

Abstract
Sepsis remains the leading cause of death in critically ill patients despite modern advances in critical care. Intestinal barrier dysfunction may lead to secondary bacterial translocation and the development of the multiple organ dysfunction syndrome during sepsis. Cyclooxygenase-2 (COX-2) is highly upregulated in the intestine during sepsis and we hypothesized that it may be critical in the maintenance of intestinal epithelial barrier function during peritonitis-induced polymicrobial sepsis. COX-2−/− and COX-2+/+ BALB/c mice underwent cecal ligation and puncture (CLP) or sham surgery. Mice chimeric for COX-2 were derived by bone marrow transplantation and underwent CLP. C2BBe1 cells, an intestinal epithelial cell line, were treated with the COX-2 inhibitor NS-398, PGD2, or vehicle and stimulated with cytokines. COX-2−/− mice developed exaggerated bacteremia and increased mortality compared with COX-2+/+ mice following CLP. Mice chimeric for COX-2 exhibited the recipient phenotype suggesting that epithelial COX-2 expression in the ileum attenuates bacteremia following CLP. Absence of COX-2 significantly increased epithelial permeability of the ileum and reduced expression of the tight junction proteins zonula occludens-1 (ZO-1), occludin, and claudin-1 in the ileum following CLP. Furthermore, PGD2 attenuated cytokine-induced hyperpermeability and ZO-1 downregulation in NS-398-treated C2BBe1 cells. Our findings reveal that absence of COX-2 is associated with enhanced intestinal epithelial permeability and leads to exaggerated bacterial translocation and increased mortality during peritonitis-induced sepsis. Taken together, our results suggest that epithelial expression of COX-2 in the ileum is a critical modulator of tight junction protein expression and intestinal barrier function during sepsis.
Introduction
Sepsis is a complex illness resulting from a systemic inflammatory response to infection and is the leading cause of death in critically ill patients (1). Intra-abdominal infection, often leading to polymicrobial sepsis, accounts for 20% of cases of sepsis and has substantial mortality of up to 60% (2). In this study, we investigated the role of cyclooxygenase-2 (COX-2), the inducible isoform of COX, in a murine model of polymicrobial sepsis. The COX enzymes (COX-1 and COX-2) catalyze the conversion of arachidonic acid to PGH2 which is then converted to a series of prostanoids by cell-specific synthases (3). COX-2 is upregulated by many pro-inflammatory stimuli including LPS, peptidoglycan, and high mobility group box 1 (HMGB1), as well as pro-inflammatory cytokines (eg. TNF-α and IL-1β) (4). COX-2-derived prostanoids mediate acute inflammation by increasing vascular permeability and recruiting inflammatory cells to sites of infection, however these mediators are also key regulators in the resolution of inflammation (5).
In the gastrointestinal (GI) tract, COX-2-derived prostaglandins play a key role in mucosal repair and defense (6, 7). As COX-1 is constitutively expressed, initial studies suggested that COX-2 activity contributed minimally to homeostatic prostaglandin production in the GI tract. Accumulating evidence suggests, however, that COX-2 plays a critical role in both prostaglandin production and mucosal defense in the GI tract in the setting of mucosal injury (6, 7). Selective inhibition of COX-2 led to significantly increased gastric injury in an ischemia-reperfusion injury model (8), while inhibition of both COX-1 and COX-2 was necessary for inducing gastric ulceration in rats (9). In the colon, selective inhibition of COX-2 led to increased colonic ulceration and injury in a trinitrobenzene sulfonic acid (TNBS) colitis model in rats (10), while deficiency of COX-2 led to significantly increased colonic injury following dextran sulfate sodium (DSS) compared with COX-1 deficient and wild-type mice (11). Additionally, COX-2 deficient mice have been reported to develop spontaneous peritonitis (11, 12), further supporting the role of COX-2-derived prostaglandins in preserving mucosal integrity in the GI tract. In the small bowel, COX-1-derived prostaglandins protect against epithelial cell injury following radiation (13, 14), however the contribution of COX-2-derived prostanoids to mucosal defense in the small intestine is less well characterized.
Increasing evidence suggests that epithelial cell barrier dysfunction may play a significant role in the development of the multiple organ dysfunction syndrome (MODS) during sepsis (15). Furthermore, studies suggest that increased intestinal permeability is common in critically ill patients (16) and that it is independently associated with the development of MODS in patients with critical illness (17). Tight junctions are critical for the maintenance of normal epithelial barrier function and are the key regulators of epithelial paracellular permeability. Tight junctions are located at the most apical region of the paracellular space between adjacent epithelial cells and form a selectively permeable barrier that controls paracellular flux of ions, hydrophilic solutes, and water (18). In the intestine, tight junctions also serve as the principal barrier against the paracellular leak of toxic luminal antigens and translocation of enteric microbial products. Tight junctions are assembled from at least three transmembrane proteins including occludin, the claudins, and the junctional adhesion molecule (JAM) that are anchored to the cytoskeleton via peripheral membrane proteins such as zonula occludens-1 (ZO-1) (18). ZO-1 plays a key role in the assembly of the tight junction complex as it has multiple binding domains that allow it to interact with the intracellular domains of the integral membrane proteins occludin and the claudins, as well as bind to actin, thereby anchoring the complex into the perijunctional actomyosin ring (18).
Many extracellular stimuli, including cytokines and bacterial infection, have been shown to disrupt epithelial tight junction integrity and compromise paracellular barrier function (18, 19). In the intestine, this can lead to increased paracellular translocation of normally excluded luminal antigens and enteric bacteria. Furthermore, penetration of luminal microbial products through the paracellular leak pathway can lead to mucosal immune activation with recruitment of inflammmatory cells and secretion of pro-inflammatory mediators which further increase barrier permeability and compromise intestinal barrier function. LPS, HMGB1, and several pro-inflammatory cytokines, including TNF-α, IFN-γ, IL-1β, and IL-6, have been shown to increase transepithelial permeability in intestinal epithelial monolayers (20) and alter expression of tight junction proteins (21) in vitro. In addition, tight junction dysfunction leading to intestinal barrier disruption and bacterial translocation has been demonstrated in animal models of endotoxemia (22, 23) and common bile duct ligation (24). We hypothesized that COX-2 plays an essential role in maintaining intestinal epithelial barrier function during sepsis. While COX-derived prostaglandins have a well-recognized protective role in the GI tract (6, 7), the significance of COX-2-derived prostanoids in the intestinal epithelium during sepsis has yet to be elucidated. Here we demonstrate that COX-2 deficiency is detrimental in a murine model of peritonitis-induced polymicrobial sepsis with increased bacterial translocation, exaggerated inflammation, severe hypotension, and increased mortality following cecal ligation and puncture (CLP). Our results suggest that COX-2 induction in the ileal epithelium is critical to the regulation of tight junction proteins and maintenance of intestinal barrier function during polymicrobial sepsis.
Materials and Methods
Reagents and Antibodies
NS-398, PGD2, SC560, and BW245C were obtained from Cayman Chemical (Ann Arbor, MI). Fluorescein-5-(and-6)-sulfonic acid, trisodium salt (FS) was obtained from Invitrogen (Carlsbad, CA). Abs used include: rabbit anti-ZO-1 (Z-R1), anti-occludin (Z-T22), and anti-claudin-1 (JAY.8) polyclonal Abs (Zymed/Invitrogen); mouse anti-ZO-1 (1/ZO-1) and CD45 (RA3-6B2) and rat Ly-6G (RB6-8C5) mAbs (BD Biosciences, San Jose, CA); mouse β-actin (AC-15) and α-tubulin (B-5-1-2) mAbs (Sigma, St. Louis, MO); rabbit anti-COX-2 and anti-COX-1 polyclonal Abs (Cayman); control mouse IgG1 Ab (Invitrogen); control rabbit IgG Ab and peroxidase-conjugated goat anti-rabbit IgG (Santa Cruz, Santa Cruz, CA); secondary Alexa Fluor® 488-conjugated goat anti-mouse IgG and Alexa Fluor® 546-conjugated goat anti-rabbit IgG Abs (Molecular Probes/Invitrogen); rhodamine TRITC donkey anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA); and peroxidase-conjugated goat anti-mouse IgG (ThermoScientific, Waltham, MA). All cell culture reagents were obtained from Cellgro (Herndon, VA) unless otherwise specified. All other reagents were obtained from Sigma-Aldrich unless specified.
Animals
Mice that were wild-type (+/+) or homozygous null (−/−) for targeted disruption of COX-2 (B6:129S7-Ptgs2tm1Jed, Jackson Laboratories, Bar Harbor, ME) were backcrossed 10 generations to a pure BALB/c background. All experiments were performed on 8-12 week old male and female BALB/c mice (unless otherwise specified) and littermate controls generated by heterozygous breeding. All animals were housed in a pathogen-free barrier facility at Harvard Medical School and all animal experiments performed in compliance with the relevant laws and guidelines as set forth by the Harvard Medical Area Standing Committee on Animals.
CLP model of polymicrobial sepsis
The murine CLP model of polymicrobial sepsis was performed as described (25, 26). Anesthesia was induced by i.p. administration of 100 mg/kg ketamine HCl and 43 mg/kg xylazine HCl. A 1.5-cm midline abdominal incision was made under sterile conditions and 50% of the cecum was ligated below the ileocecal valve thereby preserving intestinal continuity (25, 26). Once ligated, the cecum was punctured once with a 19-gauge (g) needle for mortality experiments or twice with a 23-g needle for everted gut sac experiments to allow for improved survival in COX-2−/− mice. The cecum was repositioned and the abdominal incision closed in layers with 6-0 sutures. Sham-operated mice underwent the same procedure, including opening of the peritoneum and exposing the bowel, but without ligation and needle perforation of the cecum. Following abdominal closure, sham and CLP-operated mice were administered 1 ml of sterile saline i.p.. No antibiotics were administered to the mice after surgery in order to assess the effect of COX-2 on bacterial levels in blood and organs. Survival rates were determined over an 8 d period, with assessment every 12 h. All mice had ad libitum access to food and water.
Blood pressure analysis
Systolic blood pressure (SBP) was measured 24 h before and every 24 h following CLP using a tail-cuff method as described (27).
Histological and cytokine analysis
Ileums and colons were harvested, fixed in 10% formalin or Methyl Carnoy’s at 4°C, and embedded in paraffin. Sections were stained with H&E or Alcian blue and immunostaining performed with an anti-CD45 Ab (1:1000), anti-Ly-6G Ab (1:200), and anti-COX-2 Ab (1:250) (28, 29). Histologic scoring of H&E-stained ileum sections was performed in a blinded manner by a pathologist. The sections were scored for mucosal injury using the Chiu/Park scoring system (30, 31). Mucosal damage was graded from 0 to 8 according to the following criteria: grade 0, normal mucosal villi; grade 1, development of subepithelial space; grade 2, extension of the subepithelial space with moderate lifting of the epithelial layer from the lamina propria; grade 3, massive epithelial lifting down the sides of villi, possibly with a few denuded tips; grade 4, denuded villi with lamina propria and dilated capillaries exposed; grade 5, digestion and disintegration of the lamina propria, hemorrhage, and ulceration; grade 6, crypt layer injury; grade 7, transmucosal infarction; and grade 8, transmural infarction. Quantitative assessment of immunostaining was performed using FRIDA Software (FRamework for Image Dataset Analysis, http://bui2.win.ad.jhu.edu/frida/) (32, 33). This software provides a pixel color threshold mask (hue, saturation, and brightness) through which a range of positive immunohistochemistry color signal is specified and the software quantitates all pixels with the selected range of colors within a field. The same pixel color mask was applied to all samples being analyzed for a given Ab marker. Cytokines were measured in serum by SearchLight multiplex immunoassay (Aushon, Billerica, MA).
Flow cytometry analysis of lamina propria
Twenty-four h following sham and CLP, ileums were isolated and cells from the lamina propria were isolated as described (34) with modification. Ileums were flushed with PBS to remove fecal contents, inverted and shaken in PBS containing 5% DTT and 0.5 M EDTA for 30 min at 37° C. After removing epithelial cells and fat tissue, the intestines were washed in PBS, cut into small pieces, and incubated in RPMI containing 5% FBS, 1.5 mg/ml collagenase type II (GIBCO), and 0.5 mg/ml dispase (GIBCO) for 1 h at 37° C under constant horizontal shaking (250 rpm). Cells from digested intestinal tissues were isolated by centrifugation, washed three times, and resuspended in PBS containing 2% FBS. The isolated cell suspensions were incubated with the mAb 2.4G2 (10 μg/ml) for 20 min at 4° C to block Fcγ receptors. The cells were stained with a combination of the following directly conjugated fluorescent anti-mouse mAb for 30 min at 4° C: CD45-PE-Cy7, F4/80-PE, rat IgG2b-PerCP-Cy5.5, rat IgG2b-APC-Cy7, rat IgG2a-FITC, rat IgG2a-PE-Cy7, rat IgG2a-PE, and rat IgG2b-PE (eBioscience); Ly-6C-FITC, Ly-6G-PE, and CD11b-PerCP-Cy5.5 (BD Biosciences); and CD11b-APC-Cy7 and F4/80-PerCP-Cy5.5 (BioLegend). The cells were analyzed with a BD Biosciences Canto II flow cytometer and Flow Jo software (Tree Star). Live cell and CD45+ cell gating was used to exclude dead and non-hematopoietic cells. Neutrophils were quantified as Ly-6G+Ly-6C+ gated on CD45+CD11b+ cells and macrophages were quantified as F4/80+CD11b+ gated on CD45+ cells.
Bacterial cultures
Serial log10 dilutions of whole blood and homogenized tissue were made and aliquots cultured on LB agar plates as described (25). Mesenteric lymph nodes (MLN) were isolated and weighed, homogenized in 250 μl of PBS as described (35), and aliquots of serial log10 dilutions cultured on LB agar plates. The number of CFU were counted following overnight incubation at 37°C (25).
Bone marrow transplantation (BMT)
Animals chimeric for COX-2 were derived as previously described (29, 36). Recipient COX-2+/+ or COX-2−/− mice (6-8 weeks old) received total body irradiation of 900 cGy (60Co source) in two separate doses 4 h apart. One hour following the second dose of irradiation, 2 × 106 donor cells were delivered to the recipient animal via tail vein injection. Following BMT, mice remained in a pathogen-free facility with ad libitum access to food and water. Six weeks following BMT, chimeric animals for COX-2 (COX-2+/+ donor cells into COX-2−/− recipients [COX-2+/+ → COX-2−/−] and COX-2−/− donor cells into COX-2+/+ recipients [COX-2−/− → COX-2+/+]) were subjected to CLP. Non-chimeric transplanted controls were similarly generated by BMT (COX-2+/+ donor cells into COX-2+/+ mice [COX-2+/+ → COX-2+/+] and COX-2−/− donor cells into COX-2−/− mice [COX-2−/−→ COX-2−/−]) and subjected to CLP as described above.
FITC labeling of bacteria and phagocytosis assay
E. coli (25) and a clinical isolate of S. aureus (37) were heat inactivated and labeled with FITC as described (25, 38). FITC-labeled E. coli (6 × 108 CFU) or S. aureus (3 × 108 CFU) were injected into the peritoneum of COX-2+/+ and COX-2−/− mice and peritoneal neutrophils isolated after 24 h as described (25). Following a 2 h incubation at 37°C, adherent cells were washed and treated with 0.2% trypan blue to quench extracellular fluorescence. The cells were washed, treated with 5 mM EDTA in PBS, gently scraped, and then centrifuged (25). The pellet was resuspended in 400 μl of PBS containing 4% FBS and 0.009% sodium azide. Total cells were counted and 1 × 106 cells were scanned by flow cytometry.
In vivo permeability assay
In vivo intestinal permeability was measured using the FITC-labeled dextran (FD-4) method as described (39, 40) with modification. Mice were administered 100 μl of FD-4 (50 mg/ml) by oral gavage 18 h following CLP and sham. Six h later, blood was harvested, and serum isolated. Serial dilutions of FD-4 were made to generate a standard curve and serum concentrations of FD-4 were determined using a BioTek FLx800 Fluorescence Microplate Reader (BioTek, Winooski, VT) with an excitation wavelength of 490 nm and emission wavelength of 530 nm. Serum concentrations of FD-4 following CLP were normalized to FD-4 concentrations following sham.
Everted gut sac
Ileums were harvested 24 h following CLP and sham surgeries and permeability to FD-4 measured ex vivo by the everted gut sac method as described (41). Briefly, segments of ileum were ligated at one end, everted over a 18-g Jelco catheter (Smiths, Dublin, OH), and sutured to a 20-g Jelco catheter attached to a 1 ml syringe containing Krebs-Henseleit bicarbonate buffer (KHBB, pH 7.4). The everted gut sac was distended with 500 μl of KHBB and suspended in a beaker containing FD-4 (40 mg/ml) in KHBB. A sample was aspirated from the beaker to measure the initial concentration of FD-4 (mucosal surface) and the sac was incubated in the beaker for 30 min. Following incubation, fluid was aspirated from the inside of the sac (serosal surface) and the length of the gut sac measured. The serosal and mucosal samples were centrifuged and the supernatant diluted with PBS. Fluorescence was measured with a F-2500 fluorescence spectrophotometer (Hitachi, Pleasanton, CA). Permeability is expressed as the mucosal-to-serosal clearance (nL/min/cm2) of FD-4 as described (41).
Cell culture
Human intestinal epithelial cells (C2BBe1 CRL-2102, clone of Caco-2) were purchased from ATCC (Manassas, VA) and cultured in DMEM containing 10% FBS, 10,000 IU/ml penicillin, 10,000 mg/ml streptomycin, 29.2 mg/ml L-glutamine, 2 mM sodium pyruvate, and 1× MEM non-essential amino acids in a humidified incubator (21% O2, 5% CO2) at 37° C. Cells were grown on collagen-I coated Biocoat tissue culture dishes (BD) and passages 7-10 were used for all experiments.
In vitro epithelial permeability assays
C2BBe1 cells (5 × 104 cells/well) were plated in collagen-coated transwell inserts (3 μm pore size) in 24 well plates (COSTAR, Corning, NY) and permeability studies performed as described (22, 42) using confluent monolayers 21-24 d after plating. In specific experiments, cells were pre-treated with NS-398 at 10 or 50 μM (doses selective for inhibition of COX-2 (43)) or SC-560 (10 or 50 nM) (doses selective for inhibition of COX-1 (44)) in the basolateral media. NS-398 was dissolved in 25% DMSO/75% PBS while SC-560 was dissolved in 50% DMSO/50% PBS; equal volumes of vehicle were administered as controls. In other experiments, cells were pre-treated with PGD2 (10 μM), or BW245C (10 μM) in the basolateral media. After 24 h, HEPES-buffered DMEM (pH 6.8) containing FS (200 μg/ml) was added to the apical side of the transwells and cytomix (CM) (IFN-γ 1000 U/ml, TNF-α 10 ng/ml, IL-1β 1 ng/ml) or vehicle containing NS-398, SC-560, PGD2, or BW245C was added to the basolateral surface. Fluorescence was measured with a fluorescence microplate reader as above. Permeability is expressed as flux of FS per unit area of membrane divided by the concentration of the probe in the apical compartment (nL/h/cm2) as described (21, 22) and is normalized to the respective vehicle.
Transepithelial resistance (TER) measurements
C2BBe1 cells (1 × 105 cells/well) were plated in collagen-coated transwell inserts (3 μm pore size) in 12 well plates with a surface area of 1.12 cm2. TER was measured in Ohms (Ω) before and after CM stimulation with an epithelial voltohmmeter (World Precision Instruments, Sarasota, FL).
Western blot analysis
Mucosal protein was isolated as described (24) from ileums of COX-2+/+ and COX-2−/− mice. Protein extracts from ileums were analyzed by Western blot with anti-ZO-1 (1:500), anti-occludin (1:2000), anti-claudin-1 (1:1000), anti-COX-2 (1:200), and anti-COX-1 (1:1000) Abs. Equal loading was confirmed with an anti-β-actin Ab (1:5000) or anti-α-tubulin (1:5000) Ab. Densitometry was performed using the Gel Doc XR System with Quantity One-4.6.2 software (Bio-Rad, Hercules, CA) or Image J (NIH, Bethesda, MD).
Immunofluorescence confocal microscopy
C2BBe1 cells were treated with NS-398 (50 μM) or vehicle followed by CM stimulation. After 48 h, cells were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100 in PBS (PBST), and blocked with 3% BSA in PBST for 1 h. Cells were incubated with anti-ZO-1 (1:100), anti-occludin (1:100), or anti-claudin-1 Abs (1:100) for 1 h. Cells were washed and then incubated with secondary Abs (1:200). Cells were washed, counterstained with DAPI, and mounted with VECTASHIELD® Mounting Medium. Staining was analyzed using a laser scanning confocal imaging system (Bio-Rad Radiance 2100) attached to an Olympus 1X71 inverted microscope with an oil-immersion 60× objective.
Quantification of cell death
Trypan blue absorbance
Cells were treated with 100 μl of 10 mM KCN (positive controls) or media for 1 h at 37° C as described (22). The media was removed, 100 μl of 0.4% trypan blue solution was added to each well, and incubated for 15 min at 37° C (45). The cells were visualized under a microscope, washed, and lysed with 300 μl of 0.1% SDS. Absorbance was measured in the SDS/trypan blue solution at 590 nm as described (45) and normalized to KCN positive control wells. Lactate dehydrogenase (LDH) assay. Total LDH and LDH release into the basolateral media was measured via a commercial in vitro toxicology assay kit (TOX-7, Sigma).
RT-PCR
Total RNA was extracted from ileums using the RNeasy Mini Kit (Qiagen, Valencia, CA). RNA was treated with DNase (Invitrogen) followed by reverse transcription to cDNA using the Superscript III qRT-PCR Kit (Invitrogen). Quantitative RT-PCR (qPCR) was performed on a 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA) to determine gene expression for COX-1 (Mm00477214_m1) and COX-2 (Mm00478374_m1) using validated TaqMan gene expression assay primer/probe combinations (Applied Biosystems). All qPCR results were normalized to the expression of the endogenous positive control 18S (Hs99999901_s1). The ΔCt was calculated as the difference between the cycle threshold (Ct) for the gene of interest and the respective Ct for 18S.
ELISA
Ileums were minced in 10 mM sodium phosphate buffer (pH 7.4) as described (46). The supernatants were collected following centrifugation at 9,000 g for 1 min, extracted with acetone, and evaporated to dryness. The supernatants were resuspended in PBS and analyzed for PGD2 (Elisatech, Aurora, CO) and PGE2 (Cayman) by ELISA.
Statistical analysis
Data are presented as median ± IQR except as indicated. Statistical significance was determined by the nonparametric Mann-Whitney U test or unpaired t-test for comparisons between 2 groups or the nonparametric Kruskal-Wallis one-way ANOVA followed by Dunn’s post test for comparisons between more than 2 groups or for multiple comparisons. Categorical and ordinal variables were compared using the χ2 test for trend. Comparisons of mortality were made by analyzing Kaplan-Meier survival curves and the log-rank test was used to assess for differences in survival. The number of samples per group (n) is specified in the Figure Legend. All p values were two-tailed and statistical significance was accepted at p<0.05.
Results
COX-2 deficient mice have increased mortality and exaggerated bacteremia following peritonitis-induced polymicrobial sepsis
To examine the role of COX-2 in a polymicrobial model of sepsis, COX-2+/+ male BALB/c mice underwent CLP using a 19-g needle and a single cecal puncture. COX-2, but not COX-1, expression was induced 3-fold in the ileum of COX-2+/+ mice at the mRNA level (Fig. 1A-B) and more than 20-fold at the protein level (Fig. 1C-D) following CLP. Given the dramatic induction of COX-2 in the ileum following CLP, we hypothesized that COX-2 was playing a critical role in the ileum during peritonitis-induced sepsis. To investigate the significance of COX-2 induction in the ileum on mortality during polymicrobial sepsis, COX-2 deficient (COX-2−/−) and COX-2+/+ male BALB/c mice underwent CLP. Absence of COX-2 was detrimental during polymicrobial sepsis with a significant increase in mortality in COX-2−/− mice following CLP compared with COX-2+/+ mice. COX-2−/− mice had 80% mortality by day 4 following CLP compared with 30% mortality in COX-2+/+ mice by day 5 (Fig. 2A). In addition, COX-2−/− mice developed significant hypotension following CLP compared with COX-2+/+ mice. At baseline, there was no significant difference in SBP between COX-2−/− mice (SBP 113 ± 3 mm Hg) and COX-2+/+ mice (SBP 119 ± 1.5 mm Hg), however, COX-2−/− mice developed significant hypotension (SBP 57 ± 4 mm Hg) compared with COX-2+/+ mice (SBP 73 ± 2 mm Hg, p<0.05) 72 h following CLP.
FIGURE 1.

COX-2, but not COX-1, is upregulated in the ileum following peritonitis-induced polymicrobial sepsis. RNA was isolated from ileums of COX-2+/+ mice following sham or CLP (n=2-3), reverse-transcribed, and qPCR performed for COX-2 (A) and COX-1 (B). Results were normalized to endogenous control 18S (p<0.05 by the Mann-Whitney test). (C) Protein was harvested from ileums of COX-2+/+ mice (n=2-4) following sham or CLP and Western blot performed for COX-2 and COX-1. (D) Bar graphs represent protein levels (normalized for α-tubulin) as determined by densitometry. Protein levels are expressed as fold induction compared with COX-2+/+ mice following sham (p<0.05 by the Kruskal-Wallis one-way ANOVA test followed by Dunn’s post test). (E) Representative COX-2 immunostaining of ileums harvested from COX-2+/+ and COX-2−/− mice 24 h following sham or CLP. Arrows represent positively stained inflammatory cells in the lamina propria and arrowheads represent positively stained epithelial cells in the ileum.
FIGURE 2.

COX-2 deficient mice have increased mortality and exaggerated bacteremia following peritonitis-induced polymicrobial sepsis. (A) Male BALB/c COX-2+/+ (■ n=10) and COX-2−/− (□ n=9) underwent CLP with a 19-g needle (1 hole) in two independent experiments. Survival was monitored over 8 d. Data are expressed as percent of mice alive at each time point (p<0.05 by the log-rank test). (B) Blood was drawn from COX-2+/+ (■ n=11) and COX-2−/− (□ n=11) mice via right ventricular puncture and (C) MLN (COX-2+/+ ■ n=7; COX-2−/− □ n=12) and (D) organs (COX-2+/+ ■ n=11; COX-2−/− □ n=12) harvested 24 h following CLP. Blood and homogenized tissue were serially diluted and cultured on LB plates. Horizontal bars represent the median of each group and are from at least four independent experiments (p<0.05 by the Mann-Whitney test).
To investigate the mechanism of increased mortality in COX-2−/− mice during polymicrobial sepsis, blood, MLN, and organs (lung, liver, and spleen) were cultured 24 h following CLP. COX-2−/− mice demonstrated increased incidence of bacteremia following CLP in addition to higher numbers of circulating bacteria and translocation to MLN compared with COX-2+/+ mice (Fig. 2B-C). Similarly, organ cultures from COX-2−/− mice showed significantly higher levels of bacteria in the lung, liver, and spleen following CLP compared with COX-2+/+ mice (Fig. 2D). Increased bacteremia and exaggerated bacterial seeding of organs was also observed in COX-2−/− mice 48 h following CLP (data not shown).
Increased intestinal inflammation in COX-2 deficient mice following peritonitis-induced polymicrobial sepsis
COX-2 deficient mice demonstrated increased ileal inflammation and pro-inflammatory cytokine production following peritonitis-induced polymicrobial sepsis. Examination of the intestinal tissue revealed no significant difference in necrosis of the cecum between COX-2−/− and COX-2+/+ mice following CLP. Furthermore, although there was marked ileal inflammation and injury in both COX-2+/+ and COX-2−/− mice following CLP (Fig 3A, c, d respectively) compared with sham (Fig 3A, a, b respectively), there was no histologic difference in intestinal mucosal injury between COX-2−/− (Fig. 3A, d) and COX-2+/+ mice (Fig. 3A, c) following CLP. Histologic scoring of H&E-stained ileums by a pathologist in a blinded fashion demonstrated a significant increase in mucosal injury in COX-2−/− and COX-2+/+ mice 48 h following CLP compared with sham, however there was no significant difference in mucosal damage between COX-2−/− and COX-2+/+ mice following CLP (Fig 3B). Similar findings were observed at 24 h following CLP (data not shown). In addition, quantitation of Alcian blue staining demonstrated no difference in goblet cell number per crypt-villus in the ileums of COX-2−/− mice (Fig. 3A, h) following CLP compared with COX-2+/+ mice (Fig. 3A, g; Fig 3C).
FIGURE 3.

COX-2 deficient mice have increased ileal inflammation following peritonitis-induced polymicrobial sepsis. COX-2+/+ and COX-2−/− mice underwent CLP with a 19-g needle (1 hole). Ileums were harvested 48 h following CLP. (A) Representative H&E staining (a-d), Alcian blue staining (e-h), CD45 immunostaining (i-l), and Ly-6G immunostaining (m-p) in COX-2+/+ and COX-2−/− mice following sham surgery (left) and CLP (right). Arrows indicate cells staining positive for CD45 (l) and Ly-6G (p) in COX-2−/− mice following CLP. (B) Histologic scoring of mucosal injury in H&E-stained ileums following sham (COX-2−/− n=4; COX-2+/+ n=5) and CLP (COX-2−/− n=7; COX-2+/+ n=9) from two independent experiments (*p<0.05 versus COX-2−/− sham and **p<0.05 versus COX-2+/+ sham by the χ2 test for trend). (C) Quantitation of goblet cell number (3 mice per condition) per crypt-villus in COX-2−/− and COX-2+/+ mice following sham and CLP. Quantitation of CD45 (D) and Ly-6G (E) immunostaining in ileums of COX-2−/− and COX-2+/+ mice (2-5 mice per condition) demonstrates that COX-2−/− mice have significantly increased neutrophilic inflammation in the ileum compared with COX-2+/+ mice following CLP (*p<0.05 for COX-2−/− mice after CLP versus COX-2−/− mice after sham and versus COX-2+/+ mice after CLP by the Mann-Whitney test).
However, H&E staining and immunostaining for CD45 and Ly-6G revealed enhanced inflammation in the ileums of COX-2−/− mice (Fig. 3A, d, l, p) following CLP compared with COX-2+/+ mice (Fig. 3A, c, k, o). While there was no apparent histologic difference in intestinal mucosal integrity following CLP (Fig. 3A, c, d; Fig. 3B), ileums of COX-2−/− mice demonstrated significantly increased numbers of inflammatory cells in the lamina propria and submucosa (Fig. 3A, l) compared with COX-2+/+ mice (Fig. 3A, k; Fig. 3D). Furthermore, immunostaining for Ly-6G demonstrated an increase in the number of neutrophils in the ileums of COX-2+/+ and COX-2−/− mice following CLP with a significantly higher influx of neutrophils into the ileums of COX-2−/− mice (Fig. 3A, p) compared with COX-2+/+ mice (Fig. 3A, o; Fig. 3E). Similarly, H&E staining and immunostaining for CD45 demonstrated that colons harvested from COX-2−/− mice had increased inflammation compared with COX-2+/+ mice following CLP (Supplemental Fig. 1). In addition, COX-2−/− mice had significantly higher levels of circulating pro-inflammatory cytokines including IL-6, TNF-α, and IFN-γ, following CLP compared with COX-2+/+ mice (Supplemental Table I).
To further characterize the inflammatory cell infiltration into the ileum of COX-2−/− mice following CLP, flow cytometry of the lamina propria of ileums from COX-2+/+ and COX-2−/− mice following sham and CLP was performed. Absence of COX-2 was associated with an increased percentage of neutrophils in the lamina propria of the ileum following CLP (Fig. 4A) and significantly increased numbers of neutrophils in the ileum (Fig. 4B) compared with COX-2+/+ mice following CLP. In addition, COX-2−/− mice had significantly increased numbers of macrophages (Fig. 4C) infiltrating the ileum compared with COX-2+/+ mice following CLP.
FIGURE 4.

COX-2 deficient mice have increased neutrophil infiltration of lamina propria of the ileum following peritonitis-induced polymicrobial sepsis. COX-2+/+ and COX-2−/− mice underwent CLP and ileums were harvested 24 h following sham (COX-2−/− n=5; COX-2+/+ n=6) and CLP (COX-2−/− n=11; COX-2+/+ n=11). (A) Representative flow cytometry plots from COX-2+/+ and COX-2−/− mice following sham and CLP. Neutrophils were quantified as Ly-6G+Ly-6C+ gated on CD45+CD11b+ cells. Flow cytometry of cells isolated from the lamina propria of the ileum demonstrates significantly increased numbers of neutrophils (Ly-6G+Ly-6C+) (B) and macrophages (F4/80+CD11b+) (C) in COX-2−/− mice compared with COX-2+/+ mice following CLP (p<0.05 by the Mann-Whitney test).
Absence of COX-2 in ileal parenchymal cells correlates with increased circulating bacterial counts and bacterial seeding of vital organs during polymicrobial sepsis
To investigate the mechanism of increased mortality and bacteremia in COX-2−/− mice following peritonitis-induced polymicrobial sepsis, we performed immunostaining for COX-2 to determine which cell type upregulated COX-2 expression in the ileum following CLP. COX-2 immunostaining demonstrated localization of COX-2 expression to both epithelial cells within the crypts of the ileum as well as to inflammatory cells within the lamina propria of the ileum of COX-2+/+ mice following CLP (Fig. 1E). As COX-2 was highly expressed in both epithelial cells and inflammatory cells of the ileum following CLP, we generated mice chimeric for COX-2 to determine whether COX-2 expression in inflammatory cells or parenchymal cells was critical for the development of bacteremia and shock following peritonitis. BMT was used to derive animals chimeric for COX-2 (COX-2−/− bone marrow-derived cells into COX-2+/+ recipients [COX-2−/− → COX-2+/+] and COX-2+/+ cells into COX-2−/− recipients [COX-2+/+ → COX-2−/−]). Using our current protocol, we have previously shown via hematopoietic reconstitution experiments (47) that >90% of circulating bone marrow-derived cells in the recipient originate from donor marrow. Six weeks following BMT, these chimeric mice were subjected to CLP as described. COX-2+/+ → COX-2−/− mice demonstrated higher levels of circulating bacteria following CLP compared with COX-2−/−→ COX-2+/+ mice (Fig. 5A). Similarly, organ cultures from COX-2+/+ → COX-2−/− mice showed significantly higher levels of bacteria in the lung, liver, and spleen compared with COX-2−/−→ COX-2+/+ mice following CLP (Fig. 5B). To control for the effects of irradiation, non-chimeric irradiated control mice were also generated by BMT (COX-2+/+ donor cells into COX-2+/+ mice [COX-2+/+ → COX-2+/+] and COX-2−/− donor cells into COX-2−/− mice [COX-2−/− → COX-2−/−]). Following CLP, bacterial levels in the blood, lung, liver, and spleen of these non-chimeric mice (Fig. 5C-D) were similar to untransplanted COX-2+/+ and COX-2−/− mice (Fig. 2B, D). Furthermore, bacterial levels in COX-2−/− → COX-2+/+ mice were similar to COX-2+/+ → COX-2+/+ mice following CLP. These findings demonstrate that mice chimeric for COX-2 exhibit the phenotype of the recipient and suggest that COX-2 expression by infiltrating inflammatory cells does not influence the phenotype of the chimeric animal. Furthermore, these results suggest that epithelial COX-2 expression in the ileum protects against bacteremia and death from polymicrobial sepsis.
FIGURE 5.

Absence of COX-2 in ileal parenchymal cells correlates with increased circulating bacterial counts and bacterial seeding of vital organs during polymicrobial sepsis. (A-B) Mice chimeric for COX-2 (COX-2−/− → COX-2+/+ (■ n=7), COX-2+/+ → COX-2−/− (□ n=7)) and (C-D) non-chimeric irradiated control mice (COX-2+/+ → COX-2+/+ (■ n=4), COX-2−/− → COX-2−/− (□ n=4)) were generated via BMT. Six weeks following BMT, mice underwent CLP with a 19-g needle (1 hole) in two independent experiments. Blood was drawn via right ventricular puncture and organs harvested 24 h following CLP. (A,C) Blood and (B,D) homogenized tissue were serially diluted and cultured on LB plates (p<0.05 by the Mann-Whitney test).
COX-2 deficiency does not impair bacterial phagocytosis by peritoneal neutrophils
Previous work has demonstrated that PGE2 may impair phagocytosis and bactericidal activity of macrophages (48-51) and that COX-2 deficiency may decrease bacterial burden in murine models of invasive Streptococcus pyogenes (50) and Pseudomonas aeruginosa pneumonia (51). Based on these studies and to further substantiate our BMT findings that COX-2 expression by inflammatory cells does not determine the phenotype of chimeric mice during peritonitis-induced polymicrobial sepsis, we performed in vivo phagocytosis assays using FITC-labeled E. coli or S. aureus in COX-2−/− and COX-2+/+ mice. Total peritoneal neutrophil counts were not different between COX-2−/− (3.88 ± 1.94 × 104 cells/ml) and COX-2+/+ (3.77 ± 1.88 × 104 cells/ml) mice following i.p. injection of bacteria. Furthermore, COX-2−/− and COX-2+/+ mice had no significant difference in the percent of FITC-positive cells with either E. coli or S. aureus (Supplemental Fig. 2). These findings suggest that the mechanism of increased bacteremia and bacterial seeding of organs in COX-2−/− mice is not due to impaired phagocytosis by peritoneal neutrophils during CLP.
Inhibition or deficiency of COX-2 increases intestinal epithelial permeability following peritonitis-induced polymicrobial sepsis
Based on these findings, we next investigated whether COX-2 deficient mice had intestinal epithelial cell dysfunction leading to hyperpermeability of the ileum and enhanced secondary bacterial translocation following CLP. As our results suggested that inflammatory cell COX-2 expression did not play a role in the development of bacteremia after CLP and since COX-2 expression was highly upregulated in the ileal epithelium, we hypothesized that upregulation of COX-2 in the epithelium of the ileum plays a protective role in intestinal barrier function during CLP. Although histological evaluation of the ileum and colon in COX-2−/− mice demonstrated preservation of mucosal integrity, this does not preclude epithelial barrier dysfunction resulting in exaggerated paracellular leak of microbial products from the intestinal lumen as the etiology of increased mortality in COX-2−/− mice. To determine whether absence of COX-2 leads to exaggerated intestinal epithelial barrier dysfunction, intestinal permeability to FD-4 was examined in COX-2−/− and COX-2+/+ mice following CLP. COX-2−/− mice had more than a 4-fold increase in serum FD-4 concentration following CLP compared with a 1.2 fold increase in COX-2+/+ mice following CLP (Fig. 6A).
FIGURE 6.

Deficiency of COX-2 increases intestinal epithelial permeability following peritonitis-induced polymicrobial sepsis. (A) COX-2+/+ and COX-2−/− mice underwent CLP with a 19-g needle (1 hole). Eighteen h following CLP (COX-2+/+ n=7, COX-2−/− n=8) and sham (COX-2+/+ n=6, COX-2−/− n=6), mice were gavaged with oral FD-4 and blood harvested after 6 h. Serum was isolated, FD-4 concentration measured, and normalized to sham FD-4 concentration. Data represent the median of three independent experiments (p<0.05 by the Mann-Whitney test). (B) COX-2+/+ and COX-2−/− mice underwent CLP with a 23-g needle (2 holes). Ileums were harvested 24 h following sham (COX-2+/+ n=8, COX-2−/− n=7) or CLP (COX-2+/+ n=16, COX-2−/− n=8) and permeability to FD-4 measured by the everted gut sac method. Data represent the median of at least three independent experiments (p<0.05 for COX-2−/− mice vs. COX-2+/+ mice following CLP, **p<0.05 for COX-2−/− mice following CLP vs. sham, ‡p<0.05 for COX-2+/+ mice following CLP vs. sham by the Mann-Whitney test).
We further investigated the intestinal permeability defect at the level of the ileum using the everted gut sac method (41). This ex vivo method measures alterations in mucosal barrier function that result in intestinal epithelial hyperpermeability and allows for the measurement of intestinal permeability in the clinically relevant mucosal-to-serosal direction. There was no difference in ileal permeability between COX 2−/− and COX-2+/+ mice following sham surgery, however following CLP, ileums harvested from COX 2−/− mice had significantly increased epithelial permeability compared with ileums from COX-2+/+ mice and sham controls (Fig. 6B).
To examine the mechanism of exaggerated epithelial barrier dysfunction in COX-2−/− mice during sepsis, we performed in vitro permeability assays in C2BBe1 cells, an intestinal epithelial cell line that forms a polarized monolayer. Selective inhibition of COX-2, but not COX-1, led to enhanced epithelial permeability following CM stimulation (Fig. 7A, C). Treatment with NS-398, a selective COX-2 inhibitor, prior to CM stimulation significantly augmented apical-to-basolateral clearance of FS (Fig. 7A) compared with vehicle-treated CM-stimulated cells. Furthermore, selective COX-2 inhibition led to an exaggerated decrease in TER compared with vehicle-treated CM-stimulated cells (Fig. 7B). Selective inhibition of COX-1, however, led to similar increases in epithelial permeability at 48 h as vehicle-treated CM-stimulated cells (Fig. 7C). To determine if the mechanism of enhanced epithelial permeability was due to decreased cell viability, LDH release and trypan blue uptake were measured spectrophotometrically. There was no significant difference in cell death or LDH release between NS-398-treated and vehicle-treated CM-stimulated cells (Supplemental Fig. 3) demonstrating preserved epithelial cell viability.
FIGURE 7.

Selective inhibition of COX-2 increases epithelial permeability in cytomix-stimulated intestinal epithelial cells. C2BBe1 cells were treated with vehicle, NS-398 (A), or SC-560 (C) and stimulated with CM in collagen-coated transwells. Epithelial permeability was assessed by measuring apical-to-basolateral clearance of FS and results normalized to vehicle. (B) TER was measured before and 48 h following CM stimulation. Data represent the median of at least two independent experiments performed in triplicate (p<0.05 by the Mann-Whitney test).
Absence of COX-2 leads to reduced expression of tight junction proteins in the ileum following CLP
To elucidate the mechanism of enhanced epithelial permeability leading to exaggerated intestinal barrier dysfunction in COX-2 deficient mice following peritonitis-induced sepsis, we investigated whether the expression of key tight junction proteins was altered in the ileum following CLP. As tight junctions are essential for sealing the paracellular space between adjacent epithelial cells and maintaining intestinal barrier function (18, 52), we hypothesized that expression of tight junction proteins might be reduced in the ileum of COX-2 deficient mice leading to severe intestinal barrier dysfunction following peritonitis-induced sepsis. Western blot analysis of ileal mucosal protein from COX-2−/− and COX-2+/+ mice revealed a significant reduction in occludin and claudin-1 expression in the ileums of COX-2−/− mice compared with COX-2+/+ mice following sham surgery (Fig. 8A-B). Furthermore, following CLP, there was a dramatic reduction in the expression of ZO-1, occludin, and claudin-1 in the ileums of COX-2−/− mice compared with COX-2+/+ mice (Fig. 8A-B).
FIGURE 8.

Absence of COX-2 leads to reduced expression of tight junction proteins in the ileum following CLP. (A-B) Ileums were harvested from COX-2+/+ and COX-2−/− mice 48 h following sham or CLP and total protein was isolated from ileal mucosa. Western blot analysis was performed for ZO-1, occludin, and claudin-1. Bar graphs represent protein levels (normalized for β-actin) as determined by densitometry and averaged for 2-4 mice per condition. Protein levels are expressed as the percentage of the levels in COX-2+/+ mice following sham. Data are shown as the mean ± SD (p<0.05 as indicated and *p<0.05 for COX-2+/+ mice after CLP vs. sham control, **p<0.05 for COX-2−/− mice after CLP vs. sham control by unpaired t-test). (C) C2BBe1 cells were treated with NS-398 or vehicle and stimulated with CM in collagen-coated 8 well chamber slides. After 48 h, cells were fixed, permeabilized, and immunostained for ZO-1, occludin, and claudin-1. Confocal fluorescent microscopy was performed and photographs are representative of three independent experiments.
In addition, selective inhibition of COX-2 with NS-398 in CM-stimulated C2BBe1 cells led to significant reduction in immunostaining for ZO-1 (Fig. 8C, d), occludin (Fig. 8C, h), and claudin-1 (Fig. 8C, l) by confocal microscopy compared with vehicle-treated CM-stimulated cells (Fig. 8C, b, f, j). While vehicle-treated CM-stimulated cells demonstrated ruffling of immunostaining for ZO-1 (Fig. 8C, b), cells treated with NS-398 prior to CM had profound loss of membrane staining for both ZO-1 (Fig. 8C, d) and claudin-1 (Fig. 8C, l). Notably, many cells had disruption of circumferential staining with the remaining staining distributed in a punctate manner along the cell membrane. Selective inhibition of COX-2 in CM-stimulated C2BBe1 cells also led to significant disruption in membrane staining for occludin (Fig. 8C, h) as well as cytoplasmic staining suggestive of internalization, while occludin immunostaining remained relatively intact in vehicle-treated CM-stimulated cells (Fig. 8C, f). These findings suggest that COX-2-derived prostanoids may modulate expression of tight junction proteins in the intestinal epithelium and consequently limit intestinal barrier dysfunction during peritonitis-induced sepsis.
PGD2 attenuates epithelial hyperpermeability and downregulation of ZO-1 expression induced by selective inhibition of COX-2
Given the protective role of prostanoids in the gastrointestinal (GI) tract and the importance of COX-2 derived PGD2 in regulating inflammation and epithelial secretion following colonic injury, we investigated whether PGD2 could attenuate intestinal barrier dysfunction following CLP. While PGE2 was not induced in the ileum following CLP (data not shown), PGD2 was highly upregulated in the ileum of COX-2+/+ mice following CLP (Fig. 9). Furthermore, levels of PGD2 were significantly reduced in the ileum of COX-2−/− mice compared with COX-2+/+ mice following CLP (Fig. 9). Based on these findings, we next investigated whether PGD2 or the PGD2 receptor DP1 would alter epithelial cell permeability associated with COX-2 inhibition in CM-stimulated intestinal epithelial cells. PGD2 treatment in the presence of NS-398 attenuated apical-to-basolateral clearance of FS in CM-stimulated C2BBe1 cells (Fig. 10A) compared with NS-398-treated CM-stimulated cells. Furthermore, PGD2 significantly abrogated the decrease in TER (Fig. 10B) and attenuated the downregulation in ZO-1 expression associated with selective inhibition of COX-2 in CM-stimulated C2BBe1 cells (Fig. 10C). In addition, treatment of C2BBe1 cells with BW245C, an agonist of the PGD2 receptor DP1, significantly attenuated hyperpermeability (Fig. 10D) of NS-398-treated CM-stimulated cells.
FIGURE 9.
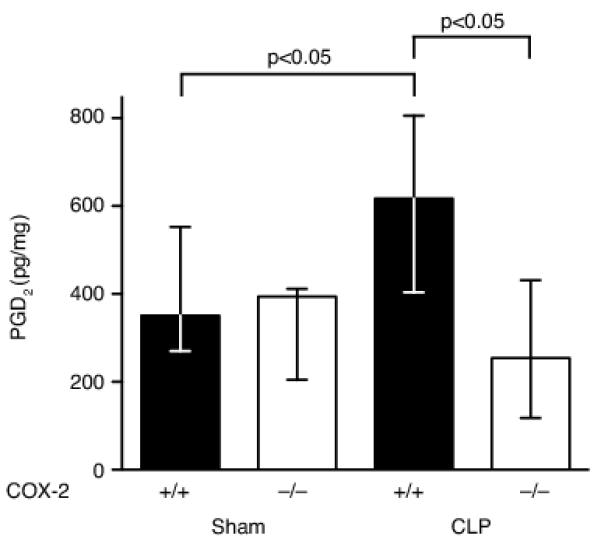
PGD2 is upregulated in the ileum during peritonitis-induced polymicrobial sepsis. Ileums were harvested from COX-2+/+ and COX-2−/− mice 24 h following sham or CLP (n=4-8 per group), homogenized, and ELISA performed for PGD2 in two independent experiments. PGD2 is significantly upregulated in the ileum of COX-2+/+ mice following CLP and COX-2−/− mice have significantly reduced expression of PGD2 in the ileum following CLP compared with COX-2+/+ mice (p<0.05 by the Mann-Whitney test).
FIGURE 10.

PGD2 attenuates epithelial hyperpermeability and downregulation of ZO-1 expression induced by selective inhibition of COX-2. C2BBe1 cells were pre-treated with (A) PGD2 or (D) BW245C (an agonist of the PGD2 receptor DP1) prior to NS-398 or vehicle and stimulated with CM in collagen-coated transwells. Epithelial permeability was assessed by measuring apical-to-basolateral clearance of FS and results normalized to vehicle. (B) TER was measured before and 48 h following CM stimulation. Data represent the median of at least two independent experiments performed in triplicate (p<0.05 by the Mann-Whitney test). (C) Total protein was harvested from C2BBe1 cells 48 h following CM and Western blot analysis performed for ZO-1. Loading was quantified with an anti-α-tubulin Ab.
Discussion
Sepsis remains the leading cause of death in critically ill patients despite modern advances in critical care. In this study, we examined the role of COX-2 in bacterial sepsis using a murine model of peritonitis-induced polymicrobial sepsis. Our results highlight four important new concepts. First, deficiency of COX-2 is deleterious during polymicrobial sepsis leading to significantly increased levels of circulating bacteria and seeding of vital organs, profound vascular collapse, and decreased survival. Second, animals chimeric for COX-2 demonstrate the phenotype of the recipient suggesting that parenchymal COX-2 expression is critical to attenuate bacterial translocation during sepsis. Third, deficiency or pharmacologic inhibition of COX-2 leads to disruption of epithelial tight junctions and loss of intestinal barrier function during bacterial sepsis. Fourth, COX-2-derived PGD2 attenuates intestinal epithelial hyperpermeability by regulating expression of tight junction proteins in intestinal epithelial cells.
Prostanoids have long been recognized as mediating many of the physiologic derangements of the systemic inflammatory response syndrome (SIRS). Early investigation into non-selective inhibition of COX with aspirin or nonsteroidal anti-inflammatory drugs (NSAIDs) showed hemodynamic improvement and improved survival in several animal models of endotoxemia (53). However, although administration of ibuprofen to patients with sepsis improved certain physiologic parameters, it did not improve survival nor prevent the development of the acute respiratory distress syndrome (ARDS) (54). More recently, studies from our laboratory demonstrated that COX-2−/− mice had attenuated inflammation and improved survival during endotoxemia (55), while work from Reddy et al. showed that pharmacologic inhibition of COX-2 improved early survival following endotoxin injection, but not following bacterial peritonitis (56). In this study, we now demonstrate that COX-2 deficiency leads to enhanced intestinal bacterial translocation, increased inflammation, and decreased survival during peritonitis-induced polymicrobial sepsis. Furthermore, we found that COX-2 expression in intestinal epithelial cells, not inflammatory cells, is critical to attenuate bacteremia and bacterial seeding of organs following CLP. Taken together, these prior studies and our present work reinforce that CLP represents a more clinically relevant model of sepsis than endotoxemia (57) and further highlight that disruption of the COX-2 pathway has distinct effects following a pure inflammatory stimulus versus during a microbial infection.
COX-2 is inducible in intestinal macrophages (58) and neutrophils (59) and is upregulated in intestinal epithelial cells following cytokine stimulation (60-62), in response to invasive bacteria (63), and in inflammatory colitis (64). However, the relative contribution and significance of COX-2-derived prostanoid production by inflammatory versus epithelial cells in the ileum during sepsis was heretofore unknown. Using mice chimeric for COX-2, we demonstrated that COX-2 expression in parenchymal cells, not inflammatory cells, influences the development of bacteremia and bacterial seeding of organs during peritonitis-induced sepsis. Furthermore, we showed both in vivo and ex vivo that COX-2 null mice had exaggerated intestinal epithelial cell permeability following CLP and we demonstrated in vitro that selective COX-2 inhibition in intestinal epithelial cells significantly enhanced cytokine-induced transepithelial hyperpermeability. Interestingly, we found no significant difference in phagocytosis by peritoneal neutrophils between mice deficient in COX-2 compared with COX-2+/+ mice. While previous studies have shown that absence of COX-2 may enhance bacterial clearance by alveolar and bone-marrow derived macrophages following intratracheal and intravenous administration of bacteria (50, 51), our findings suggest that intestinal epithelial expression of COX-2 plays a dominant role in attenuating bacterial translocation in a peritonitis-induced sepsis model. Although COX-2 deficient mice had increased recruitment of inflammatory cells to the intestine and higher levels of circulating pro-inflammatory cytokines following CLP, our results suggest that this enhanced inflammatory response is in response to exaggerated bacterial translocation from the disruption of epithelial barrier function. In light of these findings, we propose that an initial breakdown in barrier function in COX-2 null mice leads to enhanced bacterial translocation, followed by recruitment of inflammatory cells and exaggerated production of pro-inflammatory cytokines and chemokines, which in turn leads to further disruption in epithelial barrier integrity and ongoing bacterial translocation. However, as COX-2 deficient mice developed more severe hypotension following CLP and increased intestinal permeability has been observed in animal models of hemorrhagic shock (65), we cannot completely exclude the possibility that hypotension may have contributed to enhanced bacterial translocation and increased mortality in COX-2−/− mice following CLP. Overall, our findings suggest that epithelial COX-2 expression in the ileum protects against bacteremia and death from polymicrobial sepsis.
While prostaglandins have long been recognized as providing mucosal protection in the upper GI tract (66), the significance of COX-2-derived prostanoids in the lower GI tract has been less clear. In the colon, both COX-1 and COX-2 appear to be critical following intestinal injury. While both isoforms were protective during intestinal mucosal injury in a murine model of DSS colitis (11), COX-2-derived PGD2 attenuated colonic injury and inflammation following TNBS (46). In addition, both PGE2 and PGD2 play key roles in regulating epithelial chloride secretion (67) in the colon. The role of COX-2-derived prostanoids in the small intestine is less well characterized, however COX-1 appears to regulate epithelial cell homeostasis following radiation injury (13, 14). Our findings now implicate COX-2 as a key modulator of epithelial tight junction integrity and intestinal barrier function in the small bowel during peritonitis-induced sepsis.
Tight junctions are critical in regulating paracellular permeability between epithelial cells and disruption of their integrity may lead to increased intestinal permeability and translocation of microbial products into the bloodstream during sepsis. COX-2 is upregulated in the epithelium of the ileum by pro-inflammatory stimuli and our data provide evidence that COX-2-derived prostanoids play a protective role in maintaining intestinal epithelial barrier integrity during intra-abdominal sepsis. Our study demonstrates that absence of COX-2-derived prostanoids in the ileum leads to a dramatic reduction in the expression of key tight junction proteins resulting in enhanced epithelial permeability of the ileum during peritonitis-induced sepsis. Our findings suggest that in addition to attenuating inflammation, PGD2 also plays a critical role in preserving intestinal barrier function by regulating epithelial expression of tight junction proteins in the small intestine during peritonitis-induced polymicrobial sepsis.
PGD2 is the major eicosanoid released by mucosal mast cells during allergic inflammation (68), however, PGD2 is also produced by intestinal epithelial cells (69). Although PGD2 is highly expressed in the intestine compared with other murine tissues and PGD2 synthase has one of the highest activities in intestinal tissue (70), the importance of PGD2 expression in the intestine is incompletely understood. The biological effects of PGD2 are mediated via binding to one of two G protein-coupled receptors, DP1 and DP2, or via its metabolite 15-deoxy-Δ12,14-PGJ2, a ligand for PPAR-γ. DP1 modulates dendritic cell function (71) and DP2 enhances allergic inflammation via eosinophils, basophils, and Th2 lymphocytes (72). Interestingly, the mouse ileum has the most abundant expression of the DP1 receptor compared with other murine tissues (73) and the human DP1 receptor is detectable only in the retina and small intestine (74). Furthermore, in situ hybridization has demonstrated that the DP1 receptor localizes to epithelial cells and goblet cells in the small intestine (75). Signal transduction through the DP1 receptor primarily leads to GS-coupled activation of adenylate cyclase and subsequent increases in cAMP, but can also lead to rises in [Ca2+]i (74). The findings of our study now suggest a new mechanism by which COX-2-derived PGD2 or an agonist of the DP1 receptor attenuates cytokine-induced intestinal hyperpermeability in intestinal epithelial cells.
In summary, our results have revealed a novel role for COX-2-derived PGD2 in mediating protection against intestinal barrier dysfunction during polymicrobial sepsis. Epithelial tight junction dysfunction may result in loss of intestinal barrier function and lead to translocation of luminal microbial products into the bloodstream during sepsis. Furthermore, maintenance of epithelial integrity may be critical to prevent the development of ARDS and MODS during sepsis. Our findings have significant implications for our understanding of sepsis and suggest that strategies that modulate epithelial cell expression of COX-2 and PGD2 secretion may yield innovative therapeutic approaches to combat this frequently fatal disease process.
Supplementary Material
Acknowledgements
The authors thank Dr. Augustine M.K. Choi for helpful discussions and scientific input.
The authors thank Bonna Ith for technical assistance and Dr. Runkuan Yang for assistance in developing the everted gut sac model.
4. Abbreviations used in this paper
- BMT
bone marrow transplantation
- CLP
cecal ligation and puncture
- CM
cytomix
- COX
cyclooxygenase
- FD-4
FITC-labeled dextran
- FS
Fluorescein-5-(and-6)-sulfonic acid, trisodium salt
- g
gauge
- GI
gastrointestinal
- KHBB
Krebs-Henseleit bicarbonate buffer
- MODS
multiple organ dysfunction syndrome
- SBP
systolic blood pressure
- TER
transepithelial resistance
- ZO-1
zonula occludens-1
Footnotes
This work was supported by a Parker B. Francis Fellowship (LEF), an American Lung Association Biomedical Research Grant (LEF), and an American Thoracic Society Research Grant (LEF), as well as grants from the NIH including K08 GM083207 (LEF), R01 GM053249 (MAP), R01 HL060788 (MAP), R01 AI061246 (MAP), R37 DK044319 (RSB), R01 DK051362 (RSB), R01 DK053056 (RSB) and R01 DK088199 (RSB).
References
- 1.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 2.Anaya DA, Nathens AB. Risk factors for severe sepsis in secondary peritonitis. Surg. Infect. 2003;4:355–362. doi: 10.1089/109629603322761418. [DOI] [PubMed] [Google Scholar]
- 3.Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. J. Lipid Res. 2009;50(Suppl):S423–428. doi: 10.1194/jlr.R800094-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klein T, Shephard P, Kleinert H, Komhoff M. Regulation of cyclooxygenase-2 expression by cyclic AMP. Biochim. Biophys. Acta. 2007;1773:1605–1618. doi: 10.1016/j.bbamcr.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 6.Wallace JL. Prostaglandin biology in inflammatory bowel disease. Gastroenterol. Clin. North Am. 2001;30:971–980. doi: 10.1016/s0889-8553(05)70223-5. [DOI] [PubMed] [Google Scholar]
- 7.Wallace JL, Devchand PR. Emerging roles for cyclooxygenase-2 in gastrointestinal mucosal defense. Br. J. Pharmacol. 2005;145:275–282. doi: 10.1038/sj.bjp.0706201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maricic N, Ehrlich K, Gretzer B, Schuligoi R, Respondek M, Peskar BM. Selective cyclo-oxygenase-2 inhibitors aggravate ischaemia-reperfusion injury in the rat stomach. Br. J. Pharmacol. 1999;128:1659–1666. doi: 10.1038/sj.bjp.0702966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallace JL, McKnight W, Reuter BK, Vergnolle N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterol. 2000;119:706–714. doi: 10.1053/gast.2000.16510. [DOI] [PubMed] [Google Scholar]
- 10.Reuter BK, Asfaha S, Buret A, Sharkey KA, Wallace JL. Exacerbation of inflammation-associated colonic injury in rat through inhibition of cyclooxygenase-2. J. Clin. Invest. 1996;98:2076–2085. doi: 10.1172/JCI119013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morteau O, Morham SG, Sellon R, Dieleman LA, Langenbach R, Smithies O, Sartor RB. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. J. Clin. Invest. 2000;105:469–478. doi: 10.1172/JCI6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA, Smithies O. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 13.Cohn SM, Schloemann S, Tessner T, Seibert K, Stenson WF. Crypt stem cell survival in the mouse intestinal epithelium is regulated by prostaglandins synthesized through cyclooxygenase-1. J. Clin. Invest. 1997;99:1367–1379. doi: 10.1172/JCI119296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houchen CW, Stenson WF, Cohn SM. Disruption of cyclooxygenase-1 gene results in an impaired response to radiation injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2000;279:G858–865. doi: 10.1152/ajpgi.2000.279.5.G858. [DOI] [PubMed] [Google Scholar]
- 15.Fink MP, Delude RL. Epithelial barrier dysfunction: a unifying theme to explain the pathogenesis of multiple organ dysfunction at the cellular level. Crit. Care Clin. 2005;21:177–196. doi: 10.1016/j.ccc.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Harris CE, Griffiths RD, Freestone N, Billington D, Atherton ST, Macmillan RR. Intestinal permeability in the critically ill. Intensive Care Med. 1992;18:38–41. doi: 10.1007/BF01706424. [DOI] [PubMed] [Google Scholar]
- 17.Doig CJ, Sutherland LR, Sandham JD, Fick GH, Verhoef M, Meddings JB. Increased intestinal permeability is associated with the development of multiple organ dysfunction syndrome in critically ill ICU patients. Am. J. Respir. Crit. Care Med. 1998;158:444–451. doi: 10.1164/ajrccm.158.2.9710092. [DOI] [PubMed] [Google Scholar]
- 18.Turner JR. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 19.Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 2008;8:411–420. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- 20.Al-Sadi R, Boivin M, Ma T. Mechanism of cytokine modulation of epithelial tight junction barrier. Front. Biosci. 2009;14:2765–2778. doi: 10.2741/3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han X, Fink MP, Delude RL. Proinflammatory cytokines cause NO*-dependent and -independent changes in expression and localization of tight junction proteins in intestinal epithelial cells. Shock. 2003;19:229–237. doi: 10.1097/00024382-200303000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Sappington PL, Han X, Yang R, Delude RL, Fink MP. Ethyl pyruvate ameliorates intestinal epithelial barrier dysfunction in endotoxemic mice and immunostimulated Caco-2 enterocytic monolayers. J. Pharmacol. Exp. Ther. 2003;304:464–476. doi: 10.1124/jpet.102.043182. [DOI] [PubMed] [Google Scholar]
- 23.Moriez R, Salvador-Cartier C, Theodorou V, Fioramonti J, Eutamene H, Bueno L. Myosin light chain kinase is involved in lipopolysaccharide-induced disruption of colonic epithelial barrier and bacterial translocation in rats. Am. J. Pathol. 2005;167:1071–1079. doi: 10.1016/S0002-9440(10)61196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang R, Miki K, Oksala N, Nakao A, Lindgren L, Killeen ME, Mennander A, Fink MP, Tenhunen J. Bile high-mobility group box 1 contributes to gut barrier dysfunction in experimental endotoxemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009;297:R362–369. doi: 10.1152/ajpregu.00184.2009. [DOI] [PubMed] [Google Scholar]
- 25.Chung SW, Liu X, Macias AA, Baron RM, Perrella MA. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J. Clin. Invest. 2008;118:239–247. doi: 10.1172/JCI32730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiesel P, Patel AP, Carvajal IM, Wang ZY, Pellacani A, Maemura K, DiFonzo N, Rennke HG, Layne MD, Yet SF, Lee ME, Perrella MA. Exacerbation of chronic renovascular hypertension and acute renal failure in heme oxygenase-1-deficient mice. Circ. Res. 2001;88:1088–1094. doi: 10.1161/hh1001.091521. [DOI] [PubMed] [Google Scholar]
- 28.Fredenburgh LE, Liang OD, Macias AA, Polte TR, Liu X, Riascos DF, Chung SW, Schissel SL, Ingber DE, Mitsialis SA, Kourembanas S, Perrella MA. Absence of cyclooxygenase-2 exacerbates hypoxia-induced pulmonary hypertension and enhances contractility of vascular smooth muscle cells. Circulation. 2008;117:2114–2122. doi: 10.1161/CIRCULATIONAHA.107.716241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fredenburgh LE, Baron RM, Carvajal IM, Mouded M, Macias AA, Ith B, Perrella MA. Absence of heme oxygenase-1 expression in the lung parenchyma exacerbates endotoxin-induced acute lung injury and decreases surfactant protein-B levels. Cell. Mol. Biol. 2005;51:513–520. [PubMed] [Google Scholar]
- 30.Chiu CJ, McArdle AH, Brown R, Scott HJ, Gurd FN. Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Arch. Surg. 1970;101:478–483. doi: 10.1001/archsurg.1970.01340280030009. [DOI] [PubMed] [Google Scholar]
- 31.Park PO, Haglund U, Bulkley GB, Falt K. The sequence of development of intestinal tissue injury after strangulation ischemia and reperfusion. Surgery. 1990;107:574–580. [PubMed] [Google Scholar]
- 32.Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, Hicks JL, Morgan J, Cornish TC, Sutcliffe S, Isaacs WB, Luo J, De Marzo AM. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 2008;21:1156–1167. doi: 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cornish T, Morgan J, Gurel B, DeMarzo AD. FRIDA: An open-source framework for image data set analysis. Arch. Pathol. Lab Med. 2008;132:856. [Google Scholar]
- 34.Van der Heijden PJ, Stok W. Improved procedure for the isolation of functionally active lymphoid cells from the murine intestine. J. Immunol. Methods. 1987;103:161–167. doi: 10.1016/0022-1759(87)90285-7. [DOI] [PubMed] [Google Scholar]
- 35.Yang R, Gallo DJ, Baust JJ, Uchiyama T, Watkins SK, Delude RL, Fink MP. Ethyl pyruvate modulates inflammatory gene expression in mice subjected to hemorrhagic shock. Am. J. Physiol. Gastrointest. Liver Physiol. 2002;283:G212–221. doi: 10.1152/ajpgi.00022.2002. [DOI] [PubMed] [Google Scholar]
- 36.Baron RM, Carvajal IM, Fredenburgh LE, Liu X, Porrata Y, Cullivan ML, Haley KJ, Sonna LA, De Sanctis GT, Ingenito EP, Perrella MA. Nitric oxide synthase-2 down-regulates surfactant protein-B expression and enhances endotoxin-induced lung injury in mice. FASEB J. 2004;18:1276–1278. doi: 10.1096/fj.04-1518fje. [DOI] [PubMed] [Google Scholar]
- 37.Houghton AM, Hartzell WO, Robbins CS, Gomis-Ruth FX, Shapiro SD. Macrophage elastase kills bacteria within murine macrophages. Nature. 2009;460:637–641. doi: 10.1038/nature08181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henneke P, Takeuchi O, Malley R, Lien E, Ingalls RR, Freeman MW, Mayadas T, Nizet V, Akira S, Kasper DL, Golenbock DT. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J. Immunol. 2002;169:3970–3977. doi: 10.4049/jimmunol.169.7.3970. [DOI] [PubMed] [Google Scholar]
- 39.Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, Podolsky DK, Colgan SP. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J. Exp. Med. 2001;193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furuta GT, Nieuwenhuis EE, Karhausen J, Gleich G, Blumberg RS, Lee JJ, Ackerman SJ. Eosinophils alter colonic epithelial barrier function: role for major basic protein. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;289:G890–897. doi: 10.1152/ajpgi.00015.2005. [DOI] [PubMed] [Google Scholar]
- 41.Wattanasirichaigoon S, Menconi MJ, Delude RL, Fink MP. Effect of mesenteric ischemia and reperfusion or hemorrhagic shock on intestinal mucosal permeability and ATP content in rats. Shock. 1999;12:127–133. doi: 10.1097/00024382-199908000-00006. [DOI] [PubMed] [Google Scholar]
- 42.Liu S, Stolz DB, Sappington PL, Macias CA, Killeen ME, Tenhunen JJ, Delude RL, Fink MP. HMGB1 is secreted by immunostimulated enterocytes and contributes to cytomix-induced hyperpermeability of Caco-2 monolayers. Am. J. Physiol. Cell Physiol. 2006;290:C990–999. doi: 10.1152/ajpcell.00308.2005. [DOI] [PubMed] [Google Scholar]
- 43.Barnett J, Chow J, Ives D, Chiou M, Mackenzie R, Osen E, Nguyen B, Tsing S, Bach C, Freire J, Chana H, Sigalb E, Ramesha C. Purification, characterization and selective inhibition of human prostaglandin G/H synthase 1 and 2 expressed in the baculovirus system. Biochim. Biophys. Acta. 1994;1209:130–139. doi: 10.1016/0167-4838(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 44.Smith CJ, Zhang Y, Koboldt CM, Muhammad J, Zweifel BS, Shaffer A, Talley JJ, Masferrer JL, Seibert K, Isakson PC. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc. Natl. Acad. Sci. USA. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uliasz TF, Hewett SJ. A microtiter trypan blue absorbance assay for the quantitative determination of excitotoxic neuronal injury in cell culture. J. Neurosci. Methods. 2000;100:157–163. doi: 10.1016/s0165-0270(00)00248-x. [DOI] [PubMed] [Google Scholar]
- 46.Ajuebor MN, Singh A, Wallace JL. Cyclooxygenase-2-derived prostaglandin D2 is an early anti-inflammatory signal in experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2000;279:G238–244. doi: 10.1152/ajpgi.2000.279.1.G238. [DOI] [PubMed] [Google Scholar]
- 47.Layne MD, Patel A, Chen YH, Rebel VI, Carvajal IM, Pellacani A, Ith B, Zhao D, Schreiber BM, Yet SF, Lee ME, Storch J, Perrella MA. Role of macrophage-expressed adipocyte fatty acid binding protein in the development of accelerated atherosclerosis in hypercholesterolemic mice. FASEB J. 2001;15:2733–2735. doi: 10.1096/fj.01-0374fje. [DOI] [PubMed] [Google Scholar]
- 48.Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J. Immunol. 2004;173:559–565. doi: 10.4049/jimmunol.173.1.559. [DOI] [PubMed] [Google Scholar]
- 49.Ballinger MN, Aronoff DM, McMillan TR, Cooke KR, Olkiewicz K, Toews GB, Peters-Golden M, Moore BB. Critical role of prostaglandin E2 overproduction in impaired pulmonary host response following bone marrow transplantation. J. Immunol. 2006;177:5499–5508. doi: 10.4049/jimmunol.177.8.5499. [DOI] [PubMed] [Google Scholar]
- 50.Goldmann O, Hertzen E, Hecht A, Schmidt H, Lehne S, Norrby-Teglund A, Medina E. Inducible cyclooxygenase released prostaglandin E2 modulates the severity of infection caused by Streptococcus pyogenes. J. Immunol. 2010;185:2372–2381. doi: 10.4049/jimmunol.1000838. [DOI] [PubMed] [Google Scholar]
- 51.Sadikot RT, Zeng H, Azim AC, Joo M, Dey SK, Breyer RM, Peebles RS, Blackwell TS, Christman JW. Bacterial clearance of Pseudomonas aeruginosa is enhanced by the inhibition of COX-2. Eur. J. Immunol. 2007;37:1001–1009. doi: 10.1002/eji.200636636. [DOI] [PubMed] [Google Scholar]
- 52.Blikslager AT, Moeser AJ, Gookin JL, Jones SL, Odle J. Restoration of barrier function in injured intestinal mucosa. Physiol. Rev. 2007;87:545–564. doi: 10.1152/physrev.00012.2006. [DOI] [PubMed] [Google Scholar]
- 53.Fletcher JR, Collins JN, Graves EDI, Luterman A, Williams MD, Izenberg SD, Rodning CB. Tumor necrosis factor-induced mortality is reversed with cyclooxygenase inhibition. Ann. Surg. 1993;217:668–674. doi: 10.1097/00000658-199306000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bernard GR, Wheeler AP, Russell JA, Schein R, Summer WR, Steinberg KP, Fulkerson WJ, Wright PE, Christman BW, Dupont WD, Higgins SB, Swindell BB. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N. Engl. J. Med. 1997;336:912–918. doi: 10.1056/NEJM199703273361303. [DOI] [PubMed] [Google Scholar]
- 55.Ejima K, Layne MD, Carvajal IM, Kritek PA, Baron RM, Chen YH, Vom Saal J, Levy BD, Yet SF, Perrella MA. Cyclooxygenase-2-deficient mice are resistant to endotoxin-induced inflammation and death. FASEB J. 2003;17:1325–1327. doi: 10.1096/fj.02-1078fje. [DOI] [PubMed] [Google Scholar]
- 56.Reddy RC, Chen GH, Tateda K, Tsai WC, Phare SM, Mancuso P, Peters-Golden M, Standiford TJ. Selective inhibition of COX-2 improves early survival in murine endotoxemia but not in bacterial peritonitis. Am. J. Physio. Lung Cell Mol. Physiol. 2001;281:L537–L543. doi: 10.1152/ajplung.2001.281.3.L537. [DOI] [PubMed] [Google Scholar]
- 57.Remick DG, Ward PA. Evaluation of endotoxin models for the study of sepsis. Shock. 2005;24(Suppl 1):7–11. doi: 10.1097/01.shk.0000191384.34066.85. [DOI] [PubMed] [Google Scholar]
- 58.Hull MA, Faluyi OO, Ko CW, Holwell S, Scott DJ, Cuthbert RJ, Poulsom R, Goodlad R, Bonifer C, Markham AF, Coletta PL. Regulation of stromal cell cyclooxygenase-2 in the ApcMin/+ mouse model of intestinal tumorigenesis. Carcinogenesis. 2006;27:382–391. doi: 10.1093/carcin/bgi236. [DOI] [PubMed] [Google Scholar]
- 59.Smalley-Freed WG, Efimov A, Burnett PE, Short SP, Davis MA, Gumucio DL, Washington MK, Coffey RJ, Reynolds AB. p120-catenin is essential for maintenance of barrier function and intestinal homeostasis in mice. J. Clin. Invest. 2010;120:1824–1835. doi: 10.1172/JCI41414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jobin C, Morteau O, Han DS, Sartor R. Balfour. Specific NF-kappaB blockade selectively inhibits tumour necrosis factor-alpha-induced COX-2 but not constitutive COX-1 gene expression in HT-29 cells. Immunol. 1998;95:537–543. doi: 10.1046/j.1365-2567.1998.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheng H, Shao J, Hooton EB, Tsujii M, DuBois RN, Beauchamp RD. Cyclooxygenase-2 induction and transforming growth factor beta growth inhibition in rat intestinal epithelial cells. Cell Growth Differ. 1997;8:463–470. [PubMed] [Google Scholar]
- 62.Hobbs SS, Goettel JA, Liang D, Yan F, Edelblum KL, Frey MR, Mullane MT, Polk DB. Tnf transactivation of Egfr stimulates cytoprotective COX-2 expression in gastrointestinal epithelial cells. Am. J. Physiol Gastrointest. Liver Physiol. 2011;301:G220–9. doi: 10.1152/ajpgi.00383.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eckmann L, Stenson WF, Savidge TC, Lowe DC, Barrett KE, Fierer J, Smith JR, Kagnoff MF. Role of intestinal epithelial cells in the host secretory response to infection by invasive bacteria. Bacterial entry induces epithelial prostaglandin H synthase-2 expression and prostaglandin E2 and F2alpha production. J. Clin. Invest. 1997;100:296–309. doi: 10.1172/JCI119535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singer II, Kawka DW, Schloemann S, Tessner T, Riehl T, Stenson WF. Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterol. 1998;115:297–306. doi: 10.1016/s0016-5085(98)70196-9. [DOI] [PubMed] [Google Scholar]
- 65.Yang R, Han X, Uchiyama T, Watkins SK, Yaguchi A, Delude RL, Fink MP. IL-6 is essential for development of gut barrier dysfunction after hemorrhagic shock and resuscitation in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2003;285:G621–629. doi: 10.1152/ajpgi.00177.2003. [DOI] [PubMed] [Google Scholar]
- 66.Brzozowski T, Konturek PC, Konturek SJ, Brzozowska I, Pawlik T. Role of prostaglandins in gastroprotection and gastric adaptation. J. Physiol. Pharmacol. 2005;56(Suppl 5):33–55. [PubMed] [Google Scholar]
- 67.Keenan CM, Rangachari PK. Contrasting effects of PGE2 and PGD2: ion transport in the canine proximal colon. Am. J. Physiol. 1991;260:G481–488. doi: 10.1152/ajpgi.1991.260.3.G481. [DOI] [PubMed] [Google Scholar]
- 68.Fujitani Y, Kanaoka Y, Aritake K, Uodome N, Okazaki-Hatake K, Urade Y. Pronounced eosinophilic lung inflammation and Th2 cytokine release in human lipocalin-type prostaglandin D synthase transgenic mice. J. Immunol. 2002;168:443–449. doi: 10.4049/jimmunol.168.1.443. [DOI] [PubMed] [Google Scholar]
- 69.Longo WE, Panesar N, Mazuski J, Kaminski DL. Contribution of cyclooxygenase-1 and cyclooxygenase-2 to prostanoid formation by human enterocytes stimulated by calcium ionophore and inflammatory agents. Prostaglandins Other Lipid Mediat. 1998;56:325–339. doi: 10.1016/s0090-6980(98)00058-6. [DOI] [PubMed] [Google Scholar]
- 70.Ujihara M, Urade Y, Eguchi N, Hayashi H, Ikai K, Hayaishi O. Prostaglandin D2 formation and characterization of its synthetases in various tissues of adult rats. Arch. Biochem. Biophys. 1988;260:521–531. doi: 10.1016/0003-9861(88)90477-8. [DOI] [PubMed] [Google Scholar]
- 71.Trottein F, Faveeuw C, Gosset P, Angeli V. Role of the D prostanoid receptor 1 in the modulation of immune and inflammatory responses. Crit. Rev. Immunol. 2004;24:349–362. doi: 10.1615/critrevimmunol.v24.i5.30. [DOI] [PubMed] [Google Scholar]
- 72.Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, Nagata K. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J. Exp. Med. 2001;193:255–261. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hirata M, Kakizuka A, Aizawa M, Ushikubi F, Narumiya S. Molecular characterization of a mouse prostaglandin D receptor and functional expression of the cloned gene. Proc. Natl. Acad. Sci. USA. 1994;91:11192–11196. doi: 10.1073/pnas.91.23.11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boie Y, Sawyer N, Slipetz DM, Metters KM, Abramovitz M. Molecular cloning and characterization of the human prostanoid DP receptor. J. Biol. Chem. 1995;270:18910–18916. doi: 10.1074/jbc.270.32.18910. [DOI] [PubMed] [Google Scholar]
- 75.Wright DH, Nantel F, Metters KM, Ford-Hutchinson AW. A novel biological role for prostaglandin D2 is suggested by distribution studies of the rat DP prostanoid receptor. Eur. J Pharmacol. 1999;377:101–115. doi: 10.1016/s0014-2999(99)00358-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.