Lithium diisopropylamide (4.15 ml of a 2 m solution in tetrahydrofuran/heptane/ethylbenzene (Sigma), 8.3 mmol) was slowly added to a solution of anhydrous 2,3,6,2′,3′,4′,6′-hepta-O-acetyl-d-maltose (2) (3.1 g, 4.9 mmol), in the minimum volume of absolute tetrahydrofuran required to solubilize it (220 ml) at −80 °C under a N2 atmosphere. After 10 min of stirring, a solution of tetrabenzyl pyrophosphate (3.7 g, 6.9 mmol) in tetrahydrofuran (20 ml) was slowly added with cooling. The mixture was stirred with continued cooling for 40 min before being allowed to slowly warm to 4 °C followed by stirring for 16 h. An off-white precipitate of LiOP(O)(OBn)2 was removed by filtration. The resulting solution was evaporated to dryness under reduced pressure, and the product was re-dissolved in ethyl acetate (20 ml). The organic layer was washed with saturated aqueous NaHCO3 (20 ml) followed by saturated aqueous NaCl (20 ml) and dried over Na2SO4. After filtration, the solution was evaporated to dryness. The residue was purified by flash chromatography on silica gel (50–70% ethyl acetate gradient in n-hexane). The overall isolated yield of the anomeric mixture 3 was 51%, β/α ratio ∼2:1 according to 31P NMR spectroscopy, m/z (HR ESI+) 919.2397 ([M + Na]+; C40H49NaO21P requires 919.2396). The α and β phosphate anomers, 3a and 3b, were partially separated with the β anomer 3b eluting first. Fractions containing a given anomer were enriched by repeating the chromatographic step twice. Anomer-enriched samples were evaporated to dryness under reduced pressure. Each anomer was further purified by HPLC using a Phenomenex semi-preparative silica column (Luna 250 × 10 mm, 10 μm) fitted to a Dionex Ultimate 3000. Compounds were eluted with 20% ethyl acetate in n-hexane followed by a 60–67% ethyl acetate gradient and monitored by UV absorbance at 265 nm giving the α anomer 3a, δH (600 MHz; CDCl3) 7.40–7.36 (10 H, m, 2 C6H5), 5.82 (1 H, dd, J1,2 3.4, J1,P 7.0, 1-H), 5.54 (1 H, dd, J2,3 10.0, J3,4 10.0, 3-H), 5.42 (1 H, d, J1′,2′ 4.0, 1′-H), 5.37 (1 H, dd, J2′,3′ 10.0, J3′,4′ 10.0, 3′-H), 5.13 (1 H, m, 4′-H), 5.09 (4 H, m, 2 CH2C6H5), 4.88 (1 H, dd, J1,2 3.4, J2,3 10.0, 2-H), 4.84 (1 H, dd, J1′,2′ 4.0, J2′,3′ 10.0, 2′-H), 4.30 (1 H, dd, J5,6a 2.4, J6a,6b 12.4, 6a-H), 4.24 (1 H, dd, J5′,6′a 3.5, J6′a,6′b 12.4, 6′a-H), 4.11 (1 H, dd, J5,6b 3.5, J6a,6b 12.4, 6b-H), 4.02 (1 H, dd, J5′,6′b 3.5, J6′a,6′b 12.4, 6′b-H), 4.00 (1 H, dd, J3,4 10.0, J4,5 10.0, 4-H), 3.95 (1 H, m, 5-H), 3.90 (1 H, m, 5′-H), 2.10 (3 H, s, CH3), 2.08 (3 H, s, CH3), 2.07 (3 H, s, CH3), 2.04 (3 H, s, CH3), 2.021 (3 H, s, CH3), 2.016 (3 H, s, CH3), 1.88 (3 H, s, CH3); δC (150 MHz; CDCl3) 170.6 (COCH3), 170.5 (COCH3), 170.3 (COCH3), 169.9 (2 C, COCH3), 169.8 (COCH3), 169.4 (COCH3), 128.8–128.0 (C6H5), 95.7 (1′-C), 93.8 (1-C), 71.8 (4-C), 71.7 (3-C), 70.0 (2-C), 70.0 (2′-C), 69.74 (CH2C6H5), 69.69 (5-C), 69.3 (3′-C), 68.6 (5′-C), 67.9 (4′-C), 62.1 (6-C), 61.3 (6′-C), 29.7 (CH3), 20.9 (CH3), 20.72 (CH3), 20.69 (CH3), 20.64 (CH3), 20.61 (CH3), 20.3 (CH3); δP (161 MHz; CDCl3) −2.8 (dd, J1H,P 7.0, JBn,P ∼7.8); m/z (ESI+) 919.4 ([M + Na]+, 100%), 641.5 (9), MS2 641.1 (−(BnO)2PO2H), MS3 581.0 (−CH3CO2H); [α]D20 + 70.7 (c 0.92, CHCl3); Rf (TLC; ethyl acetate/petroleum ether, 2:1, v/v) 0.70, and the β anomer 3b, δH (600 MHz; CDCl3) 7.36–7.29 (10 H, m, 2 C6H5), 5.41 (1 H, d, J1′,2′ 4.0, 1′-H), 5.38 (1 H, dd, J1,2 7.4, J1,P 7.4, 1-H), 5.35 (1 H, dd, J2′,3′ 10.0, J3′,4′ 10.0, 3′-H), 5.26 (1 H, dd, J2,3 9.0, J3,4 9.0, 3-H), 5.06 (1 H, m, 4′-H), 5.02 (4 H, m, 2 CH2C6H5), 4.96 (1 H, dd, J1,2 7.4, J2,3 9.0, 2-H), 4.86 (1 H, dd, J1′,2′ 4.0, J2′,3′ 10.0, 2′-H), 4.50 (1 H, dd, J5,6a 2.3, J6a,6b 12.2, 6a-H), 4.25 (1 H, dd, J5′,6′a 3.7, J6′a,6′b 12.4, 6′a-H), 4.20 (1 H, dd, J5,6b 4.5, J6a,6b 12.2, 6b-H), 4.05 (1 H, dd, J5′,6′b 4.0, J6′a,6′b 12.4, 6′b-H), 4.04 (1 H, m, 4-H), 3.94 (1 H, m, 5′-H), 3.78 (1 H, m, 5-H), 2.10 (3 H, s, CH3), 2.049 (3 H, s, CH3), 2.047 (3 H, s, CH3), 2.03 (3 H, s, CH3), 2.004 (3 H, s, CH3), 2.002 (3 H, s, CH3), 1.87 (3 H, s, CH3); δC (150 MHz; CDCl3) 170.6 (COCH3), 170.5 (COCH3), 170.3 (COCH3), 170.0 (COCH3), 169.9 (COCH3), 169.6 (COCH3), 169.4 (COCH3), 128.7–127.8 (C6H5), 95.89 (1-C), 95.86 (1′-C), 74.8 (3-C), 73.0 (5-C), 72.3 (4-C), 70.1 (2-C), 70.1 (2′-C), 70.0 (CH2C6H5), 69.3 (3′-C), 68.6 (5′-C), 68.0 (4′-C), 62.2 (6-C), 61.4 (6′-C), 20.8 (CH3), 20.7 (CH3), 20.62 (CH3), 20.61 (CH3), 20.59 (CH3), 20.58 (CH3), 20.4 (CH3); δP (161 MHz; CDCl3) −3.2 (dd, J1H,P 7.4, JBn,P ∼7.4); m/z (ESI+) 919.3 ([M + Na]+, 100%), 641.5 (21), MS2 641.1 (−(BnO)2PO2H), MS3 581.1 (−CH3CO2H); [α]D20 + 44.7 (c 1.1, CHCl3); Rf TLC; ethyl acetate/petroleum ether, 2:1, v/v) 0.71.
α-d-Maltose 1-Phosphate, Disodium Salt (1a)
Pd/C catalyst (10 wt. %, ∼15 mg) was added to a solution of dibenzyl 2,3,6,2′,3′,4′,6′-hepta-O-acetyl-α-d-maltosyl phosphate (3a) (57 mg, 64 μmol) in methanol (20 ml). The atmosphere within the flask was replaced with H2 gas, and the reaction mixture was stirred vigorously for 24 h at ambient temperature. The reaction went to completion according to TLC. The mixture was filtered, and three drops of triethylamine were added. The solution was evaporated to dryness to yield a thick oil of 2,3,6,2′,3′,4′,6′-hepta-O-acetyl-α-d-maltosyl phosphate, bis(triethylammonium) salt (4a), Rf (TLC; dichloromethane/methanol/water, 6:3:1, v/v) 0.94, which was used directly in the next steps. Compound 4a was dissolved in methanol/water/triethylamine (7:3:1; 10 ml) and stirred at ambient temperature for 24 h. The reaction went to completion according to TLC and the mixture was evaporated to dryness to yield α-d-maltosyl phosphate, bis(triethylammonium) salt (5a) as a white solid. The sample was dissolved in H2O (1 ml), applied to a Dowex Marathon C column (Na+ form), eluted with water (10 ml) and freeze-dried to yield 1a as a white solid as follows: δH (600 MHz; D2O) 5.36 (1 H, dd, J1,2 3.0, J1,P 7.0, 1-H), 5.33 (1 H, d, J1′,2′ 3.4, 1′-H), 3.94 (1 H, m, 3-H), 3.94 (1 H, m, 5-H), 3.79 (1 H, d, J6a,6b 12.8, 6a-H), 3.76 (1 H, dd, J5′,6′a 2.2, J6′a,6′b 12.6, 6′a-H), 3.70 (1 H, d, J6a,6b 12.8, 6b-H), 3.67 (2 H, m, 5′, 6′b-H), 3.61 (1 H, m, 3′-H), 3.55 (1 H, m, 4-H), 3.47 (1 H, dd, J1′,2′ 3.4, J2′,3′ 9.8, 2′-H), 3.41 (1 H, m, 2-H), 3.32 (1 H, m, 4′-H); δC (150 MHz; D2O) 99.5 (1′-C), 93.3 (J1,P 5.1, 1-C), 76.8 (4-C), 73.6 (3-C), 72.8 (3′-C), 72.6 (5′-C), 72.1 (J2,P 6.8, 2-C), 71.8 (2′-C), 70.4 (5-C), 69.3 (4′-C), 60.7 (6-C), 60.4 (6′-C); δP (161 MHz; D2O) 2.1 (JP,H1 7.0); m/z (HR ESI+) 467.0536 ([R-OPO3Na2 + H]+; C12H22Na2O14P requires 467.0537); m/z (ESI−) 421.2 ([R-OPO3H2-H]−, 100%), MS2 259.0 (−C6H10O5), MS3 241.0 (−H2O); [α]D20 +217.3 (c 0.109, H2O); Rf (TLC; dichloromethane/methanol/water, 6:3:1, v/v) 0.31. The NMR spectra was indistinguishable from those reported for material isolated from natural sources (3) and from an undisclosed source (19).
β-d-Maltose 1-Phosphate, Disodium Salt (1b)
The β anomer 1b was prepared as described for the α anomer 1a except that the corresponding β anomer starting material 3b was used. Compound 1b was obtained as a white solid as follows: δH (600 MHz; D2O) 5.30 (1 H, d, J1′,2′ 3.9, 1′-H), 4.42 (1 H, dd, J1,2 7.7, J1,P 7.7, 1-H), 3.84 (1 H, dd, J5,6a 2.0, J6a,6b 12.2, 6a-H), 3.76 (1 H, dd, J5′,6′a 2.2, J6′a,6′b 12.2, 6′a-H), 3.72 (1 H, m, 3-H), 3.65 (1 H, m, 5′-H), 3.62 (1 H, m, 6b-H), 3.60 (1 H, m, 3′-H), 3.54 (1 H, d, J4,5 9.4, 5-H), 3.49 (1 H, d, J 9.0, 4-H), 3.48 (1 H, d, J2′,3′ 9.6, 2′-H), 3.40 (1 H, m, 6′b-H), 3.31 (1 H, d, J3′,4′9.6, 4′-H), 3.25 (1 H, dd, J1,2 7.7, J2,3 10.0, 2-H); δC (150 MHz; D2O) 99.5 (1′-C), 96.8 (J1,P 4.3, 1-C), 77.1 (4-C), 75.9 (3-C), 74.8 (5-C), 74.2 (J2,P 6.6, 2-C), 72.8 (3′-C), 72.6 (5′-C), 71.7 (2′-C), 69.3 (4′-C), 61.1 (6-C), 60.4 (6′-C); δP (161 MHz; D2O) 2.1 (JP,H1 7.7); m/z (HR ESI+) 467.0538 ([R-OPO3Na2 + H]+; C12H22Na2O14P requires 467.0537); m/z (ESI−) 421.2 ([R-OPO3H2-H]−, 100%), MS2 259.0 (−C6H10O5), MS3 241.0 (−H2O); [α]D20 +110.7 (c 0.136, H2O); Rf (TLC; dichloromethane/methanol/water, 6:3:1, v/v) 0.31.
Expression and Purification of GlgE
The genes for both isoforms of GlgE from S. coelicolor strain M145 were each subcloned into a pET15b vector using BamHI and NdeI restriction sites to allow the expression of the enzyme with an N-terminal His tag and thrombin cleavage site. Both glgE genes in the final expression plasmids were confirmed by DNA sequencing. Protein expression was carried out as described previously (3) except that Escherichia coli BL21(DE3) pLysS was used. Selenomethionine-labeled GlgE isoform I was obtained by the metabolic inhibition method (20). The method used to express GlgE from M. tuberculosis (3) was also used to express Mycobacterium smegmatis GlgE. The enzymes were purified using nickel affinity and size exclusion chromatographies (3).
Assay of GlgE Activity
GlgE activity was monitored using a quantitative stopped assay to determine Pi release with malachite green (3). Reaction mixtures comprised enzyme, substrates, and 100 mm Bistris propane,2 pH 7.0, containing 50 mm NaCl at 30 °C. Reactions were monitored over an 8-min period and progressed linearly with time for at least 4 min when donor consumption was typically <5%. Acceptor preferences were determined in triplicate using 7.5 mm maltooligosaccharide, 5 mm α-maltose 1-phosphate, and between 22 and 80 nm enzyme. Activity was also monitored qualitatively using MALDI-TOF MS to detect extension of maltooligosaccharides (3). Product oligosaccharide linkage analysis was carried out using reaction mixtures that were quenched by heating to 99 °C for 15 min before being subjected to 1H NMR spectroscopy at 600 MHz (3). Pi was removed from buffers using a “Pi mop” consisting of bacterial purine nucleotide phosphorylase and 1 mm 7-methylguanosine (21). α-Maltose 1-phosphate was generated from α-maltosyl fluoride (10 mm) in the presence of Pi (50 mm) and enzyme and identified using MALDI-TOF MS, m/z 499 ([R-OPO3K2 + H]+ with 499 expected).
Protein Size Determination
Size exclusion chromatography was carried out using a Superdex 200 10/300 column (GE Healthcare) using 100 mm Bistris propane buffer, pH 7.0, containing 50 mm NaCl. Dynamic light scattering was carried out using a DynaPro Titan molecular sizing instrument at 298 K (Wyatt Technology) with an enzyme concentration of 2 mg ml−1 in the buffer described above. Data were analyzed using the DYNAMICS software package (Wyatt Technology). Analytical ultracentrifugation experiments were performed using a Beckman Optima XL-I analytical ultracentrifuge (High Wycombe, United Kingdom) equipped with absorbance optics and an An-50 Ti rotor. Experiments were performed at 20 °C and 10,000 rpm with a protein concentration of 1 mg ml−1 in 20 mm Bistris propane, pH 7.0, containing 100 mm NaCl. The partial specific volume of GlgE was calculated from the amino acid sequence using SEDNTERP. UltraScan II was used to fit the experimental sedimentation equilibrium profiles to a single species model.
Protein Crystallization and Cryoprotection
Crystallization screens and optimizations were performed using a protein concentration of ∼5 mg ml−1 and a temperature of 20 °C. Crystals of GlgE (both apo- and selenomethionine-labeled) were obtained from 15% (w/v) polyethylene glycol 3350, 0.2 m sodium citrate, and 15% ethylene glycol. Ligand-bound structures of GlgE were obtained by co-crystallization under the same conditions with ligand concentrations of 5 mm.
Structure Determination and Refinement
All crystals were flash-cooled in Litholoops (Molecular Dimensions) by plunging into liquid nitrogen and transported in Unipuck cassettes before being robotically mounted onto the goniostat on either station I02, I03, or I04 at the Diamond Light Source (Oxford, UK), whereupon they were maintained at −173 °C with a Cryojet cryocooler (Oxford Instruments). Diffraction data were recorded using an ADSC Quantum 315 CCD detector. The resultant data were integrated using MOSFLM (22) and scaled with SCALA (23). Analysis in POINTLESS (23) suggested that the space group was P41212/P43212, although statistical tests in TRUNCATE (24) indicated that the crystals were usually hemihedrally twinned (operator, k, h, −l), and must therefore belong to a lower symmetry space group. Nevertheless, it proved to be more tractable to determine experimental phases and build a preliminary model in P41212/P43212.
A three-wavelength anomalous dispersion data set was collected to 2.8 Å resolution from a single crystal of selenomethionine-substituted protein (supplemental Table S1). The data were processed in space group P422 with approximate cell parameters of a = b = 113.8 Å, c = 316.7 Å. Experimental phases were determined using the SHELX suite (25). The two possible enantiomorphs gave comparable statistics; however, P41212 was ultimately chosen based on superior electron density map quality. SHELXD located 15 selenium sites, being consistent with two copies of the GlgE protomer (based on eight methionines per subunit) in the asymmetric unit, with a corresponding solvent content of 63% (based on a subunit molecular mass of 75,290 Da). After phasing with SHELXE and density modification, with 2-fold noncrystallographic symmetry averaging in DM (26), the figure-of-merit was 0.794 to 2.8 Å resolution. After automated building with BUCCANEER (27) and several iterations of (i) rebuilding in COOT (28), (ii) restrained refinement against the selenomethionine peak data set with REFMAC5 (29), (iii) combination of experimental and model phases using SIGMAA (30), and (iv) 2-fold averaging in DM, a model comprising 1062 residues with corresponding Rwork and Rfree values of 36.8 and 40.0%, respectively, at 2.8 Å resolution, was produced.
Several data sets were collected from crystals obtained by co-crystallization with potential ligands. Of these, only the complex with α-cyclodextrin alone yielded a data set that was essentially untwinned and therefore could be justifiably treated as belonging to space group P41212. This data set was collected to 2.3 Å resolution and was used to complete the building and refinement of the first GlgE model. This was performed with REFMAC5 using 2-fold noncrystallographic symmetry restraints, and TLS parameters (four domains per monomer). The final model consisted of 1298 residues in two subunits and two α-cyclodextrin molecules, having final Rwork and Rfree values of 17.3 and 20.1%, respectively (Table 2). From inspection, the biological unit of GlgE is a homodimer. However, the two subunits in the asymmetric unit of this model represent halves of two separate dimers, with individual dimers being completed through the application of 2-fold crystallographic symmetry.
TABLE 2.
Summary of GlgE x-ray data for refinement and model parameters
r.m.s.d. is root mean square deviation and NA is not applicable.
| Data set | Apo-GlgE | αCD-GlgE | mal-GlgE | αCD-mal-GlgE | βCD-mal-GlgE |
|---|---|---|---|---|---|
| Data collection | |||||
| Space groupa | P212121 | P41212 | P212121 | P212121 | P212121 |
| Cell parameters | a = 113.1, b = 113.0, c = 314.2 Å | a = b = 113.2, c = 314.5 Å | a = 113.8, b = 113.6, c = 315.7 Å | a = 113.9, b = 114.2, c = 315.6 Å | a = 113.3, b = 113.4, c = 315.0 Å |
| Beamlineb | I03 | I02 | I04 | I04 | I02 |
| Wavelength | 0.9709 Å | 0.9795 Å | 0.9763 Å | 0.9763 Å | 0.9795 Å |
| Resolution rangec | 53.15 to 1.80 Å | 71.35 to 2.30 Å | 56.89 to 2.10 Å | 63.99 to 2.20 Å | 71.42 to 2.50 Å |
| (1.90 to 1.80 Å) | (2.42 to 2.30 Å) | (2.21 to 2.10 Å) | (2.32 to 2.20 Å) | (2.64 to 2.50 Å) | |
| Unique reflectionsc | 351,739 | 85,772 | 236,917 | 206,349 | 140,547 |
| Completenessc | 95.0% (72.9%) | 93.7% (66.0%) | 99.5% (96.7%) | 98.9% (93.2%) | 99.8% (99.7%) |
| Redundancyc | 7.6 (5.5) | 14.3 (13.2) | 7.3 (4.7) | 5.4 (4.1) | 4.9 (5.0) |
| Rmergec,d | 0.110 (0.762) | 0.088 (0.327) | 0.121 (0.426) | 0.151 (0.602) | 0.143 (0.688) |
| Rmeasc,e | 0.118 (0.840) | 0.092 (0.340) | 0.130 (0.482) | 0.167 (0.692) | 0.160 (0.767) |
| Mean I/σ(I)c | 13.4 (2.1) | 23.6 (7.9) | 12.0 (3.2) | 7.7 (2.1) | 10.0 (2.3) |
| Wilson B value | 21.6 Å2 | 33.2 Å2 | 26.1 Å2 | 33.8 Å2 | 42.4 Å2 |
| Twin fractionf | 0.39 | 0.04 | 0.36 | 0.27 | 0.17 |
| Refinement | |||||
| Reflections: working/freeg | 333,972/17,659 | 81,357/4,301 | 224,955/11,847 | 195,790/10,447 | 133,513/6,958 |
| Rworkh | 0.231 | 0.173 | 0.204 | 0.208 | 0.191 |
| Rfreeh | 0.249 | 0.201 | 0.228 | 0.236 | 0.221 |
| Ramachandran favored/allowedi | 98.6/100.0% | 98.7/100.0% | 98.8/100.0% | 98.7/100.0% | 98.5/100.0% |
| Ramachandran outliersi | 0 | 0 | 0 | 0 | 0 |
| r.m.s.d. bond distances | 0.013 Å | 0.016 Å | 0.015 Å | 0.015 Å | 0.014 Å |
| r.m.s.d. bond angles | 1.34° | 1.53° | 1.44° | 1.46° | 1.45° |
| Twin fractionj | 0.48 | NA | 0.49 | 0.45 | 0.49 |
| Contents of model | |||||
| Protein residues | 4 × 649 | 2 × 649 | 4 × 649 | 4 × 649 | 4 × 649 |
| Glucans | 0 | 2 × αCD | 4 × mal | 4 × αCD; 4 × mal | 4 × βCD; 4 × mal |
| Ethylene glycol | 0 | 1 | 0 | 0 | 0 |
| Water molecules | 540 | 731 | 486 | 459 | 369 |
| Average atomic displacement parameters (Å2) | |||||
| Main chain atoms | 25.0 | 47.1 | 28.6 | 32.7 | 31.8 |
| Side chain atoms | 25.6 | 49.2 | 29.1 | 33.9 | 32.5 |
| Glucans | 78.5 | 25.1 | αCD: 69.3; mal: 28.3 | βCD: 41.9; mal: 37.5 | |
| Ethylene glycol | 49.8 | ||||
| Water molecules | 20.2 | 44.9 | 21.7 | 24.4 | 21.4 |
| Overall | 25.2 | 48.3 | 28.7 | 33.5 | 32.1 |
| PDB accession code | 3zss | 3zst | 3zt5 | 3zt6 | 3zt7 |
a Space group that was used for refinement.
b I02, I03, I04 = beamlines at the Diamond Light Source (Oxfordshire, UK).
c The figures in parentheses indicate the values for outer resolution shell.
d Rmerge = 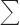 hkl i|Ii(hkl) − 〈I(hkl)〉/hkl iIi(hkl), where Ii(hkl) is the ith observation of reflection hkl, and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
hkl i|Ii(hkl) − 〈I(hkl)〉/hkl iIi(hkl), where Ii(hkl) is the ith observation of reflection hkl, and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
e Rmeas = hkl [N/(N − 1)]1/2 i|Ii(hkl) − 〈I(hkl)〉|/hkl iIi(hkl), where N is the number of observations of reflections hkl.
f Data were as calculated by TRUNCATE (24).
g The data sets were split into “working” and “free” sets consisting of 95 and 5% of the data, respectively. The free set was not used for refinement.
h The R-factors Rwork and Rfree are calculated as follows: R = (|Fobs − Fcalc|)/|Fobs| ×100, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively.
i Data were calculated using MOLPROBITY (32).
j Refined values were from REFMAC5 (29).
The highest resolution data set was collected from a crystal obtained by co-crystallization with 63-α-d-glucosyl-maltotriose (Megazyme, Bray, Ireland) and processed to 1.8 Å resolution. The structure was solved using PHASER (31) with a single GlgE subunit from the α-cyclodextrin complex as the search model. Although the expectation was that the space group would be primitive tetragonal, acceptable solutions were found in space groups P212121 and C2221, in addition to P41, in each case giving four subunits per asymmetric unit (arranged as two dimers) and very similar crystal packing. However, the log-likelihood-gain value (calculated to 3 Å resolution) was higher for the P212121 solution, and it gave a lower clash score in MOLPROBITY (32). Therefore, P212121 was chosen for refinement of the model in REFMAC5, employing intensity-based twin refinement, 4-fold noncrystallographic symmetry restraints, and TLS parameters (four domains per monomer). Unfortunately, electron density maps were heavily biased due to the high twin fraction (0.48) (33). Thus, model building was performed cautiously, and water molecules were added sparingly. No evidence was seen for the added ligand, and therefore, this model was subsequently treated as a reference apo-structure. Data sets from three further co-crystallizations yielded new complexes. These were all twinned and handled as for the apo-structure, which was also used as the starting point for refinement in each case. The x-ray data collection and refinement statistics for all structures are summarized in Table 2. Structural figures were generated using PyMOL (34).
RESULTS
Synthesis of Maltose 1-Phosphate
To assay GlgE activity, it was necessary to obtain the donor substrate, α-maltose 1-phosphate 1a. A protection-deprotection strategy was used to allow the phosphorylation of the 1-position of maltose using tetrabenzyl pyrophosphate (Fig. 2). This yielded a mixed anomer product 3, from which pure anomers were obtained using silica column chromatography. Following deprotection, this route allowed the production of both α- and β-maltose 1-phosphate, 1a and 1b. The properties of the synthetic α anomer 1a (NMR and MS spectra, and TLC Rf) were indistinguishable from those of the material obtained from M. smegmatis assigned as being α-maltose 1-phosphate (3).
FIGURE 2.

Synthesis of d-maltose 1-phosphate (1). Compound numbers in the text with suffixes a and b refer to α and β anomers, respectively.
Crystallization of GlgE
Recombinant GlgE from M. tuberculosis and M. smegmatis was subjected to crystallization trials but failed to yield protein crystals. GlgE isoforms I and II from another actinomycete, S. coelicolor, were subsequently entered into trials. Although isoform II proved to be too insoluble to obtain crystals, isoform I readily yielded crystals.
Comparison of the Catalytic and Kinetic Properties of S. coelicolor GlgE Isoform I and M. tuberculosis GlgE
Before embarking on solving the structure of S. coelicolor isoform I, it was important to determine its properties and compare them with those of GlgE from M. tuberculosis. Although it seems likely that homologous enzymes from actinomycetes would share similar properties, GlgE isoforms I and II from S. coelicolor (with 86% amino acid sequence identity between them) each share only 51% identity with GlgE from M. tuberculosis.
GlgE isoform I from S. coelicolor was heterologously expressed with an N-terminal His tag in E. coli and purified to homogeneity. According to assays based on Pi release, it possessed GlgE activity. Although the pH optimum (7.0; supplemental Fig. S1) and slight activation by NaCl (∼20% at 50 mm; supplemental Fig. S2) were common to isoform I and GlgE from M. tuberculosis, their temperature optima reflected the lifestyles of the source organisms (∼30 °C for S. coelicolor isoform I (supplemental Fig. S3) and ∼37 °C for M. tuberculosis GlgE (3)). The acceptor preferences of these two enzymes were similar such that a degree of polymerization (DP) of ≥4 gave the most significant rates of reaction (Fig. 3 and supplemental Fig. S4). The acceptor length specificities were very similar, with only a marginal shift of the optimum from DP 5 to 6 in isoform I. Isoform II from S. coelicolor behaved very similarly to isoform I as would be expected given their high sequence identities (Fig. 3).
FIGURE 3.

Acceptor specificity of GlgE. A and B show acceptor specificity of S. coelicolor isoforms I and II, respectively. Enzyme activity with maltooligosaccharide acceptor substrates with different DP was determined by monitoring Pi release in triplicate. The same trends were observed with the MALDI-TOF MS assay (e.g. supplemental Fig. S4). The bars indicate means ± S.E. For comparison, data from M. tuberculosis GlgE (3) are shown in C.
The Kmapp values for α-maltose 1-phosphate with isoform I and the M. tuberculosis enzyme were very similar (0.25 ± 0.05 and 0.30 ± 0.06 mm in the presence of 1 mm maltohexaose; Table 1 and supplemental Fig. S5). (It is noteworthy that an enzyme activity consistent with GlgE detected in M. smegmatis extracts exhibited a comparable Kmapp for the donor substrate of 0.25 mm using glycogen as the acceptor (35).) Isoform I had kcatapp values up to an order of magnitude greater rate than those of the M. tuberculosis enzyme. Both Kmapp and kcatapp for α-maltose 1-phosphate increased with increasing maltohexaose concentration with isoform I (data not shown), consistent with a ping-pong (substituted) enzyme mechanism. The Kmapp for maltohexaose in the presence of 5 mm α-maltose 1-phosphate was 23-fold lower with isoform I. In general, the Michaelis-Menten parameters for isoform II were broadly similar to those of isoform I, except that the kcatapp/Kmapp values were between ∼3- and ∼5-fold lower.
TABLE 1.
Michaelis-Menten kinetic analysis of S. coelicolor GlgE isoforms I and II
Enzyme activity was monitored by detecting Pi release in triplicate and expressed as the mean ± S.E. (see supplemental Fig. S5).
| Enzyme | Substrate | Kmappp | kcatapp | kcatapp/Kmapp |
|---|---|---|---|---|
| mm | s−1 | m−1s−1 | ||
| S. coelicolor isoform I | Maltose 1-phosphatea | 0.30 ± 0.06 | 12.3 ± 0.5 | 41,000 ± 8000 |
| Maltohexaoseb | 1.5 ± 0.3 | 53 ± 2 | 36,000 ± 7000 | |
| S. coelicolor isoform II | Maltose 1-phosphatea | 1.2 ± 0.2 | 10.0 ± 0.6 | 8,000 ± 1700 |
| Maltohexaoseb | 2.3 ± 0.4 | 23.5 ± 1.1 | 10,000 ± 2000 | |
| M. tuberculosisc | Maltose 1-phosphatea | 0.25 ± 0.05 | 1.26 ± 0.07 | 5,000 ± 1000 |
| Maltohexaoseb | 35 ± 8 | 15.4 ± 1.1 | 440 ± 100 |
a This is in the presence of 1 mm maltohexaose.
b This is in the presence of 5 mm maltose 1-phosphate.
c Data are from Ref. 3.
Isoform I formed exclusively α-1,4 linkages according to NMR spectroscopy (supplemental Fig. S6), and neither β-maltose 1-phosphate nor α-d-glucose 1-phosphate served as donor substrates. It catalyzed disproportionation reactions through maltosyl transfer between maltooligosaccharides (supplemental Fig. S7) with chain length specificities indistinguishable from those of GlgE from M. tuberculosis (donor DP ≥4 but preferentially >6 with acceptor DP ≥4 (3)). Disproportionation occurred just as efficiently in the presence of a Pi mop consisting of purine nucleoside phosphorylase and 7-methylguanosine (21), providing evidence that maltosyl transfer occurred directly from donor to acceptor rather than via a maltose 1-phosphate intermediate.
The above analyses showed that GlgE isoform I from S. coelicolor is a (1→4)-α-d-glucan:phosphate α-d-maltosyltransferase that has very similar kinetic properties to GlgE from M. tuberculosis. The only differences were in kcatapp and Kmapp for maltohexaose and temperature optimum together with a small shift in the acceptor chain length specificity.
S. coelicolor GlgE Isoform I Extends a Primer at Its Nonreducing End
In the absence of a priming acceptor, GlgE forms only very small amounts of oligomeric product after many hours of incubation with maltose 1-phosphate (data not shown). It is likely that this occurs via hydrolysis of maltose 1-phosphate and extension of the resulting maltose, both very slow processes. Therefore, self- priming, although possible, is not efficient with GlgE.
To test how GlgE extends acceptors, maltotetraitol (Fig. 4A), which has no reducing end, was exposed to isoform I and α-maltose 1-phosphate. Maltotetraitol could be detected using MALDI-TOF MS before the addition of enzyme (Fig. 4B). After the addition of enzyme, a series of products was observed with masses consistent with maltotetraitol extension by one maltosyl unit at a time (Fig. 4C). 1H NMR spectroscopy before and after the addition of maltose 1-phosphate and enzyme (Fig. 4, D and E, respectively) showed a net ∼3-fold increase in normal α-1,4 linkages consistent with extension at the nonreducing end of maltotetraitol. There was very little reducing end generated in the reaction mixture (6% reducing end α resonance at ∼5.25 ppm compared with that of the α-glucosidic link to the glucitol moiety at ∼5.08 ppm), and these were likely formed by slow hydrolytic side reactions. The Pi release assay indicated that maltose was transferred to maltotetraitol at a rate 35% that with maltotetraose implying a +4 subsite has a preference for a glycopyranose ring over the ring-opened glucitol. Overall, these observations strongly support the preference for a maltooligosaccharide acceptor that is extended at its nonreducing end by GlgE.
FIGURE 4.

Maltotetraitol is an acceptor for GlgE isoform I. The structure of maltotetraitol is shown in A. B shows MALDI-TOF MS of maltotetraitol where the mass of the starting material (m/z 691; [M + Na]+; highlighted with a star) is among the peaks from the matrix and other reaction mixture components. C shows the spectrum after incubation with enzyme and α-maltose 1-phosphate revealing a series of peaks (highlighted with arrows) with the successive addition of m/z 324 corresponding to maltosyl units. The DP of each peak (including the glucitol chain) is indicated. D and E respectively show 1H NMR spectra of maltotetraitol before and after incubation with enzyme and α-maltose 1-phosphate. Peak assignments are indicated.
α-Maltosyl Fluoride Is an Efficient Donor
It is noteworthy that α-maltosyl fluoride, which bears a better leaving group than the normal substrate, was a donor (Fig. 5A) and was able to extend maltotetraose to give longer products than α-maltose 1-phosphate under the same conditions (Fig. 5B). Products from α-maltosyl fluoride of DP >34 (well beyond the limit of aqueous solubility of DP ∼18) were conspicuous because the solution became visibly white and turbid. This donor was also capable of generating maltose 1-phosphate, according to MS, in the presence of enzyme and Pi but in the absence of an acceptor (data not shown). It could therefore be of utility in the enzymatic synthesis of α-maltose 1-phosphate and maltooligosaccharides as well as in monitoring GlgE activity using a fluoride electrode.
FIGURE 5.

Ability of GlgE isoform I to use α-maltosyl fluoride as a donor. A shows MALDI-TOF MS of a reaction mixture containing maltotetraose (5 mm) after 10 min of exposure to enzyme and α-maltosyl fluoride (5 mm). B shows a control with α-maltose 1-phosphate (5 mm). The successive addition to the acceptor of m/z 324 was observed, which corresponds to maltosyl units. The DP associated with each peak is highlighted. α-Maltosyl fluoride was an efficient donor yielding longer polymers than α-maltose 1-phosphate under these conditions.
Solving the Structure of S. coelicolor GlgE Isoform I
The structure of ligand-free apo-GlgE was determined by the multiple wavelength anomalous dispersion method using selenomethionine-substituted protein (supplemental Table S1). A number of ligand-bound structures were subsequently obtained by co-crystallization. Although the majority of data sets were hemihedrally twinned, structure solution and refinement was achievable (Table 2).
Overall Structure of GlgE
The apo-GlgE structure indicated that the enzyme forms a dimer within the crystal (Fig. 6). This appears to be the biologically relevant oligomerization state as it was also a dimer in solution (172 kDa by analytical ultracentrifugation, 120 kDa by size exclusion chromatography, and 103 kDa by dynamic light scattering with 151 kDa predicted for the His-tagged dimer; data not shown). The M. tuberculosis and M. smegmatis enzymes also formed dimers in solution according to analytical ultracentrifugation.3 Some variance in the oligomer size of the S. coelicolor enzyme, as determined using the different methods, perhaps reflects its relatively flat overall structure, where the dimer interface is relatively narrow with a buried surface area of 2150 Å2, which equates to just 7.7% of the total solvent accessible area of each subunit.
FIGURE 6.

Structure of S. coelicolor GlgE isoform I. A shows the GlgE homodimer in ribbon representation and in wall-eyed stereo highlighting domain N (residues 1–108 and 192–205), domain S (residues 109–191), domain A (residues 206–253, 300–322, 368–512, and 553–573), insert 1 (residues 254–299), domain B (residues 323–367), insert 2 (residues 513–552), and domain C (residues 574–675). B shows a space-filling representation. Various features are highlighted, including the gap that the domain B lid could potentially occupy to allow access to the donor site.
Each subunit is composed of five domains (Fig. 6), four of which have been observed before in members of the GH13 α-amylase family of enzymes in the GH-H clan (36). Domain A is a (β/α)8 barrel, typical of the catalytic domain of this family of enzymes, that forms part of the dimer interface. Domain B corresponds to an insertion after the third β-strand of domain A (β12 in supplemental Fig. S8), as has been observed in many other members of this family (37). In GlgE, domain B is fairly typical for a GH13 enzyme (38) in having a pair of anti-parallel strands and one short helix. Although domain B is responsible for binding a Ca2+ ion in some GH13 proteins, there is no evidence in the electron density maps for metal ions binding to GlgE. Indeed, neither divalent metal ions nor chelators had any effect on activity with α-maltose 1-phosphate as the donor according to the Pi release assay (5 mm CaCl2, 5 mm MgCl2, 2 mm EDTA, and 2 mm EGTA). There are two additional significant insertions within domain A of note. Insert 1 is after the second β-strand of domain A (β11) and lies adjacent to domain B. Insert 2 is after the eighth β-strand (β21) and lies adjacent to insert 1.
The C-terminal domain C has a β-sandwich fold. Domain C is thought to help stabilize domain A in other family members and could be involved in substrate binding in some cases (37). The N-terminal domain N, which also consists of a β-sandwich fold, forms the core of the dimer interface. The final domain (residues 109–191) arises from an insertion within domain N and forms a four-helix bundle where the last helix is discontinuous and slightly kinked (α4 and α5 in supplemental Fig. S8). This domain, which will henceforth be referred to as domain S, participates in the dimer interface and interacts directly with domain B of the neighboring subunit. The structure comparison tools DALI (39) and SSM (40) failed to retrieve another example of such an S domain in the context of a GH13 protein. In addition, when only the S domain was used as the query, only four-helix bundles with relatively low Z scores (≤7.1) were found, and none of these had any known role in sugar interactions. Other members of this family of enzymes also possess β-sheet domains D and E following domain A (37) with the latter often being associated with starch-degrading enzymes, but GlgE has neither of these two domains.
Donor Pocket
Because α-maltose 1-phosphate is not completely stable in the presence of GlgE over the time scale of protein crystallization, co-crystallization in the presence of maltose was attempted. A ligand-bound structure was solved to 2.1 Å resolution, which will be referred to as the mal-GlgE structure. The maltose was situated at the C-terminal ends of the β-strands making up the center of the (β/α)8 barrel of domain A, typical of active sites within the α-amylase family (36). The maltose was bound in a pocket (Fig. 7A and supplemental Figs. S9 and S10), with its reducing end solvent exposed. The edge of the nonreducing end glucose ring bearing hydroxyls at its C-2′ and C-3′ positions was also partially solvent-exposed. The mal-GlgE protein structure was essentially identical to the apo-GlgE structure except for the adoption of a different rotamer by the Ile-360 side chain within the donor pocket. A key feature of the pocket is that an entire face is composed of the pair of anti-parallel β-strands of domain B (residues 350–357), which is capped off by a turn comprising Pro-353 and Pro-354. It seems highly likely that this part of the structure forms a lid that has to open to allow access to the donor pocket. The elevated B-factors of the backbone of this loop, compared with the rest of the donor pocket, are consistent with this. There is scope for the lid to open toward domain S of the neighboring subunit, where there is a gap in the structure (Figs. 6B and 7C). The movement of loops is similarly predicted to occur in amylosucrose to allow sucrose access to its donor site (41).
FIGURE 7.

Maltose and α-cyclodextrin bound to GlgE. A shows α-maltose in mal-GlgE, and B shows α-cyclodextrin in αCD-GlgE. Difference electron density “omit” maps were generated for bound ligands using phases calculated from the final models minus the ligand coordinates after simulated annealing refinement. This was performed from a starting temperature of 5000 K after applying random shifts to the model (“shake” term set to 0.3) using PHENIX (53). The resultant maps were noncrystallographic symmetry averaged to improve connectivity. The corresponding stereo images are shown in supplemental Fig. S9. Most amino acids interacting with the ligands are highlighted, but some are omitted here for clarity (all are shown in supplemental Fig. S10). C shows the relative orientations of maltose and α-cyclodextrin in the αCD-mal-GlgE structure (comparable with the lower part of Fig. 6B). GlgE is shown in space-filling mode and colored by sequence conservation between the S. coelicolor and M. tuberculosis enzymes (using a color gradient, conserved amino acids are depicted in dark blue, similar amino acids in colors through green, and dissimilar amino acids in red). The donor pocket is highly conserved; the linear cleft is well conserved, and some of the cyclodextrin binding patch is well conserved except for a variable loop as indicated.
Maltose was bound such that the C-2′ hydroxyl of the nonreducing end sugar ring was close in space (3.9 Å) to the C-3 hydroxyl of the reducing end sugar ring (Fig. 7A). The conformation was similar to that of the major species found in solution (42), indicating a low energy conformation of maltose bound to GlgE. Despite maltose being present as a mixture of α and β anomers in solution (with an α/β ratio of ∼1:2 at equilibrium according to NMR spectroscopy),4 the enzyme bound the α anomer, consistent with this pocket being tailored to bind, break, and make α-1-linked bonds. The orientation of the maltose, compared with other ligand-bound structures of the GH13 family, is consistent with it being the donor pocket comprising −1 and −2 sugar-binding subsites (37). The reducing end of maltose sits between Asp-394 and Glu-423 (Fig. 7A). Using sequence and structural comparisons with other family members, these residues of GlgE are predicted to be the nucleophile/base and proton donor, respectively (Fig. 8), associated with the typical double displacement mechanism of such retaining enzymes (43). The mean distance between the carboxyl side chain oxygen atoms of Asp-394 and Glu-423 was 4.9 Å. This is within the range observed in other retaining glycosidases of 4.8–5.3 Å and contrasts with that of inverting glycosidases of 9.0–9.5 Å (43).
FIGURE 8.

Proposed mechanism of GlgE. The extension of a maltooligosaccharide acceptor by α-maltose 1-phosphate is shown. The reversibility of the second step allows disproportionation reactions to occur.
The −1 subsite is lined with amino acid side chains that include Asp-480, which forms hydrogen bonding interactions with the C-2 and C-3 hydroxyls of the reducing end sugar of maltose (Fig. 7 and supplemental Fig. S10). A carboxylate side chain in this position is highly conserved within this enzyme family and is thought to assist in catalysis by stabilizing the oxocarbenium ion-like transition state and also for maintaining the Glu base in the correct protonation state (37). The maltose molecule is sandwiched between the hydrophobic side chains of Trp-281 from insert 1 and Tyr-357 from the domain B lid. Thus domain A, domain B, and to a lesser extent insert 1 have a role in defining subsite −1.
Subsite −2 is defined by domain B and inserts 1 and 2 (supplemental Fig. S10). There is no subsite −3 because of the presence of domain B and insert 1 within this region of the protein providing a reason why GlgE is specific for maltose as the donor. Overall donor specificity is therefore defined by domains A and B and inserts 1 and 2, a typical arrangement that determines specificity in GH13 enzymes (37, 38).
The location of the +1 subsite can be predicted to be adjacent to the −1 subsite, projecting from the reducing end anomeric α-hydroxyl of maltose and by analogy with other family members. This site must be able to bind the phosphate of α-maltose 1-phosphate, promote its cleavage, and yet also be able to bind and deprotonate the nonreducing end of an acceptor maltooligosaccharide without activating water. Polar residues likely to define the phosphate-binding site include Asn-352 and Tyr-357 of the domain B lid as well as other candidates from domain A (supplemental Fig. S10).
Acceptor Site
To define the site where an acceptor binds, the protein was crystallized in the presence of maltooligosaccharides and analogues thereof. However, no extra density was observed in structures solved from co-crystallizations with either maltotriose, 63-α-d-glucosyl-maltotriose (which yielded the apo-GlgE structure), or acarbose, for example. This is perhaps not surprising given that they are neither acceptors nor inhibitors. Maltotetraose, maltopentaose, and maltohexaose each gave ligand-bound structures (data not shown), but they were all indistinguishable from the mal-GlgE structure. It would appear that over the time scale of the crystallization, GlgE hydrolyzed these oligomers to generate sufficient maltose to occupy the donor pocket.
The interaction of cyclodextrins (cyclic maltooligosaccharides) with GlgE was then explored. According to MALDI-TOF MS, cyclodextrins were not converted to any products by GlgE. However, α-cyclodextrin was shown to inhibit the extension of 1 mm maltohexaose with an IC50 of ∼19 mm, according to the Pi release assay (supplemental Fig. S11A). Both β- and γ-cyclodextrins were also inhibitory, each with an IC50 value of ∼6 mm (supplemental Fig. S11, B and C). Their lower IC50 values suggest slightly more favorable protein contacts with the larger diameter cyclodextrins. The dependence of inhibition by α-cyclodextrin on donor and acceptor concentrations was then tested. The percentage inhibition almost halved when the acceptor concentration was increased 4-fold (supplemental Fig. S12). This is consistent with α-cyclodextrin competing with the acceptor for a common binding site on GlgE. Inhibition was more pronounced when the donor concentration was increased 4-fold. This is consistent with an increase in Kmapp for the acceptor when the donor concentration increases in a ping-pong reaction, allowing the inhibitor to compete with the donor more effectively. These observations strongly suggest that the acceptor-binding site overlaps with the α-cyclodextrin-binding site of the S. coelicolor enzyme. Interestingly, the M. tuberculosis GlgE enzyme was not significantly inhibited by the cyclodextrins in the concentration range tested (data not shown).
Co-crystallization of GlgE with α-cyclodextrin yielded a ligand-bound structure, αCD-GlgE, that was solved to 2.3 Å resolution. There were no significant changes within the protein compared with the apo-GlgE and mal-GlgE structures. The α-cyclodextrin was bound to a largely hydrophobic ridge near the donor pocket (Fig. 7B). This ridge comprises largely nonpolar side chains of domain A and Gly-84 of domain N of the neighboring subunit (Fig. 7B and supplemental Figs. 9B and S10B). Thus, domain N not only participates in enzyme dimerization but also appears to be involved in specificity. Similar roles for domain N have been identified in a maltogenic amylase from Thermus sp. (44), despite its role in other enzymes being unclear (37). The orientation of the cyclodextrin-GlgE interaction was close to and parallel to the linear binding cleft, near the predicted +1 subsite and roughly orthogonal to the orientation of the maltose (Figs. 6B and 7C).
There are two additional features on either side of the cyclodextrin binding patch worthy of note. There is a linear cleft that extends from the exit of the donor pocket and through what is predicted to be the +1 subsite (Figs. 6B and 7C). It is defined by domains A and B at its origin and extends between domains N and S of the neighboring subunit. There is also a diagonal cleft that runs across both subunits of the dimer and intersects both of the linear clefts at the points where they exit the protein (Fig. 6B). These clefts could therefore be involved in binding a growing α-glucan chain.
Co-crystallization of GlgE with α-cyclodextrin and maltose yielded a structure showing density for both ligands consistent with both individual ligand-bound structures (data not shown). However, the highest resolution structure with both of these ligands bound, αCD-mal-GlgE, happened to be obtained from a co-crystallization with α-cyclodextrin and maltohexaose that was solved to 2.2 Å resolution. Co-crystallization of GlgE with β-cyclodextrin yielded a ligand-bound structure, βCD-mal-GlgE, that was solved to 2.5 Å resolution. The β-cyclodextrin interacted with GlgE in a manner very similar to that of α-cyclodextrin (supplemental Fig. S9C). Electron density within the donor pocket was consistent with the presence of maltose, which presumably was a contaminant from the β-cyclodextrin.
DISCUSSION
Relationship between GlgE Activity, GlgE Structure, and GH13_3 Membership
We have determined the structure of GlgE isoform I from S. coelicolor, which is the first example from the GH13_3 subfamily (4). There are a large number of structures of other GH13 subfamily members in the PDB data base (145 nonredundant structures similar to GlgE with a Z score of ≥10 according to DALI (39)). The S domain is a novel feature of GlgE, and the particular configuration of domain B and inserts 1 and 2 is specific to GlgE. For example, the protein with the most similar structure to GlgE according to both DALI (39) and SSM (40) is annotated as an α-amylase from Lactobacillus plantarum (PDB code 3dhu). Despite it possessing a B domain and inserts 1 and 2 at the same junctions of domain A, they are different in length, sequence, and conformation such that there is almost no conservation of the residues defining the maltose-binding site. When only domains A and B together with inserts 1 and 2 of GlgE were used as the query, DALI gave the same top hit. SSM gave Thermatoga maritima 4-α-glucanotransferase (PDB code 1lwj (45)), which again has significantly different elaborations of its active site. Inspection of other relevant and high scoring hits revealed even greater structural diversity in and around their active sites, e.g. maltogenic amylase, which binds maltose in its −1 and −2 subsites (GH13_20; PDB code 1gvi (46)) and amylosucrase, which generates an α-1,4 glucan polymer (GH13_4; PDB code 1g5a (47)).
We have failed to detect GlgE activity in other GH13 enzymes that are capable of disproportionating maltooligosaccharides,3 such as T. maritima maltosyltransferase (48). Therefore, the ability to use maltose 1-phosphate as a donor may be restricted to members of the GH13_3 subfamily. The majority of glgE genes are clustered with either one or all of the other genes of the GlgE pathway (6). In addition, there is substantial overlap between the set of proteins encoded by these genes and those defined as GH13_3 members by the CAZy database. This lends weight to the likelihood that most, if not all, GH13_3 subfamily members have GlgE activity.
Catalytic Center
There was some doubt about the presence of the entire catalytic machinery in the GH13_3 subfamily of proteins (4) before GlgE was discovered (3). However, the key side chains can now be clearly identified in the structure of GlgE, whereby Asp-394 and Glu-423 are well placed to carry out the roles of nucleophile/base and proton donor, respectively (Figs. 7A and 8). This arrangement is consistent with the evidence for extension of acceptors at their nonreducing ends and the ability of GlgE to use α-maltosyl fluoride as a donor.
GlgE catalyzes glycosyl transfer reactions to acceptors other than water despite it being a GH class member. Although some phosphorylases are members of the GT class (e.g. GT35 glycogen phosphorylase with a distinct GT_B fold), there are examples of others in the GH class (49), such as GH13_18 sucrose phosphorylase (4, 50). The way in which the sucrose phosphorylase +1 subsite is tailored to utilize phosphate as a leaving group and yet to also accept sugar acceptors involves local conformational changes (51). Whether this is also the case with GlgE remains to be seen because the +1 subsites of these two enzymes are very different. Although it is not clear from our structures how GlgE kinetically suppresses hydrolytic reactions, the way other phosphorylase-type enzymes achieve this appears equally elusive at this time.
It is noteworthy that phosphorylases are generally associated with phosphorolysis rather than saccharide polymerization primarily due to a relatively high cytosolic concentration of Pi. However, flux through the GlgE pathway has been demonstrated (3), and this is presumably driven principally by the ATP-requiring maltose kinase step preceding GlgE (see the supplemental “Discussion” for a more extensive consideration of equilibria within the GlgE pathway).
Binding of Substrates
Although the donor pocket of GlgE is well conserved and highly tailored to bind maltose, it is less clear what defines acceptor specificity. The general location of the +1 subsite can be identified by inspection of the trajectory of the reducing end of maltose as it emerges from the donor pocket (Fig. 7C and supplemental Fig. S10). The observation that cyclodextrins compete with linear maltooligosaccharide acceptors provides strong evidence that their binding sites overlap. Thus some of the +n subsites are likely to be located in or very near the cyclodextrin binding patch, which is ∼12 Å (the equivalent of ∼3 subsites) away from the +1 subsite. It is possible that linear acceptors bind in the same orientation as cyclodextrins, with identical sugar-protein interactions. This orientation is certainly consistent with acceptors being extended at their nonreducing ends. However, to connect the donor and acceptor subsites, there would have to be a significant bend in the acceptor, for which there is some precedence in GH13 glucan-binding sites (45). Alternatively, it is possible that the cyclodextrins, which are conformationally restricted, bind in a different orientation to that of acceptors. For example, linear acceptors could bind in an orientation orthogonal to that of cyclodextrins, removing the need for a bend. Other GH13 enzymes have binding sites in such an orientation (e.g. porcine pancreatic α-amylase isozyme II complexed with trestatin A-derived pseudo-octasaccharide V-1532 (52)). Some support for this possibility comes from the observation that the M. tuberculosis enzyme is not inhibited by cyclodextrins and yet its acceptor specificity is quite similar to that of S. coelicolor GlgE. Although most of the cyclodextrin binding patch is well conserved (Fig. 7C and supplemental Fig. S8), its end distal to the donor site is likely to be different in the mycobacterial enzyme due to the presence of a variable loop (Fig. 7C). This loop bears the Gly-84 backbone that interacts with cyclodextrins in the structures (Fig. 7B) but includes an insertion of nine amino acid residues in the M. tuberculosis enzyme (supplemental Fig. S8). The 23-fold lower Kmapp for maltohexaose and an order of magnitude higher kcatapp with the S. coelicolor enzyme likely reflect the effect of the variable loop. Nevertheless, despite such a significant amino acid insertion, it remains likely that the conserved elements of the patch form part of the acceptor-binding site and help define acceptor length specificity.
The GlgE pathway ultimately generates a branched α-glucan, so it is conceivable that GlgE also extends Y-shaped branched glucans. The arms of such acceptors could occupy conspicuous diagonal and/or linear clefts (Figs. 6B and 7C), the latter being more highly conserved and partly defined by the novel domain S. Further work is clearly required to fully identify the acceptor site.
Relevance to GlgEs from Other Organisms
The glgE gene is widespread among bacteria (6). Most of these genes are similar in length to that of S. coelicolor, making it possible to generate homology models of GlgE proteins based on our structure. Interestingly, there are some examples that are about 60% longer, such as BPSL2074 of the human pathogen Burkholderia pseudomallei K96243. Inspection of the protein sequence encoded by this gene showed that it has an N-terminal extension that results from a partial duplication. This extension is unlikely to exhibit GlgE activity, however, because it lacks most of the catalytic machinery, most of the residues defining the maltose-binding pocket, and domains N and S. Whether it serves some other function remains to be seen.
The GlgEs from S. coelicolor and M. tuberculosis are very similar in length and share very similar properties, allowing one to be used as a structural model for the other. Indeed, the very high degrees of conservation between the maltose-binding site residues of these enzymes (supplemental Fig. S8) and their similar Kmapp values for maltose 1-phosphate (Table 1) illustrate this. This allows the structure of the S. coelicolor enzyme to be used to guide inhibitor design for the M. tuberculosis enzyme, which has been genetically validated as a potential novel drug target (3). Although the maltose site is largely hydrophilic, it includes two aromatic residues that sandwich maltose and provide potential hydrophobic surfaces to enhance the binding of inhibitors to the GlgE of M. tuberculosis and of other animal and plant pathogens (6). Importantly, the distinct configuration of the donor site of this GH13_3 enzyme provides the opportunity to develop inhibitors that do not target the many other GH13 subfamily enzymes present in mammals and plants.
Supplementary Material
Acknowledgments
We thank the beamline scientists at the Diamond Light Source for assistance with x-ray data collection; Lionel Hill for recording mass spectra; Tom Clarke for running the analytical ultracentrifuge; Ainhoa Fernandez for preparing α-maltosyl fluoride, and Rainer Kalscheuer, Stephen G. Withers, and Bernd Nidetzky for helpful discussions.
This work was supported by United Kingdom Biotechnology and Biological Sciences Research Council Grant BB/I012850/1 and the Metabolism Institute Strategic Programme Grant to the John Innes Centre.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S12, Table S1, “Experimental Procedures,” “Discussion,” and additional references.
The atomic coordinates and structure factors (codes 3zss, 3zst, 3zt5, 3zt6, and 3zt7) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
K. Syson and S. Bornemann, unpublished observations.
F. Miah and S. Bornemann, unpublished observations.
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- PDB
- Protein Data Bank
- DP
- degrees of polymerization
- mal
- maltose
- CD
- cyclodextrin.
REFERENCES
- 1. Koul A., Arnoult E., Lounis N., Guillemont J., Andries K. (2011) Nature 469, 483–490 [DOI] [PubMed] [Google Scholar]
- 2. Dye C. (2006) Lancet 367, 938–940 [DOI] [PubMed] [Google Scholar]
- 3. Kalscheuer R., Syson K., Veeraraghavan U., Weinrick B., Biermann K. E., Liu Z., Sacchettini J. C., Besra G., Bornemann S., Jacobs W. R., Jr. (2010) Nat. Chem. Biol. 6, 376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stam M. R., Danchin E. G., Rancurel C., Coutinho P. M., Henrissat B. (2006) Protein Eng. Des. Sel. 19, 555–562 [DOI] [PubMed] [Google Scholar]
- 5. Kalscheuer R., Jacobs W. R., Jr. (2010) Drug News Perspect. 23, 619–624 [DOI] [PubMed] [Google Scholar]
- 6. Chandra G., Chater K. F., Bornemann S. (2011) Microbiology 157, 1565–1572 [DOI] [PubMed] [Google Scholar]
- 7. Preiss J. (2009) in The Encyclopedia of Microbiology (Schaechter M. ed) Vol. 5, 3rd Ed., pp. 145–158, Elsevier, Oxford, UK [Google Scholar]
- 8. Dinadayala P., Lemassu A., Granovski P., Cérantola S., Winter N., Daffé M. (2004) J. Biol. Chem. 279, 12369–12378 [DOI] [PubMed] [Google Scholar]
- 9. Gagliardi M. C., Lemassu A., Teloni R., Mariotti S., Sargentini V., Pardini M., Daffé M., Nisini R. (2007) Cell. Microbiol. 9, 2081–2092 [DOI] [PubMed] [Google Scholar]
- 10. Kaur D., Guerin M. E., Skovierová H., Brennan P. J., Jackson M. (2009) Adv. Appl. Microbiol. 69, 23–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sambou T., Dinadayala P., Stadthagen G., Barilone N., Bordat Y., Constant P., Levillain F., Neyrolles O., Gicquel B., Lemassu A., Daffé M., Jackson M. (2008) Mol. Microbiol. 70, 762–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jackson M., Brennan P. J. (2009) J. Biol. Chem. 284, 1949–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Plaskitt K. A., Chater K. F. (1995) Philos. Trans. R. Soc. Lond. B Biol. Sci. 347, 105–121 [Google Scholar]
- 14. Schneider D., Bruton C. J., Chater K. F. (2000) Mol. Gen. Genet. 263, 543–553 [DOI] [PubMed] [Google Scholar]
- 15. Yeo M., Chater K. (2005) Microbiology 151, 855–861 [DOI] [PubMed] [Google Scholar]
- 16. Damager I., Numao S., Chen H., Brayer G. D., Withers S. G. (2004) Carbohydr. Res. 339, 1727–1737 [DOI] [PubMed] [Google Scholar]
- 17. Jünnemann J., Thiem J., Pedersen C. (1993) Carbohydr. Res. 249, 91–94 [Google Scholar]
- 18. Genghof D. S., Brewer C. F., Hehre E. J. (1978) Carbohydr. Res. 61, 291–299 [Google Scholar]
- 19. de Waard P., Vliegenthart J. F. (1989) J. Magn. Reson. 81, 173–177 [Google Scholar]
- 20. Doublié S. (1997) Methods Enzymol. 276, 523–530 [PubMed] [Google Scholar]
- 21. Brune M., Hunter J. L., Corrie J. E., Webb M. R. (1994) Biochemistry 33, 8262–8271 [DOI] [PubMed] [Google Scholar]
- 22. Leslie A. G. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 23. Evans P. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 24. French S., Wilson K. (1978) Acta Crystallogr. A 34, 517–525 [Google Scholar]
- 25. Sheldrick G. M. (2008) Acta Crystallogr. A 64, 112–122 [DOI] [PubMed] [Google Scholar]
- 26. Cowtan K. (1994) Joint CCP4 + ESF-EACBM Newsletter on Protein Crystallography 31, 34–38 [Google Scholar]
- 27. Cowtan K. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 28. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 29. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 30. Read R. J. (1986) Acta Crystallogr. A 42, 140–149 [Google Scholar]
- 31. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) Nucleic Acids Res. 35, W375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. MacRae I. J., Doudna J. A. (2007) Acta Crystallogr. D Biol. Crystallogr. 63, 993–999 [DOI] [PubMed] [Google Scholar]
- 34. DeLano W. L. (2002) The PyMOL User's Manual, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
- 35. Elbein A. D., Pastuszak I., Tackett A. J., Wilson T., Pan Y. T. (2010) J. Biol. Chem. 285, 9803–9812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Søgaard M., Abe J., Martin-Eauclaire M. F., Svensson B. (1993) Carbohydr. Polym. 21, 137–146 [Google Scholar]
- 37. MacGregor E. A., Janecek S., Svensson B. (2001) Biochim. Biophys. Acta 1546, 1–20 [DOI] [PubMed] [Google Scholar]
- 38. Janecek S., Svensson B., Henrissat B. (1997) J. Mol. Evol. 45, 322–331 [DOI] [PubMed] [Google Scholar]
- 39. Holm L., Rosenström P. (2010) Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Krissinel E., Henrick K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 41. Skov L. K., Mirza O., Sprogøe D., Dar I., Remaud-Simeon M., Albenne C., Monsan P., Gajhede M. (2002) J. Biol. Chem. 277, 47741–47747 [DOI] [PubMed] [Google Scholar]
- 42. Damager I., Engelsen S. B., Blennow A., Møller B. L., Motawia M. S. (2010) Chem. Rev. 110, 2049–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McCarter J. D., Withers S. G. (1994) Curr. Opin. Struct. Biol. 4, 885–892 [DOI] [PubMed] [Google Scholar]
- 44. Kim J. S., Cha S. S., Kim H. J., Kim T. J., Ha N. C., Oh S. T., Cho H. S., Cho M. J., Kim M. J., Lee H. S., Kim J. W., Choi K. Y., Park K. H., Oh B. H. (1999) J. Biol. Chem. 274, 26279–26286 [DOI] [PubMed] [Google Scholar]
- 45. Roujeinikova A., Raasch C., Sedelnikova S., Liebl W., Rice D. W. (2002) J. Mol. Biol. 321, 149–162 [DOI] [PubMed] [Google Scholar]
- 46. Lee H. S., Kim M. S., Cho H. S., Kim J. I., Kim T. J., Choi J. H., Park C., Lee H. S., Oh B. H., Park K. H. (2002) J. Biol. Chem. 277, 21891–21897 [DOI] [PubMed] [Google Scholar]
- 47. Skov L. K., Mirza O., Henriksen A., De Montalk G. P., Remaud-Simeon M., Sarçabal P., Willemot R. M., Monsan P., Gajhede M. (2001) J. Biol. Chem. 276, 25273–25278 [DOI] [PubMed] [Google Scholar]
- 48. Roujeinikova A., Raasch C., Burke J., Baker P. J., Liebl W., Rice D. W. (2001) J. Mol. Biol. 312, 119–131 [DOI] [PubMed] [Google Scholar]
- 49. Lairson L. L., Withers S. G. (2004) Chem. Commun. 2243–2248 [DOI] [PubMed] [Google Scholar]
- 50. Henrissat B., Sulzenbacher G., Bourne Y. (2008) Curr. Opin. Struct. Biol. 18, 527–533 [DOI] [PubMed] [Google Scholar]
- 51. Goedl C., Schwarz A., Mueller M., Brecker L., Nidetzky B. (2008) Carbohydr. Res. 343, 2032–2040 [DOI] [PubMed] [Google Scholar]
- 52. Machius M., Vértesy L., Huber R., Wiegand G. (1996) J. Mol. Biol. 260, 409–421 [DOI] [PubMed] [Google Scholar]
- 53. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.