

Abstract
Herpesvirus genomic DNA is cleaved from concatemers that accumulate in infected cell nuclei. Genomic DNA is inserted into preassembled capsids through a unique portal vertex. Extensive analyses of viral mutants have indicated that intact capsids, the portal vertex, and all components of a tripartite terminase enzyme are required to both cleave and package viral DNA, suggesting that DNA cleavage and packaging are inextricably linked. Because the processes have not been functionally separable, it has been difficult to parse the roles of individual proteins in the DNA cleavage/packaging reaction. In the present study, a virus bearing the deletion of codons 400 to 420 of UL15, encoding a terminase component, was analyzed. This virus, designated vJB27, failed to replicate on noncomplementing cells but cleaved concatemeric DNA to ca. 35 to 98% of wild-type levels. No DNA cleavage was detected in cells infected with a UL15-null virus or a virus lacking UL15 codons 383 to 385, comprising a motif proposed to couple ATP hydrolysis to DNA translocation. The amount of vJB27 DNA protected from DNase I digestion was reduced compared to the wild-type virus by 6.5- to 200-fold, depending on the DNA fragment analyzed, thus indicating a profound defect in DNA packaging. Capsids containing viral DNA were not detected in vJB27-infected cells, as determined by electron microscopy. These data suggest that pUL15 plays an essential role in DNA translocation into the capsid and indicate that this function is separable from its role in DNA cleavage.
INTRODUCTION
Like all herpesviruses, herpes simplex virus 1 (HSV-1) packages genomic viral DNA into preassembled two-shelled capsids in the nuclei of infected cells (reviewed in reference 9). Individual genomes are generated by virally encoded machinery that recognizes packaging signals in concatemeric DNA and endonucleolytically cleaves the DNA between these signals (12, 22, 42). The cleaved DNA is pumped into the capsid through a unique vertex, termed the portal vertex, which is composed of 12 copies of the UL6 gene product (7, 24, 41). As the DNA is packaged, it replaces the internal proteinaceous shell or scaffold, and the outer shell undergoes a dramatic and stabilizing conformational change (23, 40). Capsids containing DNA are termed nucleocapsids or type C capsids (15). In a default reaction that occurs in the absence of terminase components, immature two shelled capsids (termed procapsids or large cored B capsids) undergo the conformational change and retain the internal shell (23, 40). These forms are believed to represent dead end products and are termed B capsids or small cored B capsids. In instances in which the scaffold is expelled but DNA is not inserted, type A capsids are produced. Such capsids are believed to result when the DNA packaging process initiates but is subsequently aborted and the DNA slips out of the capsid.
In many DNA viral systems, genomic cleavage and packaging requires a terminase enzyme that harbors the requisite DNA recognition, endonuclease, and ATPase activities (6). Unlike bacteriophage terminases that consist of two subunits, mounting evidence suggests that the herpes simplex virus terminase consists of three, encoded by the UL15, UL28, and UL33 genes (5, 46). The terminase complex can assemble in the cytoplasm separately from capsids, eventually interacting with the portal vertex in infected cell nuclei (47). The UL28 protein (pUL28) has been shown to bind viral DNA packaging sequences (2), while the UL33 gene product (pUL33) stably associates with pUL28 and enhances the interaction between the complex of pUL28 and pUL33 (pUL28/pUL33) and the UL15 protein (pUL15) (46). The UL15 gene family of herpesviruses predicts a highly conserved ATPase motif that, at least in herpes simplex virus, is required for viral DNA cleavage and packaging (11, 49). Biochemical data and the structure of a C-terminal domain of the human cytomegalovirus homolog of pUL15 (designated UL89) indicate that this family of herpesvirus proteins also carries an RNase H-like nuclease domain that may be important for cleavage of concatemeric DNA into unit length genomes (21, 30).
Herpes simplex virus genomes comprise two components, long and short, each of which are flanked by inverted repeats (16). Two DNA cleavage events are required to release genomic viral DNA from the concatemer (42). The first of these cleavages generates the long component terminus, while the second generates the terminus of the S component and frees the cleaved genome from the concatemer (31). These separate cleavage events require different elements of the packaging sequences (39).
Thus far, mutations in DNA packaging proteins that allow DNA cleavage but preclude DNA packaging have not been identified. The absence of such observations has been interpreted to indicate that DNA cleavage requires competent packaging machinery and occurs on or about the time of the initiation of DNA packaging. Lethal mutations in the portal or terminase subunits preclude DNA cleavage, further suggesting that the cleavage and packaging activities are tightly linked (3, 25, 28, 37). These observations have led to models in which DNA is cleaved after insertion into the capsid, a possibility consistent with the observation that DNA cleavage has also not been detected in cells infected with viral mutants containing terminase but lacking portal-bearing capsids (13, 25). In the absence of UL25, genomes containing an incorrect S terminus are generated but are not retained in capsids. In this case, the scaffold is lost and A capsids accumulate in infected cell nuclei (8, 19, 31). These observations suggest that UL25 is involved late in the packaging reaction to retain DNA and to ensure accuracy of the second DNA cleavage event.
Regardless of the packaging model proposed, it is clear that energy from ATP hydrolysis must be harnessed to drive viral DNA translocation (10). Translocation would also likely require an interaction between the terminase and the portal, as has been observed in substantial detail in the T4 bacteriophage system (32, 33). The UL15 gene product is a likely candidate to couple the energy from ATP hydrolysis to DNA translocation because it contains a likely ATPase motif essential for DNA packaging (49), interacts with both the portal and other terminase subunits (1, 17, 44, 46, 47), and may be more intimately associated with the capsid than pUL28 or pUL33 (4, 45). It has also been suggested that pUL15, like the terminase subunits of tailed bacteriophages, encodes a coupling motif (C motif) that helps to harness the ATPase activity for DNA translocation (14). The present work was initiated to test whether (i) the putative C motif and/or nearby regions in pUL15 link DNA translocation to ATPase activity and (ii) DNA cleavage is inextricably linked to DNA packaging.
MATERIALS AND METHODS
Cells, viruses, antibodies, and plasmids.
CV1, CV1-derived CV15 cells expressing UL15, and rabbit skin cells were maintained as described previously (46). The F strain of HSV-1 [HSV-1(F)] was used as the wild-type strain. The UL15-null mutant virus was described previously (3). Recombinant virus vJB27, vJB28, and the restored viruses vJB27R and vJB28R, derived from vJB27 and vJB28, respectively, are described herein. Rabbit polyclonal antibodies to pUL6, the N or C termini of pUL15, pUL28, or pUL33 were described previously (26, 29, 35, 36). Plasmids pBAD-I-SceI, containing the gene encoding the yeast I-SceI endonuclease, and pEPkan-S, containing aphAI (encoding kanamycin resistance) were gifts from Klaus Osterrieder, Cornell University. Plasmid pCAGGS-nlsCre, expressing Cre recombinase, was a gift from Michael Kotlikoff, Cornell University. Plasmid pRB4192 contains HSV-1(F) DNA from position 31097 to 36202 according to the scheme of McGeoch (18). The bacterial artificial chromosome (BAC) containing the entire HSV-1(F) genome has been described (34).
Construction of recombinant virus.
Recombinant viruses were constructed by two-step Red-mediated recombination system described previously by Tischer et al., using a BAC containing the entire HSV-1(F) genome (34, 38). Procedures for virus construction, transfection of BAC DNA, and virus propagation were as described previously (47). The primers used to delete codons 400 to 420 of pUL15 were 5′-ACC AAC ACC GGG AAG GCCA GTA CGAGC TTT TTG TAC AAC CTC GTG GTG ACG CAC ACA AAC GCC TAG GGA TAA CAG GGTA ATC GATTT-3′ and 5′-GAT ATA ACA AGA ACA GGCC GTG GCG TTTG TGTG CGT CAC CAC GAG GTTG TAC AAA AAG CTCG TGC CAG TGT TAC AAC CAA TTA ACC-3′, whereas the primers used to delete codons 383 to 385 were 5′-ATT ATG GGCT TTC TCAA CCA GGCC AACT GCA AGA TTA TCTT CAC CAA CACC GGG AAG GCCA GTTA GGG ATAA CAG GGT AATC GAT TT-3′ and 5′-GAG GTTG TAC AAA AAG CTC GTA CTG GCC TTCC CGG TGTT GGT GAA GAT AAT CTTG CAG TTGG CGC CAG TGTT ACA ACC AAT TAACC-3′. To restore the deletion in UL15, CV15 cells were cotransfected with linearized plasmid pRB4192 and the viral DNA of either of the respective UL15 deletion viruses vJB27 and vJB28. UL15 DNA of the respective viruses was sequenced to verify the mutations were inserted as designed. The reconstituted viruses from lysed cells were plaque purified on CV1 cells, and the virus from a single plaque was designated vJB27R or vJB28R, respectively.
Immunoprecipitation and immunoblotting.
These procedures were performed as described previously (47). Briefly, approximately 8 × 106 CV1 cells were infected with 5.0 PFU per cell of UL15-null, vJB27, vJB28, or HSV-1(F). At 18 h after infection, cells were washed with cold phosphate-buffered saline (PBS) and lysed in cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA, and 1× protease inhibitor cocktail). The lysates were clarified at 14,000 rpm at 4°C for 5 min and reacted with anti-pUL15 antibodies. Immune complexes were denatured in sodium dodecyl sulfate (SDS) sample buffer (2% SDS, 62.5 mM Tris-HCl [pH 6.8], 12.5% glycerol, 10% β-mercaptoethanol [0.7 M]), separated on SDS-12% polyacrylamide gels, and transferred to a nitrocellulose membrane for immunoblotting. Immunoblotting was performed as described previously (47).
Southern blotting.
CV1 cells (approximately 2 × 106) were infected with UL15-null, HSV-1(F), vJB27, vJB28, or the restored viruses vJB27R and vJB28R. At 15 h postinfection, DNA was extracted as described previously (37). Briefly, this involved lysis of nuclei in SDS, followed by treatment with proteinase K and phenol-chloroform extraction. Approximately 10 μg of DNA was digested with BamHI and electrophoretically separated on 0.8% agarose gels. The DNA fragments were denatured, neutralized, and transferred to nylon membrane as described previously (48). The bound DNA was UV cross-linked to nylon membrane and hybridized with [32P]dCTP-labeled BamHI P or S fragments of HSV-1(F) DNA. The bound probe was visualized by exposure to X-ray film at −80°C in the presence of intensifying screens.
Pulsed-field gel electrophoresis.
The procedure was described previously (19). Briefly, 3.2 × 106 CV1 cells in a 60-mm-diameter dish were infected with the indicated virus at a multiplicity of infection (MOI) of 5 PFU per cell. At 15 h after infection, cells were scraped into PBS and pelleted. The cell pellet was resuspended in 150 μl of prewarmed PBS, followed by the addition of 300 μl of 1.2% agarose in PBS, and poured into blocks on the bottom of a 10-mm-diameter dish. Prior to electrophoresis, the agarose blocks were incubated for 20 h at 37 in 2 ml of lysis buffer (100 mM EDTA, 10 mM Tris [pH 8.0], 1% [wt/vol] N-lauroylsarcosine sodium salt [Sarkosyl], and 100 μg of proteinase K/ml), and washed three times with storage buffer (10 mM Tris-HCl [pH 8.0], 10mM EDTA [pH 8.0]). Roughly equal sized blocks were cut and sealed into the wells of a 0.8% agarose gel made in 0.5× TBE (1× TBE is 89 mM Tris, 89 mM boric acid, and 2 mM EDTA [pH 8.0]). The gel was run at 6 V/cm for 24 h at 14°C; the angle was 120° with a pulse time of 45 to 70 s with a Bio-Rad CHEF-DR III pulsed-field electrophoresis system. The gel was then soaked in 0.25 M HCl for 45 min to depurinate the DNA fragments, denatured in 1.5 M NaCl-0.5 M NaOH for 45 min, and neutralized in 1.5 M NaCl-0.5 M Tris HCl (pH 7.4) for 45 min. The DNA was transferred to nylon membrane in 10× SSC (1.5 M NaCl, 0.15 M sodium citrate [pH 7.0]), UV cross-linked, and hybridized with denatured 32P-labeled BamHI P fragment as described previously (48). The bound probe was revealed by exposure to X-ray film at −80°C in the presence of intensifying screens.
DNase protection assay.
CV1 cells (8 × 106) were infected with HSV-1(F) or various UL15 mutants at an MOI of 5 PFU per cell. At 18 h postinfection at 37°C, the cells were removed, washed with cold PBS, and pelleted. The cell pellets were resuspended in 800 μl of TE buffer (pH 8.0) and incubated on ice for 15 min to swell the cells. NP-40 was added to a final concentration of 0.5%, and the mixture was vortexed briefly and incubated on ice for an additional 15 min. The mixture was then subjected to centrifugation for 5 min at 5,000 rpm; the supernatant, containing both nuclear and cytoplasmic material, was then removed and split equally into two portions. One portion of the supernatant was incubated on ice, and the other was digested with 15 U of DNase I (Fermentas) for 4 h at 37°C. Both samples were then treated with proteinase K for 1 h at 50°C. Viral DNA was extracted with phenol-chloroform, precipitated with ethanol, digested with BamHI, and electrophoretically separated on a 0.8% agarose gel. The separated viral DNA was denatured by soaking the gel in 1.5 M NaCl-0.5 M NaOH, neutralized with 1.5 M NaCl-0.5 M Tris-Cl (pH 8.0), and transferred to a nylon membrane (Whatman) via upward capillary action. The DNA was cross-linked to the membrane by UV irradiation and hybridized with a 32P-labeled BamHI P fragment of HSV-1 genome representing the terminus of the short component of the viral genome. After extensive washing, the blot was exposed to X-film at −80°C, and then screened with a Molecular Dynamic PhosphorImager at room temperature. The X-ray film was developed for documentation, and the screen was scanned with a Molecular Dynamics Storm 860, followed by quantification with ImageQuant software.
Electron microscopy.
Electron microscopy was performed as described previously (27). Briefly, CV1 cells were infected with 5.0 PFU per cell UL15 mutant virus vJB27, vJB28, or the genetically restored virus vJB27R or vJB28R. At 16 hours after infection, the cells were fixed with 2.5% glutaraldehyde in 100 mM sodium cacodylate buffer (pH 7.4) for 30 min at room temperature and then for 90 min at 4°C. After a thorough washing with cacodylate buffer, the cells were fixed in 2% OsO4 in cacodylate buffer at room temperature for 2 h. Cells were washed and dehydrated through a series of increasing ethanol concentrations, followed by fixation in acetone and embedding in Epon-Araldite.
RESULTS
We were initially interested in the coupling (C) motif: a motif of terminase subunits believed to confer energy derived from ATP hydrolysis into mechanical energy for DNA translocation (14). In HSV-1 pUL15, the C motif comprises amino acids 383 to 385. Molecular modeling (data not shown) of the pUL15 Walker box using the structure of the T4 GP17 terminase subunit as a framework (32) also suggested an adjacent basic α-helix unique to pUL15 comprising amino acids 400 to 420. We reasoned that this region might conceivably interact with other proteins or DNA during DNA packaging. To investigate the function of these regions we constructed, respectively, two recombinant viruses lacking UL15 codons 383 to 385 (vJB28) or codons 400 to 420 (vJB27) using a BAC system containing the entire HSV-1(F) genome as described in Materials and Methods. Viruses derived from these recombinant viruses but containing genetically restored UL15 genes were generated and designated vJB27R and vJB28R, respectively. A schematic diagram of the corresponding viral genomes are shown in Fig. 1A. To propagate vJB27 and vJB28, the BACs encoding these viruses were transfected into a cell line CV15, derived from CV1 cells and containing the UL15 cDNA. The ability of the recombinant viruses to replicate in various cell lines was assessed and the results are shown in Table 1.
Fig. 1.

Recombinant viruses and coimmunoprecipitation of terminase and portal components. (A) Schematic representations of sequence arrangements of HSV-1 (F) and recombinant viruses relevant to this report. Line 1, HSV-1 (F) DNA. The repeats flanking the unique long (UL) and unique short (US) sequences are indicated by open rectangles. Line 2, gene cluster including UL15 exon I and exon II, UL16, and UL17. Line 3, representation of wild-type pUL15 domains. The solid line represents exon I, and the dashed line represents exon II. Functional domains are indicated in the boxes. Line 4, vJB27 DNA lacking UL15 codons 400 to 420. Line 5, vJB28 DNA lacking UL15 codons 383 to 385. Lines 6 and 7, diagram of UL15 genes from viruses designated vJB27R and vJB28R that were genetically derived from vJB27 and vJB28, respectively, but bear restored UL15 genes. (B) CV1 cells were infected with 15 null, HSV-1(F), mutant virus vJB27 or vJB28 at an MOI of 5 PFU/cell. At 18 h after infection, the cells were washed with cold PBS, lysed in RIPA buffer containing 1 M NaCl2, and immunoprecipitated with anti-pUL15 N antibodies. The immunoprecipitated proteins were eluted in SDS sample buffer, separated on a denaturing 12% polyacrylamide gel, and transferred to a nitrocellulose membrane. The transferred proteins were probed with anti-pUL6, pUL28, pUL33, and pUL15 antibodies as indicated to the right of the figure. The dark band at the bottom of panels A, B, and D is from the IgG heavy chain.
Table 1.
Replication of UL15 mutants on various cell lines
| Virus | Genotype of UL15a | Cell lines usedb | Mean titer ± SD (PFU/ml)c |
|---|---|---|---|
| vJB27 | Del 400-420aa | CV1/CV15 | <102 |
| vJB27 | CV15/CV15 | (3.2 ± 0.76) × 106 | |
| vJB28 | Del 383-385aa | CV1/CV15 | <102 |
| vJB28 | CV15/CV15 | (2 ± 0.45) × 106 | |
| vJB27R | Restored 400-420aa deletion | CV1/CV1 | (3.3 ± 1.5) × 107 |
| vJB28R | Restored 383-385aa deletion | CV1/CV1 | (2.9 ± 0.96) × 107 |
| HSV-1(F) | Wild type | CV1/CV1 | (2.5 ± 1.32) × 107 |
aa, amino acids.
Expressed as the used cells for testing virus replication/the cells used for virus quantification.
Virus titers were determined in triplicate by plaque assay as described in Materials and Methods. The data represent means for three independent experiments.
Viral replication was not detected within the limits of the assay in CV1 cells infected with vJB27 and vJB28, whereas infectious titers of >107 PFU/ml were obtained from CV1 cells infected with the wild-type virus HSV-1(F) and the genetically restored viruses vJB27R and vJB28R. Limited rescue of vJB27 and vJB28 replication was observed in CV15 cells inasmuch as these cells produced approximately 2 × 106 to 3 × 106 PFU/ml. These data indicate that the mutations in the UL15 genes of vJB27 and vJB28 precluded detection of virus replication and that lesions in UL15 were responsible for the defects.
Because deletions within vJB27 and vJB28 precluded viral propagation, experiments were performed to characterize the respective phenotypes. To investigate whether the deletions in vJB27 and vJB28 precluded interaction between terminase subunits pUL15, pUL28, and pUL33 and the portal protein pUL6, CV1 cells were infected with wild-type HSV-1(F), a UL15-null virus, vJB27, or vJB28. Lysates of the infected cells were reacted with antibody directed against the N terminus of pUL15. The presence of terminase subunits and portal protein in immunoprecipitated material was then determined on immunoblots probed with monospecific antibodies. As shown in Fig. 1B, the pUL15-specific antibody immunoprecipitated the portal protein encoded by UL6, and all terminase subunits from lysates of cells infected with HSV-1(F), but not from lysates of cells infected with the UL15-null virus. Most importantly for the purposes of this experiment, pUL28, pUL33, and the portal protein pUL6 were all coimmunoprecipitated from vJB27- and vJB28-infected cell lysates with the pUL15-specific antibody. We conclude that the deletions in vJB27 and vJB28 did not preclude interactions between terminase subunits or the interaction between portal protein and the terminase subunit encoded by pUL15.
To date, lethal mutations in terminase subunits or capsid proteins have precluded DNA cleavage and packaging. To determine whether vJB27 and vJB28 exhibited a similar phenotype, cells were infected with HSV-1(F), UL15-null, vJB27, vJB28, vJB27R, or vJB28R. DNA was then purified from the infected cells, digested with BamHI, electrophoretically separated, transferred to nylon, and probed with radiolabeled DNA representing the terminus of the short component (BamHI P fragment) of the viral genome or, in a separate experiment, the terminus of the long component (BamHI S fragments). As shown in Fig. 2C and D, the two probes also recognized the junction fragments (designated S-P) in concatemeric viral DNA linking the long and short components in all samples tested. Multiple BamHI S fragments (Fig. 2D) were detected inasmuch as the terminal a sequence is duplicated at the L terminus in a subset of HSV-1 genomes (43). Unlike DNA from cells infected with wild-type HSV-1(F), DNA from cells infected with the UL15-null virus or vJB28 did not yield the BamHI P or S fragments, indicating that neither genomic terminus was generated. Thus, the lethality of the UL15 deletions in vJB28 or UL15-null viruses correlated with a failure to cleave concatemeric genomes into genomic lengths. That the lesion in UL15 accounted for the inability of vJB28 to cleave DNA was shown by restoration of the presence of terminal fragments in DNA isolated from cells infected with vJB28R.
Fig. 2.

Analysis of viral DNA. (A and B) Schematic diagram of the HSV-1 genome showing the positions of the BamHI P or S or S-P joint fragment in HSV-1 DNA. (C) CV1 cells were infected with the indicated viruses and lysed at 15 h after infection, and DNA was purified, digested with BamHI, electrophoretically separated, transferred to a nylon membrane, and probed with radiolabeled DNA representing the terminus of the short component of the viral genome (BamHI P). Fluorographic images captured on X-ray film are shown. Bands were quantified by densitometry, and the ratios of P to S/P fragments are shown below each lane. The positions of the terminal BamHI P fragment and junction fragment S-P are also shown. (D) A different blot from that shown in panel C was hybridized with radiolabeled DNA derived from the long component terminus (BamHI S). The ratios of the S to S-P fragments are shown below each lane. (E) Southern blot analysis of viral DNA separated by pulsed-field gel electrophoresis. CV1 cells were infected with the indicated virus and, at 15 h after infection, were placed in agarose plugs as described in Materials and Methods. After pulsed-field gel electrophoresis, the DNA was denatured, transferred onto a nylon membrane, and probed with 32P-labeled BamHI P fragment of HSV-1 genome. The positions of size standards are indicated to the left.
In contrast to these results, vJB27 DNA contained both genomic termini, as indicated by readily detectable BamHI P and S fragments. The detection of end-specific fragments in vJB27 DNA was highly reproducible (e.g., see Fig. 3 and 4). Quantification with ImageJ from the digitally scanned film in Fig. 2C revealed that the ratio of the BamHI P fragment to the S-P fragment in vJB27 DNA was ca. 35% of this ratio from HSV-1(F) DNA. The ratio of the BamHI S to S-P fragments in vJB27 DNA (Fig. 2D) was ca. 80% of this ratio derived from HSV-1(F) DNA. In seven different experiments, vJB27 usually showed lower levels of terminal fragments with the ratio of end fragments to internal fragments varying from 0.14 to 1.0. For example, the ratio of P to S-P in vJB27 DNA was 0.22, 0.98, and 0.28 in Fig. 2, 3, and 4, respectively. The terminal fragments of vJB27 DNA had electrophoretic mobilities similar to those of the wild-type BamHI P and S fragments. These data indicate that vJB27 viral DNA was cleaved (albeit less efficiently) by the viral cleavage/packaging machinery despite the fact that the mutation completely precluded viral propagation.
Fig. 3.
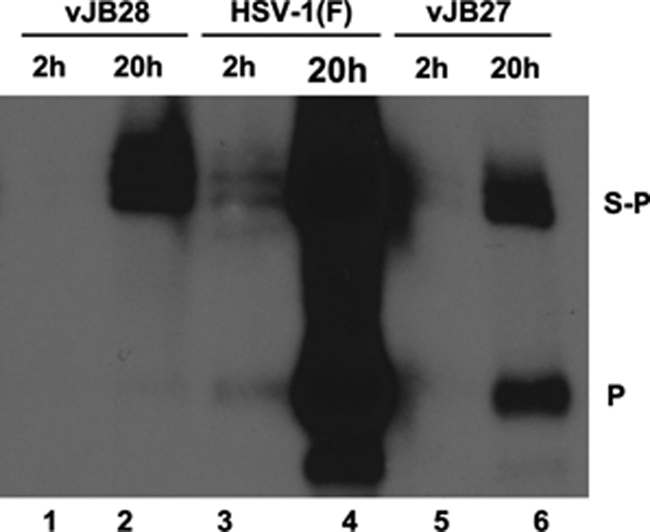
Analysis of input and replicated DNA. Vero cells were infected with 5.0 PFU per cell of the indicated viruses. Cells were lysed at 2 or 20 h postinfection, and DNA was purified, digested with BamHI, electrophoretically separated, denatured, and transferred by capillary action to a nylon membrane. The membranes were probed with radiolabeled BamHI P fragment, representing the terminus of the short component of the HSV genome.
Fig. 4.

DNase protection assay. (A) Fluorographic image of HSV-1 DNA probed with BamHI P. CV1 cells were infected with HSV-1(F) or UL15 mutants at an MOI of 5 PFU/cell. Cells were lysed at 18 h and were incubated in the presence or absence of DNase I. Nuclease treatment was stopped by treatment with proteinase K, and viral DNA was purified and analyzed by Southern blotting as described in the legend to Fig. 2. The blot was analyzed in a Molecular Dynamics Storm 860 PhosphorImager, followed by quantification with ImageQuant software. The ratios of the amounts of BamHI P to S-P fragments are shown below each lane. (B) The percentages of S-P (filled) and P-specific (empty) radioactivity protected from DNase I digestion in the different viral DNAs (i.e., the ratio of the activity of the corresponding bands in treated versus untreated samples) are indicated.
To further characterize the viral DNAs of the mutant viruses, cells were infected with UL15-null, vJB27, vJB28, or HSV-1(F) and immobilized in agarose plugs. The agarose plugs were then digested with proteinase K in the presence of SDS and incorporated into wells of agarose gels, and DNA was separated by pulsed-field electrophoresis. The electrophoretically separated viral DNA was transferred to nylon membranes and probed with radiolabeled HSV-1 viral DNA. As shown in Fig. 2E, two prominent bands hybridized with the HSV-1(F) DNA probe. One band barely entered the gel, whereas the other migrated broadly, but mostly faster than a size standard of 225 kbp. The migration of the upper band is consistent with that of concatemeric viral DNA, whereas the migration of the lower band is consistent with that of monomeric HSV-1 genomic DNA of 152 kbp. The smaller DNA band was not detected in DNA from cells infected with UL15-null and vJB28 viruses, further indicating that these viruses did not cleave concatemeric viral DNA into genomic lengths. In contrast, genomic length viral DNA was readily detected in cells infected with vJB27. Moreover, the size of vJB27 monomeric genomic DNA, as approximated from its electrophoretic mobility, resembled that of the wild-type virus.
Because vJB27 was grown on complementing cells to generate infectious virions and these contain linear genomes, it is possible that some of the cleaved genomes detected in vJB27-infected cells were due to input viral DNA. To test this possibility, cells were infected with HSV-1(F), vJB27, or vJB28, and viral DNA was purified at 2 h and 20 h after infection. The DNA was then digested with BamHI, followed by electrophoretic separation, and Southern blots were probed with radiolabeled BamHI P DNA. As shown in Fig. 3, substantially more viral DNA was produced at 20 h in cells infected with all three viruses than at 2 h, as expected, and substantially more viral DNA was detected in the HSV-1(F) samples compared to the other viruses. At 2 h postinfection, a time before viral DNA replication begins, the BamHI P fragment was detected in HSV-1(F) samples, whereas none was detected in the vJB28 or vJB27 samples. Thus, some input DNA was detectable in the HSV-1(F) samples. Importantly, the BamHI P fragment was readily detected in lysates of cells harvested at 20 h after infection with vJB27 and HSV-1(F) but not in vJB28-infected lysates. These data indicate that cleaved genomes detected in vJB27 cells were derived from replicated viral DNA and not from input DNA. Taken together, the data indicate that vJB27 is able to cleave viral DNA despite its inability to produce infectious virus in noncomplementing cells.
The presence of monomeric viral DNA in cells infected with vJB27 was surprising inasmuch as all previous mutations in UL15 have precluded viral DNA cleavage. To determine whether the cleaved vJB27 viral DNA was packaged, lysates of cells infected with vJB27, vJB28, and HSV-1(F) were incubated in the presence or absence of DNase before viral DNA purification. It was expected that encapsidated viral DNA would be protected from DNase digestion, whereas unpackaged DNA would be susceptible to digestion. After DNase treatment, viral DNA was purified and digested with BamHI, and Southern blots of electrophoretically separated fragments were probed with radiolabeled BamHI P DNA. The results shown in Fig. 4A were quantified with a Molecular Dynamics PhosphorImager (Fig. 4B). The ratio of radioactivity from the BamHI P fragment to that of the BamHI S-P fragments in undigested HSV-1(F) DNA was ∼2.9-fold higher than the ratio of these DNA fragments in untreated vJB27 DNA. In contrast, the BamHI P fragment was not detected in untreated vJB28 DNA. Moreover, none of the uncleaved DNA from vJB28-infected cells was packaged inasmuch as all of the available DNA was digested by DNase treatment. HSV-1(F) DNA was almost completely protected from digestion with DNase, with 80.9 and 90.1% of the available BamHI S-P and P fragments protected from digestion, respectively. In contrast to these results, only 0.4% of the BamHI S-P fragment and 14% of the BamHI P fragment of vJB27 DNA were protected from DNase digestion, indicating that only small amounts of vJB27 viral DNA was encapsidated. Probing the same Southern blot with total HSV-1 DNA revealed that no particular fragment was over- or under-represented in the electrophoretic profile of DNase-protected vJB27 DNA (data not shown). Thus, at least within the limits of agarose gel electrophoresis the small amount of vJB27 DNA that was packaged represented full-length genomes.
The increased susceptibility of cleaved vJB27 genomes to digestion with DNase could result from a failure to retain viral DNA in vJB27 capsids. If this were the case, we would expect to detect abundant A capsids inasmuch as these represent aborted packaging events in which DNA is not retained in capsids, as in the case of UL25-null viruses (19). To test this possibility, CV1 cells were infected with HSV-1(F), vJB27, or vJB28 and the corresponding genetically restored viruses. The cells were then fixed at 14 to 16 h after infection and embedded in Epon, and thin sections were prepared and analyzed by electron microscopy. As shown in Fig. 5, nuclei of cells infected with restored viruses vJB27R and vJB28R contained capsids with internal structures that were either small hollow circles (small cored B capsids) or filled electron-dense circles, representing viral DNA. Many cytoplasmic particles also contained densely staining cores, representing viral DNA. Occasional intranuclear capsids lacking any discernible internal structure were also observed and were most likely A capsids. Quantification from five different intranuclear sections (Table 2) indicated that vJB27R-infected nuclei contained ca. 6.9% A capsids, whereas HSV-1(F) contained ca. 4.7%. Of the intranuclear capsids within these same sections, ca. 5.9% were type C capsids.
Fig. 5.

Ultrastructural examination of CV1 cells infected with UL15 mutant and restored viruses. CV1 cells were infected with the indicated viruses, and 16 h later the cells were fixed, embedded in Epon-Araldite, sectioned, and examined by electron microscopy. Arrows in the upper left and lower left panels indicate enveloped B capsids in the cytoplasm. Arrowheads in these panels indicate B capsids in nuclei. Arrows in upper right and lower right panels indicate enveloped C capsids in the cytoplasm. Arrowheads in the upper and lower right panels indicate C capsids in the nuclei. Nuc, nucleus; Cyto, cytoplasm. As a size standard, capsid diameter is ∼125 nm.
Table 2.
Capsids examined in this studya
| Virus | No. of capsids (%) type: |
|||
|---|---|---|---|---|
| Type A (%) | Type B (%) | Type C (%) | Total | |
| vJB27 | 3 (1.1) | 258 (98.9) | 0 (0) | 261 |
| vJB27 repair | 21 (6.9) | 264 (87) | 18 (5.9) | 303 |
| HSV-1(F) | 16 (4.7) | 301 (89.3) | 20 (5.9) | 337 |
Capsids were characterized by morphology based on electron microscopic examination of thin sections of infected cell nuclei. The results from five nuclear sections containing at least 25 capsids each are shown.
In contrast to these observations, the nuclei of cells infected with only vJB28 contained capsids with hollow circular cores surrounded by an outer shell, i.e., an appearance consistent with small cored B capsids lacking viral DNA. Thus, neither A nor C capsids were detected. This was consistent with the appearance of sucrose gradients bearing vJB28 capsids which contained only a single light refracting band, as opposed to a similar gradient from lysates of cells infected with HSV-1(F) that contained three bands, each ascribable to the three capsid types (Fig. 6). Viral particles were also observed in thin sections in the cytoplasm, but such capsids lacked DNA and were surrounded by an additional circular layer. These cytoplasmic particles were reminiscent of the enveloped type B capsids previously observed in cells infected with a UL15-null mutant (3).
Fig. 6.
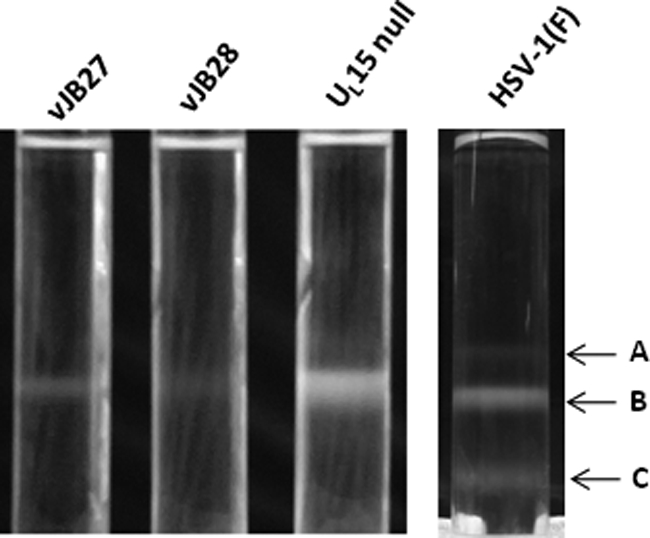
Photograph of light-refracting capsid-specific bands in continuous sucrose gradients. Cells infected with the indicated viruses were lysed, and capsids were purified by centrifugation on continuous sucrose gradients as detailed in Materials and Methods. The positions of the A, B, and C capsids in the gradient containing wild-type HSV-1(F) capsids are indicated.
In vJB27-infected cells, no C capsids were detected, and A capsids were relatively rare, with only three A capsids found in five nuclear sections, yielding a percentage of 1.1%. We did not detect A or C capsids in the cytoplasm of cells infected with vJB27 or as separate light-scattering bands in sucrose gradients containing vJB27-infected cell lysates (Fig. 6).
DISCUSSION
The most important result of these studies is that the mutation in vJB27 UL15 prevents most packaging of cleaved genomes in infected cells. This represents the first mutation in a herpesvirus terminase subunit with this phenotype. We note that the mutation in vJB27 is not expected to alter the nuclease domain of pUL15 (21), perhaps explaining why cleaved genomes can be detected. Only a small portion (between 14 and 0.4%) of these cleaved genomes were protected from DNase, indicating that most genomes were not stably packaged. If all 0.4% of packaged genomes were able to generate infectious progeny, we would expect to detect viral infectivity in noncomplementing cells, inasmuch as our plaque assay should readily detect virus reduced in titer by 250-fold. Given the observation that the mutation in vJB27 reduced viral infectivity by >30,000-fold (Table 1), it is likely that either the protected DNA is not infectious or vJB27 has defects in addition to inefficient DNA packaging.
A key consideration in the field concerns why genomes are not cleaved indiscriminately in infected cells. The fact that any detectable cleavage of genomic DNA requires intact capsids with portals argued that DNA cleavage and packaging were inextricably linked. Thus, DNA cleavage might occur after DNA packaging has initiated (or even reached completion). The phenotype of vJB27 argues against this hypothesis and suggests that DNA cleavage can occur independently of packaging. An interesting alternative possibility is that the region deleted in vJB27 acts as an intramolecular regulatory domain to inhibit the nuclease function of pUL15 until packaging begins. We do not favor this possibility because removal of such a domain should increase cleavage of packaging sequences, whereas reduced amounts of cleavage were observed in cells infected with vJB27 (e.g., Fig. 2).
We also note that despite readily detectable cleaved genomes in vJB27-infected cells, mostly small cored B capsids were observed, with a relative rarity of A capsids. Moreover, an A capsid-specific light-diffusing band was absent from sucrose gradients containing vJB27 capsids (Fig. 6). Given the modest reduction of DNA cleavage in vJB27-infected cells, if DNA was frequently packaged and then lost from the capsid as in the case of the UL25 mutant described previously (19), we would have expected to detect more abundant A capsids in cells infected with vJB27. Because scaffold expulsion/degradation is a prerequisite for DNA packaging, it follows that DNA packaging of cleaved genomes had mostly failed to initiate or was aborted very early in the majority of cases in vJB27-infected cells.
The mechanism responsible for the vJB27 phenotype (i.e., DNA cleavage without packaging) may involve an aberrant interaction with the portal vertex, a failure to interact properly with cleaved genomic termini, a direct failure to translocate DNA, or removal of the link between the energy-generating Walker box and the nuclease domain at the C terminus (Fig. 1) (11, 21). Of these possibilities, complete failure to interact with portal is unlikely because pUL15 and pUL6 can be coimmunoprecipitated from vJB27-infected cell lysates (Fig. 1B), and at least some stably packaged DNA was detected. A role for pUL15 in translocating DNA into the capsid is consistent with several previous observations placing it closest to the capsid and portal vertex, whereas its interacting partners, pUL28 and pUL33, are located more peripherally (4, 45).
We also note that a motif common to several viral terminases, including pUL15, was proposed to link ATPase activity to DNA translocation (20). This C motif formally corresponds to Val-Ser-Ser in pUL15 and starts at amino acid 383. In the present study, deletion of amino acids 383 to 385 in vJB28 precluded both DNA cleavage and DNA packaging, indicating that the motif plays roles beyond those of DNA translocation per se. In contrast, deletion of a nearby region, amino acids 400 to 420 in vJB27, conferred a phenotype similar to that expected of C-motif mutants, suggesting an analogous function.
ACKNOWLEDGMENTS
We thank Theodore Clark at Cornell University for help with the pulsed-field gel electrophoresis.
This study was supported by NIH grant GM52341 to J.D.B.
Footnotes
Published ahead of print on 31 August 2011.
REFERENCES
- 1. Abbotts A. P., Preston V. G., Hughes M., Patel A. H., Stow N. D. 2000. Interaction of the herpes simplex virus type 1 packaging protein UL15 with full-length and deleted forms of the UL28 protein. J. Gen. Virol. 81 (Pt. 12): 2999–3009 [DOI] [PubMed] [Google Scholar]
- 2. Adelman K., Salmon B., Baines J. D. 2001. Herpes simplex DNA packaging sequences adopt novel structures that are specifically recognized by a component of the cleavage and packaging machinery. Proc. Natl. Acad. Sci. U. S. A. 98: 3086–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baines J. D., Cunningham C., Nalwanga D., Davison A. J. 1997. The UL15 gene of herpes simplex virus type 1 contains within its second exon a novel open reading frame that is translated in frame with the UL15 gene product. J. Virol. 71: 2666–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beard P. M., Duffy C., Baines J. D. 2004. Quantification of the DNA cleavage and packaging proteins UL15 and UL28 in A and B capsids of herpes simplex virus type 1. J. Virol. 78: 1367–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beard P. M., Taus N. S., Baines J. D. 2002. The DNA cleavage and packaging proteins encoded by genes UL28, UL15, and UL33 of herpes simplex virus 1 form a complex in infected cells. J. Virol. 76: 4785–4791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Black L. W. 1989. DNA packaging in dsDNA bacteriophages. Annu. Rev. Microbiol. 43: 267–292 [DOI] [PubMed] [Google Scholar]
- 7. Cardone G., et al. 2007. Visualization of the herpes simplex virus portal in situ by cryo-electron tomography. Virology 361: 426–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cockrell S. K., Sanchez M. E., Erazo A., Homa F. L. 2009. Role of the UL25 protein in herpes simplex virus DNA encapsidation. J. Virol. 83: 47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conway J. F., Homa F. L. 2011. Nucleocapsid structure, assembly and DNA packaging of herpes simplex virus, p. 175–193 In Weller S. K. (ed.), Alphaherpesviruses. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 10. Dasgupta A., Wilson D. W. 1999. ATP depletion blocks herpes simplex virus DNA packaging and capsid maturation. J. Virol. 73: 2006–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davison A. J. 1992. Channel catfish virus: a new type of herpesvirus. Virology 186: 9–14 [DOI] [PubMed] [Google Scholar]
- 12. Deiss L. P., Chou J., Frenkel N. 1986. Functional domains within the a sequence involved in the cleavage-packaging of herpes simplex virus DNA. J. Virol. 59: 605–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Desai P., DeLuca N. A., Glorioso J. C., Person S. 1993. Mutations in herpes simplex virus type 1 genes encoding VP5 and VP23 abrogate capsid formation and cleavage of replicated DNA. J. Virol. 67: 1357–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Draper B., Rao V. B. 2007. An ATP hydrolysis sensor in the DNA packaging motor from bacteriophage T4 suggests an inchworm-type translocation mechanism. J. Mol. Biol. 369: 79–94 [DOI] [PubMed] [Google Scholar]
- 15. Gibson W., Roizman B. 1972. Proteins specified by herpes simplex virus VIII. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J. Virol. 10: 1044–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hayward G. S., Jacob R. J., Wadsworth S. C., Roizman B. 1975. Anatomy of herpes simplex virus DNA: evidence for four populations of molecules that differ in the relative orientations of their long and short segments. Proc. Natl. Acad. Sci. U. S. A. 72: 4243–4247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koslowski K. M., et al. 1999. Physical and functional interactions between the herpes simplex virus UL15 and UL28 DNA cleavage and packaging proteins. J. Virol. 73: 1704–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McGeoch D. J., et al. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69: 1531–1574 [DOI] [PubMed] [Google Scholar]
- 19. McNab A. R., et al. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated DNA. J. Virol. 72: 1060–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mitchell M. S., Matsuzaki S., Imai S., Rao V. B. 2002. Sequence analysis of bacteriophage T4 DNA packaging/terminase genes 16 and 17 reveals a common ATPase center in the large subunit of viral terminases. Nucleic Acids Res. 30: 4009–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nadal M., et al. 2010. Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc. Natl. Acad. Sci. U. S. A. 107: 16078–16083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nasseri M., Mocarski E. S. 1988. The cleavage recognition signal is contained within sequences surrounding an a-a junction in herpes simplex virus DNA. Virology 167: 25–30 [DOI] [PubMed] [Google Scholar]
- 23. Newcomb W. W., et al. 1996. Assembly of the herpes simplex virus capsid: characterization of intermediates observed during cell-free capsid formation. J. Mol. Biol. 263: 432–446 [DOI] [PubMed] [Google Scholar]
- 24. Newcomb W. W., et al. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 75: 10923–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Patel A. H., Rixon F. J., Cunningham C., Davison A. J. 1996. Isolation and characterization of herpes simplex virus type 1 mutants defective in the UL6 gene. Virology 217: 111–123 [DOI] [PubMed] [Google Scholar]
- 26. Reynolds A. E., Fan Y., Baines J. D. 2000. Characterization of the UL33 gene product of herpes simplex virus 1. Virology 266: 310–318 [DOI] [PubMed] [Google Scholar]
- 27. Reynolds A. E., Wills E. G., Roller R. J., Ryckman B. J., Baines J. D. 2002. Ultrastructural localization of the HSV-1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76: 8939–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salmon B., Cunningham C., Davison A. J., Harris W. J., Baines J. D. 1998. the herpes simplex virus 1 UL17 gene encodes virion tegument proteins that are required for cleavage and packaging of viral DNA. J. Virol. 72: 3779–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Salmon B., Nalwanga D., Fan Y., Baines J. D. 1999. Proteolytic cleavage of the amino terminus of the UL15 gene product of herpes simplex virus 1 UL15 protein is coupled with maturation of viral DNA into unit length genomes. J. Virol. 73: 8338–8348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scheffczik H., Savva C. G., Holzenburg A., Kolesnikova L., Bogner E. 2002. The terminase subunits pUL56 and pUL89 of human cytomegalovirus are DNA-metabolizing proteins with toroidal structure. Nucleic Acids Res. 30: 1695–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stow N. D. 2001. Packaging of genomic and amplicon DNA by the herpes simplex virus type 1 UL25-null mutant KUL25NS. J. Virol. 75: 10755–10765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun S., Kondabagil K., Gentz P. M., Rossmann M. G., Rao V. B. 2007. The structure of the ATPase that powers DNA packaging into bacteriophage T4 procapsids. Mol. Cell 25: 943–949 [DOI] [PubMed] [Google Scholar]
- 33. Sun S., et al. 2008. The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell 135: 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanaka M., Kagawa H., Yamanashi Y., Sata T., Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77: 1382–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Taus N. S., Baines J. D. 1998. Herpes simplex virus DNA cleavage and packaging: the UL28 gene product is a minor component of B capsids. Virology 252: 443–449 [DOI] [PubMed] [Google Scholar]
- 36. Taus N. S., Salmon B., Baines J. D. 1998. The herpes simplex virus 1 UL17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology 252: 115–125 [DOI] [PubMed] [Google Scholar]
- 37. Tengelsen L. A., Pedersen N. E., Shaver P. R., Wathen M. W., Homa F. L. 1993. Herpes simplex virus type 1 DNA cleavage and capsidation require the product of the UL28 gene: isolation and characterization of two UL28 deletion mutants. J. Virol. 67: 3470–3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tischer B. K., von Einem J., Kaufer B., Osterrieder N. 2006. Two-step RED-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40: 191–197 [DOI] [PubMed] [Google Scholar]
- 39. Tong L., Stow N. D. 2010. Analysis of herpes simplex virus type 1 DNA packaging signal mutations in the context of the viral genome. J. Virol. 84: 321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Trus B. L., et al. 1996. The herpes simplex virus procapsid: structure, conformational changes upon maturation, and roles of the triplex proteins VP19C and VP23 in assembly. J. Mol. Biol. 263: 447–462 [DOI] [PubMed] [Google Scholar]
- 41. Trus B. L., et al. 2004. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J. Virol. 78: 12668–12671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Varmuza S. L., Smiley J. R. 1985. Signals for site-specific cleavage of HSV DNA: maturation involves two separate cleavage events at sites distal to the recognition sequences. Cell 41: 793–802 [DOI] [PubMed] [Google Scholar]
- 43. Wagner M. J., Summers W. C. 1978. Structures of the joint region and the termini of the DNA of herpes simplex virus type 1. J. Virol. 27: 374–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. White C. A., Stow N. D., Patel A. H., Hughes M., Preston V. G. 2003. Herpes simplex virus type 1 portal protein UL6 Interacts with the putative terminase subunits UL15 and UL28. J. Virol. 77: 6351–6358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wills E., Scholtes L., Baines J. D. 2006. Herpes simplex virus 1 DNA packaging proteins encoded by UL6, UL15, UL17, UL28, and UL33 are located on the external surface of the viral capsid. J. Virol. 80: 10894–10899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang K., Baines J. D. 2006. The putative terminase subunit of herpes simplex virus 1 encoded by UL28 is necessary and sufficient to mediate interaction between pUL15 and pUL33. J. Virol. 80: 5733–5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang K., Homa F., Baines J. D. 2007. Putative terminase subunits of herpes simplex virus 1 form a complex in the cytoplasm and interact with portal protein in the nucleus. J. Virol. 81: 6419–6433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang K., Wills E., Baines J. D. 2009. The putative leucine zipper of the UL6-encoded portal protein of herpes simplex virus 1 is necessary for interaction with pUL15 and pUL28 and their association with capsids. J. Virol. 83: 4557–4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu D., Weller S. K. 1998. Genetic analysis of the UL 15 gene locus for the putative terminase of herpes simplex virus type 1. Virology 243: 32–44 [DOI] [PubMed] [Google Scholar]