

Abstract
There is pharmacological evidence that group II and III metabotropic glutamate receptors (mGluRs) function as activity-dependent autoreceptors, inhibiting transmission in supraspinal sites. These receptors are expressed by peripheral nociceptors. We investigated whether mGluRs function as activity-dependent autoreceptors inhibiting pain transmission to the rat CNS, particularly transient receptor potential vanilloid 1 (TRPV1)-induced activity. Blocking peripheral mGluR activity by intraplantar injection of antagonists LY341495 [(2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid] (LY) (20, 100 μm, group II/III), APICA [(RS)-1-amino-5-phosphonoindan-1-carboxylic acid] (100 μm, group II), or UBP1112 (α-methyl-3-methyl-4-phosphonophenylglycine) (30 μm, group III) increased capsaicin (CAP)-induced nociceptive behaviors and nociceptor activity. In contrast, group II agonist APDC [(2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate] (0.1 μm) or group III agonist l-(+)-2-amino-4-phosphonobutyric acid (l-AP-4) (10 μm) blocked the LY-induced increase. Ca2+ imaging in dorsal root ganglion (DRG) cells confirmed LY enhanced CAP-induced Ca2+ mobilization, which was blocked by APDC and l-AP-4. We hypothesized that excess glutamate (GLU) released by high intensity and/or prolonged stimulation endogenously activated group II/III, dampening nociceptor activation. In support of this, intraplantar GLU + LY produced heat hyperalgesia, and exogenous GLU + LY applied to nociceptors produced enhanced nociceptor activity and thermal sensitization. Intraplantar Formalin, known to elevate extracellular GLU, enhanced pain behaviors in the presence of LY. LY alone produced no pain behaviors, no change in nociceptor discharge rate or heat-evoked responses, and no change in cytosolic Ca2+ in DRG cells, demonstrating a lack of tonic inhibitory control. Group II/III mGluRs maintain an activity-dependent autoinhibition, capable of significantly reducing TRPV1-induced activity. They are endogenously activated after high-frequency and/or prolonged nociceptor stimulation, acting as built-in negative modulators of TRPV1 and nociceptor function, reducing pain transmission to the CNS.
Introduction
l-Glutamic acid hydrochloride (GLU) is the major excitatory transmitter in the nervous system. Much is known concerning GLU action at ionotropic glutamate receptors (iGluRs) (NMDA, AMPA, and KA), but we are at the early stages of understanding mechanisms of action of metabotropic GLU receptors (mGluRs). mGluRs are seven transmembrane domain, G-protein-coupled receptors (Pin and Duvoisin, 1995; Conn and Pin, 1997). The mGluR groups are classified by sequence homology, similarities in signal transduction mechanisms, and pharmacological profiles (Pin and Duvoisin, 1995; Conn and Pin, 1997). Activation of group I (mGluR1, mGluR5) results in neuronal excitation, but activation of groups II (mGluR2, mGluR3) and III (mGluR4, mGluR6, mGluR7, mGluR8) cause neuronal inhibition. Groups II/III are coupled to Gi/o-proteins and can lead to inhibition of adenylate cyclase, decreasing cAMP formation and cAMP-dependent protein kinase A (Schoepp and Conn, 1993).
Group II and III mGluRs are expressed by primary sensory neurons (Carlton et al., 2001a; Carlton and Hargett, 2007), and peripheral injection of a group II agonist inhibits peripheral nociceptor activity (Du et al., 2008), in particular transient receptor potential vanilloid 1 (TRPV1)-induced pain behaviors and TRPV1-induced excitation and sensitization of peripheral nociceptors (Carlton et al., 2009a). Thus, exogenous activation of peripheral mGluRs modifies pain transmission. However, in the CNS, it has been demonstrated that group II and III mGluRs can modulate neurotransmission through activity-dependent autoinhibition/depression. In these instances, the mGluRs remain “silent” unless/until high-frequency stimulation occurs. The enhanced neuronal activity releases excess GLU and only then, in this GLU-enriched environment, are the mGluRs endogenously activated. This process has been described in the hippocampus (Ugolini and Bordi, 1995; Macek et al., 1996; Scanziani et al., 1997; Kilbride et al., 1998; Kew et al., 2001) and locus ceruleus (Dube and Marshall, 2000) and is important in regulation of synaptic transmission and plasticity. It is unknown whether mGluR activity-dependent autoinhibition modulates primary afferent nociceptor function.
Anatomical evidence indicates that group II (Carlton et al., 2009a) and III (our unpublished data) mGluRs colocalize with TRPV1 receptors. TRPV1 is a Ca2+-permeable, nonselective cation channel involved in nociceptive processing, integrating multiple pain-producing stimuli (Tominaga et al., 1998). TRPV1 activation provides a one–two punch in the pain experience because activation on sensory neurons causes a burning pain and also results in GLU and neuropeptide release, causing neurogenic inflammation (Szolcsányi, 1996). Because of its critical role in nociception (Caterina and Julius, 2001), TRPV1 is an important pharmaceutical target for pain control (Premkumar and Sikand, 2008; Wong and Gavva, 2009). Because TRPV1 receptors contribute to inflammatory, neuropathic, cancer, migraine, and burn injury pain (Caterina et al., 2000; Davis et al., 2000; Akerman et al., 2004; Bölcskei et al., 2005; Ghilardi et al., 2005; Joshi et al., 2009), it is critical that the mechanisms underlying endogenous modulation of this receptor be elucidated.
The present study provides in vivo and in vitro evidence for group II/III mGluR activity-dependent inhibition of TRPV1-expressing nociceptors. Some of the physiological conditions under which these mGluRs are endogenously activated are defined.
Materials and Methods
Animal use and care
All experiments were approved by the University Animal Care and Use Committee at the University of Texas Medical Branch (Galveston, TX) and followed the International Association for the Study of Pain guidelines for the ethical care and use of laboratory animals (Zimmermann, 1983). Adult male Sprague Dawley rats (250–300 g unless otherwise noted; Harlan) were used in all experiments. Steps were taken to minimize both the number of animals used and their discomfort.
Drug preparations
A 10% capsaicin (CAP) (Sigma-Aldrich) stock solution was made as described previously (Carlton et al., 2009a). The stock solution was diluted with CAP vehicle (7% Tween 80 in saline) to make working dilutions of CAP (0.05%). The following drugs were dissolved in 1N NaOH and made up as a 100 mm stock solution: LY341495 [(2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid] (LY) (Tocris Biosciences), which antagonizes both groups II and III mGluRs (Kingston et al.,1998); (RS)-1-amino-5-phosphonoindan-1-carboxylic acid (APICA) (Tocris Bioscience), a selective group II mGluR antagonist; UBP1112 (α-methyl-3-methyl-4-phosphonophenylglycine (Tocris Bioscience), a selective group III mGluR antagonist; (2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate (APDC) (Tocris Bioscience), a selective mGluR2 agonist; l-(+)-2-amino-4-phosphonobutyric acid (l-AP-4) (Tocris Bioscience), a selective group III agonist; dizocilpine (MK801) (Tocris Bioscience), an NMDA antagonist; and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (Tocris Bioscience), a non-NMDA antagonist. GLU (RBI/Sigma) was dissolved in PBS, pH 7.4, and made up as a 100 mm stock solution. The 2% Formalin (FM) was prepared from a 10% FM stock solution. All stock solutions were diluted with PBS (vehicle) unless otherwise noted and the pH was readjusted to 7.4.
Drug injections
Intraplantar injection of drugs was performed using a 28-gauge needle attached to a 50 μl Hamilton syringe with polyethylene 20 tubing. For measurements of spontaneous behaviors (flinching, lifting/licking), rats were not anesthetized during drug injections. For measurement of evoked behavior such as paw withdrawal latency (PWL), rats were briefly (2–3 min) anesthetized with 3% isoflurane during the drug injection. The needle punctured the plantar skin distal to the pads and was guided into the subcutaneous space near the center of the hindpaw where the drug(s) were injected. Drugs must diffuse through the plantar connective tissue, dermis, and epidermis to reach the C-fiber nociceptor terminals; thus, drug concentrations are diluted 10- to 100-fold by the time they reach their target. Each rat was used only once, and the experimenter was unaware of which drug was injected into each animal.
Habituation for CAP and FM testing
Rats were housed in groups of three in plastic cages with soft bedding under a reversed 12 h light/dark cycle. All behavioral testing was done between 8:00 A.M. and 5:00 P.M., thus during the animals dark cycle. Care was taken to keep the animals in low lighting conditions during transport, habituation, and testing. After arrival at the animal care facility, they were acclimated for at least 3 d before any behavioral testing was initiated. Rats were habituated for CAP or FM testing by placing them on a wire-screen platform in Plexiglas cages (8 × 8 × 18 cm) for 1 h. Each rat was habituated two times before being placed in an experimental group.
Habituation for thermal testing
To determine PWL to a heat stimulus, rats were habituated in Plexiglas cages (8 × 8 × 18 cm) on a glass plate that was ¼ inch thick and maintained at 23°C for 1 h sessions on each of 5 d. Each hindpaw was tested twice with a 10 min interval during habituation to better acclimate them to the testing paradigm. The method described by Hargreaves et al. (1988) was used; however, the radiant heat source was replaced with a solid-state laser system. The laser system (custom-made in house) consisted of a microprocessor-controlled 980 nm (near infrared) continuous-wave, solid-state laser (4 W). The laser spot size (measured with Kentek laser alignment paper) was 2 mm in diameter. A PWL of ∼9 s was obtained with parameters of 4.35 amps and 3.5 W. Measured at the glass surface, the laser reached 40°C within 0.5 s and a maximum of 52°C in 8 s. The cutoff time was 15 s. A red (670 nm) sighting beam was coupled to the invisible laser to allow positioning of the laser on the plantar surface of the hindpaw. The sighting beam was controlled independently from the laser so it did not interfere with the thermal testing. On the day of testing, animals remained on the glass for at least 20 min before testing was initiated.
Electrophysiological recordings
In vitro skin–nerve preparation.
Male Sprague Dawley rats (250–300 g) were killed with an overdose of CO2. The glabrous skin from the ankle to the tips of the toes was dissected from the hindpaw. The medial and lateral plantar nerves were dissected free and kept intact with the glabrous skin. This preparation was used to record from C-mechanoheat (CMH)-sensitive fibers innervating the glabrous skin in a skin–nerve preparation described previously (Carlton et al., 2001b). Conduction velocity of each fiber was determined from the latency of the action potential and the distance from the stimulation electrode to the recording site. Based on measurements (n = 3) made using our in vitro chamber (34°C), the average conduction velocity of C fibers was ≤1.2 m/s, and this agreed with previously published reports in rats (Koltzenburg et al., 1992; Kress et al., 1992).
Thermal and chemical testing procedures.
Only units responding to mechanical probing of the glabrous skin with a blunt glass rod with a clearly defined receptive field were studied in detail. For thermal stimulation, radiant heat was applied to each receptive field by a feedback-controlled lamp placed beneath the organ bath. The beam was focused through the bottom of the bath onto the epidermal surface of the skin. A thermocouple was placed into the corium above the light beam to measure intracutaneous temperature. A standard heat ramp starting from an adapting temperature of 34°C and rising to 51°C in 10 s was applied to each unit from the epidermal side and 51°C on the epidermal side was equivalent to 47°C on the corium side. To investigate unit responses to chemicals, a small ring (5 mm diameter) was placed over the receptive field of each unit. The synthetic interstitial fluid (SIF) in the ring was replaced with SIF containing drugs (buffered to pH 7.40 ± 0.05). As with the in vivo plantar injections, drugs must diffuse through the plantar connective tissue, dermis, and epidermis to reach the C-fiber nerve terminals; thus, drug concentrations are diluted 10- to 100-fold by the time they reach their target. As described previously (Reeh, 1986), the majority of units from normal skin had no spontaneous activity, but the mechanical search stimulus (probing with a glass rod) or thermal stimulus sometimes resulted in ongoing activity of a very low frequency in some units. This background activity was subtracted from the activity evoked by drug or thermal stimulation.
Ca2+ imaging of dissociated DRG cells
Cell culture.
Naive Sprague Dawley rats (80–120 g) were killed with CO2. The ipsilateral and contralateral L4–L6 DRGs were removed, placed in ice-cold oxygenated dissecting solution [in mm: 130 NaCl, 5 KCl, 2 KH2PO4, 6 MgCl2, 1.5 CaCl2, 10 glucose, 10 HEPES, pH adjusted to 7.2 with NaOH base (osmolality adjusted with sucrose to 300 mOsm) (Xu and Huang, 2002)], and minced into smaller pieces. The DRG fragments were then digested in 1 mg/ml collagenase D (Roche Diagnostics) and 0.5 mg/ml trypsin (Sigma-Aldrich) in a 37°C water bath for 1 h and then washed three times with extracellular solution (ES) containing the same components listed for the dissecting solution with the following adjustments: 1 mm MgCl2 and 2.5 mm CaCl2. Fire-polished glass pipettes were used to dissociate the DRG cells mechanically. The dissociated cells (0.1 ml) were plated on poly-lysine-coated glass coverslips inside 35 mm culture dishes. The ES (2 ml) was added to the wells 15 min after plating the cells, and the wells were maintained for 1 h at room temperature, before adding fura-2. DRGs from three to four rats were used for each treatment paradigm.
Cytosolic Ca2+ imaging.
The DRG cells were incubated in 3 μm fura-2 AM (Invitrogen) in ES containing 0.2% pluronic acid F-127 at room temperature for 1 h, washed with ES, and left in this solution at room temperature in the dark for 1 h so the cells could stabilize. Ratiometric Ca2+ imaging was performed using an inverted fluorescent Nikon microscope equipped with a 340 and 380 nm excitation filter changer (Sutter Instruments), a 510 nm long-pass emission filter, and a 12-bit digital monochrome cooled CCD camera (Roper Scientific Coolsnap HQ) controlled by Metafluor software version 6.5 (Molecular Devices). Fluorescent images and ratios were acquired every 5 s and processed using MetaMorph software (Molecular Devices). Cells were considered responders if their F340/F380 ratio increased by >20% during the drug application. All experiments were performed at room temperature.
Data analysis
Data were expressed as means ± SEM unless otherwise noted and evaluated using SigmaStat (Systat Software). Parametric (one-way ANOVA followed by Student–Newman–Keuls test) or nonparametric (Kruskal–Wallis one-way ANOVA on ranks followed by a Tukey's test; Mann–Whitney U rank-sum test) analyses were used when appropriate, depending on the passing of a normality test. When time and group were factors, a two-way repeated-measures ANOVA followed by a Tukey's test was used. p < 0.05 was considered significant.
Results
Evidence for activity-dependent inhibition of TRPV1 receptors
CAP-induced behaviors are enhanced during blockade of group II/III mGluRs
If there is endogenous group II/III mGluR activity dampening the response to a noxious stimulus, this could be discerned by blocking the activity of the mGluRs during a noxious event. An enhancement of nociceptive behaviors would result if this type of modulation was occurring. To determine whether group II/III mGluRs endogenously modulated CAP-induced nociceptive behaviors, rats received an intraplantar injection of one of the following drug combinations with a 30 min interval between the first and second drug injection: vehicle (30 μl), 0.05% CAP (20 μl, n = 8); 100 μm LY (30 μl), 0.05% CAP (20 μl, n = 9). To show that the effect of LY was local and not systemic, a group of rats received 100 μm LY (30 μl) in one hindpaw, vehicle (30 μl) in the contralateral paw, followed 30 min later by 0.05% CAP (20 μl, n = 5) in the contralateral paw. To show specificity of drug action, either APCD or l-AP-4, a group II- and group III-specific agonist, respectively, was coinjected with the LY + CAP. Using the paradigm described above, rats received an intraplantar injection of one of the following drug combinations with a 30 min interval between the first and second drug injection: 100 μm LY + 0.1 μm APDC (30 μl), 0.05% CAP (20 μl, n = 9); 100 μm LY + 10.0 μm l-AP-4 (30 μl), 0.05% CAP (20 μl, n = 8). All drugs were delivered into the same place in the chosen hindpaw, and all injected volumes were kept constant within a paradigm. The number of flinches and the time spent lifting/licking the injected hindpaw were observed.
Intraplantar injection of 0.05% CAP alone produced flinching (Fig. 1A,B) and lifting/licking (Fig. 1C,D) behaviors that continued for 30–40 min. When 100 μm LY (a concentration that would antagonize both group II and III mGluRs) was coinjected with the CAP, a significant enhancement of both CAP-induced behaviors resulted. The effect of LY was on local receptors and not systemic because injection of CAP in one hindpaw and LY in the contralateral hindpaw resulted in behavior that was no different from CAP alone. Evidence that 100 μm LY had an effect at both group II and group III mGluRs was demonstrated by the fact that injection of LY + APDC (group II agonist, Fig. 1E,F) or LY + l-AP-4 (group III agonist, Fig. 1G,H) significantly attenuated the LY-induced enhancement of CAP behaviors. In the absence of LY, activation of group II or group III mGluRs by injection of the agonists APDC or l-AP-4, respectively, had a profound inhibitory effect on CAP-induced behaviors (Fig. 1E–H), significantly more so than when LY was present, suggesting that the presence of LY was counteracting or interfering with the inhibitory action of these agonists.
Figure 1.

Involvement of group II/III mGluRs in activity-dependent inhibition. Intraplantar 0.05% CAP alone produced nociceptive behaviors such as flinching (A, total flinches in 30 min; B, time course of behavior) and lift/licking behavior (C, total time spent lift/licking in 30 min; D, time course of behavior). Injection of 100 μm LY + CAP resulted in a significant increase in both behaviors. CAP in the ipsilateral (ipsil) hindpaw and LY in contralateral (contra) hindpaw resulted in behavior no different from CAP alone (*p < 0.005 compared with all other groups, one-way ANOVA followed by a Student–Newman–Keuls test). The LY enhancement of CAP-induced flinching (FL) and lifting/licking (L/L) was blocked with the addition of the group II agonist APDC (0.1 μm, E, F) or the group III agonist l-AP-4 (10.0 μm, G, H). Data in E–H are normalized to responses from the CAP alone group (dashed line). Injection of CAP + APDC or CAP + l-AP-4 in the absence of LY results in an even greater inhibition of nociceptive behaviors (*p < 0.001 compared with the 3 other groups; +p < 0.001 compared with the APDC, CAP group or the l-AP-4,CAP group; one-way ANOVA, followed by a Student–Newman–Keuls test).
CAP-induced nociceptor activity is enhanced during blockade of group II/III mGluRs
To confirm that the behavioral results reflected what was happening at the single-fiber level, we repeated experiments similar to those above while recording from nociceptors using our in vitro glabrous skin–nerve preparation. Receptive fields of nociceptors were exposed to one of the following for 2 min and activity was recorded: 0.05% CAP; CAP + 100 μm LY, CAP + APICA (selective group II antagonist), CAP + UBP1112 (selective group III antagonist), CAP + LY + 0.1 μm APDC (group II agonist), or CAP + LY + 10 μm l-AP-4 (group III agonist).
We identified and recorded from CMH units in 43 normal rats. The mean background activity for all units was 0.04 ± 0.01 impulses per second (imp/s). Their mean conduction velocity was 0.84 ± 0.18 m/s. Responses of units over time are shown in Figure 2A–E, either as individual units (first column) or as population data (second column). The mean population responses during the 2 min drug application are shown in Figure 2F. Compared with mean background activity, application of CAP to the receptive field of nociceptors resulted in an increase in mean discharge rate (0.33 ± 0.16 imp/s, Fig. 2A,F). When group II/III mGluRs were blocked by 100 μm LY, the discharge rate in response to CAP almost doubled (0.62 ± 0.2 imp/s), showing an 88% increase compared with CAP alone (Fig. 2B,F). When the group II-selective antagonist APICA (100 μm) or the group III-selective antagonist UPB1112 (30 μm) was used, these also increased the CAP-induced discharge rates and resulted in a 127 and 160% increase, respectively, compared with CAP alone (Fig. 2F). The LY-induced enhancement of the CAP activity was attenuated when either 0.1 μm APDC (Fig. 2C,F) or 10 μm l-AP-4 (Fig. 2D,F) was added to the receptive field. Importantly, LY applied alone (1 or 100 μm) had no effect on unit activity (Fig. 2E, inset in F).
Figure 2.

Single-unit analyses confirm group II/III mGluR activity-dependent inhibition of nociceptors. Unfiltered traces from representative nociceptive units. Each trace represents activity of a different unit. The standard paradigm includes testing the heat response (H1) of the unit (the standard heat ramp is shown in the inset), followed by a 5 min wait. Then the drug(s) of interest is place in the receptive field for 2 min, followed by a second heat test (H2). The responses (mean ± SD) of the units during the 2 min drug application are shown in the right column. Application of 0.05% CAP resulted in a robust activity (A). When CAP was applied in the presence of 100 μm LY, the activity was greatly increased in magnitude and duration, suggesting release from the autoinhibition (B). If the group II agonist 0.1 μm APDC (C) or group III agonist 10 μm l-AP-4 (D) was applied, the LY effects were reduced. LY at 100 μm had no effect on its own (E). Population responses of nociceptors recorded after exposure to LY alone, CAP alone, CAP + LY, CAP + APICA (100 μm), CAP + UBP (30 μm), CAP + LY + APDC, or CAP + LY + l-AP-4 (F). Dashed line represents the CAP response normalized to 100%. LY alone (1 or 100 μm) did not excite nociceptors above background (BK, inset) (*p < 0.05, significantly different from CAP alone, Kruskal–Wallis one-way ANOVA on ranks followed by Tukey's test).
CAP-induced Ca2+activity in DRG neurons is enhanced after blockade of group II/III mGluRs
To confirm that primary sensory cell bodies are under a similar control, we repeated experiments similar to those above using acutely dissociated DRG cells and Ca2+ imaging. A 2 min baseline of Ca2+ spiking was obtained, and then cells were exposed for 2 min to one of the following solutions: 0.1 μm CAP; 1, 20 or 100 μm LY + 0.1 μm CAP; 0.1 μm ADPC + 100 μm LY + 0.1 μm CAP; or 10 μm l-AP-4 + 100 μm LY + 0.1 μm CAP.
Compared with baseline, 0.1 μm CAP resulted in significant mobilization of Ca2+ in DRG neurons (Fig. 3B, compare D,E). Addition of 1 μm LY had no effect on the CAP response, 20 μm LY moderately increased the Ca2+ activity (Fig. 3A). However, 100 μm LY produced the most robust effect on CAP-induced Ca2+ mobilization (Fig. 3A–C, compare E,H). Figure 3, F and I, demonstrates the viability of the cells in each well by addition of 50 mm KCl. Application of LY + CAP produced a peak change in ratio that was significantly increased compared with CAP alone (Fig. 3C). Addition of 0.1 μm APDC or 10 μm l-AP-4 prevented the increased Ca2+ activity.
Figure 3.
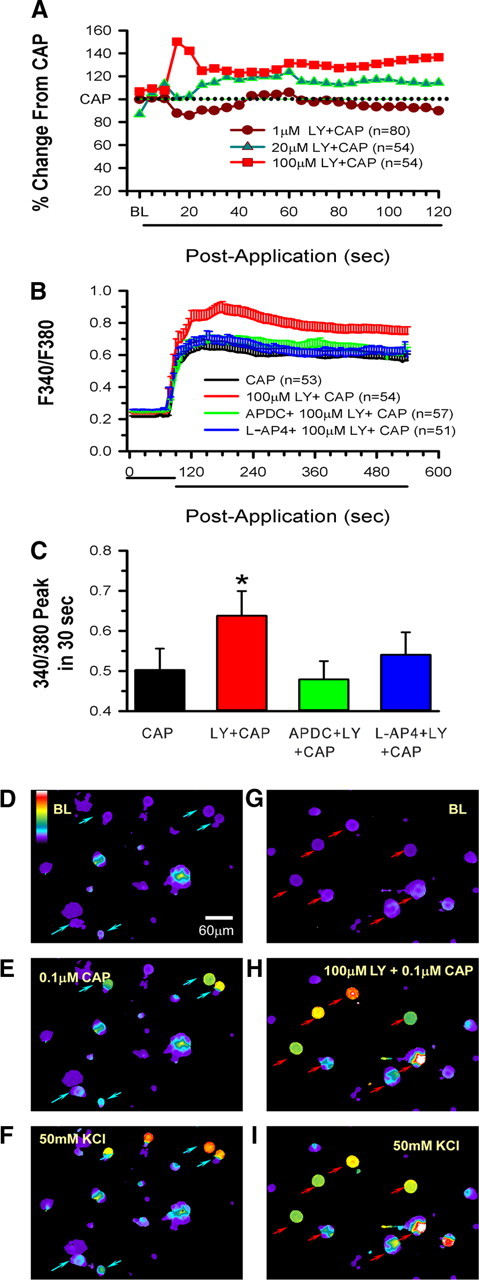
Group II/III mGluR activity-dependent inhibitory mechanisms function in acutely dissociated DRG cells. A, Dose-dependent enhancement by LY of CAP-induced Ca2+ mobilization. LY + CAP responses are normalized to the CAP alone response, which is represented by the dotted line. B, Application of 0.1 μm CAP to acutely dissociated DRG cells increases cytosolic Ca2+ that is further enhanced by 100 μm LY. If 0.1 μm APDC or 10 μm l-AP-4 is added to the CAP + LY, the LY effect is eliminated (n indicates number of cells responding). C, Peak changes in fluorescence occurring within 30 s after addition of the agents to the wells. For ease of presentation, means are plotted in C, but medians and 25th and 75th percentiles, respectively, are as follows: CAP, 0.030, 0.22, 0.77; LY + CAP, 0.49, 0.30, 0.86; APDC + LY + CAP, 0.30, 0.24, 0.67; l-AP-4 + LY + CAP, 0.34, 0.28, 0.65 (*p = 0.015, significantly different from CAP alone; the other groups are not different from CAP alone, Kruskal–Wallis one-way ANOVA on ranks followed by Dunn's test). D–I, Series of panels showing representative responses of individual DRG cells after various drug treatments (D–F are the same wells; G–I are the same wells). D and G represent baseline conditions. Addition of 0.1 μm CAP results in activation of some cells (blue arrows in E), but a more robust response is seen in wells in which CAP is applied with 100 μm LY (red arrows in H). Cell viability is tested in F and I with addition of 50 mm KCl. The color bar in D indicates magnitude of the Ca2+ response ranging from lowest (purple) to highest (white) response. BL, Baseline.
Thus, at the behavioral, nociceptor single-fiber and DRG cell levels, the data provide strong evidence of activity-dependent modulation of TRPV1 receptors by group II/III mGluRs.
Evidence that excess GLU plays a role in activity-dependent inhibition by group II/III mGluRs
We predicted that excess GLU in the microenvironment was necessary to involve the mGluRs in activity-dependent inhibition. This prediction was borne out in experiments in which either exogenous or endogenous GLU was made available.
In the presence of excess exogenous GLU, enhanced nociceptive behaviors occur during blockade of group II/III mGluRs
To determine whether excess extracellular GLU played a role in recruiting group II/III mGluRs, rats received an intraplantar injection of one of the following drug combinations with each drug given in a 10 μl volume with a 10 min interval between the first and second drug injection: 20 μm LY, 0.3 mm GLU (n = 11); vehicle, 0.3 mm GLU (n = 11); vehicle, vehicle (n = 8); 20 μm LY, vehicle (n = 6). All drugs were delivered into the same place in the hindpaw and sensitivity to heat (PWL) was measured. After baseline PWLs were obtained, each hindpaw was tested once every 10 min for 1 h after drug injections.
Intraplantar injection of either 0.3 mm GLU or 20 μm LY alone did not produce any change in thermal sensitivity because the PWLs of these groups were not different compared with baseline or the vehicle group (Fig. 4). However, coinjection of 0.3 mm GLU + 20 μm LY resulted in robust thermal hyperalgesia evidenced by a significant decrease in PWL compared with preinjection baseline and compared with all other groups after injection. This enhanced thermal sensitivity was still present 1 h after injection.
Figure 4.

Exogenous GLU evokes activity-dependent inhibition of behavioral responses to heat. Intraplantar injection of 0.3 mm GLU alone, 20 μm LY alone, or vehicle does not produce any change in hindpaw thermal sensitivity. If 0.3 mm GLU is injected in the presence of 20 μm LY, there is a significant and prolonged effect on PWL (*p < 0.001, compared with 3 other groups; +p < 0.05, compared with baseline, two-way repeated-measures ANOVA followed by Tukey's test).
In the presence of excess exogenous GLU, enhanced nociceptor activity occurs during blockade of group II/III mGluRs
To determine whether excess extracellular GLU played a role in recruiting group II/III mGluRs, receptive fields of nociceptors were exposed for 2 min to the following: 1, 10, 30, or 100 μm LY in the presence of 1 mm GLU; 0.3 mm GLU; 1.0 mm GLU; 0.3 mm GLU + 100 μm LY; 1.0 mm GLU + 100 μm LY. Background activity as well as responses to thermal stimulation (threshold and discharge rate) were recorded.
The dose–response study showed that LY produced a dose-dependent increase in nociceptor activity in the presence of 1 mm GLU with 100 μm LY, causing a significant increase compared with 1 μm LY (Fig. 5A). To understand the magnitude of the inhibitory effect of mGluRs on GLU-induced excitation, GLU alone was applied to receptive fields of nociceptors. Application of 0.3 mm GLU resulted in excitation, raising the mean discharge rate from 0.03 ± 0.03 to 0.25 ± 0.10 imp/s (Fig. 5B). When 1.0 mm GLU was applied to the receptive fields, the mean response was not further increased but slightly depressed to 0.16 ± 0.09 imp/s. However, in the presence of 100 μm LY, application of 0.3 mm GLU resulted in an increase in discharge rate to 0.41 ± 0.1 imp/s, a 64% increase from that produced by 0.3 mm GLU alone. In the presence of 100 μm LY, application of 1.0 mm GLU resulted in a significant increase (475%) in discharge rate (0.92 ± 0.17 imp/s) compared with 1.0 mm GLU alone (Fig. 5B). The data provide evidence that high concentrations of exogenous GLU promote activation of group II/III, which in turn reduces nociceptor firing. The presence of the antagonist (LY) prevents group II/III activation, and the nociceptors show significantly enhanced activity. We assumed that iGluRs were involved, initially exciting the nociceptors, which resulted in enhanced GLU release. Application of MK801 (1, 10, or 100 μm), an NMDAR antagonist, counteracted the GLU effect in a dose-dependent manner (Fig. 6A) and, in doing so, reduced the LY effect. A non-NMDAR antagonist, CNQX (10 or 100 μm), also reduced the LY effect (but this did not reach significance, Fig. 6B). These data confirm that activation of iGluRs contribute to the GLU-induced nociceptor activity that the mGluRs modulate.
Figure 5.

Exogenous GLU evokes activity-dependent inhibition of unit responses to heat. A, A dose–response study demonstrates that blocking mGluRs with increasing doses of LY (1, 10, 30, 100 μm) significantly enhanced 1 mm GLU-induced nociceptor discharge rate. B, Application of 0.3 or 1.0 mm GLU to the receptive field results in a slight increase in discharge rate. Addition of 100 μm LY + 0.3 mm GLU increases activity 64% over predrug levels. However, 100 μm LY + 1 mm GLU results in a fourfold increase (*p < 0.05, significantly different from predrug levels; +p < 0.05, significantly different from 1 mm GLU alone, two-way ANOVA followed by Tukey's test). C, Compared with vehicle, application of 100 μm LY to the receptive fields of nociceptors does not change heat-induced discharge rate (inset in C) or threshold to firing (inset in D). Application of 1.0 mm GLU alone produces a response no different from vehicle (Veh). However, blockade of group II/III mGluRs by 100 μm LY significantly enhances the heat-induced discharge rate (C) and lowers the threshold to activation (D) (*p < 0.05 significantly different compared with GLU alone, Mann–Whitney rank sum test).
Figure 6.

Evidence that iGluRs contribute to the excitatory response. A, Application of the NMDAR antagonist MK801 (1, 10, 100 μm), counteracted the GLU-induced excitation, curtailing GLU release. This reduced the probability of mGluR activation and in turn reduced the inhibitory effect of LY. B, A non-NMDAR antagonist CNQX (10 or 100 μm) also reduced the GLU-induced excitation and thus the LY effect, but this did not reach significance (*p < 0.05, Kruskal–Wallis one-way ANOVA on ranks followed by Tukey's test).
In the presence of excess exogenous GLU, enhanced thermal sensitivity occurs during blockade of group II/III mGluRs
In addition to enhancing GLU-induced unit activity, the presence of LY enhanced GLU-induced thermal responses. Exposure to LY alone did not produce any change in the discharge rate or threshold to activation to a heat stimulus (Fig. 5C,D, insets). In contrast, nociceptors exposed to 1.0 mm GLU + 100 μm LY had increased discharge rates (p < 0.05) and lowered thresholds (p < 0.05) compared with 1.0 mm GLU alone (Fig. 5D,E). The discharge rate increased from 2.82 ± 0.55 to 4.38 ± 0.85 after the GLU + LY mixture (Fig. 5C). In the presence of 1.0 mm GLU, the threshold to heat activation was 41.34 ± 0.49°C; in the presence of 1.0 mm GLU + 100 μm LY, the threshold was significantly decreased to 39.97 ± 0.42°C (Fig. 5D).
In the presence of excess endogenous GLU, enhanced nociceptive behaviors occur during blockade of group II/III mGluRs
One way to induce release of endogenous GLU is to inject FM into the hindpaw (Omote et al., 1998). To determine whether endogenous GLU recruited group II/III mGluRs and thus modulated FM-induced nociceptive behaviors, rats received an intraplantar injection of one of the following drug combinations with a 30 min interval between the first and second drug injection: vehicle (30 μl), 2% FM (20 μl, n = 6); 100 μm LY (30 μl), 2% FM (20 μl, n = 6); 100 μm LY (30 μl), mixture of 0.1 μm APDC + 2% FM (20 μl, n = 6); vehicle (30 μl), mixture of 0.1 μm APDC + 2% FM (20 μl, n = 6). All drugs were delivered into the same place in the hindpaw. The number of flinches and the time spent lifting/licking the injected hindpaw were observed.
Injection of 2% FM produced lifting/licking behavior with a classic biphasic response with phase 1 lasting 10 min and phase 2 lasting ∼10–40 min (data not shown). Injection of FM + 100 μm LY resulted in significantly elevated lifting/licking behavior in phase 1 compared with FM alone (Fig. 7). Phase 2 was not affected (data not shown). In the presence of APDC, the LY enhancement of FM behavior was reduced. In the absence of LY, activation of group II mGluRs by injection of APDC had a profound inhibitory effect on FM-induced behaviors (Fig. 7), more so than when LY was present, suggesting again that the presence of LY was counteracting or interfering with the inhibitory action of the agonist. Thus, the data infer that release of endogenous GLU played a pivotal role in engaging group II/III inhibition, which dampened FM-induced pain behaviors.
Figure 7.

Endogenous release of GLU evokes group II/III mGluR activity-dependent inhibition. Intraplantar injection of 2% FM produces lifting/licking (L/L) behavior in phase 1 (baseline FM-induced behavior is indicated by the dashed line). Addition of 100 μm LY + FM significantly enhances phase 1 pain behaviors. This LY-induced enhancement is blocked by addition of the group II agonist 0.1 μm APDC. FM + APDC in the absence of LY results in an even greater inhibition of nociceptive behaviors (*p < 0.001, significantly different compared with all other groups, Kruskal–Wallis one-way ANOVA followed by a Student–Newman–Keuls test).
There is no evidence that peripheral nociceptors are tonically inhibited by group II/III mGluRs
There are several regions in the CNS in which group II/III mGluRs maintain tonic inhibitory control over neuronal function (Grassi et al., 2002; Xi et al., 2002; Acuna-Goycolea et al., 2004; Linden et al., 2005). Hence, we wanted to determine whether this was true for primary afferents. If Group II/III mGluRs were continually activated (producing neuronal depression), injection of LY alone would be expected to relieve this inhibition.
Nociceptive behaviors do not change after blockade of group II/III mGluRs
To determine whether group II/III mGluRs maintained tonic inhibition of peripheral nociception in the absence of excess neuronal activation, naive rats received 20 μl intraplantar injections of 20 μm LY, 100 μm LY, or vehicle (n = 5–6 per group). Two spontaneous behaviors were monitored after injection: the number of flinches and the time spent lifting/licking the injected hindpaw. In separate groups of rats, PWL to heat was measured over a 60 min period after injection (n = 3–4 per group).
There was no change in PWL to a heat stimulus after LY alone (Fig. 8A). Nocifensive behaviors such as flinching or lifting/licking were not generated after LY alone (Fig. 8B,C). The flinching and lifting/licking that did occur appeared to be related more to the needle stick and possibly drug-induced irritation because it occurred immediately after the injection and quickly dissipated within 60 s.
Figure 8.

Group II/III mGluRs do not maintain tonic inhibitory control over nociceptors. Compared with vehicle, intraplantar LY at 20 or 100 μm does not produce any changes in sensitivity to heat (A) or produce any spontaneous pain behaviors such as flinching (B) or lifting/licking (C) (p = 0.459, two-way repeated-measures ANOVA). Application of 1 μm (D) or 100 μm (E) LY to the receptive field of nociceptors for 2 min does not change background activity (BK), heat threshold, or heat discharge frequency. (Kruskal–Wallis one-way ANOVA on ranks followed by Tukey's test). F, Addition of 100 μm LY had no effect on the Ca2+ activity of dissociated DRG cells; however, responses to 50 mm KCl confirmed that the cells were viable.
Nociceptor activity does not change after blockade of group II/III mGluRs
To determine whether group II/III mGluRs maintained a tonic inhibition of peripheral nociceptors, in naive rats the receptive fields of nociceptors were exposed to either 1 or 100 μm LY for 2 min, and changes in background activity and thermal responses (threshold and discharge rate) were recorded.
Exposure of nociceptors to either concentration had no effect on mean background activity (0.05 ± 0.02 compared with 0.08 ± 0.03 imp/s after drug for 1 μm LY; 0.048 ± 0.01 compared with 0.046 ± 0.01 imp/s after drug for 100 μm LY) (Fig. 8D,E). Responses to noxious thermal stimulation were not changed by LY alone because the mean heat threshold before 1 μm LY was 40.02 ± 0.84°C compared with 40.80 ± 0.67°C after LY; heat threshold before 100 μm LY was 42.88 ± 0.63°C compared with 42.41 ± 0.54°C after LY. The mean frequency of firing to a heat stimulus was also unchanged with 2.63 ± 0.63 imp/s before drug compared with 2.23 ± 0.32 imp/s after 1 μm LY and 1.52 ± 0.29 imp/s before drug compared with 1.78 ± 0.40 imp/s after 100 μm LY (Fig. 8D,E).
DRG activity does not change after blockade of group II/III mGluRs
To determine whether group II/III mGluRs tonically inhibited DRG cells, a 2 min baseline of Ca2+ spiking was recorded and then cells were exposed to 100 μm LY (2 min) followed by exposure to 50 mm KCl (to ensure cell viability) while recording effects on cytosolic Ca2+.
Application of 100 μm LY did not produce any change in baseline Ca2+ mobilization in dissociated DRG cells (Fig. 8F). However, application of 50 mm KCl resulted in activation of most cells in the dish, indicating cell viability.
Thus, at the behavioral, single-fiber, and DRG cellular levels, there was no evidence of tonic inhibitory control of nociceptors by group II/III mGluRs.
Discussion
We provide several lines of evidence that group II/III mGluRs exert a use-dependent modification of nociceptor transmission. These receptors do not influence nociceptive primary afferents when at rest (in the absence of stimulation), and they do not modulate responses to brief activation (i.e., 10 s heat stimulus). Their influence is only apparent after higher-frequency and/or prolonged stimulation (as occurs in response to algogenic substances).
Group II/III mGluRs are built-in negative modulators of peripheral nociceptor activity
We have shown that group II mGluR2s colocalize with TRPV1 (Carlton et al., 2009a), so it was not surprising to see an interaction between these receptor families at the behavioral, single-fiber, and cellular levels. Groups II and III are colocalized in DRG (Carlton and Hargett, 2007), so there is a high probability of overlap between the TRPV1-, group II-, and group III-expressing DRG populations, and our preliminary data confirm colocalization of group III with TRPV1 (our unpublished data).
We showed previously that a group II agonist can reduce peripheral sensitization of nociceptors, but the presence of endogenous activity-dependent inhibition evoked by group II/III was not explored. Endogenous, negative modulation of TRPV1 is undoubtedly important given its critical role in pain transmission (Caterina and Julius, 2001). If group II/III activation is prevented, prolonged enhancement of TRPV1 function occurs. For example, blockade of group II/III results in exaggerated and prolonged CAP-induced pain (involving TRPV1 activation). A similar phenomenon is observed at the single-fiber level in which enhanced nociceptor activity persists for many minutes after the CAP + LY are removed (Fig. 2). Use of group II- and III-selective antagonists (APICA and UBP1112, respectively) confirms modulation of CAP responses by both subtypes. Thus, without the activity-dependent modulation of group II/III mGluRs, the TRPV1-induced peripheral sensitization is prolonged and the nociceptive signal transmitted to the cord (or brainstem through the trigeminal ganglion) is amplified (Medvedeva et al., 2008). These augmented peripheral signals would impact dorsal horn neurons, most likely promoting central sensitization.
LY is a broad-spectrum mGluR antagonist when used at concentrations from 1.0 to 100 μm, with reported binding to groups I–III mGluRs (Fitzjohn et al., 1998; Kingston et al., 1998; Johnson et al., 1999; Wright et al., 2000). The potency order is mGluR3 ≥ mGluR2 > mGluR8 > mGluR7 ≫ mGluR1 = mGluR5 > mGluR4 for 0.1–100 μm LY (Kingston et al., 1998). Unlike group II/III, group I activation enhances neuronal excitation. Thus, blocking activation with LY would cause inhibition, not enhancement, of neuronal responses. Used at 100 μm, LY does not bind to any iGluRs (Kingston et al., 1998). Thus, in the present study, the LY effects are most likely attributable to blockade of group II/III mGluRs.
Tonic versus activity-dependent inhibition by mGluRs
We found no evidence for tonic inhibition by mGluRs in the periphery, but there are reports that groups II/III maintain tonic inhibitory tone in several CNS regions. For example, LY alone significantly increased c-Fos expression in cortex, hippocampus, amygdala, and other subcortical nuclei, suggesting tonic inhibition by group II/III mGluRs (Linden et al., 2005). Perfusion of LY in nucleus accumbens produced a dose-dependent increase in extracellular GLU that was reversed by coperfusion of APDC (Xi et al., 2002). The authors concluded there was tonic stimulation of group II by endogenous GLU. A group III antagonist increased synaptic activity in hypothalamic hypocretin neurons, suggesting that group III maintained tonic presynaptic inhibition of these neurons (Acuna-Goycolea et al., 2004). Finally, GLU release was limited in vestibular nuclei as a result of tonic activity of group II/III mGluRs (Grassi et al., 2002).
The parameters necessary to evoke activity-dependent inhibition in nociceptive afferents are unknown. Stimulus duration is most likely a factor because prolonged activity induced by CAP, FM, or GLU evokes the phenomenon but a 10 s heat pulse does not (Fig. 4D,E, insets). An increase in firing frequency may also play an important role. For example, Kew et al. (2001) demonstrated in hippocampal CA1 neurons that the “autoinhibitory” effects of LY were apparent only after burst stimulation of the perforant pathway but not after a single conditioning stimulus. Similarly, in locus ceruleus, group III activation provided a negative feedback during high-frequency stimulation (Dube and Marshall, 2000). The present data demonstrate that group II/III mGluRs on nociceptors are activated in response to increased firing frequency and/or prolonged stimulation duration. Either could result in enhanced GLU release, an event that appears critical to evoking this inhibitory phenomenon.
Endogenous GLU release activates mGluRs, curtailing additional GLU release
Several lines of evidence document activity-dependent release of GLU from nociceptors. Activation of C fibers electrically or by CAP results in increased GLU in the hindpaw (deGroot et al., 2000; Jin et al., 2009). Hindpaw FM injection raises GLU levels in the dialysate (Omote et al., 1998). Release of GLU from DRG somata has also been reported (Roberts, 1974; Jeftinija et al., 1991). Given that we used similar paradigms (CAP, FM), a plausible scenario might be that high-frequency stimulation of nociceptors by a TRPV1 agonist results in excess GLU release, which saturates the normal mechanisms by which GLU is removed (neuronal and glial GLU transporters). Thus, GLU may quickly pool and/or diffuse to axon terminal regions expressing mGluRs. This GLU spillover activates mGluRs producing reduction in any additional GLU release and curtails nociceptive transmission. Basal GLU levels in hindpaw skin range from 1.2 to 1.9 μm (Omote et al., 1998; deGroot et al., 2000; Jin et al., 2009). The reported range of IC50 values of group II/III mGluRs for GLU is as high as 5400 μm but as low as 0.3 μm (Schoepp et al., 1999); thus, we cannot rule out other scenarios. However, data supporting this proposed scenario come from studies demonstrating that group II/III mGluRs can work through a presynaptic mechanism to reduce GLU release from various CNS regions (Kilbride et al., 1998; Anwyl, 1999; Cartmell and Schoepp, 2000; Gerber et al., 2000; Taverna and Pennartz, 2003). These autoreceptors are activated when GLU concentrations are increased, either exogenously or endogenously, and GLU readily diffuses away from synaptic release sites (Cartmell and Schoepp, 2000). There are no “presynaptic” and “postsynaptic” components and no “synaptic” release of GLU per se in relation to peripheral nociceptors because they are, structurally, blind terminal endings. Nonetheless, a similar mechanism appears to be at work in which activation of group II/III mGluRs expressed on afferent terminals can reduce GLU release. Other peripheral cells release GLU, including keratinocytes (Genever et al., 1999), Schwann cells (Jeftinija and Jeftinija, 1998), and macrophages (Piani et al., 1991), but data indicate that primary afferents are a main source of peripheral GLU (deGroot et al., 2000).
In addition to GLU release, other mechanisms may contribute to this activity-dependent inhibition. Group II/III activation will downregulate cAMP/PKA activity (Prezeau et al., 1992; Schoepp and Johnson, 1993; Tanabe et al., 1993; Pin and Duvoisin, 1995), which could in turn inhibit TRPV1 function (De Petrocellis et al., 2001; Bhave et al., 2002; Carlton et al., 2009a). However, we cannot rule out that the reduced excitation reported here may be related to modulation of other receptors or channels in the membrane.
Conclusions and speculations
There is clearly a relationship between nociceptor activity and regulation of nociceptor function. Under basal conditions, peripheral group II/IIIs are not activated and thus have little or no effect on quiescent axons or axons responding to brief stimuli. However, group II/III mGluRs regulate TRPV1-expressing nociceptors with increased magnitude and/or duration of activity. This use-dependent modification is important, serving to prevent runaway excitation of nociceptors. In the CNS, excess GLU release leading to hyperexcitability is linked to numerous psychiatric and neurological disorders (Danysz et al., 1995; Schoepp, 2001; Rotge et al., 2010). In the PNS, our data are consistent with the hypothesis that excessive activation of nociceptors releases excess GLU that finds its way to mGluRs, thereby reducing additional excitation (activity-dependent autoinhibition). The data are compelling, indicating that activation of group II/III mGluRs is critical for keeping the “lid” on nociceptor firing and perceived pain. Without proper functioning of mGluRs on primary afferents, pain transmission would be exacerbated. It is unknown whether group II/III mGluRs are downregulated in chronic pain states. If the system fails, this could enhance both peripheral and/or central sensitization that accompanies many pain states (Carlton et al., 1995, 2009b; Du et al., 2003, 2006; Banik and Brennan, 2004, 2008; Shim et al., 2005; Ji et al., 2007, 2008). It is unknown whether group II or III knock-out mice have altered sensory thresholds because detailed sensory testing has not been reported (Linden et al., 2005; Morishima et al., 2005; Corti et al., 2007; Linden et al., 2009), but such data would elucidate the phenomenon of group II/III endogenous inhibitory control. This dynamic modulation of nociceptors reflects a heretofore unknown form of nonsynaptic, activity-dependent control of nociceptors that presumably changes the way we perceive the outside world.
Footnotes
This work was supported by NIH Grants NS027910 and NS054765 (S.M.C.).
The authors declare no competing financial interests.
References
- Acuna-Goycolea C, Li Y, Van Den Pol AN. Group III metabotropic glutamate receptors maintain tonic inhibition of excitatory synaptic input to hypocretin/orexin neurons. J Neurosci. 2004;24:3013–3022. doi: 10.1523/JNEUROSCI.5416-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman S, Kaube H, Goadsby PJ. Anandamide acts as a vasodilator of dural blood vessels in vivo by activating TRPV1 receptors. Br J Pharmacol. 2004;142:1354–1360. doi: 10.1038/sj.bjp.0705896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Banik RK, Brennan TJ. Spontaneous discharge and increased heat sensitivity of rat C-fiber nociceptors are present in vitro after plantar incision. Pain. 2004;112:204–213. doi: 10.1016/j.pain.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Banik RK, Brennan TJ. Sensitization of primary afferents to mechanical and heat stimuli after incision in a novel in vitro mouse glabrous skin-nerve preparation. Pain. 2008;138:380–391. doi: 10.1016/j.pain.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., 4th cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. doi: 10.1016/s0896-6273(02)00802-4. [DOI] [PubMed] [Google Scholar]
- Bölcskei K, Helyes Z, Szabó A, Sándor K, Elekes K, Németh J, Almási R, Pintér E, Petho G, Szolcsányi J. Investigation of the role of TRPV1 receptors in acute and chronic nociceptive processes using gene-deficient mice. Pain. 2005;117:368–376. doi: 10.1016/j.pain.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Hargett GL. Colocalization of metabotropic glutamate receptors in rat dorsal root ganglion cells. J Comp Neurol. 2007;501:780–789. doi: 10.1002/cne.21285. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Hargett GL, Coggeshall RE. Localization and activation of glutamate receptors in unmyelinated axons of rat glabrous skin. Neurosci Lett. 1995;197:25–28. doi: 10.1016/0304-3940(95)11889-5. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Hargett GL, Coggeshall RE. Localization of metabotropic glutamate receptors 2/3 on primary afferent axons in the rat. Neuroscience. 2001a;105:957–969. doi: 10.1016/s0306-4522(01)00238-x. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Du J, Zhou S, Coggeshall RE. Tonic control of peripheral cutaneous nociceptors by somatostatin receptors. J Neurosci. 2001b;21:4042–4049. doi: 10.1523/JNEUROSCI.21-11-04042.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton SM, Du J, Zhou S. Group II metabotropic glutamate receptor activation on peripheral nociceptors modulates TRPV1 function. Brain Res. 2009a;1248:86–95. doi: 10.1016/j.brainres.2008.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton SM, Du J, Tan HY, Nesic O, Hargett GL, Bopp AC, Yamani A, Lin Q, Willis WD, Hulsebosch CE. Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain. 2009b;147:265–276. doi: 10.1016/j.pain.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75:889–907. doi: 10.1046/j.1471-4159.2000.0750889.x. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci. 2001;24:487–517. doi: 10.1146/annurev.neuro.24.1.487. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Corti C, Battaglia G, Molinaro G, Riozzi B, Pittaluga A, Corsi M, Mugnaini M, Nicoletti F, Bruno V. The use of knock-out mice unravels distinct roles for mGlu2 and mGlu3 metabotropic glutamate receptors in mechanisms of neurodegeneration/neuroprotection. J Neurosci. 2007;27:8297–8308. doi: 10.1523/JNEUROSCI.1889-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danysz W, Parsons CG, Bresink I, Quack G. Glutamate in CNS disorders. Drug News Perspect. 1995;8:261–277. doi: 10.1358/dnp.1998.11.9.863689. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- deGroot J, Zhou S, Carlton SM. Peripheral glutamate release in the hindpaw following low and high intensity sciatic stimulation. Neuroreport. 2000;11:497–502. doi: 10.1097/00001756-200002280-00014. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Harrison S, Bisogno T, Tognetto M, Brandi I, Smith GD, Creminon C, Davis JB, Geppetti P, Di Marzo V. The vanilloid receptor (VR1)-mediated effects of anandamide are potently enhanced by the cAMP-dependent protein kinase. J Neurochem. 2001;77:1660–1663. doi: 10.1046/j.1471-4159.2001.00406.x. [DOI] [PubMed] [Google Scholar]
- Du J, Zhou S, Coggeshall RE, Carlton SM. N-methyl-d-aspartate-induced excitation and sensitization of normal and inflamed nociceptors. Neuroscience. 2003;118:547–562. doi: 10.1016/s0306-4522(03)00009-5. [DOI] [PubMed] [Google Scholar]
- Du J, Zhou S, Carlton SM. Kainate-induced excitation and sensitization of nociceptors in normal and inflamed rat glabrous skin. Neuroscience. 2006;137:999–1013. doi: 10.1016/j.neuroscience.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Du J, Zhou S, Carlton SM. Group II metabotropic glutamate receptor activation attenuates peripheral sensitization in inflammatory states. Neuroscience. 2008;154:754–766. doi: 10.1016/j.neuroscience.2008.03.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubé GR, Marshall KC. Activity-dependent activation of presynaptic metabotropic glutamate receptors in locus coeruleus. J Neurophysiol. 2000;83:1141–1149. doi: 10.1152/jn.2000.83.3.1141. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Bortolotto ZA, Palmer MJ, Doherty AJ, Ornstein PL, Schoepp DD, Kingston AE, Lodge D, Collingridge GL. The potent mGlu receptor antagonist LY341495 identifies roles for both cloned and novel mGlu receptors in hippocampal synaptic plasticity. Neuropharmacology. 1998;37:1445–1458. doi: 10.1016/s0028-3908(98)00145-2. [DOI] [PubMed] [Google Scholar]
- Genever PG, Maxfield SJ, Kennovin GD, Maltman J, Bowgen CJ, Raxworthy MJ, Skerry TM. Evidence for a novel glutamate-mediated signaling pathway in keratinocytes. J Invest Dermatol. 1999;112:337–342. doi: 10.1046/j.1523-1747.1999.00509.x. [DOI] [PubMed] [Google Scholar]
- Gerber G, Zhong J, Youn D, Randic M. Group II and group III metabotropic glutamate receptor agonists depress synaptic transmission in the rat spinal cord dorsal horn. Neuroscience. 2000;100:393–406. doi: 10.1016/s0306-4522(00)00269-4. [DOI] [PubMed] [Google Scholar]
- Ghilardi JR, Röhrich H, Lindsay TH, Sevcik MA, Schwei MJ, Kubota K, Halvorson KG, Poblete J, Chaplan SR, Dubin AE, Carruthers NI, Swanson D, Kuskowski M, Flores CM, Julius D, Mantyh PW. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J Neurosci. 2005;25:3126–3131. doi: 10.1523/JNEUROSCI.3815-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi S, Frondaroli A, Pettorossi VE. Different metabotropic glutamate receptors play opposite roles in synaptic plasticity of the rat medial vestibular nuclei. J Physiol. 2002;543:795–806. doi: 10.1113/jphysiol.2002.023424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Jeftinija SD, Jeftinija KV. ATP stimulates release of excitatory amino acids from cultured Schwann cells. Neuroscience. 1998;82:927–934. doi: 10.1016/s0306-4522(97)00310-2. [DOI] [PubMed] [Google Scholar]
- Jeftinija S, Jeftinija K, Liu F, Skilling SR, Smullin DH, Larson AA. Excitatory amino acids are released from rat primary afferent neurons in vitro. Neurosci Lett. 1991;125:191–194. doi: 10.1016/0304-3940(91)90025-o. [DOI] [PubMed] [Google Scholar]
- Ji G, Zhou S, Kochukov MY, Westlund KN, Carlton SM. Plasticity in intact A delta- and C-fibers contributes to cold hypersensitivity in neuropathic rats. Neuroscience. 2007;150:182–193. doi: 10.1016/j.neuroscience.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Zhou S, Carlton SM. Intact Adelta-fibers up-regulate transient receptor potential A1 and contribute to cold hypersensitivity in neuropathic rats. Neuroscience. 2008;154:1054–1066. doi: 10.1016/j.neuroscience.2008.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YH, Yamaki F, Takemura M, Koike Y, Furuyama A, Yonehara N. Capsaicin-induced glutamate release is implicated in nociceptive processing through activation of ionotropic glutamate receptors and group I metabotropic glutamate receptor in primary afferent fibers. J Pharmacol Sci. 2009;109:233–241. doi: 10.1254/jphs.08262fp. [DOI] [PubMed] [Google Scholar]
- Johnson BG, Wright RA, Arnold MB, Wheeler WJ, Ornstein PL, Schoepp DD. [3H]-LY341495 as a novel antagonist radioligand for group II metabotropic glutamate (mGlu) receptors: characterization of binding to membranes of mGlu receptor subtype expressing cells. Neuropharmacology. 1999;38:1519–1529. doi: 10.1016/s0028-3908(99)00053-2. [DOI] [PubMed] [Google Scholar]
- Joshi SK, Honore P, Hernandez G, Schmidt R, Gomtsyan A, Scanio M, Kort M, Jarvis MF. Additive antinociceptive effects of the selective Nav1.8 blocker A-803467 and selective TRPV1 antagonists in rat inflammatory and neuropathic pain models. J Pain. 2009;10:306–315. doi: 10.1016/j.jpain.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Kew JN, Ducarre JM, Pflimlin MC, Mutel V, Kemp JA. Activity-dependent presynaptic autoinhibition by group II metabotropic glutamate receptors at the perforant path inputs to the dentate gyrus and CA1. Neuropharmacology. 2001;40:20–27. doi: 10.1016/s0028-3908(00)00118-0. [DOI] [PubMed] [Google Scholar]
- Kilbride J, Huang LQ, Rowan MJ, Anwyl R. Presynaptic inhibitory action of the group II metabotropic glutamate receptor agonists, LY354740 and DCG-IV. Eur J Pharmacol. 1998;356:149–157. doi: 10.1016/s0014-2999(98)00526-3. [DOI] [PubMed] [Google Scholar]
- Kingston AE, Ornstein PL, Wright RA, Johnson BG, Mayne NG, Burnett JP, Belagaje R, Wu S, Schoepp DD. LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology. 1998;37:1–12. doi: 10.1016/s0028-3908(97)00191-3. [DOI] [PubMed] [Google Scholar]
- Koltzenburg M, Kress M, Reeh PW. The nociceptor sensitization by bradykinin does not depend on sympathetic neurons. Neuroscience. 1992;46:465–473. doi: 10.1016/0306-4522(92)90066-b. [DOI] [PubMed] [Google Scholar]
- Kress M, Koltzenburg M, Reeh PW, Handwerker HO. Responsiveness and functional attributes of electrically localized terminals of cutaneous C-fibers in vivo and in vitro. J Neurophysiol. 1992;68:581–595. doi: 10.1152/jn.1992.68.2.581. [DOI] [PubMed] [Google Scholar]
- Linden AM, Bergeron M, Schoepp DD. Comparison of c-Fos induction in the brain by the mGlu2/3 receptor antagonist LY341495 and agonist LY354740: evidence for widespread endogenous tone at brain mGlu2/3 receptors in vivo. Neuropharmacology. 2005;49(Suppl 1):120–134. doi: 10.1016/j.neuropharm.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Linden AM, Johnson BG, Trokovic N, Korpi ER, Schoepp DD. Use of MGLUR2 and MGLUR3 knockout mice to explore in vivo receptor specificity of the MGLUR2/3 selective antagonist LY341495. Neuropharmacology. 2009;57:172–182. doi: 10.1016/j.neuropharm.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Macek TA, Winder DG, Gereau RW, 4th, Ladd CO, Conn PJ. Differential involvement of group II and group III mGluRs as autoreceptors at lateral and medial perforant path synapses. J Neurophysiol. 1996;76:3798–3806. doi: 10.1152/jn.1996.76.6.3798. [DOI] [PubMed] [Google Scholar]
- Medvedeva YV, Kim MS, Usachev YM. Mechanisms of prolonged presynaptic Ca2+ signaling and glutamate release induced by TRPV1 activation in rat sensory neurons. J Neurosci. 2008;28:5295–5311. doi: 10.1523/JNEUROSCI.4810-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima Y, Miyakawa T, Furuyashiki T, Tanaka Y, Mizuma H, Nakanishi S. Enhanced cocaine responsiveness and impaired motor coordination in metabotropic glutamate receptor subtype 2 knockout mice. Proc Natl Acad Sci U S A. 2005;102:4170–4175. doi: 10.1073/pnas.0500914102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omote K, Kawamata T, Kawamata M, Namiki A. Formalin-induced release of excitatory amino acids in the skin of the rat hindpaw. Brain Res. 1998;787:161–164. doi: 10.1016/s0006-8993(97)01568-0. [DOI] [PubMed] [Google Scholar]
- Piani D, Frei K, Do KQ, Cuénod M, Fontana A. Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett. 1991;133:159–162. doi: 10.1016/0304-3940(91)90559-c. [DOI] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Sikand P. TRPV1: a target for next generation analgesics. Curr Neuropharmacol. 2008;6:151–163. doi: 10.2174/157015908784533888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prezeau L, Manzoni O, Homburger V, Sladeczek F, Curry K, Bockaert J. Characterization of a metabotropic glutamate receptor: direct negative coupling to adenylyl cyclase and involvement of a pertussis toxin-sensitive G protein. Proc Natl Acad Sci U S A. 1992;89:8040–8044. doi: 10.1073/pnas.89.17.8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeh PW. Sensory receptors in mammalian skin in an in vitro preparation. Neurosci Lett. 1986;66:141–146. doi: 10.1016/0304-3940(86)90180-1. [DOI] [PubMed] [Google Scholar]
- Roberts PJ. Amino acid release from isolated rat dorsal root ganglia. Brain Res. 1974;74:327–332. doi: 10.1016/0006-8993(74)90586-1. [DOI] [PubMed] [Google Scholar]
- Rotge JY, Aouizerate B, Tignol J, Bioulac B, Burbaud P, Guehl D. The glutamate-based genetic immune hypothesis in obsessive-compulsive disorder. An integrative approach from genes to symptoms. Neuroscience. 2010;165:408–417. doi: 10.1016/j.neuroscience.2009.10.043. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther. 2001;299:12–20. [PubMed] [Google Scholar]
- Schoepp DD, Conn PJ. Metabotropic glutamate receptors in brain function and pathology. Trends Pharmacol Sci. 1993;14:13–20. doi: 10.1016/0165-6147(93)90107-u. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Johnson BG. Pharmacology of metabotropic glutamate receptor inhibition of cyclic AMP formation in the adult rat hippocampus. Neurochem Int. 1993;22:277–283. doi: 10.1016/0197-0186(93)90056-b. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Shim B, Kim DW, Kim BH, Nam TS, Leem JW, Chung JM. Mechanical and heat sensitization of cutaneous nociceptors in rats with experimental peripheral neuropathy. Neuroscience. 2005;132:193–201. doi: 10.1016/j.neuroscience.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Szolcsányi J. Capsaicin-sensitive sensory nerve terminals with local and systemic efferent functions: facts and scopes of an unorthodox neuroregulatory mechanism. Prog Brain Res. 1996;113:343–359. doi: 10.1016/s0079-6123(08)61097-3. [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Nomura A, Masu M, Shigemoto R, Mizuno N, Nakanishi S. Signal transduction, pharmacological properties, and expression patterns of two rat metabotropic glutamate receptors, mGluR3 and mGluR4. J Neurosci. 1993;13:1372–1378. doi: 10.1523/JNEUROSCI.13-04-01372.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S, Pennartz CM. Postsynaptic modulation of AMPA- and NMDA-receptor currents by Group III metabotropic glutamate receptors in rat nucleus accumbens. Brain Res. 2003;976:60–68. doi: 10.1016/s0006-8993(03)02676-3. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Ugolini A, Bordi F. Metabotropic glutamate group II receptors are responsible for the depression of synaptic transmission induced by ACPD in the dentate gyrus. Eur J Pharmacol. 1995;294:403–410. doi: 10.1016/0014-2999(95)00560-9. [DOI] [PubMed] [Google Scholar]
- Wong GY, Gavva NR. Therapeutic potential of vanilloid receptor TRPV1 agonists and antagonists as analgesics: recent advances and setbacks. Brain Res Rev. 2009;60:267–277. doi: 10.1016/j.brainresrev.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Wright RA, Arnold MB, Wheeler WJ, Ornstein PL, Schoepp DD. Binding of [3H](2S,1'S,2'S)-2-(9-xanthylmethyl)-2-(2′-carboxycyclopropyl) glycine ([3H]LY341495) to cell membranes expressing recombinant human group III metabotropic glutamate receptor subtypes. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:546–554. doi: 10.1007/s002100000305. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Baker DA, Shen H, Carson DS, Kalivas PW. Group II metabotropic glutamate receptors modulate extracellular glutamate in the nucleus accumbens. J Pharmacol Exp Ther. 2002;300:162–171. doi: 10.1124/jpet.300.1.162. [DOI] [PubMed] [Google Scholar]
- Xu GY, Huang LY. Peripheral inflammation sensitizes P2X receptor-mediated responses in rat dorsal root ganglion neurons. J Neurosci. 2002;22:93–102. doi: 10.1523/JNEUROSCI.22-01-00093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]