

Abstract
The mechanism by which K+ channels select for K+ over Na+ ions has been debated for the better part of a century. The prevailing view is that K+ channels contain highly conserved sites that selectively bind K+ over Na+ ions through optimal coordination. We demonstrate that a series of alternating sites within the KcsA channel selectivity filter exists, which are thermodynamically selective for either K+ (cage made from two planes of oxygen atoms) or Na+ ions (a single plane of four oxygen atoms). By combining Bennett free energy perturbation calculations with umbrella sampling, we show that when K+ and Na+ are both permitted to move into their preferred positions, the thermodynamic preference for K+ over Na+ is significantly reduced throughout the entire selectivity filter. We offer a rationale for experimental measures of thermodynamic preference for K+ over Na+ from Ba2+ blocking data, by demonstrating that the presence of Ba2+ ions exaggerates K+ over Na+ thermodynamic stability due to the different binding locations of these ions. These studies reveal that K+ channel selectivity may not be associated with the thermodynamics of ions in crystallographic K+ binding sites, but requires consideration of the kinetic barriers associated with the different multi-ion permeation mechanisms.
Keywords: ion selectivity, potassium ion channel, BAR-US method
Potassium (K+) channels are membrane-spanning pores that catalyze rapid and selective transport of K+ ions across cell membranes (1), essential to electrical and chemical activity in the body. The ability of K+ channels to pass K+ ions at near diffusion-limited rates, while deftly selecting against the similar and physiologically abundant Na+ ions, is essential for controlling the membrane resting potential and repolarization during electrical signal propagation (1, 2), and has been at the center of experimental and theoretical investigation for decades.
The high conductance of K+ channels has been explained in terms of a multi-ion “knock-on” mechanism (3, 4), where interionic repulsions balance attractions to the protein to yield near barrierless permeation of K+ ions (5–9). But how do these channels so efficiently discriminate against the similar (only ∼0.4 Å smaller) Na+ ions? In 1972, Bezanilla and Armstrong carried out electrophysiological recordings on squid giant axons to conclude, based on a simplified single binding site model (2, 4), that K+ channels discriminate against Na+ ions by increased entry barriers for Na+ (a mechanism termed “selective exclusion”) (10). However, in 1988, Neyton and Miller used Ba2+ blocking studies in BK channels to propose that a K+ channel pore contains multiple binding sites, and showed that there exists an external “lock-in” site (hampering exit of Ba2+ block to the outside solution), that preferentially binds K+ ions over Na+ ions (11, 12). This selective binding by a factor of ∼1,000 corresponds to a free energy difference of 4–5 kcal/mol for K+ over Na+, and remains the most concrete evidence that the channel pore provides a better home for K+ than Na+ ions.
The hypothesis of selective permeation via selective binding of K+ ions became widely accepted after the emergence of the first high resolution structure of the K+ channel, KcsA (13). This structure revealed that, in the narrow pore region forming the selectivity filter (see Fig. 1), the backbone carbonyl oxygen atoms (as well as threonine hydroxyls) of the TVGYG sequence form a series of cage-like K+ binding sites, termed S1–S4 (Fig. 1, right), and a non-cage-forming S0 site [as well as Sext, not coordinated by protein and nonselective (7, 8)]. These coordination cages consist of two planes of four oxygen atoms that compensate for the energetic cost of dehydration as K+ ions enter the narrow pore. It has been suggested that these oxygen cages provide a “snug fit” for the K+ ions, while being too distant to effectively coordinate the smaller Na+ ion (13). However, the precise, sub-Å positioning of ligands is not likely the origin of selective binding, because the flexibility of the protein backbone allows it to easily deform to accommodate the smaller Na+ ion (14–16). In this study we do not aim to explain the underlying causes of selective binding to the crystallographic cage-of-oxygen sites, which has been the subject of much discussion in recent years (17). What we will show is that the thermodynamic stability of K+ and Na+ ions in those sites is perhaps not what underpins selective permeation through K+ channels.
Fig. 1.

Fully atomistic simulation system (Left) and possible crystallographic K+ (S) and planar Na+ sites (B) for K+ (pink) and Na+ (green), respectively, in the KcsA selectivity filter (Right).
The coordination cage of eight oxygen atoms has been examined intensely in recent years using reduced models (15, 18–21), as well as KcsA itself (e.g., refs. 22–24). These studies showed that the cage-of-oxygen sites provide selective binding for K+ over Na+, by up to 5 kcal/mol, reinforcing the view that K+ channels exclude Na+ ions due to differing thermodynamic stabilities in the crystallographic sites. However, while K+ may bind to any one of the crystallographic cage sites, this may not be where Na+ ions actually bind (23–30), owing to the preference for the smaller Na+ ion to bind to fewer ligands (31, 32). We have previously shown that the S4 and S3 K+ sites are separated by a plane-of-carbonyl oxygen atoms site for Na+ ions, where Na+ ions have been proposed to cause slow blocking from the intracellular side (24) (labeled B34 in Fig. 1), supported by electrophysiological studies and an X-ray crystal structure in the presence of Li+ (24), as well as structures of MthK in the presence of Na+ (30). This planar arrangement of carbonyls has been shown to yield close packing around Na+, whereas the cage arrangement leads to a seemingly unfavorable partial-coordination by the eight ligands (e.g., refs. 24, 27). We demonstrate here that such planar sites exist between all crystallographic K+ sites, and that each exhibits favorable binding for Na+.
Our observation of distinct binding sites for K+ and Na+ presents a challenge for calculating relative thermodynamic stabilities, requiring special methods to ensure each ion is permitted to achieve its preferred multiple ion configurations. To achieve this sampling, we have developed an approach for efficient and convenient combination of alchemical mutation calculations, using Bennett Acceptance Ratio [BAR, (33)] together with Umbrella Sampling [US, (34)], to calculate relative free energies of binding to the selectivity filter. We will show that when K+ is allowed to bind to its free energy minimum position, and Na+ is allowed to bind to its free energy minimum position, all sites in the selectivity filter lose much of their selective binding ability. We will explain how this outcome remains consistent with experimental results, by carrying out free energy calculations that model the Ba2+ blocking experiments of Neyton and Miller. These observations lead us to question the hypothesis of selectivity via selective binding and to consider alternatives originating from the different multi-ion conduction mechanisms of K+ and Na+.
Results and Discussion
We first report molecular dynamics (MD) simulations of the fully atomistic membrane protein system (Fig. 1) to demonstrate distinct K+ and Na+ selective binding sites. We then explore the equilibrium distributions of ion positions and use this information to obtain net measures of selective binding.
Selective Binding to Distinct Filter Sites.
When the channel was initially occupied with K+ ions in one of the two low-lying free energy configurations, S1/S3/Cavity or S0/S2/S4 (6–8), the K+ ions all remained in their respective cage sites (see Fig. S1). Now consider the configuration S0/S2/S4, when the central S2 K+ ion was transformed into a Na+ ion. This configuration was stable for several hundred ps, after which Na+ left the cage site for a neighboring site formed by a plane of four carbonyl oxygens between S2 and S3 (B23). The ion remained there for the rest of the simulation, suggesting a stable Na+ site does exist, even in this central S2 region.
Such a movement, from the crystallographic site to the adjacent planar site was also observed when K+ was transformed into Na+ in the S0 and S4 sites of the S0/S2/S4 configuration, as well as in the S1 and S3 sites of the S1/S3/Cavity configuration. From such trajectories alone, it is clear that one cannot correctly compare stabilities of K+ and Na+ by looking only at the crystallographic sites; by comparing these ions only at the position known to be the free energy minimum for K+, one is biasing the outcome.
The free energy of transformation from K+ to Na+ within a particular site (ΔG), relative to the same transformation in bulk water (ΔGbulk), has been computed to yield a measure of selective thermodynamic binding (ΔΔG) to that site. We first carried out constrained free energy perturbation (FEP) calculations by holding the ion relative to the carbonyl groups of a site. Calculations within the S2 site, surrounded by a cage of eight carbonyl oxygen atoms, yield a familiar K+-selective ΔΔG value of +5.42 ± 0.07 kcal/mol. However, the lower plane of S2 (B23), yields a ΔΔG value of -3.7 ± 0.6 kcal/mol, while the upper plane of S2 (B12) gives -2.2 ± 0.4 kcal/mol, clearly being Na+ selective sites. Table 1 reveals that each cage site (S1, S2, S3, and S4) is K+-selective, but that each planar site (B01, B12, B23, and B34) is Na+-selective. We remark that the plane between S3 and S4 (B34) is the most selective for Na+, by -4.7 kcal/mol. This observation may explain why only that site was inferred from the X-ray structure in the presence of Li+ (24), and for density at that site in the presence of Na+ (35).
Table 1.
Relative thermodynamic stability in selectivity filter sites, ΔΔG(K+ → Na+), from BAR-US (or BAR for unconstrained simulations)
| ΔΔG(kcal/mol) | S0 | S1 | S2 | S3 | S4 |
| Upper plane | −1.4 ± 0.3 (B01) | −2.2 ± 0.4 (B12) | −3.9 ± 0.3 (B23) | −4.7 ± 0.4 (B34) | |
| Crystal/cage | +1.7 ± 0.3 (S0) | +2.6 ± 0.2 (S1) | +5.42 ± 0.07 (S2) | +4.96 ± 0.07 (S3) | +3.97 ± 0.09 (S4) |
| Lower plane | −2.0 ± 0.1 (B01) | −2.8 ± 0.4 (B12) | −3.7 ± 0.6 (B23) | −3.4± 0.5 (B34) | |
| Unconstrained | −0.9 ± 0.1 | −0.3± 0.3 | +0.5 ± 0.3 | +2.0 ± 0.4 | −1.3 ± 0.4 |
| BAR-US | −0.7 ± 0.1 (1D) | −0.56 ± 0.07 (1D) | +0.32 ± 0.06 (1D) | +1.54 ± 0.07 (1D) | −1.3± 0.2 (1D) |
| −0.74 ± 0.07 (2D) | +2.27± 0.05 (3D) | +1.89 ± 0.05 (2D) |
Thus, each site in the selectivity filter is comprised of a K+-selective cage and one or two adjacent Na+-selective planar sites. One might predict from this outcome that if one transforms a K+ ion into a Na+ ion inside the selectivity filter, the Na+ ion would (eventually) move from the cage to an adjacent plane site, lowering its free energy. However, previous studies have tended to either simulate for too short a time, or have held the ion such that only the cage was sampled (7, 15, 22, 26), missing this effect.
Equilibrium Distributions of K+ and Na+.
In order to quantify the relative stability of K+ and Na+ ions, we must know their equilibrium distributions across the cage and adjacent planar sites. Consider first the (most selective) central S2 site. The potential of mean force (PMF) across S2 for K+ (pink curve) is compared to that for Na+ (green curve) in Fig. 2C. These results have used multidimensional US simulations, to be explained below, and represent the free energy of the system as a function of the position of the S2 ion, when all other ions have obtained an equilibrium distribution of their positions. The free energy minimum locations for K+ and Na+ are also indicated in Fig. 2C (left). The K+ PMF exhibits a deep minimum in the cage of S2, and shows that K+ ions would be unstable in both the upper (B12) and lower (B23) planar sites by 2 ∼ 3 kcal/mol. The Na+ PMF across S2 shows a deep minimum in the lower plane (B23), an elevated (by ∼4 kcal/mol) shallow minimum in the cage (explaining why it was trapped there for some time), and an elevated shallow minimum in the upper plane (B12).
Fig. 2.

Free energy minimum ion positions (Left) and PMFs (Right) of K+ (pink) and Na+ (green) across all sites (A, S0; B, S1; C, S2; D S3; E, S4; using different horizontal scales for clarity). For S1, S2, and S3, the PMFs have been derived from multi-ion US. The Na+ PMF has been shifted such that the net relative free energy reproduces the overall ΔΔG for this site.
The PMFs across all filter sites, S0–S4, shown in Fig. 2 A–E (with convergence for the S2 site shown in Fig. S2), similarly reveal a minimum at the crystallographic site location for K+, and wells for Na+ in the adjacent planar sites. In addition to the observation of distinct K+ and Na+ binding sites, it is interesting that the variation in free energy across each site is somewhat similar for K+ and Na+. i.e., although this is not a complete PMF across the filter, it is evident that K+ would encounter a barrier as it passes each plane, and Na+ would encounter a barrier as it passes the cage, and they are not so different. This similarity suggests that once the K+ or Na+ ion enters the selectivity filter (in the presence of other K+ ions in a conducting-state conformation), rates of crossing the filter may be comparable.
We note that the PMFs in Fig. 2 are all consistent with the ΔΔG values for individual sites in Table 1. i.e., ΔΔGK+→Na+(plane) = ΔΔGK+→Na+(cage) + [(WNa+(plane) - WNa+(cage)) - (WK+(plane) - WK+(cage))]. We also note that the ΔΔG calculations in each planar site are approximately consistent between neighboring sites. e.g., ΔΔG for B23 is -3.7 ± 0.6 kcal/mol when computed as the lower plane of S2 in a S0/S2/S4 configuration, and is -3.9 ± 0.3 kcal/mol when computed for the upper plane of S3 in a S1/S3/Cavity configuration. We do observe in Fig. 2, however, differences in both the K+ and Na+ free energies between the upper and lower planes of each site. Sites away from the center of the filter have obvious inherent asymmetry, due to the differing orientations of the carbonyls above and below the ion, different accessibility to water above and below, and the locality of other ions. In addition, the central S2 site may be asymmetric owing to the difference in valine 76 and glycine 77 interactions with the surrounding residues, the weak constraint used to prevent distortion of the filter to a non-conducting-state, as well as movements of K+ ions in other sites (S0 and S4) which differ when the ion moves to the lower or upper plane (Fig. S4). The results in Fig. 2 have already accounted for the movements of other ions in the filter, as we now describe.
The Challenge of Multi-Ion Movements.
The configuration resulting from a cage-plane movement as a result of a K+ → Na+ transformation can be unstable. Given time, other ions can adjust to accommodate this movement, as is especially the case for the S2 site due to the presence of two peripheral K+ ions (in S0 and S4). The sequence of events during unconstrained simulations (Fig. S1) reveals that, after Na+ moved from S2 to B23, the K+ in S0 eventually moved down to S1, and subsequently, the ion in S4 moved deeper into the S4 site, approaching the cavity. The adjustment corresponds to the shift, S0(K+)/S2(K+)/S4(K+) → S0(K+)/B23(Na+)/S4(K+) → S1(K+)/B23(Na+)/S4(K+)⇌S1(K+)/B23(Na+)/Cavity(K+), and resembles the S0/S2/S4 → S1/S3/Cavity change between the two low-lying configurations for K+ ions (6, 7). Similarly, when the Na+ ion is moved to the upper plane of S2, the K+ ion in S4 moves to S3 after several hundred ps. i.e., S0(K+)/S2(K+)/S4(K+) → S0(K+)/B12(Na+)/S4(K+) → S0(K+)/B12(Na+)/S3(K+); likely a precursor to a knock-on event with an eventual configuration resembling S1/S3/Cavity. Thus, the transformation to Na+ may lead to just a single stable configuration, with implications for the multi-ion permeation mechanism.
Fig. 3 reveals the effect on the free energy of the movement of the upper K+ ion (ion 1, originating in S0) relative to the central S2 ion (ion 2), which has settled in the cage for K+ and the lower plane for Na+. In the case where the S2 ion was K+ (pink curve), the preferred position of ion 1 remains S0. In the case where ion 2 was Na+ (green curve), the preferred position for ion 1 is S1, but with a shallow elevated minimum at the original S0 location. Therefore, when the S2 site is occupied by Na+, which moves to the lower planar (B23), the K+ in S0, should, after some time, move to the S1 site, causing a drop in free energy by more than 3 kcal/mol. Fig. S1 reveals similar movements of S1 and S3 ions during transformations within a S1/S3/Cavity configuration, suggesting the need to sample all ions inside the selectivity filter during a K+ → Na+ transformation.
Fig. 3.

Multi-ion sampling challenge. (A) Movement of K+ from S0 to S1 (ion 1) due to a K+ → Na+ transformation in S2 (ion 2). (B) PMFs along the distance between ions 1 and 2. When ion 2 is a K+ ion in the cage of S2 (λ = 0, before transformation; pink curve), ion 1 remains in S0. When ion 2 is a Na+ ion in the B23 plane (λ = 1, after transformation; green curve), ion 1 in S0 moves down to S1 (vertical black arrow). (C) PMFs of ion 1 movement for different intermediate λ values for the ion 2 transformation.
We have carried out two-dimensional (2D) and three-dimensional (3D) US simulations for the S1/S3/Cavity and S0/S2/S4 configurations, respectively. The coordinates of other ions in the 2D or 3D PMFs were then integrated out to yield one-dimensional (1D) PMFs for the S1, S2, and S3 sites, as shown in Fig. 2. These PMFs are generally consistent with 1D US simulations (Fig. S5) because the equilibrium distribution of other ions is dominated by a single low free energy multi-ion configuration, which was established prior to the 1D US simulations. However, improved multi-ion sampling will be seen to have small, though significant influence on the relative stabilities of K+ and Na+ ions.
Net Relative Stability of K+ and Na+ Ions.
We would like to generate a meaningful measure of the relative thermodynamic stability of Na+ and K+ ions within the selectivity filter. Given that K+ and Na+ reside in different locations, such an FEP calculation is not straightforward (26), requiring very long simulation times to sample ion positions during a transformation. One instead needs a method that enforces the sampling of an equilibrium distribution of the transforming ion’s position, as well as the other ions inside the filter.
To converge FEP calculations, one typically introduces a linear coupling parameter, λ, to slowly convert from one endpoint (K+) to the other (Na+), via thermodynamic integration (36) or the Weighted Histogram Analysis Method (37). However, the simulation of such hybrid ions can lead to challenges in sampling the multi-ion configuration. E.g., for intermediate coupling parameter λ, a clear double well sampling problem exists for the upper S0–S1 ion during the transformation of the S2 ion (see Fig. 3C; for λ = 0.5, the Kramer’s rate to escape S0 and move to S1 is ∼2.9 ns-1). We therefore must either enforce sampling of an equilibrium distribution of multiple ions for every λ value (through very long simulations or the use of US), or avoid ever simulating such hybrid ions in the first place.
The challenge in calculating a ΔG value for the perturbation K+ → Na+ by using only the endpoints, λ = 0 (K+) and 1 (Na+), is that their configurations are so different, involving different cage and planar coordination structures of K+ and Na+, as well as energy histograms (shown in Fig. S6A left) that do not overlap well. The inability of perturbation energy distributions to overlap is proof that simulations for K+ and Na+ are restricted to sampling different regions of configurational space leading to poor convergence of free energy estimates (see Fig. S6A middle and right). In fact, methods such as BAR rely on analysis of this distribution overlap and will fail. How can we ensure that these histograms overlap well for accurate calculations of relative stability?
Within the K+ channel selectivity filter, we’re in a fairly unique position to address this challenge, because the different coordination geometries correspond to adjacent sites, and can be transformed by simply moving the ion in the z direction by a distance of the order of 1–2 Å. Moreover, we already carried out the simulations to connect these adjacent sites when we performed US simulations. Thus, if we combine the endpoint BAR calculation with US along z, we will sample not only the coordination geometries for the endpoints, but will have equilibrium paths connecting them (see Methods).
We now report measures of relative thermodynamic stability site-by-site, meaning that a crystallographic S site will be combined with the planar B sites above and/or below. One could alternatively assign just one planar site to each cage site, but would obtain fairly similar overall measures (at worst a kT ln 2 ≈ 0.4 kcal/mol difference). Using BAR-US, we obtain good convergence (see Fig. S6B) and calculate a net thermodynamic selectivity, ΔΔG, for the S2 region of just +2.27 ± 0.05 kcal/mol (∼50 fold in relative binding), revealing a loss of thermodynamic preference in that site from ∼5 kcal/mol (> 1,000-fold) for calculations based on the cage only. This result is not surprising given that the calculation encompasses both highly K+-selective cage and Na+-selective plane sites. This reduction in thermodynamic preference is observed for all sites (Table 1), with only S2 and S3 maintaining some thermodynamic preference for K+ over Na+, and with S0, S1, and S4 actually exhibiting an overall preference for Na+ ions.
In separate simulations, the occurrence of carbonyl flipping led to a distorted filter conformation in which Na+ preferentially binds to the cage of S2 (Fig. S7), possibly due to reduced strains for the partially coordinating cage with fewer ligands. In this geometry, we find a net ΔΔG of +3.0 kcal/mol, consistent with a previous estimate for the cage alone (38). However, it is known that such a conformation leads to increased barriers for K+ permeation, suggesting a nonconducting channel (38–40). Importantly, regardless of whether or not carbonyl flipping is permitted, we have demonstrated that the KcsA selectivity filter does not select K+ over Na+ ions by the extent previously suggested. We now offer an explanation for how this reduced thermodynamic preference could remain consistent with experimental evidence.
Rationalizing Selective Binding Experiments.
The accepted experimental measurement of preferential binding of K+ over Na+ ions in the K+ channel selectivity filter comes from an analysis of Ba2+ channel blocking single channel recordings (11, 12). Ba2+ blocks the channel inside the selectivity filter (41, 42). The external binding of K+ or Na+, at a site termed the “lock-in” site, can prevent Ba2+ from escaping to the outside. By modeling the relief from Ba2+ block [exponential fitting of off-rates in the long-closure regime (12)] as a function of K+ or Na+ concentration, equilibrium constants for K+ and Na+ binding were found to have a ratio of ∼1,000 (11, 12), corresponding to an energy difference of 4–5 kcal/mol, which appears inconsistent with the above calculations.
The KcsA selectivity filter possesses two Ba2+ binding sites, in S4 and S2 (toward upper plane of S2) (42). We suggest that a filter occupied by Ba2+ ions, to a significant extent in S2, could have considerable influence on the binding of K+ and Na+ ions in the external lock-in site above S2. We have shown in this study that this site would not be one single site, but rather distinct K+ and Na+ sites that will be influenced differently by Ba2+ ions. As an aside, higher ion concentrations can promote inward dissociation of Ba2+ ions; the so-called “enhancement” effect (11). We hypothesize that, at large external concentrations, K+ may bind to S2, with Ba2+ residing in S4 only, as seen in an earlier structure (41), consistent with the stronger voltage dependence of this site (11, 41). Here we focus only on the lock-in effect and determine the effects of Ba2+ ions in S4 and S2 on relative binding of K+ and Na+.
Fig. 4C shows the PMFs for K+ and Na+ entering the filter from the outside, in the presence of K+ ions in S4 and S2 (dashed curves). It can be seen that binding of both ions is favorable, by 1–2 kcal/mol, and that K+ binds at the crystallographic site of S0, while Na+ binds in the plane site B01, between S0 and S1 (Fig. 4A). Interestingly, binding of Na+ is slightly stronger than K+ in the presence of only K+ inside the filter. Fig. 4C also shows the profiles when the filter is instead occupied by Ba2+ ions, constrained to reside in both S4 and S2 sites (solid curves; noting that Ba2+ resides in the S2 cage). The binding locations of K+ and Na+ remain S0 and B01 (Fig. 4B), respectively, consistent with the weak voltage dependence of binding to a lock-in site near the extracellular solution (41, 43).
Fig. 4.

(A) Binding locations for K+ (pink) and Na+ (green) from the outside (vertical arrows) in the presence of K+ ions in S2 and S4 (dashed arrows). (B) Binding locations in the presence of Ba2+ ions (yellow) in S2 and S4 (solid arrows). (C) PMFs for binding of K+ (pink) or Na+ (green) in the presence of K+ in S2 and S4 (dashed), Ba2+ ions in S2 and S4 (solid) or Ba2+ ions in B12 and S4 (dotted).
The comparison of solid and dashed curves (see Fig. 4C) highlights the detrimental effect that Ba2+ ions have on external binding, creating a significant bias toward K+. Because Na+ binds deeper inside the filter, it is affected more adversely by the Ba2+ ion. Remarkably, the K+ ion is stabilized by nearly 1 kcal/mol (possibly due to the alignment of coordinating water molecules), whereas the Na+ is destabilized by nearly 2 kcal/mol. i.e., the relative stabilities are influenced by 2–3 kcal/mol by the Ba2+ ions. We have calculated the relative KD values (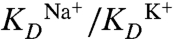 ) to observe a change from 1.26/2.19 = 0.57 to 6.64/1.00 = 6.64 when Ba2+ is introduced, corresponding to more than one order of magnitude change.
) to observe a change from 1.26/2.19 = 0.57 to 6.64/1.00 = 6.64 when Ba2+ is introduced, corresponding to more than one order of magnitude change.
Intuitively, this effect should be greater if Ba2+ were to reside near the upper plane of S2 (as in ref. 42). However, Fig. 4C (dotted curves) shows that binding of both K+ and Na+ is much weaker, due to the proximity of the Ba2+ ion, and is also less selective. Though weaker binding may be consistent with recent data (43), the precise free energies of Na+ vs. K+ may be sensitive to the model and the holding of the divalent ion in close proximity to K+ or Na+. Despite potential variations with the choice of Ba2+ locations, these calculations offer a means for reconciling the proposed reduced selective binding with experiments.
Conclusions
We have employed fully atomistic simulations to demonstrate that the KcsA selectivity filter consists of a series of crystallographic cage-of-ligand binding sites that are K+-selective, separated by a series of plane-of-ligand binding sites that are Na+-selective. Selective binding of ions to the crystallographic cage binding sites alone, which has been the focus of previous computational studies, does not capture the relative thermodynamic stability of K+ and Na+ inside the K+ channel pore. Instead, when K+ and Na+ are permitted to obtain equilibrium distributions of their positions within the selectivity filter, the net thermodynamic preference is dramatically reduced, by several kcal/mol, even in the most selective S2 region.
We have provided a rationale for how experimental evidence for selective binding may exaggerate the preference for K+ relative to the physiological case, owing to a greater impact on Na+ ions which bind closer to the Ba2+ ions. Because these experiments provide the most accepted estimate for the relative stability of K+ and Na+ ions in a K+ channel pore, we conclude that either this estimate should be downgraded (from ∼5 to ∼2 kcal/mol), remaining consistent with experimental structures that indicate some preference for K+ over Na+ (30), or perhaps even that thermodynamic stability is not the underlying cause of selective permeation in K+ channels.
Given the reduced thermodynamic preference for K+, one can consider the possibility that selective permeation may instead originate from barriers that exclude Na+ ions from the pore. We notice that the external entrance barrier (for a prearranged S2/S4 configuration) for Na+ is higher than that for K+ in Fig. 4, by as much as 2 kcal/mol. This increased entrance barrier originates from the Na+ binding site being located deeper inside the filter than K+, with no stable Na+ coordination above the B01 planar site. In contrast, K+ can remain continuously hydrated as it only has to reach S0. However, this estimate of the dehydration barrier maybe insufficient to explain selective permeation of KcsA, as well as the absence of rapid external block by Na+ (2). Perhaps higher barriers for Na+ may arise due to the different multi-ion configurations for Na+ or K+-Na+ mixtures. We previously showed, for the case of ion entry from the intracellular side, that the presence of Na+, with different (by ∼1 Å) binding site locations, disrupts the optimized knock-on conduction mechanism of KcsA, leading to large permeation barriers (24). We suggest that such an elevated barrier for Na+ may also exist for entry from the external solution.
For a long time it has been thought that there is no home for Na+ ions inside a K+ channel selectivity filter. We showed here that there exists much less preferential binding of K+ over Na+ within the selectivity filter of a K+ channel than previously thought. Unlike transporters, ion channels rely on short occupation times to facilitate rapid transit of permeant ions, and thus K+ ions cannot be too stable inside the selectivity filter. At the same time, protein backbone is known to be inherently capable of providing stable coordination of both K+ and Na+, yet with Na+ requiring fewer ligands than K+. Given that the K+ channel selectivity filter contains sites with both four or eight carbonyl ligands, perhaps it is no surprise that the K+ channel is capable of stabilizing both K+ and Na+ ions. This study has demonstrated the need to take a broader view of selectivity mechanisms, that does not focus solely on the properties of individual K+ binding sites, but which requires consideration of the multiple ion conduction process.
Methods
The fully atomistic MD simulation system, including the KcsA protein [PDB 1k4c(35)] embedded in a hydrated dipalmitoylphosphatidylcholine membrane (Fig. 1), has been simulated using the CHARMM program (44), with details provided in the SI Text. K+ ions were placed and equilibrated in one of the two low-lying free energy configurations, S1/S3/Cavity or S0/S2/S4 (6–8). To prevent flipping of valine 76 carbonyl groups in the selectivity filter [possibly associated with a non-conducting-state (35, 38, 39)], a weak dihedral constraint has been imposed, as done in previous studies [e.g., (45)]. Simulations without this constraint are presented in Fig. S7.
We have employed US (34) by combining a series of biased simulations (see SI Text), to calculate 1D PMFs across S0, S1, S2, S3, and S4. Observations of multiple ion movements led us to also carry out 2D and 3D US for multiple ions inside the selectivity filter, from which an improved 1D PMF was calculated using Eq. S10. 1D US simulations have also been used to study K+ and Na+ binding from outside the channel in the presence of either K+ or Ba2+ ions (Fig. 4).
Free energies of alchemical perturbation were computed for an ion held at a particular cage (eight carbonyl) or plane (four carbonyl) center of mass, using the S0/S2/S4 configuration to study K+ → Na+ transformations in S0, S2, or S4, as well as the S1/S3/Cavity configuration to study S1 and S3 sites. For unconstrained ΔG calculations, a flat-bottom constraint on the region, ranging from -10 to +8 Å, was imposed. The results listed in Table 1 used the BAR method, with other FEP methods described in the SI Text. We calculated unnormalized histograms, HK+(ΔU) and HNa+(ΔU), of the energy difference, ΔU = U(λ = 1) - U(λ = 0), from simulations with endpoints λ = 0 (K+) and 1 (Na+), respectively. Bennett-based methods rely on examination of the overlap of these histograms, with the optimal estimate of the free energy obtained via the iterative BAR equations (Eqs. S5 and S6) (33). However, in unconstrained simulations, following equilibration, BAR produced poor overlaps due to the slow rates of ion movement across a site (Fig. S6A). We overcame this limitation by using the BAR-US method (see SI Text and Fig. S6B; with a comprehensive study of applications of this method to be reported in future), where we introduced the conditional histogram of energy difference, given a particular value of z, Hion(ΔU|z), where the simulated ion = K+ or Na+. After computing the normalized position density, 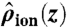 , we computed the energy histogram over the entire region, z0 to z1, by
, we computed the energy histogram over the entire region, z0 to z1, by
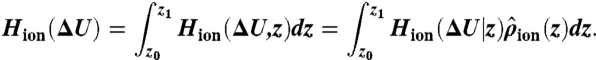 |
[1] |
Fig. S6B (left), reveals excellent histogram overlap from use of Eq. 1 allowing for more accurate free energy calculation using the BAR approach (33), involving the substitution of Eq. 1 into Eqs. S5 and S6. We remark that one could alternatively combine an FEP result for a single location (e.g., a cage) with the PMFs for K+ and Na+ across a site to obtain similar results (SI Text), but this does not make use of all perturbation data across the site, and thus we favor the more accurate BAR-US approach.
Supplementary Material
Acknowledgments.
The authors thank Crina Nimigean and Olaf Andersen for helpful discussions. We acknowledge National Science Foundation (NSF) awards MCB-0546768 and MCB-1052477, and Teragrid award MCB-050005.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1110735108/-/DCSupplemental.
References
- 1.Hille B. Ion channels of excitable membranes. 3rd ed. Sunderland, Ma: Sinauer; 2001. [Google Scholar]
- 2.Nimigean CM, Allen TW. Origins of ion selectivity in potassium channels from the perspective of channel block. J Gen Physiol. 2011;137:405–413. doi: 10.1085/jgp.201010551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodgkin AL, Keynes RD. The potassium permeability of a giant nerve fibre. J Physiol. 1955;128:61–88. doi: 10.1113/jphysiol.1955.sp005291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hille B. Potassium channels in myelinated nerve. Selective permeability to small cations. J Gen Physiol. 1973;61:669–686. doi: 10.1085/jgp.61.6.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung SH, Allen T, Hoyles M, Kuyucak S. Permeation of ions across the potassium channel: brownian dynamics studies. Biophys J. 1999;77:2517–2533. doi: 10.1016/S0006-3495(99)77087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aqvist J, Luzhkov V. Ion permeation mechanism of the potassium channel. Nature. 2000;404:881–884. doi: 10.1038/35009114. [DOI] [PubMed] [Google Scholar]
- 7.Bernèche S, Roux B. Energetics of ion conduction through the K+ channel. Nature. 2001;414:73–77. doi: 10.1038/35102067. [DOI] [PubMed] [Google Scholar]
- 8.Morais-Cabral JH, Zhou Y, MacKinnon R. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 2001;414:37–42. doi: 10.1038/35102000. [DOI] [PubMed] [Google Scholar]
- 9.Bernèche S, Roux B. A microscopic view of ion conduction through the kcsa K+ channel. Proc Nat Acad Sci USA. 2003;100:8644–8648. doi: 10.1073/pnas.1431750100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bezanilla F, Armstrong CM. Negative conductance caused by entry of sodium and cesium ions into the potassium channels of squid axons. J Gen Physiol. 1972;60:588–608. doi: 10.1085/jgp.60.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neyton J, Miller C. Discrete Ba2+ block as a probe of ion occupancy and pore structure in the high-conductance Ca2+-activated K+ channel. J Gen Physiol. 1988;92:569–586. doi: 10.1085/jgp.92.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neyton J, Miller C. Potassium blocks barium permeation through a calcium-activated potassium channel. J Gen Physiol. 1988;92:549–567. doi: 10.1085/jgp.92.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doyle D, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 14.Allen TW, Andersen OS, Roux B. On the importance of flexibility in studies of ion permeation. J Gen Physiol. 2004;124:679–690. doi: 10.1085/jgp.200409111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noskov S, Bernèche S, Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–834. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 16.Noskov SY, Roux B. Importance of hydration and dynamics on the selectivity of the KcsA and NaK channels. J Gen Physiol. 2007;129:135–143. doi: 10.1085/jgp.200609633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen OS. Perspectives on: ion selectivity. J Gen Physiol. 2011;137:393–395. doi: 10.1085/jgp.201110651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varma S, Rempe SB. Tuning ion coordination architectures to enable selective partitioning. Biophys J. 2007;93:1093–1099. doi: 10.1529/biophysj.107.107482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas M, Jayatilaka D, Corry B. The predominant role of coordination number in potassium channel selectivity. Biophys J. 2007;93:2635–2643. doi: 10.1529/biophysj.107.108167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu H, Noskov SY, Roux B. Two mechanisms of ion selectivity in protein binding sites. Proc Natl Acad Sci USA. 2011;107:20329–20334. doi: 10.1073/pnas.1007150107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bostick DL, Brooks CL. Selective complexation of K+ and Na+ in simple polarizable ion-ligating systems. J Am Chem Soc. 2010;132:13185–13187. doi: 10.1021/ja106197e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luzhkov VB, Aqvist J. K(+)/Na(+) selectivity of the KcsA potassium channel from microscopic free energy perturbation calculations. Biochim Biophys Acta. 2001;1548:194–202. doi: 10.1016/s0167-4838(01)00213-8. [DOI] [PubMed] [Google Scholar]
- 23.Shrivastava IH, Tieleman DP, Biggin PC, Sansom MS. K(+) versus Na(+) ions in a K channel selectivity filter: a simulation study. Biophys J. 2002;83:633–645. doi: 10.1016/s0006-3495(02)75197-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson AN, et al. Mechanism of potassium-channel selectivity revealed by Na+ and Li+ binding sites within the KcsA pore. Nat Struct Mol Biol. 2009;16:1317–1324. doi: 10.1038/nsmb.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biggin P, Smith G, Shrivastava I, Choe S, Sansom M. Potassium and sodium ions in a potassium channel studied by molecular dynamics simulations. Biochim Biophys Acta. 2001;1510:1–9. doi: 10.1016/s0005-2736(00)00345-x. [DOI] [PubMed] [Google Scholar]
- 26.Burykin A, Kato M, Warshel A. Exploring the origin of the ion selectivity of the KcsA potassium channel. Proteins. 2003;52:412–426. doi: 10.1002/prot.10455. [DOI] [PubMed] [Google Scholar]
- 27.Miloshevsky GV, Jordan PC. Conformational changes in the selectivity filter of the open-state KcsA channel: an energy minimization study. Biophys J. 2008;95:3239–3251. doi: 10.1529/biophysj.108.136556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fowler PW, Tai K, Sansom MS. The selectivity of K+ ion channels: testing the hypotheses. Biophys J. 2008;95:5062–5072. doi: 10.1529/biophysj.108.132035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Egwolf B, Roux B. Ion selectivity of the KcsA channel: a perspective from multi-ion free energy landscapes. J Mol Biol. 2010;401:831–842. doi: 10.1016/j.jmb.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye S, Li Y, Jiang Y. Novel insights into K+ selectivity from high-resolution structures of an open K+ channel pore. Nat Struct Mol Biol. 2010;17:1019–1023. doi: 10.1038/nsmb.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varma S, Sabo D, Rempe SB. K+/Na+ selectivity in K channels and valinomycin: over-coordination versus cavity-size constraints. J Mol Biol. 2008;376:13–22. doi: 10.1016/j.jmb.2007.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bucher D, Guidoni L, Carloni P, Rothlisberger U. Coordination numbers of K and Na ions inside the selectivity filter of the KcsA potassium channel: insights from first principles molecular dynamics. Biophys J. 2010;98:L47–L49. doi: 10.1016/j.bpj.2010.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennett CH. Efficient estimation of free energy differences from Monte Carlo Data. J Comput Phys. 1976;22:245–268. [Google Scholar]
- 34.Torrie GM, Valleau JP. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J Comp Phys. 1977;23:187–199. [Google Scholar]
- 35.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 36.Kirkwood JG. Statistical mechanics of fluid mixtures. J Chem Phys. 1935;3:300–313. [Google Scholar]
- 37.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem Rev. 1992;13:1011–1021. [Google Scholar]
- 38.Berneche S, Roux B. A gate in the selectivity filter of potassium channels. Structure. 2005;13:591–600. doi: 10.1016/j.str.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 39.Zhou Y, MacKinnon R. The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J Mol Biol. 2003;333:965–975. doi: 10.1016/j.jmb.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 40.Cuello LG, Jogini V, Cortes DM, Perozo E. Structural mechanism of C-type inactivation in K(+) channels. Nature. 2010;466:203–208. doi: 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y, MacKinnon R. The barium site in a potassium channel by X-ray crystallography. J Gen Physiol. 2000;115:269–272. doi: 10.1085/jgp.115.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lockless SW, Zhou M, MacKinnon R. Structural and thermodynamic properties of selective ion binding in a K+ channel. PLoS Biol. 2007;5:e121. doi: 10.1371/journal.pbio.0050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piasta KN, Miller C. KcsA barium permeation blocked by external potassium. Biophys J. 2010;98:115a. [Google Scholar]
- 44.Brooks BR, et al. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 45.Khalili-Araghi F, Tajkhorshid E, Schulten K. Dynamics of K ion conduction through Kv1.2. Biophys J. 2006;91:L72–L74. doi: 10.1529/biophysj.106.091926. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.