

Abstract
Sensory neurons express a variety of voltage-gated Ca2+ channel subtypes, but reports differ on their proportionate representation, and the effects of painful nerve injury on each subtype are not established. We compared levels of high-voltage activated currents in medium-sized (30-40μm) dorsal root ganglion neurons dissociated from control animals and those subjected to spinal nerve ligation, using sequential application of semiselective channel blockers (nisoldipine for L-type, SNX-111 or ω-conotoxin GVIA for N-type, agatoxin IVA or ω-Conotoxin MVIIC for P/Q-type, and SNX-482 for a component of R-type) during either square wave depolarizations or action potential waveform voltage commands. Using sequential administration of multiple blockers, proportions of total Ca2+ current attributable to different subtypes and the effect of injury depended on the sequence of blocker administration and type of depolarization command. Overall, however, N-type and L-type currents comprised the dominant components of ICa in sensory neurons under control conditions, and these subtypes showed the greatest loss of current following injury (L-type 26-71% loss, N-type 0-51% loss). Further exploration of N-type current identified by its sensitivity to ω-conotoxin GVIA applied alone showed that injury reduced the peak N-type current during step depolarization by 68% and decreased the total charge entry during action potential waveform stimulation by 44%. Isolation of N-type current by blockade of all other subtypes demonstrated a 50% loss with injury, and also revealed an injury-related rightward shift in the activation curve. Nonstationary noise analyses of N-type current in injured neurons revealed unitary channel current and number of channels that were not different from control, which indicates that injury-induced loss of current is due to a decrease in channel open probability. Our findings suggest that diminished Ca2+ influx through N-type and L-type channels may contribute to sensory neuron dysfunction and pain after nerve injury.
Keywords: Ca2+ currents, nerve injury, neuropathic pain, neurotoxins, dorsal root ganglion, sensory neuron
Progress in devising successful treatments for neuropathic pain has been slow, in part due to incomplete understanding of the molecular basis of neuronal dysfunction in these conditions. Investigations in our laboratory and others have shown that peripheral nerve injury leading to neuropathic pain in animal models is accompanied by loss of voltage-gated Ca2+ influx (ICa) through the sensory neuron plasmalemma, including both low-voltage activated (LVA) currents, as well has high-voltage activated (HVA) currents (Baccei and Kocsis, 2000, Hogan et al., 2000, Abdulla and Smith, 2001, McCallum et al., 2003). Reduced ICa after injury is associated with diminished resting cytoplasmic Ca2+ levels (Fuchs et al., 2005), decreased transient elevations in cytoplasmic Ca2+ levels during neuronal activation (Fuchs et al., 2007a), and heightened neuronal excitability (Lirk et al., 2008). Because of the role of cytoplasmic Ca2+ in regulating diverse functions such as membrane currents, gene expression, neurotransmitter release, and cell death, understanding the ICa deficit following injury in greater detail may contribute to clarifying the molecular mechanisms of neuropathic pain.
Sensory neurons contain a rich variety of Ca2+ current subtypes identified according to the genetic structure of the pore-forming α1 subunit, as well as pharmacological and functional properties. Among HVA currents, L-type ICa typically requires a strong depolarization for activation, shows minimal inactivation, and is blocked by dihydropyridine antagonists. N-type and P/Q-type ICa, which also require strong depolarization for activation, are relatively unaffected by dihydropyridines but are blocked by specific polypeptide toxins from snail and spider venoms. A residual R-type current is insensitive to blockers of these other channels and is probably made up of several components in DRG neurons (Wilson et al., 2000). The LVA T-type calcium current is activated by weak depolarizations, inactivates rapidly during sustained depolarization, and is resistant to both the dihydropyridines and the peptide toxins used to define the N-, P/Q-, and R-type HVA ICa.
To some extent, the functional roles for these current subtypes are distinct. T-type current prolongs the Ca2+ entry associated with action potentials due to its slow deactivation (Scroggs and Fox, 1992a), and may influence patterns of repetitive firing (Nelson et al., 2005). L-type current is a dominant path for Ca2+ that triggers genetic events associated with neuronal activity (Dolmetsch et al., 2001). While various HVA currents may interact synergistically at certain CNS synapses to produce neurotransmitter release, the central terminals of DRG neurons demonstrate distinct roles for various ICa channel subtypes. N-type channels are the principal source for Ca2+ that triggers excitatory synaptic transmission in the dorsal horn of the spinal cord, with a lesser contribution by P/Q-type currents (Bao et al., 1998, Heinke et al., 2004, Rycroft et al., 2007). However, P/Q-type currents predominate at inhibitory synapses (Takahashi and Momiyama, 1993, Heinke et al., 2004). Subcellular colocalization may functionally link ICa subtypes with specific Ca2+-dependent processes, including opening distinct subtypes of Ca2+-activated K+ currents that regulate neuronal excitability (Marrion and Tavalin, 1998, Cordoba-Rodriguez et al., 1999).
Loss of ICa is a consistent and robust consequence of various models of primary sensory neuron injury in rats (Baccei and Kocsis, 2000, Hogan et al., 2000, Abdulla and Smith, 2001, McCallum et al., 2006). However, apart from the injury-induced depression of T-type current in medium-sized neurons (Andre et al., 2003, McCallum et al., 2003) and increased T-type current in small neurons (Jagodic et al., 2008), the specific subtypes of ICa affected by injury have not been defined. In order to identify the distribution of injury-induced ICa loss among specific HVA subtypes in a clinically relevant model of painful peripheral nerve injury, we examined currents in axotomized neurons of the fifth lumbar (L5) dorsal root ganglion (DRG) from rats after L5 spinal nerve ligation and section (SNL), compared to currents in neurons from control rats. A corresponding loss of ICa is not found in adjacent L4 neurons after SNL (McCallum et al., 2006), so the present study examines only the directly injured L5 neurons. We focussed our study on medium-sized neurons (30-40 μm diameter) since 74% of neurons with these diameters have a slow conduction velocity and long action potential (AP) duration typical of lightly myelinated A∂ neurons (intracellular recordings from intact DRGs, data not shown), which undergo the greatest ICa loss after injury (McCallum et al., 2006). N- and L-type channels conduct the bulk of inward Ca2+ flux in most DRG neurons (Regan et al., 1991, Cardenas et al., 1995, Rusin and Moises, 1995, Wu and Pan, 2004), so we hypothesized that these would be the most affected by injury. Finally, we examined N-type current in greater detail as indirect measures have highlighted its role in plasticity of dorsal horn sensory transmission after injury (Matthews and Dickenson, 2001), and because N-type blockade has shown efficacy as analgesics in animal and human subjects (Staats et al., 2004).
1. Experimental Procedures
1.1 Animal Preparation
Adult male Sprague Dawley rats (Taconic, Hudson, NY) weighing 125-150 g were studied under approval of the Medical College of Wisconsin Animal Care and Use Committee. Rats in the neuropathic group were subjected to SNL modified from the original description (Kim and Chung, 1992), specifically ligation and section of the right L5 and L6 spinal nerves approximately 5mm distal to the DRG, while control rats received skin incision and closure only. Following recovery from isoflurane anesthesia, rats were returned to individual cages under climate and light controlled conditions. Sensory function was evaluated as previously described (Hogan et al., 2004). Briefly, a pin was applied to the plantar skin with pressure adequate to indent but not puncture it. In response to this noxious stimulus, rats either briefly withdraw their paw as a reflexive flinch or display a sustained (> 2 s), complex lifting, shaking, and grooming of the paw. This later response is rarely seen in control animals but increases in frequency following nerve injury, and is selectively associated with conditioned place avoidance (Wu et al., 2010). These behavior types were tabulated by a blinded observer while rats rested on a 0.25 in. wire grid in clear plastic enclosures. Five needle applications were made in turn to the plantar skin of each hindpaw at 2s intervals and repeated 3 min later, allowing calculation of the frequency of the complex hyperalgesia-type response averaged over 3 separate tests between 10 and 17 days postoperatively.
1.2 Neuronal Dissociation
After a postsurgical interval of 23.5±2.0 days for control rats (n=47) and 19.2±0.7d for SNL animals (n=56), L5 ganglia were removed following decapitation under isoflurane anesthesia and placed into a 35 mm Petri dish containing Ca2+/Mg2+-free, iced HBBS (Gibco) for mincing with iris scissors. Cleaned ganglia were incubated in 0.05% blendzyme 2 (Roche) in DMEM/F12 with glutaMAX (Invitrogen, Carlsbad, CA) for 30 min, washed, vortexed and resuspended in 0.0625% trypsin (Sigma-Aldrich Corp., St. Louis, MO) and 0.0125% Dnase (Sigma) for another 30 min. Dissociated neurons were placed in Trypsin inhibitor (Sigma, Type II), centrifuged, lightly triturated and resuspended in adult neural basal media (1X) (Invitrogen) containing 2% (v:v) B27 supplement (50x) (Life Technologies), 0.5 mM glutamine, 0.02 mg/ml gentamicin and 100 ng/ml nerve growth factor 7S (Alomone Labs Ltd., Jerusalem, Israel) for plating onto poly-L-lysine (70-150 kDa) coated 12 mm coverslips (Spiegelglas #2, Carolina Biological Supply, Burlington, NC) and placed in a 95:5 O2:CO2 incubator for 2 hr before study. All neurons were studied 6-8 hr after dissociation. Medium-sized (30-40 μm diameter) neurons from the L5 DRG after SNL and from uninjured control animals were compared, as previous studies have shown that ICa loss and increased excitability occurs predominantly in axotomized neuronal group after SNL (Sapunar et al., 2005, McCallum et al., 2006).
1.3 Voltage and Current Recording
Voltage and currents were recorded in the whole cell configuration of the patch-clamp technique. Patch pipettes, ranging from 2-5 MΩ resistance, were formed from borosilicate glass (Garner Glass Co., Claremont, CA) and fire polished. Coating with Sylgard (Dow Corning Corp., Midland, MI) was used in noise analysis experiments. Currents were recorded with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA), filtered at 2 kHz through a 4-pole Bessel filter, and digitized at 10 kHz with a Digidata 1320 A/D interface and pClamp 9 software (Molecular Devices) for storage on a personal computer. After achieving gigohm seal and breakthrough, membrane capacitance was recorded for current density measurements, and access resistance was 80-90% compensated. Neurons with >10 MΩ access resistance after breakthrough were discarded. Leak currents were identified by ICa block with application of Cd2+ (200 μM) at the end of each experiment, in order to measure residual ICa not blocked by selective blockers. Leak currents were not otherwise subtracted from measured currents, although their contribution to the calculation of subtype blocker sensitivities is automatically nulled by the determination of difference currents.
Neurons were first characterized in current clamp mode, using solutions designed to duplicate natural cytosolic and extracellular conditions. A modified Tyrode’s solution was used for external bath solution, consisting of the following (in mM): 140 NaCl, 4 KCl, 2 CaCl, 2 MgCl, 10 D-glucose, 10 (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) at pH of 7.4 with NaOH and an osmolarity of 300 mOsm. The internal pipette solution contained (in mM) 120 KCl, 5 Na-ATP, 0.4 Na-GTP, 5 ethylene glycol-bis(2-amino-ethylether)-N,N, N’, N’-tetra-acetic acid (EGTA), 2.25 CaCl2, 5 MgCl2, 20 HEPES at a pH of 7.4 with KOH and osmolarity of 296 mOsm. Depolarizing current pulses (100ms) were used to identify the rheobase current just adequate to generate an AP. It was then determined whether the neuron fired repetitively or singly in response to depolarization to twice the rheobase current (Harper, 1991).
After current clamp recording, a different bath solution was employed to isolate Ca2+ currents during voltage-clamp recording. Because of the difficulty of changing internal pipette solutions after seal formation, ICa was isolated by external blockade of other voltage sensitive currents, using a solution that contained (in mM): 2 BaCl2, 1 4-aminophyridine (4-AP), 10 HEPES, 160 tetraethylammonium chloride (TEA), 0.1 tetrodotoxin (TTX) at a pH of 7.4 with tetraethylammonium hydroxide and an osmolarity of 300 mOsm. TTX blocked TTX-sensitive Na+ current, and 4-AP, TEA and Ba2+ blocked K+ current. Although Ba2+ was the charge carrier, we hereafter refer to currents as ICa.
Two voltage clamp protocols were used to measure whole-cell ICa. One measured ICa in response to 200 ms step pulses from -90 to -10 mV every 10s during applications of antagonists. Another protocol used the neuron’s own AP waveform as a voltage stimulus (McCobb and Beam, 1991), generating currents that were characterized by amplitude and total charge transfer, defined as the integral of current over time in response to the AP waveform voltage command, starting at the neurons natural resting membrane potential.
In other experiments, noise analysis (Sigworth, 1980) was performed on whole-cell currents after isolation of N-type current. A holding potential of -60 mV was used to inactivate residual T-type currents. After determining the voltage that produced peak ICa, current was recorded during 4×50 ms pulses to a voltage that produced half-maximal current (between -25mV and -15mV), using a 10 kHz sampling frequency and 2 kHz 4-pole Bessel low-pass band filtering frequency. Only neurons with minimal rundown of ICa (<10% during the data collection interval) were included in data collection. Mean current (I) and variance (σ2), calculated after background noise was subtracted, was fitted to a parabola
where i is the unitary current in pA, and N is the number of channels (Sigworth, 1980). Successful isolation of the noise due to channel opening was confirmed by obtaining comparable findings when background noise was determined two different ways, specifically during either a step hyperpolarization comparable to the test depolarization, or during elimination of all ICa by cadmium (200μM). To correct for cell size, the total number of channels was divided by the cell’s capacitance in pF and this ratio was multiplied by the specific capacitance of plasma membranes (0.01 pF/μm2), resulting in N expressed as channels per μm2. Unitary current was converted to chord conductances using the formula
where Vtest was the potential at which the half-maximal amplitude of ICa was recorded, and Vrev was the reversal potential for that cell. Isolation of noise due to channel opening was confirmed by obtaining comparable findings when background noise was determined two different ways, specifically during either a step hyperpolarization comparable to the test depolarization, or during elimination of all ICa by cadmium (200μM).
1.4 Current Subtype Isolation
Isolation of HVA ICa subtypes was achieved with administration of selective HVA ICa antagonists including nisoldipine (200 nM) to block L-type current, ω-conotoxin GVIA (hereafter referred to as GVIA, 200nM) or SNX-111 (synthetic ω-conotoxin MVIIA, 200 nM) to block N-type current, and agatoxin IVA (AgaIVA, 200nM) to block P/Q-type current (Kass, 1982, Randall and Tsien, 1995, Bowersox et al., 1996, Feng et al., 2003). ω-Conotoxin MVIIC (MVIIC, 200 nM) provided blockade of both N- and P/Q type currents (Hillyard et al., 1992, Randall and Tsien, 1995). SNX-482 (200 nM) selectively blocks current through the α1E subunit that contributes to the residual R-type current (Wilson et al., 2000). We refer to the current sensitive to SNX-482 as R-type, although as much as 20% of HVA ICa in DRG neurons remains insensitive to all of these blockers, including SNX-482 (Wilson et al., 2000). With the exception of experiments in which N-type current was isolated by incubation of neurons in antagonists of all other subtypes (see below), protocols used non-saturating concentrations of antagonists to minimize non-selective effects while achieving >90% of the maximal blockade, as was determined in preliminary concentration-effect control experiments in which antagonists were applied individually (data not shown). The overall high efficacy of blockade was confirmed by our observations (see below) of a residual current sensitive to Cd2+ of 10-25% of total ICa. Although currents are thus somewhat underestimated, these concentrations limit the non-specific effects that would accompany ablative current antagonism.
Potential limitations of pharmacological separation of ICa current subtypes include overlapping non-specific blockade of current subtypes by the antagonists (Burley and Dolphin, 2000), simultaneous current rundown that may mistakenly be attributed to antagonist action during necessarily long protocols, and variable washout for antagonists with reversible effects. We addressed these challenges by employing several complementary strategies. Two different protocols employing sequential applications of blockers were used. Nisoldipine was applied first in both of these protocols (Randall and Tsien, 1995). In the first set of experiments, the other antagonists were applied in random order, to minimize a sequence effect (Rusin and Moises, 1995). Antagonists were chosen that are relatively reversible, including SNX-111 for N-type current (Olivera et al., 1987), AgaIVA for P/Q-type current (Rusin and Moises, 1995), and SNX-482 for R-type current (Arroyo et al., 2003). A period of washout followed administration of each antagonist (Wilson et al., 2000, Wu and Pan, 2004) other than nisoldipine, in order to minimize off-target effects of blockers. Current sensitive to each antagonist was determined by subtracting the current at maximal effect from the baseline current preceding the administration of that antagonist.
To address limitations due to potentially incomplete washout of blocking effects (Mould et al., 2004, Liu et al., 2006), confirmatory observations were sought in other experiments using a second protocol in which a fixed sequence of antagonist application was designed that does not require washout of blockers, in order to minimize the effect of overlapping non-specific blockade (Randall and Tsien, 1995). As above, nisoldipine was applied first and was continued throughout serial administration of other agents. We measured N-type current by determining sensitivity to GVIA, whereupon subsequent application of MVIIC, which blocks N-type as well as P/Q-type currents, selectively revealed P/Q current. SNX-482 was applied next to block R-type current, and Cd2+ was applied last to identify residual ICa. The current sensitive to each antagonist was measured as in the preceding protocol.
In a third approach, GVIA was applied to other neurons as the only blocker in order to examine N-type current without concerns of undesired effects of other agents. In a final complementary protocol, N-type current was isolated by blockade of all other HVA current subtypes through incubation for 30 min with nisoldipine (200 nM), and SNX-482 (500 nM), which were continued during recording. MVIIC (2 μM) was applied only during incubation, but was removed during recording to avoid blockade of N-type current, resulting in relief of N-type block but persistence of P/Q-type block (McDonough et al., 1996). Identity of the current remaining under these conditions as N-type was confirmed with application of SNX-111 at the end of each experiment.
All antagonists were delivered in the external Ba2+ solution with 0.1 mg/ml cytochrome C to prevent non-specific binding in delivery systems. Ca2+ channel antagonists were applied directly to the neuron through a pressure regulated microperfusion system with a single 100 μm tip and one channel for control superfusate (ALA-BP8 system, ALA Scientific). GVIA and MVIIC were purchased from Sigma. TTX was obtained from Alomone Labs (Tel Aviv, Israel). Nisoldipine was a gift from Pentex-Miles, and SNX-111 was a gift from Dr. Scott Bowersox from Neurex Corporation, while AgaIVA was a gift from Dr. Nicholas Saccomano from Pfizer (Groton, CT), and SNX-482 was purchased from Peptides International (Louisville, KY).
1.5 Analysis and Statistics
Voltage dependence of activation was expressed as the fraction of macroscopic conductance in proportion to total conductance (G/GMax). To determine total conductance, current-voltage (I-V) data from recordings during step depolarizations were fit to a Boltzmann equation in the form
where IBa is current (not leak subtracted), GMax is the maximum channel conductance, V1/2 is the voltage at which current is half maximal, k is a slope factor describing voltage dependence of conductance, VR is the reversal potential for current, and VM is the membrane potential. The calculated values of GMax and VR were then used to determine fractional conductance (G/GMax) at each VM, using the equation
where G is the total macroscopic conductance at VM. Voltage dependence of activation curve was then fitted with the Boltzmann equation in the following form
separately for each cell, using Prism v. 4 for Macintosh (GraphPad Software, Inc.). Steady state inactivation was determined using a standard two-pulse protocol, in which conditioning pulses (-60 to 50mV) were employed at a duration that achieved complete current inactivation (1s). The current measured during the following test pulse was normalized to the peak current, and fitted with the Boltzmann equation
Comparisons of V1/2 and k between groups were performed by Student’s t-test. Data from whole-cell ICa and noise protocols were evaluated post hoc with Axograph X v. 1.1 (Axograph Scientific). Peak inward currents and charge transfer were measured after each drug application reached plateau (2-3min). To correct for cell size, all inward currents were expressed as a function of cell capacitance (pA/pF). For each antagonist, the main effect of injury group was tested with standard univariate analysis of variance, followed by planned Bonferroni post hoc comparisons between groups (Statistica 7, Statsoft Inc, Tulsa, OK). Unpaired t tests were performed between surgical groups. Data are expressed and mean ± SEM. Significance was estimated a P ≤ 0.05 versus control.
2. Results
2.1 Behavior after SNL
SNL animals showed a hyperalgesia-type behavioral response to nociceptive mechanical stimulation after 41.3±3.0% of touches ipsilateral to SNL and 3.8%±0.5% contralateral to the SNL, whereas control animals showed a 0.8±0.4% hyperalgesia response rate (right and left combined, since there was no difference; P<0.001 vs. SNL ipsilateral). Recordings were obtained from 56 L5 neurons after SNL, which had a diameter of 33.9±0.7μm, and from 47 neurons from control animals, which had a diameter of 33.5±0.5μm.
2.2 Effect of Injury on Non-differentiated ICa
As in prior reports, axonal injury decreased total ICa (Hogan et al., 2000, McCallum et al., 2006). Currents generated by square wave voltage commands at baseline before application of ICa subtype antagonists were less in neurons from SNL rats (76.6±7.8 pA/pF, n=28) compared to control neurons (132.0±9.0 pA/pF, n=25; P=0.01). A loss of ICa was also evident when currents were generated by AP waveform voltage commands. Specifically, injury reduced the total charge transfer for SNL L5 neurons (-410±31 ms·pA/pF, n=38) compared to control neurons (-494±39ms·pA/pF, n=30; P<0.05). Peak ICa during AP waveform commands was not significantly affected (-165±14pA/pF for SNL L5 neurons vs. -197±18pA/pF for control neurons, P=0.08). Injury had no effect on resting membrane potential (-53.8±1.1mV for SNL L5 neurons vs. -54.7±1.5mV for control neurons, P=0.32), membrane resistance (0.65±0.12 GΩ for SNL L5 neurons vs. 0.71±0.13 GΩ for control neurons, P=0.37), or rheobase current (0.67±0.1nA for SNL L5 neurons vs. 0.45±0.11nA for control neurons, P=0.09), and there was no correlation between ICa (measured as GMax) and rheobase (r=0.22, P=0.24).
2.3 ICa Subtype Blockade in Random Sequence
Initial experiments used discontinuous application of peptide blockers with relatively reversible effects and a random sequence of application to minimize the effect of sequence order. Using square wave depolarization commands, application of selective blockers demonstrated currents sensitive to antagonists of all ICa subtypes (Figures 1A, B). Injury resulted in selective reduction of ICa subtypes. Specifically, axotomized SNL L5 neurons showed diminished L-type current (Figure 1C), since nisoldipine blocked only 29% as much current in this group (n=12) as in control neurons (n=11). N-type current subtype was also diminished by axotomy, since SNX-111 blocked only 59% as much ICa in SNL L5 neurons as in control neurons. This random sequence protocol revealed R-type current sensitive to AgaIV that was only 12% as large in SNL L5 neurons as in control neurons.
Figure 1.

ICa sensitive to blockers of specific current subtypes, given in random sequence, using a square wave voltage command (holding potential -90mV, step to -10mV for 200ms). A. Sample current traces in a control neuron. Currents are corrected for cell capacitance. B. Typical time course of blockers and current responses, with numbers indicating traces shown in A. Bars indicate time period of blocker administration, including nisoldipine (Nisol, L-type current blocker, 200 nM), agatoxin IVA (AgaIV, P/Q-type current blocker, 200nM), SNX-111 (N-type current blocker, 200nM), SNX482 (R-type current blocker, 200nM), and cadmium (Cd2+, 200μM), a nonspecific ICa blocker. Current run-down and incomplete reversal of blockade (particularly of SNX-111) is evident in this example. C. Average current sensitive to each blocker, determined by subtraction of the current during blocker administration from the preceding agent-free baseline, in control neurons and axotomized neurons after spinal nerve ligation (SNL). * P< 0.05 comparing SNL to control.
2.4 ICa Subtype Blockade in Fixed Sequence
Some cells showed incomplete washout of peptide blocking agents (Figure 1B). Because of uncertain reversal of channel blockade during washout, we obtained further observations using a fixed sequence of blockers designed to minimize the influence of overlapping current sensitivities. Serial applications of antagonists in this fashion during square wave depolarizations (Figures 2A, B) also revealed a full mix of ICa subtypes, with a dominance of N-type and L-type current (Figure 2C), whereas P/Q-type and R-type were present at lower levels, consistent with previous findings (Regan et al., 1991, Cardenas et al., 1995, Rusin and Moises, 1995, Wu and Pan, 2004). Less L-type current sensitive to nisoldipine was again found in injured neurons (74% compared to control), and N-type current sensitive to GVIA was also diminished by axotomy (blockade in SNL L5 neurons was 49% of block in control neurons). This fixed sequence protocol showed no differences due to injury in P/Q-type and R-type current, a discordance from the random sequence data that may result from the uncertain degree of washout of blockers in the that protocol.
Figure 2.
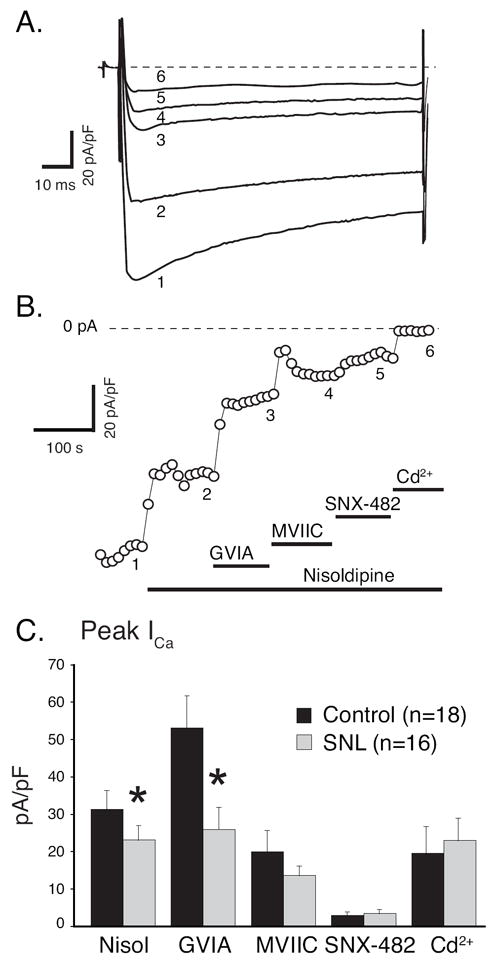
ICa sensitive to blockers of specific current subtypes, given in a fixed sequence, using a square wave voltage command (holding potential -90mV, step to -10mV for 200ms). A. Sample current traces from a control neuron. Currents are corrected for cell capacitance. B. Typical time course of blockers and current responses, with numbers indicating traces shown in A. Bars indicate time period of blocker administration, including nisoldipine (Nisol, L-type current blocker, 200 nM), ω-conotoxin GVIA (GVIA, N-type current blocker, 200nM), ω-Conotoxin MVIIC (MVIIC, N- and P/Q-type current blocker, 200nM), SNX482 (R-type current blocker, 200nM), and cadmium (Cd2+, 200μM), a nonspecific ICa blocker. C. Average current sensitive to each blocker in control neurons and axotomized neurons after spinal nerve ligation (SNL). * P< 0.05 comparing SNL to control.
Additional experiments were performed on other neurons using an AP waveform as a voltage stimulus, with the intention of identifying injury effects on various current subtypes generated in response to natural neuronal firing. ICa in response to AP waveform commands showed peaks that were delayed until the descending limb of the AP (Figure 3A), confirming previous reports (Scroggs and Fox, 1992b, McCallum et al., 2006). Using the fixed order of sequential blockade, the presence of all ICa subtypes was demonstrated in control sensory neurons, with L-type and N-type again predominating (Figures 3B, C). Compared to the previously described square wave findings, a somewhat different picture emerged from AP waveform experiments regarding the effect of injury on individual ICa subtypes. Sensitivity of ICa to antagonists was diminished by axotomy only for nisoldipine for both peak ICa (43% block in SNL L5 compared to control; Figure 3B) and charge transfer (59%; Figure 3C). In contrast to square wave data, injured and non-injured neurons depolarized with AP waveform commands responded comparably to N-type Ca2+ channel blockade with GVIA.
Figure 3.

ICa sensitive to blockers of specific current subtypes, given in a fixed sequence, using an action potential waveform voltage command. A. Sample current traces from an axotomized neuron dissociated from ganglion after spinal nerve ligation (SNL). Currents are corrected for cell capacitance. Blockers are as indicated for Figure 2. B. Voltage command for the traces in A, shown in temporal alignment with panel A. C. Average peak current (corrected for cell capacitance) that is sensitive to each blocker in control neurons and axotomized neurons after SNL. D. Average charge transfer (corrected for cell capacitance) that is sensitive to each blocker in control neurons and axotomized neurons after SNL. * P< 0.05 comparing SNL to control.
2.5 Selective action of GVIA on ICa
Because of the uncertainties produced by combined application of multiple antagonists (Burley and Dolphin, 2000), including the potential for blockade of N-type current by nisoldipine (Jones and Jacobs, 1990, Elmslie et al., 1992), we further examined N-type current using the sole application of GVIA without other antagonists. Current-voltage (I-V) relationships in response to square wave depolarizations were generated before and after application of GVIA to control (Figure 4A) and injured neurons (Figure 4B). Prior to application of the toxin, baseline GMax was less in SNL L5 neurons (2.8±0.2pS/pF, n=17) compared to control neurons 4.4±0.4pS/pF, n=16, P<0.001). The conductance sensitive to GVIA in SNL L5 neurons (0.6±0.2pS/pF) was less than in control neurons (1.9±1.8pA/pS, P<0.01; Figure 4C), and GMax no longer differed between control and SNL conditions, indicating that much of the ICa lost by injury is N-type. In the same neurons, currents were measured during AP waveform voltage commands (Figure 4D). The holding potential duplicated the neurons’ resting membrane potential, and was not different in SNL neurons (-55.1±1.3mV, n=17) compared to control neurons (-56.9±1.1mV, n=16), and duration of the AP waveform command was not different (3.6±0.3ms for SNL, 4.2±0.7 for controls, P=0.20). In contrast to findings in the context of prior nisoldipine application, sensitivity to GVIA was significantly diminished in injured neurons. Specifically, the total charge transfer sensitive to GVIA in SNL L5 neurons (-105±23ms·pA/pF; Figure 4E) was reduced compared to control neurons (-176±29ms·pA/pF, P<0.05). Similarly, the peak amplitude of the current sensitive to GVIA during AP waveform commands in SNL L5 neurons (-40±8pA/pF; Figure 4F) was reduced compared to control neurons (-72±12pA/pF, P<0.05).
Figure 4.
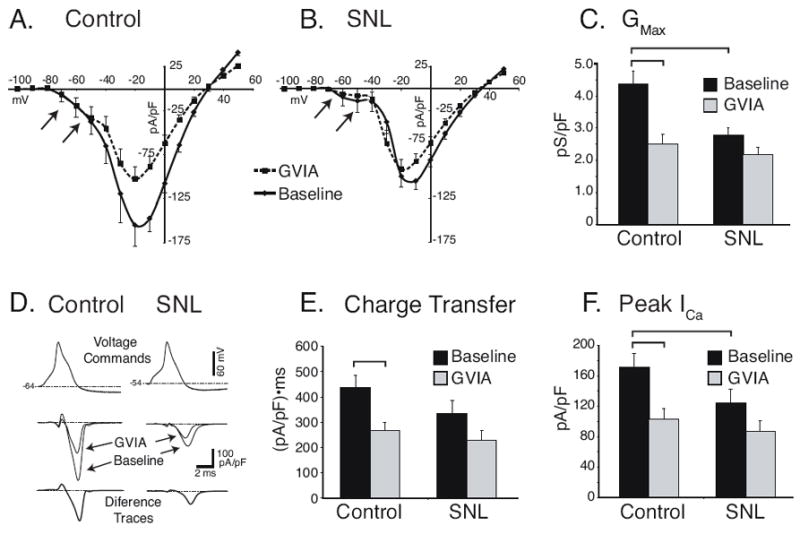
ICa sensitive to the N-type blocker ω-conotoxin GVIA (GVIA, 200nM) given alone. A. Current-voltage relationships determined with square-wave voltage commands (200ms, VH = -90mV) at baseline conditions and during GVIA application in control neurons (n=16). Arrows indicate T-type low voltage-activated current that is unaffected by GVIA. B. Current-voltage relationships after spinal nerve ligation (SNL, n=17) demonstrate loss of GVIA-sensitive current and diminished low voltage-activated current (arrows). C. Summary data show the total Ca2+ current conductance was decreased and the GVIA-sensitive component was lost after SNL. D. Action potential waveform commands (top panel) were used in the same neurons to generate currents at baseline and during GVIA application (middle panel) to allow calculation of the difference current that is sensitive to GVIA (bottom panel). The area of the GVIA-sensitive trace, equivalent to the charge transfer was reduced by SNL (E.), as was the amplitude of the GVIA-sensitive current (F.). Brackets indicate significant paired comparisons.
2.6 Mechanism of the injury effect on N-type current
Other neurons were incubated with antagonists against L-, P/Q-, and R-type currents in order to selectively expose N-type current for examination. Square wave depolarizations again revealed a diminished N-type conductance in SNL L5 neurons (1.2±0.2 pS/pF, n=18) compared to control neurons (2.4±0.4 pS/pF, n=11; P<0.05). Standard prepulse protocols showed that voltage dependence of activation (Figure 5A) shifted in a depolarizing direction from V1/2 in control neurons of -24.2±0.5 to -18.9±0.2 mV after SNL (P<0.001), while k was unchanged. Examination of steady state inactivation (Figure 5B) showed that V1/2 was unchanged (control -32.1±1.4mV vs. SNL -35.9±1.9mV, P=0.055), but the Boltzmann slope factor k is reduced (control 11.4±0.6 vs. SNL 9.2±0.9; P<0.05), indicating a narrow range of voltage at which inactivation occurs. Identity as N-type current was confirmed by complete blockade of remaining current with SNX-111.
Figure 5.

Evaluation of N-type ICa revealed by blockade of other high voltage-activated current subtypes through incubation with nisoldipine (200 nM), SNX-482 (500 nM), and MVIIC (2 μM), the last of which was removed during recording. A. Voltage dependence of activation was shifted in a depolarizing direction by spinal nerve ligation (SNL, V1/2 -18.9±0.2 mV, n=18) compared to control neurons (-24.2±0.5, n=11, P<0.001). B. The V1/2 for steady state inactivation was unchanged by SNL (-35.9±1.9mV) compared to control (-32.1±1.4mV, P=0.055), but the Boltzmann constant k was less negative (control -11.4±0.6 vs. SNL -9.2±0.9; P<0.05).
Analysis of nonstationary ensemble variance was used to examine whether decreased channel number or decreased channel conductance contributed to loss of N-type ICa after injury (Figure 6). In a subset of control and injured neurons that were incubated in blockers of all subtypes but N, depolarization to the voltage that produced half-maximal current (-21.4±1.4mV in SNL L5 neurons, n=5; -21.4±1.3mV in control neurons, n=6; not significantly different) revealed a unitary current amplitude that was unchanged in axotomized SNL L5 neurons (-0.78±0.17 pA) compared to control neurons (-0.72±0.20 pA). These single channel currents corresponded to single-channel conductance of 18.3±2.8 pS in SNL L5 and 16.9±3.5 pS in controls. Finally, channel density was not affected by axotomy SNL (0.40±0.09 /μm2 in SNL L5; 0.60±0.15 /μm2 in controls).
Figure 6.

Examples of nonstationary noise analysis in control (A.) and spinal nerve ligated (SNL) neurons, indicating variance (σ) plotted against total mean current (I) during depolarization from a holding potential of -60mV to -20mV in each case. The unitary current (i), and the number of channels recorded in the cell (N), were derived from fitting the formula .
3. Discussion
The central involvement of Ca2+ signaling in sensory neurons makes it imperative to resolve the effects of injury on the various subtypes of voltage gated currents after injury. Impediments to meeting this challenge are the diversity of ICa subtypes in sensory neurons and the imperfect specificity of pharmacologic agents for distinguishing their specific contributions. An alternative approach employing selective gene elimination might be considered for identifying subtype-specific effects of injury, but the interpretation of such findings is complicated by compensatory changes in expression of retained subtypes prior to injury and an altered influence of injury upon the retained subtypes (Wilson et al., 2000, Yang and Stephens, 2009). We therefore evaluated subtype current expression through complementary pharmacological approaches that overall indicate a loss of both N-type and L-type Ca2+ current in DRG neurons after peripheral nerve injury. Further, we employed a nerve injury model (SNL) that produces a clinically relevant incomplete sensory denervation of the extremity.
Our new findings are compatible with observations from previous studies that show loss of HVA current in sensory neurons after distal (sciatic nerve) axotomy. One of these (Abdulla and Smith, 2001), however, examined a mixed population of axotomized and intact neurons that results from the fact that only 54-57% of L5 DRG neurons project to the sciatic site of injury (Devor et al., 1985), in contrast to near 100% axotomy that results from proximal spinal nerve transection as used in the present study. Another prior study also showed decreased HVA current after injury (Baccei and Kocsis, 2000) but examined only cutaneous afferents, and L-type current was not determined. Our use of a more complete range of channel antagonists, including the recent addition of R-type selective SNX482, also allowed greater specificity in determining contribution of the HVA channel subtypes to post-injury ICa loss.
Because of the overlapping sensitivities of various HVA channel subtypes to the available peptide and dihydropyridine blockers (Burley and Dolphin, 2000), our approach incorporated alternative sequences of administration. A valuable finding of our study is the recognition that inconsistent results may result from the influence of the sequence with which blockers are applied. This is evident in the different apparent proportions of N-type and L-type currents in control neurons in our data (Figures 1C, 2C, 3C,D), and may also explain the variability in proportions of current subtypes reported by other studies of uninjured DRG neurons (Regan et al., 1991, Scroggs and Fox, 1992a, Rusin and Moises, 1995, Honma et al., 1999, Abdulla and Smith, 2001, Wu and Pan, 2004).
We further examined the response of ICa to voltage commands in the form of AP waveforms. Prior studies have pointed out that the more natural kinetics of this stimulus reveal a greater contribution of N-type relative to L-type currents (Scroggs and Fox, 1992a, b), compared to sustained square wave voltage commands, and a dominant role of N-type current is likewise evident in our AP waveform recordings from control neurons. Using AP waveform voltage commands, we observed that ICa sensitive to GVIA is substantially decreased by injury when GVIA is the only blocker applied to the neurons, but this difference vanishes if GVIA application follows nisoldipine. Since even high doses of GVIA up to 10μM have negligible effects on L-type current in DRG neurons (Regan et al., 1991), our data suggest that nisoldipine blocks a portion of N-type current, in agreement with other reports (Diochot et al., 1995, Burley and Dolphin, 2000). The conclusion that injury substantially reduces N-type current is confirmed by our experiments isolating N-type current by elimination of all other subtypes.
Despite the imperfections of pharmacological characterization of channel subtypes, our overall observations allow us to conclude that the main effect of injury is a decrease of L-type and N-type currents. As these are also the dominant current subtypes, it is evident that injury has a relatively broad effect on ICa. This parallels our previous observations that HVA channel subtypes have overlapping functional roles. Specifically, N, L, R, and P/Q-subtypes all trigger currents that repolarize the AP and generate afterhyperpolarizations that follow the AP (Lirk et al., 2008), and all contribute to the cytoplasmic Ca2+ transient that accompanies neuronal activation (Fuchs et al., 2007b).
Several features place special importance on the N-type current compared to the other subtypes of ICa found in sensory neurons. Influx of Ca2+ through N-type channels disproportionately contributes to release of stored Ca2+ by the process of Ca2+-induced Ca2+ release (Cordoba-Rodriguez et al., 1999, Tully and Treistman, 2004). N-type current in sensory neurons also provides the majority of Ca2+ influx that triggers excitatory neurotransmission of nociceptive afferent activity in the spinal cord dorsal horn (Chaplan et al., 1994, Rycroft et al., 2007), with a preferential contribution by the splice isoform containing exon 37a (Bell et al., 2004, Castiglioni et al., 2006). Additionally, N-type current is a target of current analgesic drug development. All the neurons examined in the present study showed N-type current sensitive to GVIA. Our finding that N-type current is diminished in axotomized neurons during both square wave and AP waveform stimulation is supported by previous observations that SNL severely restricts the contribution of this subtype to activity-induced cytoplasmic Ca2+ transients (Fuchs et al., 2005), and that expression of the mRNA for the N-type isoform containing exon 37a is halved after SNL (Altier et al., 2007).
In our present data, isolated N-type current showed a depolarizing shift in the activation voltage. Although this would not contribute to the diminished GMax observed after injury, it could account for part of the loss of GVIA-sensitive current noted during AP waveform depolarizations. Characterization by noise analysis identified an N-type channel conductance in close agreement with a prior report (Nowycky et al., 1985), but no injury-related change was observed in either unitary current or channel density. This suggests that injury reduces N-type current through influences on channel open probability, which is compatible with the influence of a cytosolic modulation of the channel.
Our study was not designed to directly identify the upstream signaling leading to ICa loss with injury, but several possibilities may be considered. A previous report has shown that activation of N-type current during AP waveform stimulation is greater with longer AP durations (King et al., 1999). However, the AP waveform commands used here did not differ in duration, so this is not an explanation for the diminished GVIA-sensitive N-type currents in axotomized SNL neurons. The affinity of conotoxin inhibitors for the N-type channel is reduced in the presence of the α2δ Ca2+ channel subunit (Mould et al., 2004). Since the α2δ subunit is increased in DRG neurons following SNL (Luo et al., 2001), the loss of N-type current could be illusory, as a result of diminished GVIA current blockade. However, GVIA is the peptide blocker least subject to this effect (Mould et al., 2004), and decreased GVIA affinity would not alter our findings on N-type current isolated through blockade of all other currents.
Under natural conditions, N-type current is highly regulated by multiple signaling pathways. A wide range of ligands inhibits N-type current through G protein-coupled receptors. However, since both membrane delimited/voltage-dependent G protein inhibition, as well as the voltage-independent tyrosine kinase-mediated pathway, are rapidly reversible (Bean, 1989, Bourinet et al., 1996, Schiff et al., 2000), the absence of ligands in our dissociated neuron preparation make this an unlikely mechanism to explain persistent injury effects. Similarly, although TRPV1 activation may inhibit HVA channels broadly (Wu et al., 2006), this effect washes out in the absence of a TRPV1 agonist. The N-type channel in DRG neurons constitutively interacts with the nociceptin receptor (ORL-1), even in the absence of nociceptin, to produce chronic inhibition of current (Beedle et al., 2004). Since ORL-1 is upregulated in injury (Chen and Sommer, 2006), this could contribute to loss of N-current after axotomy. Binding of ORL-1 by nociceptin, which is also upregulated (Chen and Sommer, 2006), produces internalization of channels (Altier et al., 2006), but we did not observe any loss of N-type channels.
A potential cause of post-injury ICa loss may be diminished kinase activation of channels. Our examination of voltage-dependent steady state channel inactivation revealed a diminished Boltzmann constant k, which indicates a greater sensitivity of the channel to voltage, consistent with altered phosphorylation levels. Withdrawal of glial-derived neurotrophic factor (GDNF) from sensory neurons results in loss of ICa, particularly N-type, through an extracellular signal-regulated kinase (ERK)-dependent pathway (Martin et al., 2006,Woodall et al., 2008), while replacement of GDNF reverses behavioral and histochemical consequences of nerve injury. Similarly, both L- and N-type currents are also regulated by kinases such as Ca2+/calmodulin-dependent protein kinase (CaMKII) and protein kinase C, and the pore-forming alpha subunits supporting these currents are targets of CaMKII modulation (Swartz, 1993, Hell et al., 1994, Zhu and Ikeda, 1994, Hudmon et al., 2005). We have noted decreased levels of phosphorylated CaMKII following sensory neuron injury (Kojundzic et al., 2010), and our observations of parallel losses of hyperpolarization activated H-current (Hogan and Poroli, 2008), Ca2+-activated K+ currents (Sarantopoulos et al., 2007), and ATP-sensitive K+ current (Kawano et al., 2009) after sensory neuron axotomy may also be explained by withdrawal of CaMKII activity (Kong et al., 2000, Sansom et al., 2000, Fan et al., 2005).
3.1 Conclusions
The cytoplasmic Ca2+ signal serves diverse roles in neurons that differ between neuronal groups, and also between different portions of the neuron. In the spinal cord, blockade of HVA Ca2+ channels produces analgesia through interruption of synaptic transmission between the sensory neuron and dorsal horn neurons (Sluka, 1998). However, at the sensory neuron soma, which is a site of neuronal hyperexcitability following nerve damage (Wall and Devor, 1983, Sapunar et al., 2005), exposing the neuron to conditions that increase ICa produces a correction of the injury-induced neuronal hyperexcitability (Hogan et al., 2008). It is possible that correction of ICa loss in sensory neurons at the level of the DRG may be a novel route for clinical treatment of neuropathic pain. Our present data indicate that N-type and L-type ICa are most diminished by axotomy and would be suitable targets for therapy.
Research Highlights.
We examine neuronal Ca2+ current loss that follows painful peripheral nerve injury.
Ca2+ current in DRG neurons decreases for both L- and N-channel subtypes.
This effect is evident during both square and AP waveform depolarizations.
The activation curve shifts right for N-type current after injury.
Injury does not affect N-type channel number or unitary current.
Abbreviations
- AgaIVA
agatoxin IVA
- CaMKII
Ca2+/calmodulin-dependent protein kinase
- DRG
dorsal root ganglion
- EGTA
ethylene glycol-bis(2-amino-ethylether)-N,N, N’, N’-tetra-acetic acid
- GDNF
glial-derived neurotrophic factor
- GVIA
ω-conotoxin GVIA
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HVA
high-voltage activated
- ICa
Ca2+ current
- L5
fifth lumbar vertebral level
- LVA
low-voltage activated
- MVIIC
ω-Conotoxin MVIIC
- SNL
spinal nerve ligation
- TEA
tetraethylammonium chloride
- TTX
tetrodotoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdulla FA, Smith PA. Axotomy- and autotomy-induced changes in Ca2+ and K+ channel currents of rat dorsal root ganglion neurons. Journal of neurophysiology. 2001;85:644–658. doi: 10.1152/jn.2001.85.2.644. [DOI] [PubMed] [Google Scholar]
- Altier C, Dale CS, Kisilevsky AE, Chapman K, Castiglioni AJ, Matthews EA, Evans RM, Dickenson AH, Lipscombe D, Vergnolle N, Zamponi GW. Differential role of N-type calcium channel splice isoforms in pain. J Neurosci. 2007;27:6363–6373. doi: 10.1523/JNEUROSCI.0307-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altier C, Khosravani H, Evans RM, Hameed S, Peloquin JB, Vartian BA, Chen L, Beedle AM, Ferguson SS, Mezghrani A, Dubel SJ, Bourinet E, McRory JE, Zamponi GW. ORL1 receptor-mediated internalization of N-type calcium channels. Nat Neurosci. 2006;9:31–40. doi: 10.1038/nn1605. [DOI] [PubMed] [Google Scholar]
- Andre S, Puech-Mallie S, Desmadryl G, Valmier J, Scamps F. Axotomy differentially regulates voltage-gated calcium currents in mice sensory neurones. Neuroreport. 2003;14:147–150. doi: 10.1097/00001756-200301200-00027. [DOI] [PubMed] [Google Scholar]
- Arroyo G, Aldea M, Fuentealba J, Albillos A, Garcia AG. SNX482 selectively blocks P/Q Ca2+ channels and delays the inactivation of Na+ channels of chromaffin cells. Eur J Pharmacol. 2003;475:11–18. doi: 10.1016/s0014-2999(03)02084-3. [DOI] [PubMed] [Google Scholar]
- Baccei ML, Kocsis JD. Voltage-gated calcium currents in axotomized adult rat cutaneous afferent neurons. Journal of neurophysiology. 2000;83:2227–2238. doi: 10.1152/jn.2000.83.4.2227. [DOI] [PubMed] [Google Scholar]
- Bao J, Li JJ, Perl ER. Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J Neurosci. 1998;18:8740–8750. doi: 10.1523/JNEUROSCI.18-21-08740.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- Beedle AM, McRory JE, Poirot O, Doering CJ, Altier C, Barrere C, Hamid J, Nargeot J, Bourinet E, Zamponi GW. Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat Neurosci. 2004;7:118–125. doi: 10.1038/nn1180. [DOI] [PubMed] [Google Scholar]
- Bell TJ, Thaler C, Castiglioni AJ, Helton TD, Lipscombe D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron. 2004;41:127–138. doi: 10.1016/s0896-6273(03)00801-8. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Stea A, Snutch TP. Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proc Natl Acad Sci U S A. 1996;93:1486–1491. doi: 10.1073/pnas.93.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowersox SS, Gadbois T, Singh T, Pettus M, Wang YX, Luther RR. Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. J Pharmacol Exp Ther. 1996;279:1243–1249. [PubMed] [Google Scholar]
- Burley JR, Dolphin AC. Overlapping selectivity of neurotoxin and dihydropyridine calcium channel blockers in cerebellar granule neurones. Neuropharmacology. 2000;39:1740–1755. doi: 10.1016/s0028-3908(99)00266-x. [DOI] [PubMed] [Google Scholar]
- Cardenas CG, Del Mar LP, Scroggs RS. Variation in serotonergic inhibition of calcium channel currents in four types of rat sensory neurons differentiated by membrane properties. Journal of neurophysiology. 1995;74:1870–1879. doi: 10.1152/jn.1995.74.5.1870. [DOI] [PubMed] [Google Scholar]
- Castiglioni AJ, Raingo J, Lipscombe D. Alternative splicing in the C-terminus of CaV2.2 controls expression and gating of N-type calcium channels. J Physiol. 2006;576:119–134. doi: 10.1113/jphysiol.2006.115030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. Journal of Pharmacology & Experimental Therapeutics. 1994;269:1117–1123. [PubMed] [Google Scholar]
- Chen Y, Sommer C. Nociceptin and its receptor in rat dorsal root ganglion neurons in neuropathic and inflammatory pain models: implications on pain processing. J Peripher Nerv Syst. 2006;11:232–240. doi: 10.1111/j.1529-8027.2006.0093.x. [DOI] [PubMed] [Google Scholar]
- Cordoba-Rodriguez R, Moore KA, Kao JP, Weinreich D. Calcium regulation of a slow post-spike hyperpolarization in vagal afferent neurons. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7650–7657. doi: 10.1073/pnas.96.14.7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M, Govrin-Lippmann R, Frank I, Raber P. Proliferation of primary sensory neurons in adult rat dorsal root ganglion and the kinetics of retrograde cell loss after sciatic nerve section. Somatosensory Research. 1985;3:139–167. doi: 10.3109/07367228509144581. [DOI] [PubMed] [Google Scholar]
- Diochot S, Richard S, Baldy-Moulinier M, Nargeot J, Valmier J. Dihydropyridines, phenylalkylamines and benzothiazepines block N-, P/Q- and R-type calcium currents. Pflugers Arch. 1995;431:10–19. doi: 10.1007/BF00374372. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Elmslie KS, Kammermeier PJ, Jones SW. Calcium current modulation in frog sympathetic neurones: L-current is relatively insensitive to neurotransmitters. J Physiol. 1992;456:107–123. doi: 10.1113/jphysiol.1992.sp019329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h) Nat Neurosci. 2005;8:1542–1551. doi: 10.1038/nn1568. [DOI] [PubMed] [Google Scholar]
- Feng ZP, Doering CJ, Winkfein RJ, Beedle AM, Spafford JD, Zamponi GW. Determinants of inhibition of transiently expressed voltage-gated calcium channels by omega-conotoxins GVIA and MVIIA. J Biol Chem. 2003;278:20171–20178. doi: 10.1074/jbc.M300581200. [DOI] [PubMed] [Google Scholar]
- Fuchs A, Lirk P, Stucky C, Abram SE, Hogan QH. Painful nerve injury decreases resting cytosolic calcium concentrations in sensory neurons of rats. Anesthesiology. 2005;102:1217–1225. doi: 10.1097/00000542-200506000-00023. [DOI] [PubMed] [Google Scholar]
- Fuchs A, Rigaud M, Hogan QH. Painful nerve injury shortens the intracellular Ca2+ signal in axotomized sensory neurons of rats. Anesthesiology. 2007a;107:106–116. doi: 10.1097/01.anes.0000267538.72900.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Rigaud M, Sarantopoulos CD, Filip P, Hogan QH. Contribution of calcium channel subtypes to the intracellular calcium signal in sensory neurons: the effect of injury. Anesthesiology. 2007b;107:117–127. doi: 10.1097/01.anes.0000267511.21864.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper AA. Similarities between some properties of the soma and sensory receptors of primary afferent neurones. Experimental Physiology. 1991;76:369–377. doi: 10.1113/expphysiol.1991.sp003504. [DOI] [PubMed] [Google Scholar]
- Heinke B, Balzer E, Sandkuhler J. Pre- and postsynaptic contributions of voltage-dependent Ca2+ channels to nociceptive transmission in rat spinal lamina I neurons. Eur J Neurosci. 2004;19:103–111. doi: 10.1046/j.1460-9568.2003.03083.x. [DOI] [PubMed] [Google Scholar]
- Hell JW, Appleyard SM, Yokoyama CT, Warner C, Catterall WA. Differential phosphorylation of two size forms of the N-type calcium channel alpha 1 subunit which have different COOH termini. J Biol Chem. 1994;269:7390–7396. [PubMed] [Google Scholar]
- Hillyard DR, Monje VD, Mintz IM, Bean BP, Nadasdi L, Ramachandran J, Miljanich G, Azimi-Zoonooz A, McIntosh JM, Cruz LJ, et al. A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron. 1992;9:69–77. doi: 10.1016/0896-6273(92)90221-x. [DOI] [PubMed] [Google Scholar]
- Hogan Q, Lirk P, Poroli M, Rigaud M, Fuchs A, Fillip P, Ljubkovic M, Gemes G, Sapunar D. Restoration of calcium influx corrects membrane hyperexcitability in injured rat dorsal root ganglion neurons. Anesth Analg. 2008;107:1045–1051. doi: 10.1213/ane.0b013e31817bd1f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan Q, Sapunar D, Modric-Jednacak K, McCallum JB. Detection of neuropathic pain in a rat model of peripheral nerve injury. Anesthesiology. 2004;101:476–487. doi: 10.1097/00000542-200408000-00030. [DOI] [PubMed] [Google Scholar]
- Hogan QH, McCallum JB, Sarantopoulos C, Aason M, Mynlieff M, Kwok WM, Bosnjak ZJ. Painful neuropathy decreases membrane calcium current in mammalian primary afferent neurons. Pain. 2000;86:43–53. doi: 10.1016/s0304-3959(99)00313-9. [DOI] [PubMed] [Google Scholar]
- Hogan QH, Poroli M. Hyperpolarization-activated current (I(h)) contributes to excitability of primary sensory neurons in rats. Brain Res. 2008;1207:102–110. doi: 10.1016/j.brainres.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma Y, Yamakage M, Ninomiya T. Effects of adrenergic stimulus on the activities of Ca2+ and K+ channels of dorsal root ganglion neurons in a neuropathic pain model. Brain Research. 1999;832:195–206. doi: 10.1016/s0006-8993(99)01499-7. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. The Journal of cell biology. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagodic MM, Pathirathna S, Joksovic PM, Lee W, Nelson MT, Naik AK, Su P, Jevtovic-Todorovic V, Todorovic SM. Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. Journal of neurophysiology. 2008;99:3151–3156. doi: 10.1152/jn.01031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SW, Jacobs LS. Dihydropyridine actions on calcium currents of frog sympathetic neurons. J Neurosci. 1990;10:2261–2267. doi: 10.1523/JNEUROSCI.10-07-02261.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass RS. Nisoldipine: a new, more selective calcium current blocker in cardiac Purkinje fibers. J Pharmacol Exp Ther. 1982;223:446–456. [PubMed] [Google Scholar]
- Kawano T, Zoga V, Gemes G, McCallum JB, Wu HE, Pravdic D, Liang MY, Kwok WM, Hogan Q, Sarantopoulos C. Suppressed Ca2+/CaM/CaMKII-dependent K(ATP) channel activity in primary afferent neurons mediates hyperalgesia after axotomy. Proc Natl Acad Sci U S A. 2009;106:8725–8730. doi: 10.1073/pnas.0901815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- King AP, Hall KE, Macdonald RL. kappa- and mu-Opioid inhibition of N-type calcium currents is attenuated by 4beta-phorbol 12-myristate 13-acetate and protein kinase C in rat dorsal root ganglion neurons. Journal of Pharmacology & Experimental Therapeutics. 1999;289:312–320. [PubMed] [Google Scholar]
- Kojundzic SL, Puljak L, Hogan Q, Sapunar D. Depression of Ca(2+)/calmodulin-dependent protein kinase II in dorsal root ganglion neurons after spinal nerve ligation. J Comp Neurol. 2010;518:64–74. doi: 10.1002/cne.22209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong ID, Koh SD, Bayguinov O, Sanders KM. Small conductance Ca2+-activated K+ channels are regulated by Ca2+-calmodulin-dependent protein kinase II in murine colonic myocytes. J Physiol. 2000;524(Pt 2):331–337. doi: 10.1111/j.1469-7793.2000.t01-1-00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lirk P, Poroli M, Rigaud M, Fuchs A, Fillip P, Huang CY, Ljubkovic M, Sapunar D, Hogan Q. Modulators of calcium influx regulate membrane excitability in rat dorsal root ganglion neurons. Anesth Analg. 2008;107:673–685. doi: 10.1213/ane.0b013e31817b7a73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Dai J, Dai L, Deng M, Hu Z, Hu W, Liang S. Function and solution structure of Huwentoxin-X, a specific blocker of N-type calcium channels, from the Chinese bird spider Ornithoctonus huwena. J Biol Chem. 2006;281:8628–8635. doi: 10.1074/jbc.M513542200. [DOI] [PubMed] [Google Scholar]
- Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, Yaksh TL. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci. 2001;21:1868–1875. doi: 10.1523/JNEUROSCI.21-06-01868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- Martin SW, Butcher AJ, Berrow NS, Richards MW, Paddon RE, Turner DJ, Dolphin AC, Sihra TS, Fitzgerald EM. Phosphorylation sites on calcium channel alpha1 and beta subunits regulate ERK-dependent modulation of neuronal N-type calcium channels. Cell Calcium. 2006;39:275–292. doi: 10.1016/j.ceca.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Dickenson AH. Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain. 2001;92:235–246. doi: 10.1016/s0304-3959(01)00255-x. [DOI] [PubMed] [Google Scholar]
- McCallum JB, Kwok WM, Mynlieff M, Bosnjak ZJ, Hogan QH. Loss of T-type calcium current in sensory neurons of rats with neuropathic pain. Anesthesiology. 2003;98:209–216. doi: 10.1097/00000542-200301000-00032. [DOI] [PubMed] [Google Scholar]
- McCallum JB, Kwok WM, Sapunar D, Fuchs A, Hogan QH. Painful peripheral nerve injury decreases calcium current in axotomized sensory neurons. Anesthesiology. 2006;105:160–168. doi: 10.1097/00000542-200607000-00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCobb DP, Beam KG. Action potential waveform voltage-clamp commands reveal striking differences in calcium entry via low and high voltage-activated calcium channels. Neuron. 1991;7:119–127. doi: 10.1016/0896-6273(91)90080-j. [DOI] [PubMed] [Google Scholar]
- McDonough SI, Swartz KJ, Mintz IM, Boland LM, Bean BP. Inhibition of calcium channels in rat central and peripheral neurons by omega-conotoxin MVIIC. J Neurosci. 1996;16:2612–2623. doi: 10.1523/JNEUROSCI.16-08-02612.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould J, Yasuda T, Schroeder CI, Beedle AM, Doering CJ, Zamponi GW, Adams DJ, Lewis RJ. The alpha2delta auxiliary subunit reduces affinity of omega-conotoxins for recombinant N-type (Cav2.2) calcium channels. J Biol Chem. 2004;279:34705–34714. doi: 10.1074/jbc.M310848200. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Joksovic PM, Perez-Reyes E, Todorovic SM. The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J Neurosci. 2005;25:8766–8775. doi: 10.1523/JNEUROSCI.2527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature. 1985;316:440–443. doi: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- Olivera BM, Cruz LJ, de Santos V, LeCheminant GW, Griffin D, Zeikus R, McIntosh JM, Galyean R, Varga J, Gray WR, et al. Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using omega-conotoxin from Conus magus venom. Biochemistry. 1987;26:2086–2090. doi: 10.1021/bi00382a004. [DOI] [PubMed] [Google Scholar]
- Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. Journal of Neuroscience. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan LJ, Sah DW, Bean BP. Ca2+ channels in rat central and peripheral neurons: high-threshold current resistant to dihydropyridine blockers and omega-conotoxin. Neuron. 1991;6:269–280. doi: 10.1016/0896-6273(91)90362-4. [DOI] [PubMed] [Google Scholar]
- Rusin KI, Moises HC. mu-Opioid receptor activation reduces multiple components of high-threshold calcium current in rat sensory neurons. Journal of Neuroscience. 1995;15:4315–4327. doi: 10.1523/JNEUROSCI.15-06-04315.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycroft BK, Vikman KS, Christie MJ. Inflammation reduces the contribution of N-type calcium channels to primary afferent synaptic transmission onto NK1 receptor-positive lamina I neurons in the rat dorsal horn. J Physiol. 2007;580:883–894. doi: 10.1111/j.1469-7793.2000.t01-1-02117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom SC, Ma R, Carmines PK, Hall DA. Regulation of Ca(2+)-activated K(+) channels by multifunctional Ca(2+)/calmodulin-dependent protein kinase. Am J Physiol Renal Physiol. 2000;279:F283–288. doi: 10.1152/ajprenal.2000.279.2.F283. [DOI] [PubMed] [Google Scholar]
- Sapunar D, Ljubkovic M, Lirk P, McCallum JB, Hogan QH. Distinct membrane effects of spinal nerve ligation on injured and adjacent dorsal root ganglion neurons in rats. Anesthesiology. 2005;103:360–376. doi: 10.1097/00000542-200508000-00020. [DOI] [PubMed] [Google Scholar]
- Sarantopoulos CD, McCallum JB, Rigaud M, Fuchs A, Kwok WM, Hogan QH. Opposing effects of spinal nerve ligation on calcium-activated potassium currents in axotomized and adjacent mammalian primary afferent neurons. Brain Res. 2007;1132:84–99. doi: 10.1016/j.brainres.2006.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff ML, Siderovski DP, Jordan JD, Brothers G, Snow B, De Vries L, Ortiz DF, Diverse-Pierluissi M. Tyrosine-kinase-dependent recruitment of RGS12 to the N-type calcium channel. Nature. 2000;408:723–727. doi: 10.1038/35047093. [DOI] [PubMed] [Google Scholar]
- Scroggs RS, Fox AP. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. Journal of Physiology. 1992a;445:639–658. doi: 10.1113/jphysiol.1992.sp018944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggs RS, Fox AP. Multiple Ca2+ currents elicited by action potential waveforms in acutely isolated adult rat dorsal root ganglion neurons. Journal of Neuroscience. 1992b;12:1789–1801. doi: 10.1523/JNEUROSCI.12-05-01789.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ. The variance of sodium current fluctuations at the node of Ranvier. Journal of Physiology. 1980;307:97–129. doi: 10.1113/jphysiol.1980.sp013426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluka KA. Blockade of N- and P/Q-type calcium channels reduces the secondary heat hyperalgesia induced by acute inflammation. Journal of Pharmacology & Experimental Therapeutics. 1998;287:232–237. [PubMed] [Google Scholar]
- Staats PS, Yearwood T, Charapata SG, Presley RW, Wallace MS, Byas-Smith M, Fisher R, Bryce DA, Mangieri EA, Luther RR, Mayo M, McGuire D, Ellis D. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: a randomized controlled trial. Jama. 2004;291:63–70. doi: 10.1001/jama.291.1.63. [DOI] [PubMed] [Google Scholar]
- Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G protein-mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- Tully K, Treistman SN. Distinct intracellular calcium profiles following influx through N- versus L-type calcium channels: role of Ca2+-induced Ca2+ release. Journal of neurophysiology. 2004;92:135–143. doi: 10.1152/jn.01004.2003. [DOI] [PubMed] [Google Scholar]
- Wall PD, Devor M. Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain. 1983;17:321–339. doi: 10.1016/0304-3959(83)90164-1. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Toth PT, Oh SB, Gillard SE, Volsen S, Ren D, Philipson LH, Lee EC, Fletcher CF, Tessarollo L, Copeland NG, Jenkins NA, Miller RJ. The status of voltage-dependent calcium channels in alpha 1E knock-out mice. J Neurosci. 2000;20:8566–8571. doi: 10.1523/JNEUROSCI.20-23-08566.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodall AJ, Richards MA, Turner DJ, Fitzgerald EM. Growth factors differentially regulate neuronal Cav channels via ERK-dependent signalling. Cell Calcium. 2008;43:562–575. doi: 10.1016/j.ceca.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Wu HE, Gemes G, Zoga V, Kawano T, Hogan QH. Learned avoidance from noxious mechanical simulation but not threshold semmes weinstein filament stimulation after nerve injury in rats. J Pain. 2010;11:280–286. doi: 10.1016/j.jpain.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZZ, Chen SR, Pan HL. Signaling mechanisms of down-regulation of voltage-activated Ca2+ channels by transient receptor potential vanilloid type 1 stimulation with olvanil in primary sensory neurons. Neuroscience. 2006;141:407–419. doi: 10.1016/j.neuroscience.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Wu ZZ, Pan HL. High voltage-activated Ca(2+) channel currents in isolectin B(4)-positive and -negative small dorsal root ganglion neurons of rats. Neurosci Lett. 2004;368:96–101. doi: 10.1016/j.neulet.2004.06.067. [DOI] [PubMed] [Google Scholar]
- Yang L, Stephens GJ. Effects of neuropathy on high-voltage-activated Ca(2+) current in sensory neurones. Cell Calcium. 2009;46:248–256. doi: 10.1016/j.ceca.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ikeda SR. Modulation of Ca(2+)-channel currents by protein kinase C in adult rat sympathetic neurons. Journal of neurophysiology. 1994;72:1549–1560. doi: 10.1152/jn.1994.72.4.1549. [DOI] [PubMed] [Google Scholar]