

Abstract
Following their assembly, herpesvirus capsids exit the nucleus by budding at the inner nuclear membrane. Two highly conserved viral proteins are required for this process, pUL31 and pUL34. In this report, we demonstrate that the pUL31 component of the pseudorabies virus nuclear egress complex is a conditional capsid-binding protein that is unmasked in the absence of pUL34. The interaction between pUL31 and capsids was confirmed through fluorescence microscopy and Western blot analysis of purified intranuclear capsids. Three viral proteins were tested for their abilities to mediate the pUL31-capsid interaction: the minor capsid protein pUL25, the portal protein pUL6, and the terminase subunit pUL33. Despite the requirement for each protein in nuclear egress, none of these viral proteins were required for the pUL31-capsid interaction. These findings provide the first formal evidence that a herpesvirus nuclear egress complex interacts with capsids and have implications for how DNA-containing capsids are selectively targeted for nuclear egress.
INTRODUCTION
Although the herpesviridae constitute a diverse array of viral agents, all share a related structure that begins as an icosahedral protein shell that is assembled and packaged with a linear double-stranded DNA (dsDNA) genome in the nucleus of the host cell. Capsid assembly and genome encapsidation are complex processes with a reasonably high failure rate; defective capsid species are readily purified from infected cells in culture and are visible in electron micrographs of infected cells and tissues. The modest fidelity of these processes appears to be compensated for by a quality control step operating at the nuclear membrane that selects capsids containing genomes for egress from the nucleus to the cytosol (16, 65, 79, 87). Viral proteins resident in the inner nuclear membrane constitute a nuclear egress complex (NEC). How the NEC selects for packaged capsids is not understood, and in fact, interactions between the capsid and NEC remain to be defined.
Herpesvirus assembly begins with the formation of fragile procapsid icosahedral shells that are built upon protein scaffolds (15, 51, 53, 68, 83). The encapsidation of the viral genome occurs through a dodecameric portal ring that occupies one of the 12 vertices of the procapsid shell and is encoded by the UL6 gene (11, 12, 19, 52, 84). The terminase complex, consisting of the products of the UL15, UL28, and UL33 genes, is the molecular motor that facilitates the packaging of the genome through the portal (2, 8, 60, 90, 94). The initial stabilization of the capsid occurs concomitantly with the cleavage of the internal scaffolding by the VP24 protease (32), with additional structural rigidity gained at a subsequent step that may be coincident with the release of the proteolyzed scaffold and genome packaging (70). From the progenitor procapsid, three stable capsid species that can be isolated by differential sedimentation in continuous sucrose gradients are produced: A, B, and C capsids (9, 26, 58). The production of A capsids, which lack DNA and a scaffold, requires a functional terminase (1, 3, 44). A capsids are likely failed attempts at genome packaging (59, 70). B capsids are also nonproductive structures but, unlike A capsids, retain a proteolyzed scaffold, lack DNA, and can form independently of encapsidation (1, 3, 44, 57, 60, 71, 95). During successful packaging, the scaffolding is cleaved and replaced by the incoming genomic DNA, resulting in a C capsid (9, 58, 59).
The NEC consists primarily of two conserved viral proteins, pUL31 and pUL34 in herpes simplex virus (HSV) nomenclature, that are required for primary envelopment in all herpesviruses studied (25, 40, 45, 47, 93). pUL34 is a type II transmembrane protein (63, 69) that localizes to the endoplasmic reticulum (ER) and the nuclear membrane in transfected cells (66, 75). Upon coexpression, pUL31 and pUL34 are retained within the inner nuclear membrane, which reflects the normal distribution of the proteins during infection (25, 66, 67, 91). Therefore, each of these proteins requires the presence of the other for their proper localization to the nuclear membrane. Although pUL31 and pUL34 do not have known enzymatic activities, their coexpression can produce budding events at the inner nuclear membrane (34), and they are required for the budding of capsids into the perinuclear space in infected cells (20, 25, 27, 38, 49, 66, 69, 75). Perinuclear enveloped capsids are infrequently observed during infection with wild-type viruses and are thought to represent short-lived intermediates in the nuclear egress pathway that rapidly fuse with the outer nuclear membrane to deliver the capsid to the cytosol (28). However, the NEC mechanism of action remains ambiguous, and alternative models of nuclear egress that do not involve capsid budding through the inner nuclear membrane have been proposed (37, 41, 79).
Part of the difficulty in identifying interactions between the NEC and capsids likely stems from the transience of the interactions during infection: although the NEC interacts with capsids, the NEC also disassociates from capsids following egress to the cytosol (25, 67). We considered that the blocking of nuclear egress may stabilize an NEC-capsid interaction and allow its identification. To this end, we present one strategy that proved successful: by deleting the UL34 gene from pseudorabies virus (PRV), pUL31 accumulated at intranuclear puncta during late infection. We show that these puncta overlap with capsid assemblies and demonstrate that pUL31 is a capsid-binding protein that copurifies with capsids in the absence of pUL34 in a manner that is independent of the pUL6 portal protein, pUL25 minor capsid protein, and pUL33 terminase protein.
MATERIALS AND METHODS
Cells.
Vero cells were used for live-cell imaging. Viruses were propagated in pig kidney epithelial (PK15) cells. A PK15 cell line stably expressing pUL34 (PK15-UL34), used to propagate UL34-null viruses, was previously described (17). The UL34 stable cell line was further modified to stably express either the pUL6, pUL25, or pUL33 protein. The subcloning of UL25 into pLPCX (Clontech) was previously described (17). To subclone UL6, an R6K plasmid (conditional replication origin) was PCR amplified with primers 5′-GTTGGTGGCGCGCGGACCCTCTGCGGCGGCGGCGGCGGCCGAATTCCCACATGTGGAATTCCCAT and 5′-GGCAGAGCCCGTCCGCAGCCGCCGCCGTCGCAGCCGACATGGTAGATCTGTCATCCATATCACCACG and inserted into a derivative of the pBecker3 infectious clone that was first modified to encode an EcoRI restriction site immediately downstream of the UL6 stop codon using two-step recombination (77, 81). Based on the homologies of the 5′ ends of the primers, the R6K plasmid along with the BglII and EcoRI restriction sites (underlined) were inserted upstream of the UL6 open reading frame (ORF). Additionally, a Kozak consensus sequence was introduced with the reverse primer (in boldface type). The UL6 ORF was liberated upon digestion with EcoRI and self-ligated to produce an R6K-based clone in S17-1 λpir bacteria. The UL6 ORF was subcloned into the pLPCX vector by digestion with BglII and EcoRI. UL33 was subcloned by an equivalent strategy following R6K amplification with primers 5′-GGCCGTGTACGGCGCCTCGAAGGCGTCCTCGCTCCAGCCCGTCGACGAATTCCCACATGTGGAATTCCCAT and 5′-CGCGCAGATCGACCGCCGCGCCGCCGCCGCCGCGCGCCATGGTAGATCTGTCATCCATATCACCACG.
To generate retroviral particles, the pLPCX plasmids were cotransfected with pVSV-G (Clontech) into the GP-293 packaging cell line. The harvested transducing particles were used to deliver the expression cassettes to PK15 cells and selected with 500 μg/ml G418. Clones of each lineage were saved as either PK15-UL6, PK15-UL25, or PK15-UL33 cells following the confirmation of the complementation of PRV deleted for the respective genes. The UL6, UL25, and UL33 ORFs were also transferred from the pLPCX vector to the pLNCX2 vector (Clontech) by digestion with SnaBI and ClaI. Retroviral particles were prepared and used to transduce the PK15-UL34 cell line. Three stable cell lines were selected with 500 μg/ml G418 and 1 μg/ml puromycin, and each lineage was saved as either PK15-UL6/UL34, PK15-UL25/UL34, or PK15-UL33/UL34 cells following the confirmation of the complementation of PRV deleted for the respective pairs of genes.
Viruses.
Two-step recombination was used to modify either the pBecker3 infectious clone or a pBecker3 derivative, pGS847, which encodes an mRFP1-VP26 capsid reporter (77, 78, 81). The following primers were used to fuse the c-myc epitope to the 5′ end of UL31: 5′-CCACGGCCTCGCGCTCGTCTCCCTCGAGCACACGCTCGGCAGCTACGTTTGAATAAACCATGGAGCAGAAGCTCATCTCGGAGGAGGACCTGGGCAGCAGGATGACGACGATAAGTAGGG and 5′-CTTGCGCCGCGCGGCCGACGACTTGCGCCGCAGGAGCCGCCGTCGCTCAAAGCTGCCCAGGTCCTCCTCCGAGATGAGCTTCAACCAATTAACCAATTCTGATTAG. Additionally, the primers reconstructed the 3′ end of UL32 and introduced a silent point mutation to destroy the natural UL31 initiation codon (in boldface type). To compensate for potential polar effects on the overlapping UL32 gene, an ectopic polyadenylation signal was introduced immediately downstream of the UL32 stop codon (in italic type). The PCR product was recombined into the unmodified pBecker3 bacterial artificial chromosome (BAC) clone, resulting in pGS3393, and the recombinant PRV-GS3393 virus was harvested from transfected PK15 cells.
To delete the UL34 ORF, the following primers were used to amplify the pEP-KanS2 plasmid: 5′-CTCACCCCCACCCTCACGCTCGCGACGACGACGACGACGACCATGTAAGGCCCCGACGACGCCAGGATGACGACGATAAGTAGGG and 5′-GGCGCGGTCCAGGAGGCGCTGCGTCTGCGAGGCGTCGTCGGGGCCTTACATGGTCGTCGTCGTCAACCAATTAACCAATTCTGATTAG. The regions of homology to the pEP-KanS2 template are underlined. The PCR product was used for homologous recombination into either pGS847, resulting in pGS2847, or pGS3393, resulting in pGS3406. The primers inserted a stop codon immediately following the initiation codon, resulting in the deletion of codons 2 to 155 (of 260 amino acids [aa] total). The resulting infectious clones were transfected into PK15-UL34 cells to produce PRV-GS2847 and PRV-GS3406, respectively.
The following primers were used to fuse egfp to the 5′ end of UL31: 5′-CCACGGCCTCGCGCTCGTCTCCCTCGAGCACACGCTCGGCAGCTACGTTTGACCATGGTGAGCAAGGGCGAGGAG and 5′-CCGCGCGGCCGACGACTTGCGCCGCAGGAGCCGCCGTCGCTCAAACTTGTACAGCTCGTCCATGC. The PCR product amplified from the pEP-EGFP-in plasmid was recombined into pGS2847, resulting in pGS2868. The regions of homology are underlined. Due to the overlap between the 3′ end of the UL32 ORF and the 5′ end of the UL31 ORF, the primers reconstructed the 3′ end of the UL32 ORF and introduced a silent point mutation to destroy the natural UL31 initiation codon (in boldface type). The transfection of the recombinant BAC clone into PK15 cells resulted in the production of the dual-fluorescent virus PRV-GS2868. Although this virus did not require complementation for its propagation, the inclusion of green fluorescent protein (GFP) on the amino terminus of pUL31 was debilitating.
A second viral ORF (UL6, UL33, or UL25) was removed from pGS3406 by using two-step mutagenesis to produce BAC clones encoding deletions in two viral genes. For each of these deletions, the pEP-KanS2 template was PCR amplified with the following primers. The regions of homology with the template are underlined.
The primers used to delete UL6 were 5′-GCGGAGGAGGCTAATAACCTGTTGGGCGCCCCCTCGCGGATCCTCTAAAGGCGACGACGCCTACAGGATGACGACGATAAGTAGGG and 5′-CACGTAGCGCGACTGGAAGCTGTTGGCCACGTAGGCGTCGTCGCCTTTAGAGGATCCGCGAGGGCAACCAATTAACCAATTCTGATTAG. Because UL6 overlaps upstream and downstream viral genes, the primers were designed to introduce a stop codon after codon 32 and to delete codons 33 to 482 (of 642 aa total). To further ensure that the remaining 3′ UL6 ORF was not expressed, a frameshift mutation was also introduced after the stop codon. The primers used for the deletion of UL25 were 5′-GGTGGGCGCGGTGATAAGGCGGCGCGCGGCGGCGCGCGGCCCATGTAACAGTTTGGCGTCTCCAGGATGACGACGATAAGTAGGG and 5′-GCAGAGAAAGTACAGGAGGTCGTAGTCGCTGGAGACGCCAAACTGTTACATGGGCCGCGCGCCCAACCAATTAACCAATTCTGATTAG. The primers introduced a stop codon after the endogenous start ATG codon and deleted codons 2 to 509 (of 534 aa total). This deletion design was based on a previous study (17). The primers used to delete UL33 were 5′-CTGCGCGCGGCGATCCCGGAGGCCGCGCTGCGCGACTTCGACGTGTAAACCGCGCGAGCTGGAAAGGATGACGACGATAAGTAGGG and 5′-TAGCTTGGCGTCCGTCGTGGGGAGGATCACTTCCAGCTCGCGCGGTTTACACGTCGAAGTCGCGCAACCAATTAACCAATTCTGATTAG. To maintain the promoter for the neighboring UL32 gene, the primers were designed to introduce a stop codon after codon 26 and to delete codons 27 to 46 (of 115 aa total). A frameshift mutation was also incorporated immediately following the stop codon.
Plasmids.
The pUL25-myc mammalian expression plasmid was previously described (17). To subclone UL31 into a GFP expression vector, the R6K plasmid was PCR amplified with primers 5′-GCCTCGCGCTCGTCTCCCTCGAGCACACGCTCGGCAGCTGTCGACGAATTCCCACATGTGGAATTCCCAT and 5′-CGACGACTTGCGCCGCAGGAGCCGCCGTCGCTCAAACATAGATCTGTCATCCATATCACCACG and inserted upstream of the UL31 ORF within a pBecker3 infectious clone. The primers encode a SalI site (in boldface type) and EcoRI and HindIII sites (underlined). An endogenous SalI site was located downstream of the UL31 ORF. The UL31 ORF was liberated by digestion with SalI and self-ligated, resulting in an R6K plasmid clone in S17-1 λpir bacteria. The UL31 ORF was then subcloned into pEGFP-C1 (Clontech) by digestion with EcoRI and HindIII, resulting in pGS2896.
The four conserved regions of PRV UL31 were made synthetically (42). The following oligonucleotides were used: 5′-CCAGATCTGCCCGCGATCGCTACGCGCCCTACTTTGCCTACGCGGCCGCGCAGCCCTCGGACGAGGTGACCACCGTGCGCGGCCTCTCGAACCCGCTCATCAAGACGGCCCCCGTGACGCTGCCC and 5′-CCAAGCTTAGGCGGCGGCGCACGTGGGGCAGCAGCCGCCGAGGCCCAGGTAGTAGCCCATGCCCGAGAGCGACAGGCAGTTGTCGGCCACCGCCTGGCCGAGGTCGAAGGGCAGCGTCACGGGGGC (conserved region 1 [CR1]) (nucleotides 70 to 285), 5′-CCAGATCTGAGCCGCGGCTCGGGCGCAGCGACCGCGCGGCCCTCGTGCTGGCCTACGTGCAGCAGCTCAACAGC and 5′-CCAAGCTTAGGGGTCCCGCGCGGCCACCGAGGCCAGGAACACGCGGTACTCGTAGATGCTGTTGAGCTGCTGCA (CR2) (nucleotides 286 to 399), 5′-CCAGATCTTCGGAGCGCGCCCTCGAGGAGGTGCTCGCGCACCCGGAGCTCTTTTTCGCCTACTACGTGCTGCGCGACGGGGGCCTGCGCGACGTGCGCGTGCTCTTCTTCGAGGACCCCGACGCGCAGGGCGCGCTCATGATGTACGTGGTGTTC and 5′-CCAAGCTTACCCGTCTCCCTTCTTTCGCACCACCAACACGAACATGGTCTGCCACACGTGCGCGACGATGCGGTGGCCGGCGCAGGCCCCGAGCAGGCGGTCCAGCACGCGGTGGTGCACGTGCACCGACTTCTCGGGGAACACCACGTACATCA (CR3) (nucleotides 400 to 675), and 5′-CCAGATCTAGACCCGCGGACGACGTGCCCGCCGTCAGCGCGAGCGACATTTATTGCAAGATGCGGGACATCAGCTTCGACGGGGAGC and 5′-CCAAGCTTACGGGCGAGGGGGGCGAAAGTCTTCAAACGCTGCGTACAATCTTTTGTACTCCAACAGCAGCTCCCCGTCGAAGCTG (CR4) (nucleotides 676 to 816). The oligonucleotide pairs encode each entire conserved region with redundancies in the overlapping 3′ ends (underlined). The forward primers encode BglII restriction sites, and the reverse primers encode HindIII restriction sites (in italic type). Stop codons were incorporated into the reverse primers (in boldface type). The primer pairs were annealed to each other and elongated by using Taq polymerase. The products from the oligonucleotide extension reactions were digested with BglII and HindIII and subcloned into pEGFP-C1 as in-frame fusions to GFP. The plasmids were confirmed by sequencing.
Transfection.
For transient-transfection experiments with GFP-pUL31, 250 ng of pGS2896 DNA and 5 μl of Lipofectamine 2000 (Invitrogen) were incubated together in 400 μl of Dulbecco's modified Eagle's medium (DMEM) for 20 min. The mixture was then added to Vero cells plated onto glass coverslips in a 6-well plate. Cells were infected with either PRV-GS847 or PRV-GS2847 16 h following transfection and imaged at 8 to 10 h postinfection.
Intranuclear capsid isolation.
Capsids were isolated by using a protocol adapted from that described previously by Bucks et al. (10), with slight modifications. Vero cells were grown to confluence in 850-cm2 roller bottles and infected at a multiplicity of infection (MOI) of 10. Infected cells were harvested at 18 h postinfection and pelleted at 1,000 × g for 10 min at 4°C. The cells were then washed once in phosphate-buffered saline before being lysed in 1% NP-40 lysis buffer (0.15 M NaCl, 0.01 M Tris-HCl [pH 7.2], 2 mM MgCl2, 1% NP-40) supplemented with 5 mM dithiothreitol and protease inhibitor cocktail (Sigma) for 30 min on ice. The nuclei were separated from cytoplasmic material by centrifugation at 1,000 × g for 10 min at 4°C and washed once in 1% NP-40 lysis buffer. Nuclear lysis was achieved by passage through 18-, 21-, and then 25-gauge needles. Following lysis, 100 U of DNase I (MP Biomedicals) was added to the sample, and the sample was incubated for 15 min at 37°C. Nuclear debris was removed by centrifugation at 3,000 × g for 10 min at 4°C. The resulting supernatant was overlaid onto 35% sucrose dissolved in TNE (20 mM Tris [pH 7.6], 500 mM NaCl, 1 mM EDTA) and centrifuged in an SW41 rotor at 25,000 rpm for 1 h at 4°C. The supernatant was aspirated, and the pelleted capsids were resuspended in TNE and dispersed by sonication. Capsids were then transferred onto a 20 to 50% sucrose gradient made in TNE using a Gradient Master instrument (BioComp Instruments). The sample was then centrifuged for 1 h at 4°C at 25,000 rpm in an SW41 rotor. The gradients were fractionated into 18 fractions by using a Gradient Fractionator (BioComp Instruments). Total protein was precipitated by the addition of trichloroacetic acid (TCA) to a 10% final concentration. Following a 1-h incubation on ice, samples were centrifuged for 10 min at 4°C. The supernatant was aspirated, and the pellet was resuspended in loading buffer (31.25 mM Tris [pH 6.8], 5% glycerol, 0.005% bromophenol blue, 1% SDS, 5% β-mercaptoethanol) supplemented with 0.1 N NaOH.
Coimmunoprecipitation and Western blotting.
HEK-293 cells transiently transfected with pGS2060 and pGS2896 were suspended in TEEN buffer (50 mM Tris [pH 7.4], 1 mM EDTA, 1 mM EGTA, 150 mM NaCl) and subjected to 10 1-s pulses in a cup sonicator. The samples were then rested on ice for 1 h. Ten microliters of prewashed protein A/G agarose beads (EMD) was incubated with 1 μl of anti-GFP rabbit serum (Invitrogen) for 2 h at 4°C. After three washes in TEEN buffer, the loaded beads were added to each sample and incubated overnight at 4°C. They were then washed five times and resuspended in 2× final sample buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100, 10% β-mercaptoethanol).
For Western blot analysis, samples were boiled for 5 min and separated through SDS-polyacrylamide gels. After transfer onto a polyvinylidene fluoride membrane (Pall), the membranes were blocked in 5% milk diluted in phosphate-buffered saline with 0.1% Tween. The membranes were then probed with either anti-myc antibody (clone 9E10; provided by the Northwestern Monoclonal Antibody Facility) diluted 1:2,000, anti-GFP antibody (clone B2; Santa Cruz Biotechnology) diluted 1:1,000, anti-VP16 antibody (chicken polyclonal; generously provided by Lynn Enquist) diluted 1:1,000, or anti-VP5 antibody (clone 3C10; generously provided by Lynn Enquist) diluted 1:8,000. The membranes were incubated with peroxidase-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch) diluted 1:10,000 or rabbit anti-chicken IgY secondary antibody (Sigma) diluted 1:5,000. Following incubation with a luminol-coumaric acid-H2O2 chemiluminescence solution, the membranes were exposed to film. ImageJ was used to perform densitometry analysis.
Fluorescence microscopy.
Microscopy was performed with a Nikon Eclipse TE2000-U inverted wide-field microscope with a 60×, 1.4-numerical-aperture oil objective (Nikon) and a Cascade:650 camera (Photometrics). The images were acquired and analyzed with Metamorph software (Molecular Devices).
RESULTS
GFP-pUL31 localizes to capsid assemblies in the absence of pUL34.
If the nuclear egress complex binds capsids to facilitate their egress, the interaction must be transient, as neither pUL31 nor pUL34 is detected on cytosolic capsids or in virions (25, 38, 49, 67). We hypothesized that if pUL31 was a conditional capsid-binding protein, its interaction with capsids may be stabilized if nuclear egress was blocked. To achieve this, the UL34 open reading frame was deleted from a recombinant strain of PRV that encodes two fluorescent tags: mRFP1-VP26 (monomeric red fluorescent protein 1 fused to capsids) and GFP-pUL31. The virus was propagated on a cell line, PK15-UL34, that stably expresses pUL34.
In Vero cells infected with a virus encoding an intact UL34 allele, GFP-pUL31 was enriched at the nuclear membrane, consistent with previous findings (25, 66, 67). A significant proportion of pUL31 was also distributed diffusely throughout the nucleus, which, based on a previous study, may have been associated with the nuclear matrix (13). In contrast, GFP-pUL31 was not enriched at the nuclear membrane in the absence of pUL34 but instead was frequently distributed to intranuclear capsid assemblies (Fig. 1A). This relocalization was observed for 78% (n = 59) of cells in which capsid assemblies were present, with the GFP-pUL31 emissions typically being detected at the periphery of the capsid assemblies (Fig. 1B). This result provided the first indication that pUL31 may be a conditional capsid-binding protein.
Fig. 1.

Fluorescence localization of GFP-tagged pUL31. (A) Vero cells were infected at an MOI of 10 with dual-fluorescent viruses, which encode an mRFP1 fusion to the capsid protein VP26 and a GFP fusion to the N terminus of pUL31. The virus had either an intact UL34 allele (wild type) or a UL34 deletion allele (ΔUL34). Cells were imaged between 8 and 10 h postinfection. In the absence of UL34, pUL31 localization to the nuclear membrane is lost, and pUL31 becomes enriched at capsid assemblies. (B) Magnification of the region indicated by the dotted box in the ΔUL34 merged image in panel A. (C) Vero cells were transfected with a plasmid encoding a GFP-pUL31 fusion protein and subsequently infected with PRV encoding RFP-VP26 (red capsid) with or without an intact UL34 gene. (D) Magnification of the boxed region in the ΔUL34 merged image in panel C.
Because PRV encoding the GFP-pUL31 fusion propagated poorly (Table 1), the colocalization of pUL31 and capsid assemblies in the absence of pUL34 was confirmed by using a virus encoding unmodified pUL31. In this experiment, Vero cells were first transfected with a plasmid encoding the GFP-pUL31 fusion. These cells emitted GFP fluorescence diffusely from the nucleus (not shown). The cells were then infected with an mRFP1-VP26 virus with either an intact or a deleted UL34 allele. In cells infected with PRV encoding pUL34, the GFP-pUL31 localization remained diffuse throughout the nucleus but was also enriched at the nuclear membrane. In the absence of pUL34, GFP-pUL31 enrichment at the nuclear membrane did not occur (Fig. 1C). In this paradigm, the background level of GFP fluorescence was much higher than that in cells infected with the GFP-pUL31-encoding virus, but nevertheless, GFP-pUL31 foci were observed at the periphery of capsid assemblies in the absence of pUL34, thus recapitulating the findings made with PRV encoding the GFP-UL31 allele (Fig. 1D).
Table 1.
Viral titers
| Virus | Descriptiona | Cell line used for propagation | Titer (PFU/ml) |
|---|---|---|---|
| PRV-GS999 | No tags (WT) | PK15 | 1.0 × 109 |
| PRV-GS2868 | mRFP1-VP26 GFP-UL31 ΔUL34 | PK15-UL34 | 2.2 × 106 |
| PRV-GS3393 | myc-UL31 | PK15 | 5.5 × 108 |
| PRV-GS3406 | myc-UL31 ΔUL34 | PK15 | <500 |
| PK15-UL34 | 4.8 × 108 | ||
| PRV-GS4142 | myc-UL31 ΔUL25 ΔUL34 | PK15-UL34 | <500 |
| PK15-UL25 | <500 | ||
| PK15-UL25/UL34 | 2.1 × 108 | ||
| PRV-GS3888 | myc-UL31 ΔUL6 ΔUL34 | PK15-UL34 | <500 |
| PK15-UL6 | <500 | ||
| PK15-UL6/UL34 | 2.6 × 108 | ||
| PRV-GS4143 | myc-UL31 ΔUL33 ΔUL34 | PK15-UL34 | <500 |
| PK15-UL33 | <500 | ||
| PK15-UL33/UL34 | 1.7 × 108 |
WT, wild type.
pUL31 enrichment on purified capsids.
To determine if pUL31 stably interacts with capsids, capsids were recovered from the nuclei of infected Vero cells and isolated by sedimentation through sucrose gradients. The viruses that were used for these experiments did not encode fluorescent tags but instead encoded a myc epitope on the N terminus of pUL31. We note that although the fusion of GFP to the N terminus of pUL31 resulted in a >100× reduction in PRV titers, the myc epitope fusion propagated to wild-type titers (Table 1). A derivative of this virus was also constructed, in which UL34 was deleted. Capsids were separated in a continuous 20 to 50% sucrose gradient by rate-zonal centrifugation. The entire gradient was separated into 18 0.58-ml fractions that were subsequently TCA precipitated and examined by Western blot analysis.
Three light-diffracting bands were observed in sucrose gradients following centrifugation for both viruses, consistent with previous reports of A, B, and C capsids isolated from cells infected with HSV-1 (Fig. 2) (for an example, see reference 57). Due to the close migration of A and B capsid bands from the PRV-Becker clone, these capsid species did not resolve well following fractionation. We therefore refer to capsids in fractions containing A and B capsids as A/B capsids in this report. In cells infected with PRV encoding pUL34, myc-pUL31 was present evenly throughout the gradient (Fig. 3). The presence of pUL31 across the gradient is reminiscent of terminase proteins and the VP16 tegument protein from HSV-1-infected cells (56, 94). The lack of enrichment of myc-pUL31 in PRV capsid-containing fractions indicated that its presence was not capsid specific. However, in the absence of pUL34, myc-pUL31 was enriched in A/B- and C-capsid-containing fractions. The distribution of VP16 was also probed in the pUL34-null/myc-pUL31 gradient, and no reactivity was observed, indicating that the enrichment of myc-pUL31 was not due to a nonspecific aggregation process (not shown). The specificity of the pUL31-capsid interaction was further confirmed by using PRV mutants that do not produce C capsids; in these instances, myc-pUL31 reactivity was specifically lost from the corresponding fractions, while it was retained in the A/B fractions (see below). These data indicate that the localization of GFP-pUL31 to nuclear capsid assemblies was likely due to a pUL31-capsid interaction.
Fig. 2.
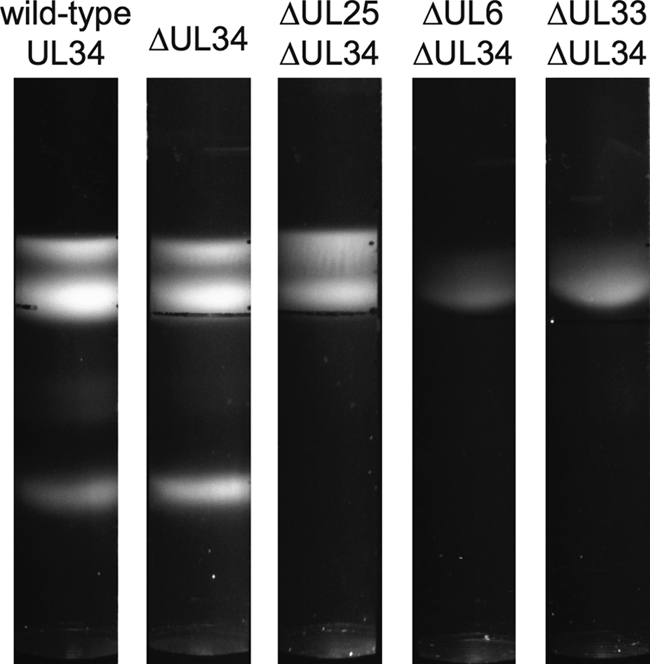
Sedimentation of PRV capsids through sucrose. Intranuclear capsids recovered from cells infected with viruses encoding a myc-pUL31 fusion were separated on continuous 20 to 50% sucrose gradients. Three light-diffracting bands are visible for the viruses with either a wild-type UL34 allele or a UL34 deletion allele. Only A and B capsid bands are apparent in the absence of pUL25, and only B capsids are visible in the absence of either pUL6 or pUL33.
Fig. 3.

Association of pUL31 with purified intranuclear capsids. Capsids were isolated from the nuclei of Vero cells infected with PRV encoding a myc-pUL31 fusion and either an intact UL34 allele (wild type) or a UL34 deletion allele (ΔUL34) and separated on a linear sucrose gradient by rate-zonal centrifugation. Western blots of gradient fractions probed for myc-pUL31 and VP5 are shown. Fractions are labeled 1 (top of the gradient) to 18 (bottom of the gradient). Densitometries of myc-pUL31 (solid lines) and VP5 (dashed lines) are presented in the bottom panels.
pUL31 association with the capsid protein pUL25.
We previously applied a genetic screen based on a library of PRV isolates that encode red fluorescent capsids and deletions of individual viral genes to identify a capsid-binding site for the pUL36 large tegument protein (17). Because the localization of pUL31 to capsid assemblies occurred only in the absence of pUL34, the use of the mutant library to identify the pUL31-capsid interaction first required the design of a pUL31 construct that was able to interact with capsids but not with pUL34. We utilized a previously reported sequence alignment of pUL31 orthologs that identified four highly conserved regions of pUL31, conserved region 1 (CR1) through CR4 (42). Each of the conserved regions was subcloned into GFP expression vectors. Unfortunately, none of the conserved-region constructs localized robustly to capsid assemblies (data not shown). We therefore instead took a targeted approach and ranked candidate capsid proteins based on their potential roles in recruiting pUL31. The minor capsid protein pUL25 was our top candidate because (i) pUL25 was identified as a pUL31-binding partner in a yeast two-hybrid screen (86); (ii) pUL25 is enriched on C capsids, which are targeted for nuclear egress (18, 31, 50, 85); and (iii) pUL25 is required for nuclear egress independent of its role in genome encapsidation (54).
To determine if pUL31 and pUL25 interact in mammalian cells, HEK-293 cells were transiently transfected with PRV myc-pUL25 and GFP-pUL31 expression plasmids. myc-pUL25 coprecipitated GFP-pUL31 but not GFP (Fig. 4). This result provided support for the two-hybrid finding in yeast performed with varicella-zoster virus (VZV) proteins (86). Several different functions of pUL31 are ascribed to separate conserved regions of the protein (42, 61, 73). To determine if one of the four conserved regions was sufficient for the pUL25 interaction, we included the four conserved-region constructs in the coimmunoprecipitation experiments. GFP-CR3 coprecipitated myc-UL25 at levels comparable to those of full-length pUL31, while GFP-CR2 and GFP-CR4 also proved capable of coprecipitation with myc-UL31 at reduced levels (Fig. 4). We cannot currently discriminate if these results indicate the presence of multiple independent pUL25-binding sites in pUL31 or if the pUL25-pUL31 interaction is nonspecific. Nevertheless, a role for pUL25 in pUL31 recruitment to capsids was next examined.
Fig. 4.
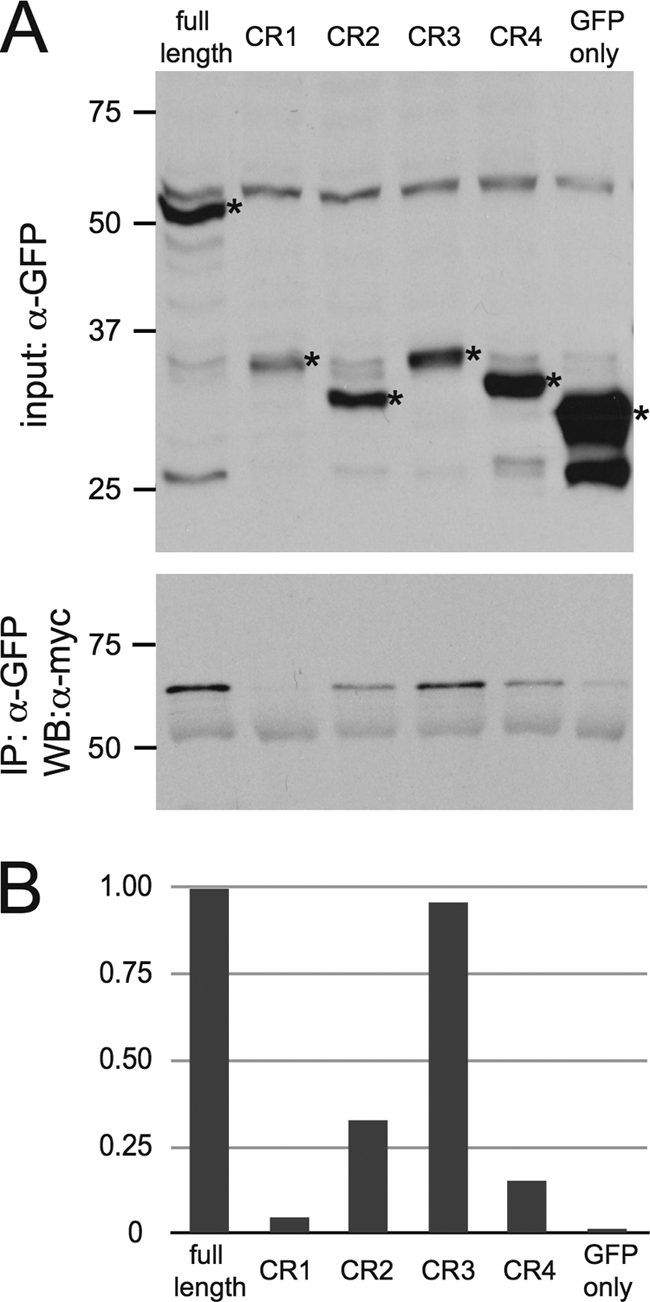
Identification of an interaction between pUL25 and pUL31. (A) Coimmunoprecipitation of myc-pUL25 and GFP-pUL31 constructs. HEK-293 cells were transfected to transiently express myc-pUL25 and either GFP alone, GFP fused to full-length UL31, or GFP fused to individual conserved regions of UL31 (CR1 to CR4). Cells were lysed at 72 h posttransfection and probed with an anti-GFP antibody (top) or immunoprecipitated (IP) with an antibody directed against GFP and probed with an anti-myc antibody to detect pUL25 (bottom). WB, Western blot. (B) The efficiency of coimmunoprecipitation is presented as the densitometry ratio of precipitated pUL25 to input pUL31, with the resulting values normalized to the full-length GFP-pUL31 sample.
pUL25 is not required for the association of pUL31 with capsids.
Because the pUL31-capsid interaction is observed only in the absence of pUL34, a recombinant form of PRV that encodes the myc-pUL31 fusion and deletions of the UL25 and UL34 open reading frames was made. The mutant virus was propagated in complementing cells that stably expressed both pUL25 and pUL34. Stocks of the mutant virus were monitored for the reversion of each gene by measuring titers from cells that expressed pUL25 and pUL34 separately (Table 1). Intranuclear capsids were then isolated from noncomplementing cells, resolved through sucrose gradients, and analyzed by Western blotting for the association of myc-pUL31. As previously described for HSV-1 lacking pUL25, the ΔUL25 ΔUL34 virus did not produce stable C capsids (Fig. 2) (44). Although C capsids were observed for a PRV UL25-null mutant by transmission electron microscopy, others have also noted the absence of PRV C capsids following sedimentation through sucrose, consistent with the requirement for pUL25 for the production of stable C capsids (39; F. Homa, personal communication). Surprisingly, the pattern of myc-pUL31 fractionation in the absence of pUL25 and pUL34 was similar to that observed in the absence of pUL34 alone; myc-pUL31 was enriched in A/B-capsid-containing fractions for both viruses (Fig. 5). While the absolute values derived from densitometry analyses of the Western blots varied between experiments, the trend of increased levels of pUL31 in capsid fractions remained consistent. Also of note, the absence of C capsids from the ΔUL25 ΔUL34 infections was accompanied by the loss of pUL31 specifically from the corresponding fractions, providing further support that the presence of pUL31 in the gradients was specifically a result of the capsid association.
Fig. 5.

pUL25 is not required for the association of pUL31 with capsids. Vero cells were infected with PRV encoding myc-UL31 and either ΔUL34 or ΔUL25/ΔUL34. Intranuclear capsids were processed and analyzed as described in the legend of Fig. 3. Densitometries of myc-pUL31 (solid lines) and VP5 (dashed lines) are presented in the bottom panels.
The capsid portal and pUL33 terminase protein are not required for the capsid-pUL31 interaction.
Two additional proteins were tested for roles in pUL31 recruitment by making double mutant viruses and corresponding dual complementing cell lines: pUL6 and pUL33. In addition to interacting with pUL25, the VZV pUL31 ortholog interacts with the terminase protein pUL33 in a yeast two-hybrid paradigm. Unlike the finding for pUL25-pUL31, the pUL31-pUL33 interaction can be observed with orthologs from the alpha-, beta-, and gammaherpesvirinae subfamilies, and the formation of a murine cytomegalovirus (MCMV) M50-M53-M51 (pUL34-pUL31-pUL33) complex was observed previously in transfected mammalian cells (22, 86). Although pUL33 is not a capsid protein, it is a component of the terminase, which is associated with the capsid during genome encapsidation and copurifies with B capsids (7). The pUL6 portal protein was included for study because it is required for the production of C capsids and nuclear egress (23, 57).
PRV encoding myc-pUL31 and either the ΔUL6 ΔUL34 or ΔUL33 ΔUL34 alleles were propagated on PK15-derived cell lines that stably expressed pUL34 together with pUL6 or pUL33. The resulting virus stocks were confirmed for a lack of reversion by a plaque assay on cell lines that stably express one of the proteins missing from the virus (Table 1). Intranuclear capsids were purified from infected noncomplementing Vero cells. Following rate-zonal centrifugation, pUL31 was enriched in B-capsid-containing fractions for both double mutant viruses (Fig. 6). Collectively, these results indicate that pUL6 (portal), pUL25 (capsid surface component), and pUL33 (terminase), all of which are essential for capsid nuclear egress, are each independently dispensable for the recruitment of pUL31 to capsids in this paradigm.
Fig. 6.

pUL31 association with capsids in the absence of the pUL6 portal and UL33 terminase component. Vero cells were infected with PRV encoding myc-UL31 and either ΔUL6/ΔUL34 or ΔUL33/ΔUL34. Intranuclear capsids were processed and analyzed as described in the legend of Fig. 3. Densitometries of myc-pUL31 (solid lines) and VP5 (dashed lines) are presented in the bottom panels.
DISCUSSION
Herpesvirus capsids are assembled and packaged in the nucleus. The herpesvirus capsid is 125 nm in diameter, which is on par with the external diameter of a nuclear pore. Therefore, herpesvirus capsids must egress from nuclei by a process not normally active in mammalian cells. Based foremost on electron microscopy data, the favored model of nuclear egress is by capsids budding into the nuclear envelope. Once capsids are in the cytosol, the final envelopment process is in part engaged by capsids interacting with tegument and envelope proteins. For HSV-1, some tegument proteins are associated with nuclear and perinuclear capsids, and viral glycoproteins are resident in the nuclear membranes (4–6, 10, 21, 48, 55, 64, 67). For PRV, few of the virion structural proteins are seen associated with the nuclear membrane or nuclear capsids (29, 33, 46). The core NEC proteins pUL31 and pUL34 are nonstructural viral proteins conserved across the herpesviridae (25, 38, 67). In the alphaherpesvirinae, the pUS3 kinase is one protein known to serve as an accessory NEC component (36, 67, 88).
Defining the NEC is complicated in that if the mutation or deletion of a gene does not cause a dramatic defect in nuclear egress, its role may go unnoticed. This is due in part to a heavy reliance on electron microscopy to study nuclear egress and the challenge in identifying small reductions in levels of cytosolic capsids by this method. While the deletion of UL31 or UL34 results in a dramatic reduction in nuclear egress, and the deletion of US3 results in easily observed accumulations of enveloped capsids between the inner and outer nuclear membranes, the role of other proteins in nuclear egress has been controversial. For example, we previously reported a partial defect in nuclear egress for PRV deleted for the UL36 gene using a fluorescence microscopy assay, while another study could not detect a nuclear egress defect with a UL36-null PRV based on electron microscopy (24, 43). We are currently preparing a manuscript describing additional data that substantiate a supporting role for UL36 in PRV nuclear egress, and it was this work that began our interest in a better understanding of NEC-capsid interactions.
The incorporation of pUL31 and pUL34 in primary enveloped capsids has led to a model that the nuclear egress complex interacts directly with capsids (61, 67). Although the VP5 major capsid protein coprecipitates with the NEC from infected cells, evidence supporting a direct link between the NEC and intact capsids has yet to be established (93). Furthermore, the observation that C capsids are enriched for nuclear egress suggests that the NEC selects for capsids containing genomes. The mechanism of C capsid selectivity is unknown and is particularly mysterious given that the genome internal to the C capsid must somehow engender a feature on the C capsid exterior that is recognized by the NEC. The pUL25 minor capsid protein, along with its binding partner pUL17, was proposed as a candidate C-capsid-specific component (CCSC) that may interact with the NEC (85). However, the CCSC was subsequently identified on A and B capsids, making it a less compelling candidate as a selectivity factor (82).
This study provides the first formal evidence that the NEC binds to capsids. Key to this finding was the requirement for the work to be carried out with a UL34-null virus, which is incapable of exporting C capsids from the nucleus. Under these circumstances, pUL31 remained competent to bind capsids, while the trigger for the dissociation of the NEC from capsids upon deposition in the cytosol was absent. We expect that this allowed the accumulation of what normally would be a short-lived pUL31-capsid intermediate complex. The interaction of pUL31 with capsids was initially identified through fluorescence microscopy and was further confirmed by the purification of capsids from the nuclei of infected cells.
Interestingly, pUL31 was located at the periphery of capsid assemblies during infection with the UL34-null virus. This is different from the pattern of colocalization seen when the C-terminal capsid-binding domain of the VP1/2 tegument protein is overexpressed in infected cells, which is centered upon capsid assemblies (17). While the meaning of these differences is unknown to us, they do merit some consideration. In these studies, capsid assemblies were observed based on a fluorescent protein fusion to the VP26 hexon component of capsids. Because VP26 is absent from procapsids, the VP1/2 capsid-binding domain presumably decorates clusters composed of A, B, or C capsids (15, 53, 74). Procapsids, seen as large-core B capsids by transmission electron microscopy, and C capsids are often observed at the periphery of capsid crystalline arrays in the nucleus (1, 16, 30, 68). Because capsid crystalline arrays and VP26 fluorescent capsid assemblies are likely one and the same, the peripheral localization of pUL31 may indicate that the NEC has a propensity to interact with procapsids undergoing maturation to C capsids at sites that are likely replication compartments (62, 66, 76, 89). This model is consistent with previous findings that pUL31 participates in DNA encapsidation (14, 61).
We were initially surprised to observe pUL31 associated with not just C capsids but also A/B capsids. This finding puts an important constraint on models of C capsid egress selectivity: selection does not occur by restricting NEC binding to packaged capsids. Several putative binding partners for pUL31 were identified previously by two-hybrid analysis, most notably pUL25 and pUL33. The interaction between pUL33 and pUL31 was identified in all three subfamilies of the herpesviridae, while the pUL25-pUL31 interaction appeared to be unique to the alphaherpesvirinae (22, 86). Both pUL25 and pUL33 are attractive candidates for links between the NEC and capsids, as both are required for capsid nuclear egress and both are capsid-associated proteins (7, 35, 54). pUL25 is a nonintegral capsid component that contributes to a structure variously referred to as the CCSC for its enrichment on C capsids and, more recently, as the capsid vertex-specific component (CVSC), because the structure was also identified on A and B capsids and is consistently observed around the icosahedral vertices (82, 85). pUL33 is a component of the viral terminase and as such is transiently associated with the capsids during encapsidation (3, 7). Evidence supporting the pUL33 interaction with the NEC comes from the colocalization of the MCMV orthologs at the nuclear membrane following cotransfection (22). A confirmation of pUL25 as an authentic binding partner of pUL31 is currently lacking. Although PRV pUL25 coimmunoprecipitated with pUL31 from transfected cells in this study, making conclusions regarding the specificity of this interaction was difficult, given that multiple nonoverlapping fragments of pUL31 bound pUL25. Furthermore, pUL25 was dispensable for the recruitment of pUL31 to capsids, indicating that if the interaction between pUL25 and pUL31 is biologically relevant, it is at best redundant for the recruitment of pUL31 to capsids. Because pUL33 and pUL6 were also dispensable for the pUL31-capsid interaction, pUL31 binding to capsids likely occurs through another protein or by multiple redundant interactions. In the above-mentioned VZV yeast two-hybrid study, pUL31 interacted with several additional proteins of interest, including the minor capsid protein pUL17 and the inner tegument protein pUL36 (86). It is also possible that integral capsid proteins may provide pUL31-binding sites. In support of the latter possibility, pUL31 coimmunoprecipitates with several proteins from infected cells, including the major capsid protein VP5 (93).
With regard to the egress trigger, the finding that pUL25 functions in nuclear egress in addition to its role in C capsid stabilization has placed pUL25 at center stage in current models of egress (54). However, it may be of interest that one of the earliest-described roles of pUL31 is in DNA cleavage and encapsidation of HSV-1, and support for this concept was more recently obtained with MCMV (14, 61). Coupling between the NEC and encapsidation is a compelling candidate as a nuclear egress trigger that may work in conjunction with pUL25. Genome encapsidation triggers the cleavage of the pUL15 component of the terminase, and at least two of the terminase components disassociate from the capsid (70, 72, 74, 95). Therefore, there are several potential signals that may be relayed from the terminase to the NEC and provide for C capsid selective egress.
While the manuscript was under review, a related study using HSV-1 was published (92). Inconsistent with our study, those authors observed that pUL31 was enriched on capsids despite the presence of pUL34. Also inconsistent was that pUL31 recruitment to HSV-1 capsids was dependent on pUL25. Although those findings were interpreted as evidence of an interaction between the NEC and the CVSC, the pUL17 component of the CVSC was dispensable for the pUL31-capsid association. Another recent study reported that in the absence of pUL17, the entire CVSC density is lost from capsids (82). However, some pUL25 is known to remain on capsids in the absence of pUL17, and it may be that this residual pUL25 could account for the pUL31 recruitment reported for HSV-1 in the absence of the CVSC (80). This would also be consistent with their finding that the level of pUL31 binding to C capsids was lower than that to A and B capsids, which would not be predicted if pUL31 was recruited specifically by the CVSC structure. The absence of this particular relationship may serve as an indication that HSV-1 pUL31 interacts with capsids through an additional capsid-associated protein, which is consistent with our model.
ACKNOWLEDGMENTS
The monoclonal antibody directed against the myc epitope was provided by the Robert H. Lurie Comprehensive Cancer Center Monoclonal Antibody Facility at Northwestern University. The PRV VP5 and VP16 antibodies were a gift from Lynn Enquist.
This work was funded by NIH grant R01 AI080658 to G.A.S.
Footnotes
Published ahead of print on 31 August 2011.
REFERENCES
- 1. Addison C., Rixon F. J., Preston V. G. 1990. Herpes simplex virus type 1 UL28 gene product is important for the formation of mature capsids. J. Gen. Virol. 71(Pt. 10): 2377–2384 [DOI] [PubMed] [Google Scholar]
- 2. Adelman K., Salmon B., Baines J. D. 2001. Herpes simplex virus DNA packaging sequences adopt novel structures that are specifically recognized by a component of the cleavage and packaging machinery. Proc. Natl. Acad. Sci. U. S. A. 98: 3086–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. al-Kobaisi M. F., Rixon F. J., McDougall I., Preston V. G. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180: 380–388 [DOI] [PubMed] [Google Scholar]
- 4. Baines J. D., Jacob R. J., Simmerman L., Roizman B. 1995. The herpes simplex virus 1 UL11 proteins are associated with cytoplasmic and nuclear membranes and with nuclear bodies of infected cells. J. Virol. 69: 825–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baines J. D., Roizman B. 1992. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J. Virol. 66: 5168–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baines J. D., Wills E., Jacob R. J., Pennington J., Roizman B. 2007. Glycoprotein M of herpes simplex virus 1 is incorporated into virions during budding at the inner nuclear membrane. J. Virol. 81: 800–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beard P. M., Baines J. D. 2004. The DNA cleavage and packaging protein encoded by the UL33 gene of herpes simplex virus 1 associates with capsids. Virology 324: 475–482 [DOI] [PubMed] [Google Scholar]
- 8. Beard P. M., Taus N. S., Baines J. D. 2002. DNA cleavage and packaging proteins encoded by genes U(L)28, U(L)15, and U(L)33 of herpes simplex virus type 1 form a complex in infected cells. J. Virol. 76: 4785–4791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Booy F. P., et al. 1991. Liquid-crystalline, phage-like packing of encapsidated DNA in herpes simplex virus. Cell 64: 1007–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bucks M. A., O'Regan J. K., Murphy M. A., Wills J. W., Courtney R. J. 2007. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 361: 316–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cardone G., et al. 2007. Visualization of the herpes simplex virus portal in situ by cryo-electron tomography. Virology 361: 426–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang J. T., Schmid M. F., Rixon F. J., Chiu W. 2007. Electron cryotomography reveals the portal in the herpesvirus capsid. J. Virol. 81: 2065–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang Y. E., Roizman B. 1993. The product of the UL31 gene of herpes simplex virus 1 is a nuclear phosphoprotein which partitions with the nuclear matrix. J. Virol. 67: 6348–6356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang Y. E., Van Sant C., Krug P. W., Sears A. E., Roizman B. 1997. The null mutant of the U(L)31 gene of herpes simplex virus 1: construction and phenotype in infected cells. J. Virol. 71: 8307–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chi J. H., Wilson D. W. 2000. ATP-dependent localization of the herpes simplex virus capsid protein VP26 to sites of procapsid maturation. J. Virol. 74: 1468–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Church G. A., Wilson D. W. 1997. Study of herpes simplex virus maturation during a synchronous wave of assembly. J. Virol. 71: 3603–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coller K. E., Lee J. I., Ueda A., Smith G. A. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 81: 11790–11797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Conway J. F., et al. 2010. Labeling and localization of the herpes simplex virus capsid protein UL25 and its interaction with the two triplexes closest to the penton. J. Mol. Biol. 397: 575–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deng B., O'Connor C. M., Kedes D. H., Zhou Z. H. 2007. Direct visualization of the putative portal in the Kaposi's sarcoma-associated herpesvirus capsid by cryoelectron tomography. J. Virol. 81: 3640–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Farina A., et al. 2005. BFRF1 of Epstein-Barr virus is essential for efficient primary viral envelopment and egress. J. Virol. 79: 3703–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Farnsworth A., et al. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. U. S. A. 104: 10187–10192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fossum E., et al. 2009. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 5: e1000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fuchs W., Klupp B. G., Granzow H., Leege T., Mettenleiter T. C. 2009. Characterization of pseudorabies virus (PrV) cleavage-encapsidation proteins and functional complementation of PrV pUL32 by the homologous protein of herpes simplex virus type 1. J. Virol. 83: 3930–3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fuchs W., Klupp B. G., Granzow H., Mettenleiter T. C. 2004. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 78: 11879–11889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fuchs W., Klupp B. G., Granzow H., Osterrieder N., Mettenleiter T. C. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 76: 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gibson W., Roizman B. 1972. Proteins specified by herpes simplex virus. 8. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J. Virol. 10: 1044–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Granato M., et al. 2008. Deletion of Epstein-Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J. Virol. 82: 4042–4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Granzow H., et al. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 75: 3675–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Granzow H., Klupp B. G., Mettenleiter T. C. 2004. The pseudorabies virus US3 protein is a component of primary and of mature virions. J. Virol. 78: 1314–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Granzow H., et al. 1997. Ultrastructural analysis of the replication cycle of pseudorabies virus in cell culture: a reassessment. J. Virol. 71: 2072–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaelin K., Dezelee S., Masse M. J., Bras F., Flamand A. 2000. The UL25 protein of pseudorabies virus associates with capsids and localizes to the nucleus and to microtubules. J. Virol. 74: 474–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kennard J., Rixon F. J., McDougall I. M., Tatman J. D., Preston V. G. 1995. The 25 amino acid residues at the carboxy terminus of the herpes simplex virus type 1 UL26.5 protein are required for the formation of the capsid shell around the scaffold. J. Gen. Virol. 76(Pt. 7): 1611–1621 [DOI] [PubMed] [Google Scholar]
- 33. Klupp B., Altenschmidt J., Granzow H., Fuchs W., Mettenleiter T. C. 2008. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J. Virol. 82: 6299–6309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Klupp B. G., et al. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. U. S. A. 104: 7241–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klupp B. G., Granzow H., Keil G. M., Mettenleiter T. C. 2006. The capsid-associated UL25 protein of the alphaherpesvirus pseudorabies virus is nonessential for cleavage and encapsidation of genomic DNA but is required for nuclear egress of capsids. J. Virol. 80: 6235–6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Klupp B. G., Granzow H., Mettenleiter T. C. 2001. Effect of the pseudorabies virus US3 protein on nuclear membrane localization of the UL34 protein and virus egress from the nucleus. J. Gen. Virol. 82: 2363–2371 [DOI] [PubMed] [Google Scholar]
- 37. Klupp B. G., Granzow H., Mettenleiter T. C. 2011. Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. J. Virol. 85: 8285–8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klupp B. G., Granzow H., Mettenleiter T. C. 2000. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 74: 10063–10073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuhn J., et al. 2008. Partial functional complementation of a pseudorabies virus UL25 deletion mutant by herpes simplex virus type 1 pUL25 indicates overlapping functions of alphaherpesvirus pUL25 proteins. J. Virol. 82: 5725–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lake C. M., Hutt-Fletcher L. M. 2004. The Epstein-Barr virus BFRF1 and BFLF2 proteins interact and coexpression alters their cellular localization. Virology 320: 99–106 [DOI] [PubMed] [Google Scholar]
- 41. Leuzinger H., et al. 2005. Herpes simplex virus 1 envelopment follows two diverse pathways. J. Virol. 79: 13047–13059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lotzerich M., Ruzsics Z., Koszinowski U. H. 2006. Functional domains of murine cytomegalovirus nuclear egress protein M53/p38. J. Virol. 80: 73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luxton G. W., Lee J. I., Haverlock-Moyns S., Schober J. M., Smith G. A. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80: 201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McNab A. R., et al. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J. Virol. 72: 1060–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Milbradt J., Auerochs S., Marschall M. 2007. Cytomegaloviral proteins pUL50 and pUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J. Gen. Virol. 88: 2642–2650 [DOI] [PubMed] [Google Scholar]
- 46. Mohl B. S., et al. 2009. Intracellular localization of the pseudorabies virus large tegument protein pUL36. J. Virol. 83: 9641–9651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Muranyi W., Haas J., Wagner M., Krohne G., Koszinowski U. H. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297: 854–857 [DOI] [PubMed] [Google Scholar]
- 48. Naldinho-Souto R., Browne H., Minson T. 2006. Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J. Virol. 80: 2582–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Neubauer A., Rudolph J., Brandmuller C., Just F. T., Osterrieder N. 2002. The equine herpesvirus 1 UL34 gene product is involved in an early step in virus egress and can be efficiently replaced by a UL34-GFP fusion protein. Virology 300: 189–204 [DOI] [PubMed] [Google Scholar]
- 50. Newcomb W. W., Homa F. L., Brown J. C. 2006. Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. J. Virol. 80: 6286–6294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Newcomb W. W., et al. 1996. Assembly of the herpes simplex virus capsid: characterization of intermediates observed during cell-free capsid formation. J. Mol. Biol. 263: 432–446 [DOI] [PubMed] [Google Scholar]
- 52. Newcomb W. W., et al. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 75: 10923–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Newcomb W. W., et al. 2000. Isolation of herpes simplex virus procapsids from cells infected with a protease-deficient mutant virus. J. Virol. 74: 1663–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. O'Hara M., et al. 2010. Mutational analysis of the herpes simplex virus type 1 UL25 DNA packaging protein reveals regions that are important after the viral DNA has been packaged. J. Virol. 84: 4252–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Padula M. E., Sydnor M. L., Wilson D. W. 2009. Isolation and preliminary characterization of herpes simplex virus 1 primary enveloped virions from the perinuclear space. J. Virol. 83: 4757–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Patel A. H., MacLean J. B. 1995. The product of the UL6 gene of herpes simplex virus type 1 is associated with virus capsids. Virology 206: 465–478 [DOI] [PubMed] [Google Scholar]
- 57. Patel A. H., Rixon F. J., Cunningham C., Davison A. J. 1996. Isolation and characterization of herpes simplex virus type 1 mutants defective in the UL6 gene. Virology 217: 111–123 [DOI] [PubMed] [Google Scholar]
- 58. Perdue M. L., Cohen J. C., Kemp M. C., Randall C. C., O'Callaghan D. J. 1975. Characterization of three species of nucleocapsids of equine herpesvirus type-1 (EHV-1). Virology 64: 187–204 [DOI] [PubMed] [Google Scholar]
- 59. Perdue M. L., Cohen J. C., Randall C. C., O'Callaghan D. J. 1976. Biochemical studies of the maturation of herpesvirus nucleocapsid species. Virology 74: 194–208 [PubMed] [Google Scholar]
- 60. Poon A. P., Roizman B. 1993. Characterization of a temperature-sensitive mutant of the UL15 open reading frame of herpes simplex virus 1. J. Virol. 67: 4497–4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Popa M., et al. 2010. Dominant negative mutants of the murine cytomegalovirus M53 gene block nuclear egress and inhibit capsid maturation. J. Virol. 84: 9035–9046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Prichard M. N., et al. 2008. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J. Virol. 82: 5054–5067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Purves F. C., Spector D., Roizman B. 1992. UL34, the target of the herpes simplex virus U(S)3 protein kinase, is a membrane protein which in its unphosphorylated state associates with novel phosphoproteins. J. Virol. 66: 4295–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Read G. S., Patterson M. 2007. Packaging of the virion host shutoff (Vhs) protein of herpes simplex virus: two forms of the Vhs polypeptide are associated with intranuclear B and C capsids, but only one is associated with enveloped virions. J. Virol. 81: 1148–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Remillard-Labrosse G., Guay G., Lippe R. 2006. Reconstitution of herpes simplex virus type 1 nuclear capsid egress in vitro. J. Virol. 80: 9741–9753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Reynolds A. E., et al. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 75: 8803–8817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Reynolds A. E., Wills E. G., Roller R. J., Ryckman B. J., Baines J. D. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76: 8939–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rixon F. J., McNab D. 1999. Packaging-competent capsids of a herpes simplex virus temperature-sensitive mutant have properties similar to those of in vitro-assembled procapsids. J. Virol. 73: 5714–5721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Roller R. J., Zhou Y., Schnetzer R., Ferguson J., DeSalvo D. 2000. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J. Virol. 74: 117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Roos W. H., et al. 2009. Scaffold expulsion and genome packaging trigger stabilization of herpes simplex virus capsids. Proc. Natl. Acad. Sci. U. S. A. 106: 9673–9678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Salmon B., Cunningham C., Davison A. J., Harris W. J., Baines J. D. 1998. The herpes simplex virus type 1 U(L)17 gene encodes virion tegument proteins that are required for cleavage and packaging of viral DNA. J. Virol. 72: 3779–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Salmon B., Nalwanga D., Fan Y., Baines J. D. 1999. Proteolytic cleavage of the amino terminus of the U(L)15 gene product of herpes simplex virus type 1 is coupled with maturation of viral DNA into unit-length genomes. J. Virol. 73: 8338–8348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Schnee M., Ruzsics Z., Bubeck A., Koszinowski U. H. 2006. Common and specific properties of herpesvirus UL34/UL31 protein family members revealed by protein complementation assay. J. Virol. 80: 11658–11666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sheaffer A. K., et al. 2001. Herpes simplex virus DNA cleavage and packaging proteins associate with the procapsid prior to its maturation. J. Virol. 75: 687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shiba C., et al. 2000. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J. Gen. Virol. 81: 2397–2405 [DOI] [PubMed] [Google Scholar]
- 76. Simpson-Holley M., Baines J., Roller R., Knipe D. M. 2004. Herpes simplex virus 1 U(L)31 and U(L)34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J. Virol. 78: 5591–5600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Smith G. A., Enquist L. W. 2000. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 97: 4873–4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Smith G. A., Pomeranz L., Gross S. P., Enquist L. W. 2004. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. U. S. A. 101: 16034–16039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stackpole C. W. 1969. Herpes-type virus of the frog renal adenocarcinoma. I. Virus development in tumor transplants maintained at low temperature. J. Virol. 4: 75–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Thurlow J. K., Murphy M., Stow N. D., Preston V. G. 2006. Herpes simplex virus type 1 DNA-packaging protein UL17 is required for efficient binding of UL25 to capsids. J. Virol. 80: 2118–2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tischer B. K., von Einem J., Kaufer B., Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40: 191–197 [DOI] [PubMed] [Google Scholar]
- 82. Toropova K., Huffman J. B., Homa F. L., Conway J. F. 2011. The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J. Virol. 85: 7513–7522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Trus B. L., et al. 1996. The herpes simplex virus procapsid: structure, conformational changes upon maturation, and roles of the triplex proteins VP19c and VP23 in assembly. J. Mol. Biol. 263: 447–462 [DOI] [PubMed] [Google Scholar]
- 84. Trus B. L., et al. 2004. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J. Virol. 78: 12668–12671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Trus B. L., et al. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol. Cell 26: 479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Uetz P., et al. 2006. Herpesviral protein networks and their interaction with the human proteome. Science 311: 239–242 [DOI] [PubMed] [Google Scholar]
- 87. Vlazny D. A., Kwong A., Frenkel N. 1982. Site-specific cleavage/packaging of herpes simplex virus DNA and the selective maturation of nucleocapsids containing full-length viral DNA. Proc. Natl. Acad. Sci. U. S. A. 79: 1423–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wagenaar F., et al. 1995. The US3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J. Gen. Virol. 76(Pt. 7): 1851–1859 [DOI] [PubMed] [Google Scholar]
- 89. Ward P. L., Ogle W. O., Roizman B. 1996. Assemblons: nuclear structures defined by aggregation of immature capsids and some tegument proteins of herpes simplex virus 1. J. Virol. 70: 4623–4631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. White C. A., Stow N. D., Patel A. H., Hughes M., Preston V. G. 2003. Herpes simplex virus type 1 portal protein UL6 interacts with the putative terminase subunits UL15 and UL28. J. Virol. 77: 6351–6358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yamauchi Y., et al. 2001. Herpes simplex virus type 2 UL34 protein requires UL31 protein for its relocation to the internal nuclear membrane in transfected cells. J. Gen. Virol. 82: 1423–1428 [DOI] [PubMed] [Google Scholar]
- 92. Yang K., Baines J. D. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc. Natl. Acad. Sci. U. S. A. 108: 14276–14281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ye G. J., Vaughan K. T., Vallee R. B., Roizman B. 2000. The herpes simplex virus 1 U(L)34 protein interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. J. Virol. 74: 1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yu D., Weller S. K. 1998. Genetic analysis of the UL 15 gene locus for the putative terminase of herpes simplex virus type 1. Virology 243: 32–44 [DOI] [PubMed] [Google Scholar]
- 95. Yu D., Weller S. K. 1998. Herpes simplex virus type 1 cleavage and packaging proteins UL15 and UL28 are associated with B but not C capsids during packaging. J. Virol. 72: 7428–7439 [DOI] [PMC free article] [PubMed] [Google Scholar]