

1. INTRODUCTION
Fascination with supramolecular chemistry over the last few decades has led to the synthesis of an ever-increasing number of elegant and intricate functional structures with sizes that approach nanoscopic dimensions. Today, it has grown into a mature field of modern science whose interfaces with many disciplines have provided invaluable opportunities for crossing boundaries both inside and between the fields of chemistry, physics, and biology. This chemistry is of continuing interest for synthetic chemists; partly because of the fascinating physical and chemical properties and the complex and varied aesthetically pleasing structures that supramolecules possess. For scientists seeking to design novel molecular materials exhibiting unusual sensing, magnetic, optical, and catalytic properties, and for researchers investigating the structure and function of biomolecules, supramolecular chemistry provides limitless possibilities. Thus, it transcends the traditional divisional boundaries of science and represents a highly interdisciplinary field.
In the early 1960s, the discovery of ‘crown ethers’, ‘cryptands’ and ‘spherands’ by Pedersen,1 Lehn,2 and Cram3 respectively, led to the realization that small, complementary molecules can be made to recognize each other through non-covalent interactions such as hydrogen-bonding, charge-charge, donor-acceptor, π-π, van der Waals, etc. Such ‘programmed’ molecules can thus be self-assembled by utilizing these interactions in a definite algorithm to form large supramolecules that have different physicochemical properties than those of the precursor building blocks. Typical systems are designed such that the self-assembly process is kinetically reversible; the individual building blocks gradually funnel towards an ensemble that represents the thermodynamic minimum of the system via numerous association and dissociation steps. By tuning various reaction parameters, the reaction equilibrium can be shifted towards the desired product. As such, self-assembly has a distinct advantage over traditional, stepwise synthetic approaches when accessing large molecules.
It is well known that nature has the ability to assemble relatively simple molecular precursors into extremely complex biomolecules, which are vital for life processes. Nature’s building blocks possess specific functionalities in configurations that allow them to interact with one another in a deliberate manner. Protein folding, nucleic acid assembly and tertiary structure, phospholipid membranes, ribosomes, microtubules, etc. are but a selective, representative example of self-assembly in nature that is of critical importance for living organisms. Nature makes use of a variety of weak, non-covalent interactions such as hydrogen–bonding, charge–charge, donor–acceptor, π-π, van der Waals, hydrophilic and hydrophobic, etc. interactions to achieve these highly complex and often symmetrical architectures. In fact, the existence of life is heavily dependent on these phenomena. The aforementioned structures provide inspiration for chemists seeking to exploit the ‘weak interactions’ described above to make scaffolds rivaling the complexity of natural systems.
The breadth of supramolecular chemistry has progressively increased with the synthesis of numerous unique supramolecules each year. Based on the interactions used in the assembly process, supramolecular chemistry can be broadly classified in to three main branches: i) those that utilize H-bonding motifs in the supramolecular architectures, ii) processes that primarily use other non-covalent interactions such as ion-ion, ion-dipole, π–π stacking, cation-π, van der Waals and hydrophobic interactions, and iii) those that employ strong and directional metal-ligand bonds for the assembly process. However, as the scale and degree of complexity of desired molecules increases, the assembly of small molecular units into large, discrete supramolecules becomes an increasingly daunting task. This has been due in large part to the inability to completely control the directionality of the weak forces employed in the first two classifications above. Coordination-driven self-assembly, which defines the third approach, affords a greater control over the rational design of 2D and 3D architectures by capitalizing on the predictable nature of the metal-ligand coordination sphere and ligand lability to encode directionality. Thus, this third strategy represents an alternative route to better execute the “bottom-up” synthetic strategy for designing molecules of desired dimensions, ranging from a few cubic angstroms to over a cubic nanometer. For instance, a wide array of 2D systems: rhomboids, squares, rectangles, triangles, etc., and 3D systems: trigonal pyramids, trigonal prisms, cubes, cuboctahedra, double squares, adamantanoids, dodecahedra and a variety of other cages have been reported. As in nature, inherent preferences for particular geometries and binding motifs are ‘encoded’ in certain molecules depending on the metals and functional groups present; these moieties help to control the way in which the building blocks assemble into well-defined, discrete supramolecules.4
Since the early pioneering work by Lehn5 and Sauvage6 on the feasibility and usefulness of coordination-driven self-assembly in the formation of infinite helicates, grids, ladders, racks, knots, rings, catenanes, rotaxanes and related species,7 several groups - Stang,8 Raymond,9 Fujita,10 Mirkin,11 Cotton12 and others13,14 have independently developed and exploited novel coordination-based paradigms for the self-assembly of discrete metallacycles and metallacages with well-defined shapes and sizes. In the last decade, the concepts and perspectives of coordination-driven self-assembly have been delineated and summarized in several insightful reviews covering various aspects of coordinationdriven self-assembly.15 In the last decade, the use of this synthetic strategy has led to metallacages dubbed as “molecular flasks” by Fujita,16 and Raymond and Bergman,17 which due to their ability to encapsulate guest molecules, allowed for the observation of unique chemical phenomena and unusual reactions which cannot be achieved in the conventional gas, liquid or solid phases. Furthermore, these assemblies found applications in supramolecular catalysis18,19 and as nanomaterials as developed by Hupp20 and others.21,22
This review focuses on the journey of early coordination-driven self-assembly paradigms to more complex and discrete 2D and 3D supramolecular ensembles over the last decade. We begin with a discussion of various approaches that have been developed by different groups to assemble finite supramolecular architectures. The subsequent sections contain detailed discussions on the synthesis of discrete 2D and 3D systems, their functionalizations and applications.
2. BACKGROUND AND DESIGN PRINCIPLES
The assembly of supramolecular ensembles depends on the information coded within the complementary building blocks that form the rigid framework of the architectures. The highly directional and predictable nature of the metal-ligand coordination sphere is a critical feature of coordination-driven self-assembly. The energies of metal-ligand bonds (15-50 kcal/mol), which are intermediate between the energies of organic covalent bonds (ca. 60-120 kcal/mol) and the weak interactions (ca. 0.5-10 kcal/mol) that nature employs so elegantly to self-assemble biomolecules, helps in modulating the coordination kinetics of the self-assembly process by introducing rigidity and reversibility. The kinetic reversibility between complementary building blocks, reaction intermediates, and self-assembled architectures provides a way for the system to self-correct (an “incorrectly” formed bond can dissociate and reassociate “correctly”), leading to a product that is thermodynamically more stable than the starting components and any kinetically formed intermediates. Transition metals, with their preferred coordination geometries, have served as acceptor units that can logically self-assemble with various rigid or flexible donors into predictable architectures. Although macrocyclization is a kinetically unfavorable process, thermodynamic conditions facilitate the formation of macrocycles at the expense of increased angle strain. This is due, among other factors, to the fact that entropy favors closed structures with a minimum number of components rather than polymeric structures, which involve a far larger number of components.
With a growing knowledge of the synthesis and characterization of large, complex molecules, the last few years have seen a tremendous proliferation of new supramolecules and strategies to achieve complex topologies. Of the various strategies developed in recent years using metal-ligand coordination, Directional Bonding,8 Symmetry Interaction,9 Molecular Paneling,10 Weak Link,11 and Dimetallic Building Block12 approaches are the most extensively used and adopted. These synthetic strategies have led to a wide variety of 2D and 3D molecular architectures of different shapes and sizes, which can be modulated through the judicious choice of metals and ligands. The metal-ligand bond serves as the cornerstone in all these design principles. Mechanistically, the first four approaches rely on thermodynamic control; the global minimum of the reaction coordinate is the desired product. The weak link approach utilizes both thermodynamic and kinetic control to access a variety of large, open and flexible 2D and 3D structures.
2.1. Directional Bonding Approach
The directional bonding approach is a general, high-yielding synthetic strategy that gives access to a wide variety of 2D and 3D supramolecular ensembles.8 Since the seminal studies on the rational synthesis of molecular squares in the early 1990s by Fujita23,24 and Stang,25 a plethora of metallamacrocycles and metallacages have been assembled using this design stretegy. This rational design stretegy allows for a combinatorial molecular library consisting of complementary building blocks that allows one to think retrosynthetically on how best to achieve the geometry of a particular discrete assembly.
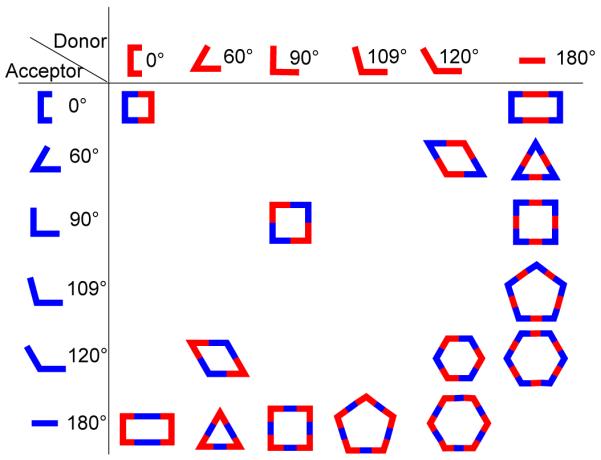
There are two basic structural requirements for the construction of supramolecular architectures by this approach. First, the complementary precursor units must be structurally rigid with predefined bite angles; and second, the appropriate stoichiometric ratio of the precursors must be used. The donor building blocks are generally organic ligands having two or more binding sites possessing angular orientations ranging from 0° to 180° (Figure 1).
Figure 1.
Combination of various building units for accessing convex polygons and canonical polyhedra.
The acceptors are metal-containing subunits that are vital in this design approach because they possess available coordination sites, which are at a fixed angle relative to one another for binding incoming ligands. The symmetry and number of binding sites within each precursor unit guide the shape of the target assembly. While the design of monocyclic ensembles requires subunits having symmetry axes no higher than a two-fold axis, polycyclic entities require one of the subunits to possess a symmetry axis higher than two-fold. In this strategy, the shapes of the resulting monocyclic entities resemble convex polygons and those of polycyclic frameworks bear a resemblance to canonical polyhedra. For example, a molecular rectangle can be designed by the combination of 0° acceptor units with 180° donor units and vice versa. A molecular triangle can be prepared by the combination of three 60° donor units and three 180° acceptors. A molecular square can be accessed in different ways: the combination of four ditopic 90° angular units and four 180° linear units or the 2:2 assembly of two different 90° angular units. 3D polyhedral architectures can be designed using a combination of angular and linear subunits with multidentate precursor that have more than two binding sites (Figure 2). For example, a truncated tetrahedron can be designed by combining four tridentate subunits, where the angles between the binding sites are 109°, with six ditopic subunits having an angular disposition of 80-90°. A trigonal bipyramid can be formed in several ways, such as by combining two tritopic units, where the binding sites are orthogonal to each other, with three 80-90° ditopic units or by combining two tridentate 60° building blocks with three ditopic units which have 90° angles between the donor sites. Similarly, eight tritopic subunits possessing 90° angles between their binding sites and twelve linear ditopic units leads to the formation of a cube. Although it is assumed that the angle subtended at the angular subunit does not change upon incorporation into the assembly, in practice the angles can distort up to several degrees. Nevertheless, due to the relatively weak nature of metal-ligand bonds compared to covalent ones, the final geometry of the supramolecular entity remains more or less intact.
Figure 2.
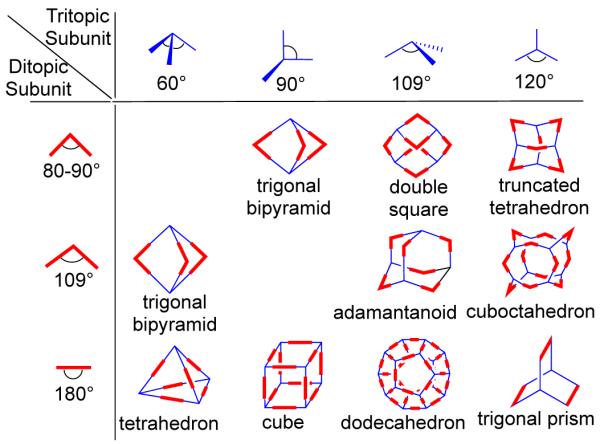
Three-dimensional architectures formed by the combination of ditopic and tritopic subunits by the directional bonding approach.
2.2. Symmetry Interaction Approach
This design strategy has been developed as a rational synthetic approach for the synthesis of high-symmetry coordination clusters using metal-ligand bonds. It is based on the geometric relationship between the chelating ligands and the metals used. The strong binding affinity and coordination mode of chelating ligands, along with the inherent symmetry of the coordination sites available on the “naked” metal center, act as the driving force for the assembly process. In general, multi-branched chelating ligands with rigid backbones are used in conjunction with transition metals or main group metals. The orientation of the multiple binding sites that are rigidly fixed is critical to the selectivity of a particular molecular geometry and helps to avoid the formation of oligomers and polymers. Similar to the directional bonding approach, it relies on the thermodynamic control and kinetic reversibility for error checking and self-correction.
Raymond and coworkers have defined the requisites of this design principle in terms of the geometric relationship between the ligand and the metal component using symmetry considerations.9 A coordinate vector represents the interaction between a ligand and metal. For chelating ligands, the plane orthogonal to the major symmetry axis of a metal complex is the chelate plane (Figure 3), which in the case of bidentate chelators holds all chelate vectors. Thus, depending on the orientation of the chelate planes, the construction of high-symmetrical coordination clusters can be realized. Furthermore, the proper organization of the multiple binding sites with respect to one another is of a paramount importance.
Figure 3.
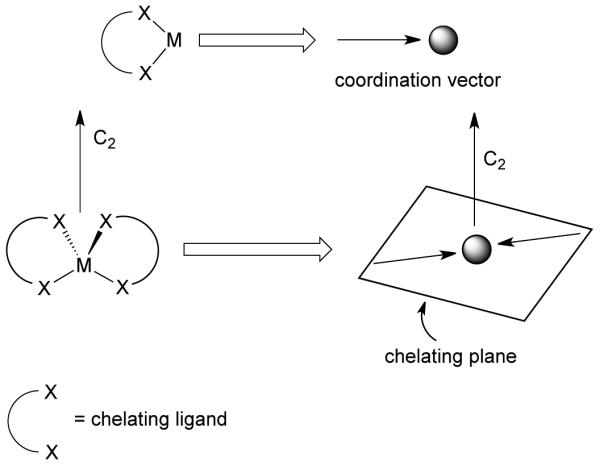
Coordinate vector and chelate plane for the symmetry interaction method.
For example, in order to design a M2L3 triple helicate having an idealized D3h symmetry, it must be ensured that both the C2 and C3 axes are orthogonal and are pre-programmed into the chelating ligand and the metal center. Since the two pseudo-octahedral metal centers share the same C3 axis, the two chelating planes must be parallel to achieve the triple helicate (Figure 4).
Figure 4.
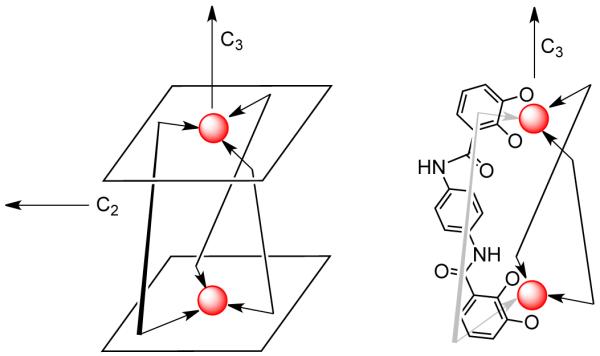
Design of a D3-symmetrical triple helicate.
A similar approach can be applied for the rational design of tetrahedral clusters. In a M4L6 tetrahedron, the four metal atoms occupy the vertices and the six ligands are disposed on the edges of the tetrahedron. This requires that the C2 axes of the tetrahedron lie within the chelate plane at each of the metal centers. Furthermore, the chelate vectors within the ligand must maintain an angle of 70.6° (Figure 5a). A tetrahedron can also be generated by the combination of four metal atoms with four ligands (M4L4). In this case, the four ligands occupy each of the four faces of the tetrahedron with four metal atoms at the vertices. Symmetry considerations thus require that both the ligands and metals possess C3 symmetry (Figure 5b).
Figure 5.

Design strategy for (a) M4L6 and (b) M4L4 tetrahedra by the symmetry interaction approach.
2.3. Paneling Approach
This design strategy, pioneered by Fujita and coworkers, has led to the formation of various functional and aesthetically elegant 3D architectures that resemble platonic solids, including equilateral triangles, squares and pentagons.10 Thus, 3D molecular architectures can, in principle, be designed by reducing these polyhedra to molecular components. For example, a tetrahedron can be designed by stitching together four triangular panels, while an octahedron can be prepared by bringing together eight such triangular panels (Figure 6). Similarly, the paneling of squares results in the formation of cubes and prisms. In this paradigm, flat, panel-like organic ligands having more than two linking sites are paneled together using appropriate structural corner units. These 2D molecular panels span all or some of the faces of the resulting platonic architectures. The corner units employed are usually cis-protected square planar Pt(II) or Pd(II) metal centers. In contrast to the “naked” metal centers used in the symmetry interaction approach, cis-protection makes the coordination geometry around the metal center convergent. This enables the efficient generation of discrete supramolecular arrays without the formation of oligomeric products.
Figure 6.
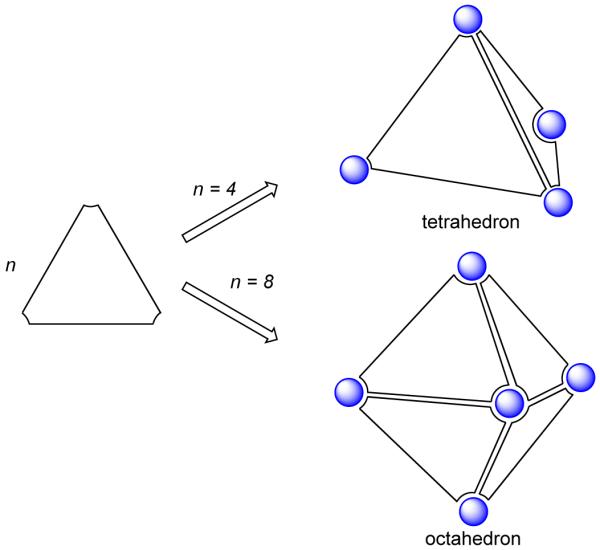
Representation for assembling a tetrahedron and an octahedron using triangular panels.
The triangular molecular panels, in conjunction with the cis-blocked corner unit, lead to varied supramolecular architectures including the M6L4 truncated tetrahedral cage, the M6L4 square pyramidal cone, M8L4 tetrahedra and cones and M18L6 hexahedra. Square and rectangular panels can be used to construct cubes, parallelopipeds and prisms of various topologies: trigonal, square, pentagonal, hexagonal, etc., by the judicious introduction of multiple binding sites into the edges and/or corners of the panels. Placement of the donor binding sites in the panel is vital and guides the final geometry of the resulting architectures. For example, an M6L4 octahedron can be designed by using triangular molecular panels having D3h symmetry with three linking sites at the vertices of the triangle (Figure 7a).26 The four triangular panels span alternate faces of the octahedron and are linked together at the vertices by cis-blocked corner units. A slightly different disposition of the donor sites in the triangular panel would results in the formation of a bowl-like M6L4 square-pyramidal cone (Figure 7b).27
Figure 7.
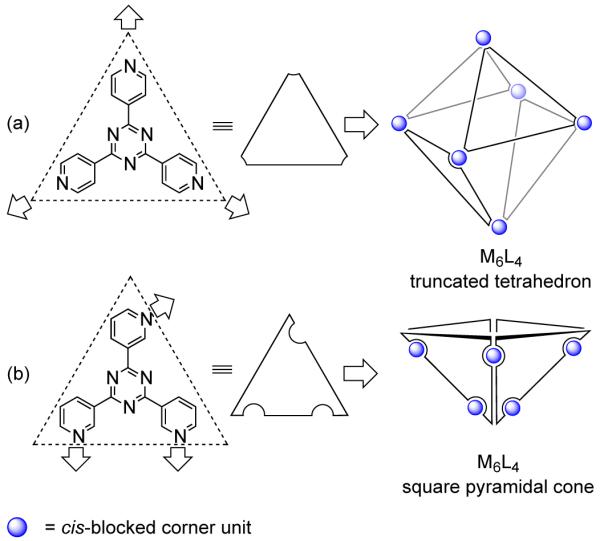
Representation for assembling: a) a M6L4 truncted tetrahedron and b) an M6L4 square pyramidal cone using triangular panels.
Similarly, tetratopic square panels having D4h symmetry can lead to different topologies depending upon the positions of the donor sites on the panel (Figure 8). While panels having linking sites on the edges of the square form M6L3 trigonal prisms,28 donor sites on the four corners give access to M12L6 hexagonal prisms.29 One of the prime advantages of this strategy is that the ensembles formed through this approach have large accessible interior cavities, which have been exploited for reversible guest inclusion, molecular recognition, and cavity-driven catalysis. However, to achieve some of the designed architectures, guest molecules need to be used as a template during the formation of the prisms.
Figure 8.
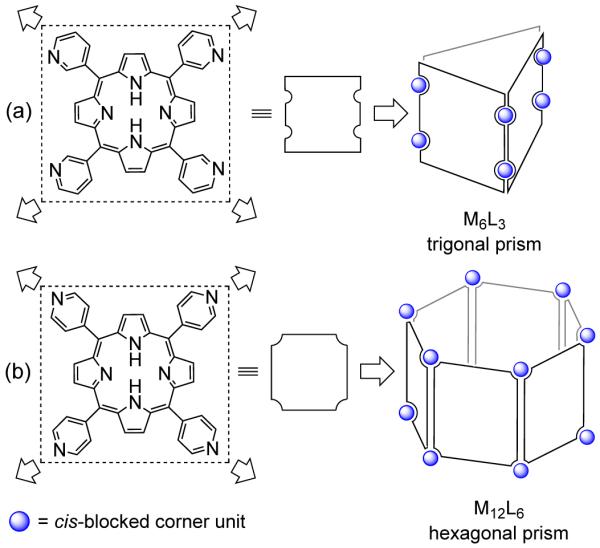
Representation for assembling: a) a M6L3 trigonal prism and b) a M12L6 hexagonal prism using tetratopic panels.
2.4. Weak Link Approach
In this design strategy, pioneered by Mirkin and coworkers, both 2D and 3D supramolecular assemblies are accessible using hemilabile ligands and transition metals.11 The hemilabile, flexible ligands coordinate in a bidentate chelating mode to the metal center such that one of the metal-ligand bonds is weaker than the other. The formation of the kinetically controlled product is driven by the chelating effect of the bidentate ligands and the π–π interaction between the two central bridging units (Scheme 1). The weak ligands of this condensed intermediate structure can be selectively displaced upon treatment with small molecules or ions that have stronger affinity for the metal center, thereby generating the thermodynamically controlled product. One important feature of this approach is the conformational flexibility of the ligand. The coordinative lability of the metal centers imparts flexibility to the supramolecular arrays, making this design strategy different from the other approaches in which the final products are rigid. The flexibility of the architectures also makes the ensembles potential candidates for applications that require conformational changes, such as in molecular sensing, catalysis and host-guest chemistry. The fact that the metal centers are free for further coordination without the supramolecular assembly being destroyed makes this approach amenable for the construction of more complex topologies.
Scheme 1.
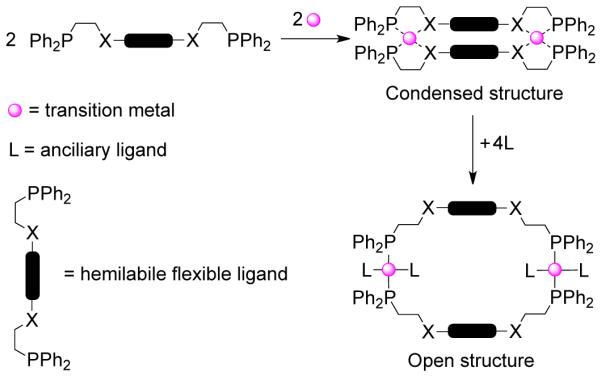
2.5. Dimetallic Building Block Approach
In Cotton’s dimetallic building block approach12 well-known paddlewheel or lantern frameworks form the basic structural units. Dimetallic units of Mo24+ and Rh24+ have been extensively used in conjunction with a variety of organic linkers to construct a wide array of neutral supramolecules. The cis-blocked dimetallic units have one or more of the edges blocked by non-labile chelating N-donor ligands such as amidinate, while the other coordination sites are occupied by labile ligands such as acetonitrile or chloride ions (Figure 9). Polycarboxylate and polypyridyl ligands serve as the equatorial and axial ligands, respectively. Depending on the choice of the dimetallic unit (Figure 9a) and linkers, the units can be joined face-to-face with an axial linker (Figure 9b), end-to-end with an equatorial linker (Figure 9c) or by a combination of both linkers (Figure 9d).
Figure 9.
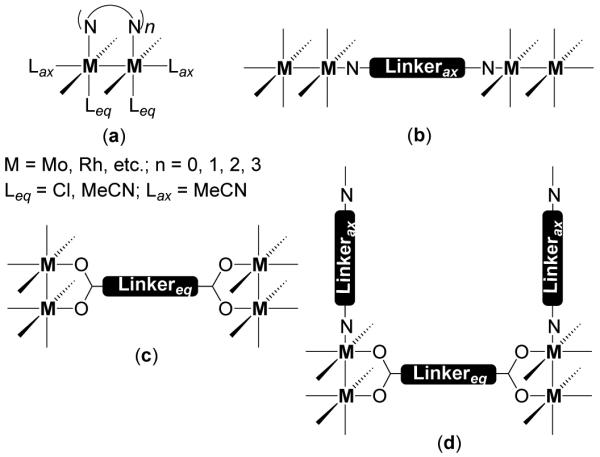
Modes of assembly of dinuclear units in dimetallic building block approach.
3. TWO-DIMENSIONAL (2D) ENSEMBLES
Since the synthesis of metal-cornered molecular squares by Fujita23 and Stang25 in early 1990s, a wide variety of metallamacrocyles consisting of varied 2D geometries and sizes ranging from simple dinuclear rhomboids to endo- and exo-functionalized polynuclear hexagons. Although most of these fascinating molecular ensembles were synthesized by employing the design principles described above (Section 2), many more have been obtained using different synthetic pathways, such as the transformation of mononuclear metal complexes or the metal-induced transformation of added reagents. Our main focus in this section will be on the synthetic and topological aspects of the self-assembled metallacycles using the design principles described above. The functional properties of these 2D architectures will be discussed in a separate sub-section.
3.1. Charged Systems
In metal-mediated, self-assembled, supramolecular metallacycles, the reactivity and coordination geometry of the metal center plays a vital role in guiding the topologies of the final architectures. These ensembles have typically been prepared utilizing transition metals as directing units, due to their well-defined coordination preferences. In particular, electron-poor square planar divalent Pt(II) and Pd(II) ions have been extensively used in conjunction with electron-rich nitrogen-containing moieties in the self-assembly process. In contrast, the use of main group metals as the directing units has found less favor with supramolecular chemists due to their rather unpredictable coordination preferences. Nevertheless, interest involving main group elements as structure-directing units is growing in recent years.30 The vast majority of these metallacycles are highly charged due the use of neutral N-donor ligands with oxidized metal ions. This has a profound impact on both the solubilities of the complexes and their potential to act as hosts for anions/cations via electrostatic interactions.
3.1.1. Molecular Rhomboids
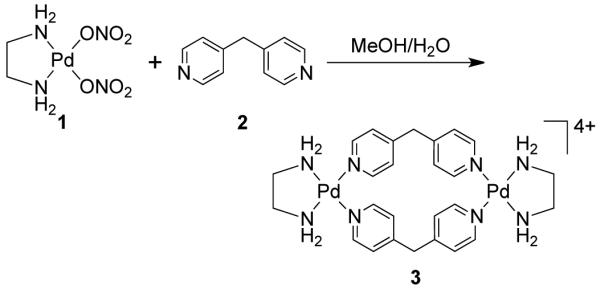
From a topological point of view, dinuclear molecular rhomboids are the simplest among the metallacycles. It has been observed that flexible bridging ligands generally favor the self-assembly of low-nuclearity macrocycles. Using the directional bonding approach, a molecular rhomboid can be assembled by a [2 + 2] combination of 120° donor and acceptor units. Fujita and coworkers reported some early examples of self-assembled dinuclear cationic molecular rhomboids.31-34 In all these assemblies, a high fidelity for the formation of the dinuclear macrocyclic cores was observed from a reaction of 90° cis-blocked [(en)Pd(NO3)2] (en = ethylenediamine) (1) with bidentate bis(pyridine) based ligands having wide range of angles between the donor sites. For example, the synthesis of rhomboid 3 involved the assembly of 90° cis-blocked [Pd(en)(NO3)2] (en = ethylenediamine) (1) with bis(4-pyridyl)methane 2 in methanol-water medium (Scheme 2).
Scheme 2.
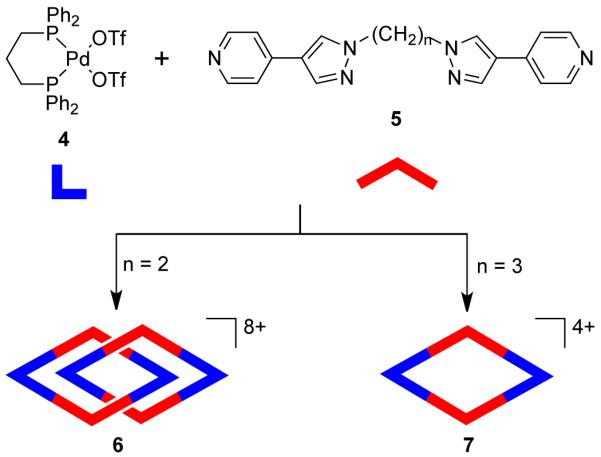
Introduction of flexible, nonlinear spacer 1,4-CH2(C6H4)CH2 in the bispyridyl bridging ligand led to the interesting observation of a dynamic equilibrium between a dinuclear rhomboid and an interpenetrating [2]catenane at ambient temperature.32,33 The Pt analogue of 1 has led to the formation of a stable interpenetrating [2]catenane, owing to the higher strength of the Pt-N.34 Batten et al.35 have demonstrated that the formation of a [2]catenane is also dependent on the length of the bridging ligand. The assembly of 90° cis-blocked [Pd(dppp)(OTf)2] (dppp = diphenylphosphinopropane) 4 with ditopic ligands 5, containing two 4-(4-pyridyl)pyrazolyl arms that are connected by ethane and propane bridges, led to the formation of [2]catenane 6 and molecular rhomboid 7, respectively (Scheme 3). The face-to-face π interaction between the aromatic rings drives the formation of 6. Disruption of this array due to an increase in the length of the bridging ligand led to the formation of only a single noncatenated rhomboid (7).
Scheme 3.
Stang et al. have reported cationic dinuclear rhomboids of carbon- and silicon-based tectons using cis-[Pt(PEt3)2(OTf)2] (8) and cis-[Pd(PEt3)2(OTf)2] (9) as the corner units.36 For example, treatment of dimethyl-bis(4-pyridyl)silane with 90° corner units 8 and 9 in a 1:1 ratio led to the formation of silicon containing rhomboids 10-11 (Scheme 4). An analogous rhomboid was also prepared from bispyridyl acetal using cis-[Pt(PEt3)2(OTf)2] as the corner unit. Multinuclear NMR, FAB-mass spectrometry and X-ray structural studies established the formation of the rhomboids. Due to the unexpected hydration of ketone, treatment of bis(4-pyridyl)ketone with 8 gave a similar rhomboid instead of the less strained trimeric assembly. It was observed that entropy plays an important role in the formation of these macrocyclic systems, favoring the formation of the smallest macrocycle even though the hydration is thermodynamically disfavored. Similarly, using a bis(pyridyl) subunit possessing a tethered C60 molecule in conjunction with 8 gave a supramolecular rhomboid containing two C60 units.37 Similarly, a series of molecular rhomboids have been prepared using flexible bispyridyl ligands to assemble molecular rhomboids.38 The combination of a 60° bent bifunctional Pt(II)-based building block (12) with a structurally rigid 120° linker, 2,6-Di(4,4′-dipyridyl)-9-thiabicyclo[3.3.1]nonane, resulted in an octacationic molecular rhomboid (13) having large cavity dimensions in excellent yields (Scheme 5).39 Several other dinuclear endo- and exo-functionalized molecular rhomboids with large cavity spaces have been designed using the same 60° bent Pt(II)-acceptor as the directing unit (vide infra).
Scheme 4.
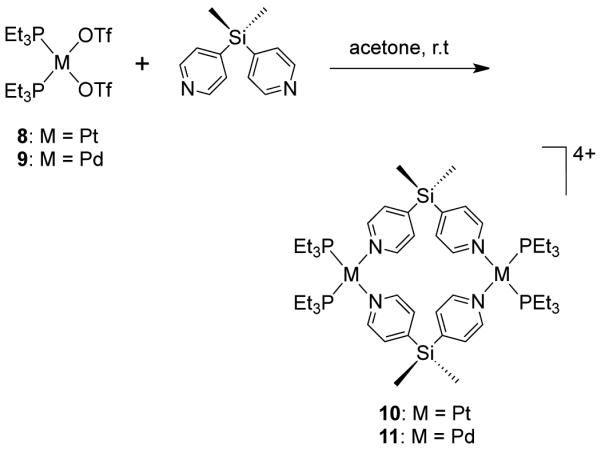
Scheme 5.

3.1.2. Molecular Triangles
A large number of molecular triangles designed by coordination-driven self-assembly have appeared in the literature in last few decades and have been reviewed.40 In principle, molecular triangles can be designed in three general ways (Figure 10). A strain-free molecular triangle can be obtained by the combination of 180° linear metal-containing acceptors with 60° angular donor moieties (Figure 10a). Though the complementary method should also lead to a triangle (Figure 10b), the lack of appropriate metallocorner acceptor units with 60° bite angles makes this assembly impractical. However, the combination of distorted metallocorner units with flexible linear donor units can also give rise to molecular triangles (Figure 10c).40
Figure 10.
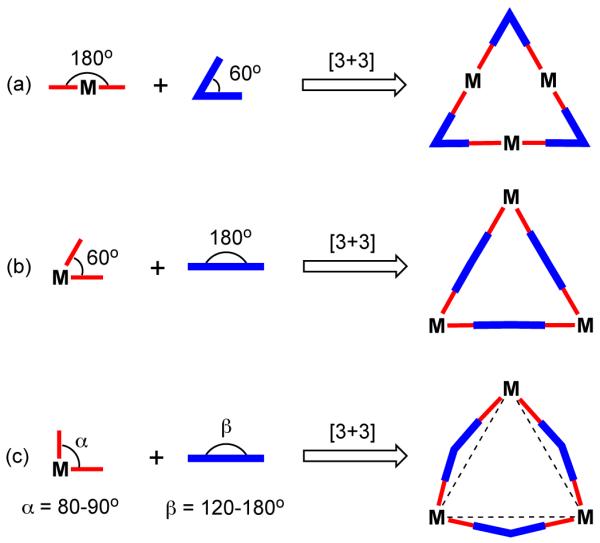
General design strategy for the synthesis of M3L3 molecular triangles.
There exist a large number of strain-free molecular triangles wherein the 60° donor units occupy the vertices and linear metal-containing acceptors form the edges. A well-known family of triangles of this type are the [M(μ-pz)]3 (14; M = Cu, Ag, Au) metallatriangles synthesized using pyrazole or substituted pyrazole and coinage metals (Figure 11).41,42 Dias and coworkers have reported a number of such triangles containing fluorinated pyrazolate ligands and Cu(I), Ag(I) or Au(I).43 These metallacycles have interesting photophysical properties with potential applications as molecular light-emitting devices. 44 Closely related Cu(I) 45 and Ag(I) 46 metallatriangles (15) of the fluorinated triazolate ligand have also been synthesized using the corresponding metal(I) oxides and the triazole. Other molecular triangles have also been reported, in which 4,7-phenanthroline acts as a rigid 60° donor occupying the corners while the linear phenyl bridged dipalladium fragment spans the sides of triangle 16.47
Figure 11.

A few examples of molecular triangles of Cu(I), Ag(I) and Pd(II).
Espinet et al.48 have reported the formation of a strain-free metallotriangle (19) via the 1:1 stoichiometric reaction of trans-[Pt(C6F5)2(AsPh3)2] (17) with 1,2-phenylene diisocyanide (18; Scheme 6). The solid-state structure showed that the phenylene diisocyanide moieties occupy the vertices while the trans-Pt(C6F5)2 fragments act as the linear linkers comprising the sides. The C6F5 rings dispose themselves perpendicular to the plane of the metallotriangle. Following this report, a heterometallic anionic Pt2Au triangle was synthesized in which trans-[Pt(PMe3)2]2+ fragments spanned two of the edges with the other edge consisting of one Au(I) edge connected by an {o-C≡C(C6Me4)C≡C}2− fragment.49
Scheme 6.

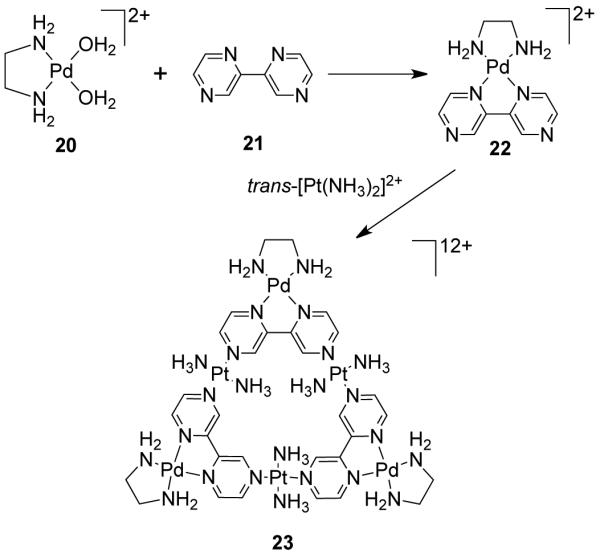
A novel approach for the formation of cationic metallotriangles of this type was demonstrated by Lippert et al. where the vertices of the triangle are metal-containing 60° ligands.50 The reaction between [(en)Pd(H2O)2]2+ (20) and 2,2′-bipyrazine (2,2′-bpz; 21) leads to the formation of a cis-blocked mononuclear complex [(en)Pd(2,2′-bpz)]2+ (22) with two available N-donor sites with coordinate vectors oriented at 60°. The subsequent combination of 22 with trans-[Pt(NH3)2]2+ in a 1:1 ratio forms the cationic metallotriangle 23 (Scheme 7).51,52 Another strategy to achieve this class of metallotriangles involves the use of predesigned terpyridine-based angular linkers that possess a 60° angle between the two terpyridine binding sites.53 For example, the 1:1 stoichiometric reaction of terpyridine-based ligand 24 with FeCl2·4H2O or RuCl2·(DMSO)4 in MeOH leads to the formation of cationic homonuclear molecular triangles [M3(24)3]4+ [M = Fe2+ (25), Ru3+] (Scheme 8).53 NMR (1H, 13C) and electrospray ionization–mass spectroscopic (ESI-MS) studies established the formation of the molecular triangles. Heteronuclear triangles can also be accessed by a stepwise construction process by first preparing a [M2(24)2]4+ species followed by treatment with a different metal ion. Similar predesigned terpyridine based 60° ligand afforded a trinuclear Fe(II) molecular triangle and a tetranuclear molecular square.54
Scheme 7.
Scheme 8.

As mentioned above, the design of molecular triangles with 60° metal-containing corner units and linear donors as edges is impractical due the unavailability of single-center acceptor units with 60° bond angles between the coordinated ligands. However, an elegant use of a 60° diplatinum(II) acceptor unit 12 has made it possible to realize such triangles through coordination-driven self-assembly.55 The [3 + 3] assembly of 12 with different linear bridging bispyridyl ligands, 4,4′-bipyridine, trans-1,2-bis(4-pyridyl)ethylene, and trans-[bis(4-pyridylethynyl)bis(triethylphosphine)] platinum(II), allows for the formation of strain-free hexacationic supramolecular triangles (26-28; Scheme 9). The exclusive formations of the supramolecular triangles were confirmed by multinuclear NMR, elemental analysis, and ESI-mass spectrometry, and in the case of 26, by X-ray crystallography, which revealeda large, accessible cavity. Although the self-assembly of 90° metal corners with linear linkers should form molecular squares, the distortions from linearity of the linkers also permits the formation of triangular structures. A decrease in rigidity of the linkers (i.e. upon increasing the length) allows the coordination sphere to maintain near 90° angles; the strain of the trinuclear metallacycle is relieved by a bending of the ligands. A large number of molecular triangles reported in the last two decades belong to this class. The self-assembly of such triangular assemblies is often accompanied by the concomitant formation of molecular squares. Several research groups have extensively investigated the dynamic equilibria observed in such systems. A discussion of such systems is presented in section 3.1.4. There are, however, but a few examples where charged molecular triangles of this class were selectively obtained through coordination-driven self-assembly of cis-protected metal corner units with linear linkers.
Scheme 9.

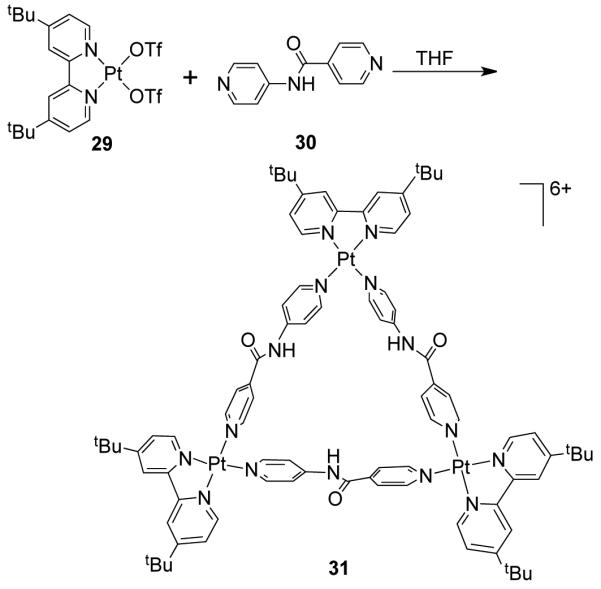
Puddephat et al. have reported the synthesis of cationic molecular triangles by a combination of cis-blocked square planar palladium(II) or platinum(II) units with linear unsymmetrical bis(pyridyl) ligands possessing bridging amides.56,57 The [3 + 3] self-assembly of [Pt(bu2bpy)(OTf)2] (29; bu2bipy = 4,4′-di-tert-butyl-2,2′-bipyridine) with N-(4-pyridinyl)isonicotinamide (30) led to the formation of molecular triangle [Pt3(bu2bpy)3(μ-N-(4-pyridinyl)isonicotinamide)3]6+ (31), as shown in Scheme 10. The analogous Pd(II) acceptor unit [Pd(bu2bpy)(thf)2][BF4]2 also forms a molecular triangle with 30. X-ray structural studies of 31 showed that the three platinum metals occupy the vertices of an approximately equilateral triangle. The unsymmetrical ligand, N-(4-pyridyl)isonicotinamide (30), bridges the metal centers and is bowed slightly outwards. Of the two possible isomers due to the relative orientation of the unsymmetrical linkers, only one of the lower symmetry isomer was isolated in the solid state.
Scheme 10.
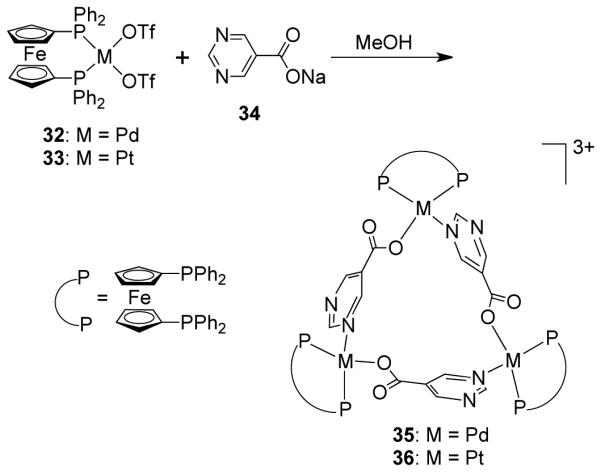
Interestingly, the use of the ambidentate ligand 5-pyrimidine carboxylate with cis-blocked Pd(II) and Pt(II) metal corners led to the formation of the higher-symmetry isomer out of two possible arrangements.58 The [3 + 3] self-assembly of [cis-(dppf)M(OTf)2] (M = Pd (32), Pt (33); dppf = 1,1′-bis(diphenylphosphino)ferrocene]) with sodium 5-pyrimidine carboxylate (5-pmc; 34) led to the formation of cationic triangles [(dppf)3Pd3(5-pmc)3](OTf)3 (35) and [(dppf)3Pt3(5-pmc)3](OTf)3 (36), as shown in scheme 11. Multinuclear NMR, mass spectrometry and X-ray structural studies established the formation of these complexes. The crystal structures show that both symmetrical heterometallic molecular triangles 35 and 36 consist of three partially distorted square-planar Pd(II) or Pt(II) centers present at the vertices, of an approximate triangle with three 5-pmc ligands bridging the metal centers in end-to-end fashion through the pyrimidyl-N on one end and the carboxylate-O on the other. The angle of ca. 100° between the two asymmetric donor sites in the 5-pyrimidine carboxylate ligand favors the exclusive formation of triangles over squares since it reduces the ring strain. A variable temperature 31P NMR study of complexes 35 and 36 established that the triangle is the only product in both cases. The steric bulkiness of the dppf ligand is also a driving force for the preferential formation of triangles. Similar self-selection for the symmetrical isomer was also observed in the self-assembly of the ambidentate isonicotinate ligand with 90° acceptor, [cis-(dppf)Pd(OTf)2] (32).59 Hor et al. have used nicotinates (Nic) as ambidentate linkers for the exclusive assembly of molecular triangles using phosphine-based Pd(II) and Pt(II) metal complexes as corner building units.60 The molecular triangles [(dppf)3Pt3(Nic)3](OTf)3 and [(PPh3)6Pt3(Nic)3](OTf)3 were synthesized by self-assembly. Use of the dimetallic gold containing unit [Au2(dppf)(MeCN)2](OTf)2 in the self-assembly with isonicotinate as the linker led to the formation of hexametallic gold cornered metallotriangles.61
Scheme 11.
Extended and flexible ditopic bispyridyl ligands ‘beaded’ with cucurbituril have been used to assemble interesting molecular necklaces by coordination-driven self-assembly.62 To design the ditopic flexible ligand, terminal pyridyl groups were attached to 1,4-diaminobutane or 1,5-diaminopentane to form short ‘strings’ that were then treated with cucurbituril ‘beads’ to form stable pseudorotaxanes. The self-assembly of the ditopic ligands with 90° cis-blocked [Pt(en)(NO3)2] (37) led to the formation of cationic molecular triangles. X-ray structural studies on the 1,5-dimaniopentane-derived ditopic ligand showed that Pt(II) metal units occupy the vertices of the triangle and are linked on the edges by the sigmoidal-shaped bispyridyl rotaxane (Figure 12). The nature of the pseudorotaxane linkers and reaction conditions significantly influences the final assembly. While refluxing conditions leads exclusively to triangular assemblies, reaction at lower temperature leads to a mixture of triangle and square.
Figure 12.
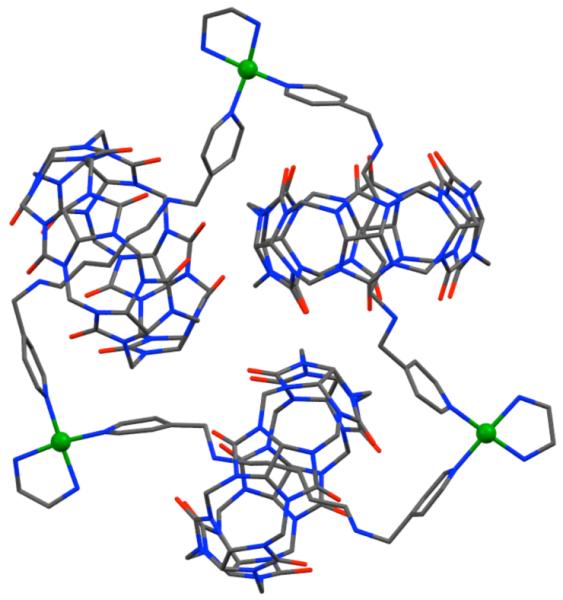
The structure of a triangular molecular necklace. Hydrogen atoms are omitted for clarity. Color code: green, Pt; red, O; blue, N; gray, C.
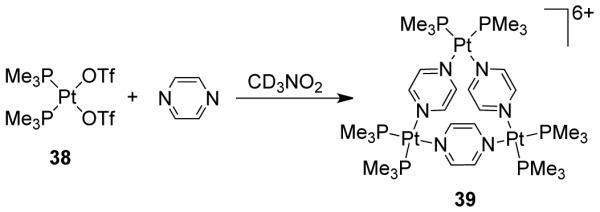
Stang et al. have established the formation of an unusual molecular triangle [(PMe3)6Pt3(μ-pyz)3](OTf)6 (39) upon treatment of [cis-(PMe3)2Pt(OTf)2] (38) with pyrazine (Scheme 12).63 Solid-state structural studies show that despite the considerable strain present in 39, the coordination geometry about the Pt atoms is not significantly distorted from ideal 90° angles to account for the triangular assembly. This result was believed to be due to a combination of entropic factors and the electronic effects of having two platinum centers per pyrazine unit. Two isostructural Pd(II) and Pt(II)-based triangles have been synthesized and structurally characterized using short 4,5-dicyanoimidazolato ligands as the linkers and [M(bu2bpy)Cl2] (M = Pd, Pt; bu2bipy = 4,4′-di-tert-butyl-2,2′-bipyridine) as the metal corners64.
Scheme 12.
A redox-active molecular triangle [(cyclen)3Ru3(bpy)3]Cl6 (cyclen = 1,4,7,10-tetraazacyclododecane), was synthesized by the 1:1 stoichiometric self-assembly of [(cyclen)Ru(DMSO)Cl]Cl and 4,4′-bpy in a 3:1 ethanol:water mixture.65 X-ray structural studies revealed a strained equilateral triangle wherein the 4,4′-bpy bridges are bowed outward, forcing the pyridine rings in each linker to be coplanar and perpendicular to the Ru3 triangle. Using [RuII([9]aneS3)] ([9]aneS3 = 1,4,7-trithiacyclononane) as corner unit, kinetically inert Ru3 molecular triangles that display well-behaved reversible electrochemical behaviour were also reported through self-assembly.66,67 For example, a reaction of [([9]aneS3)Ru(DMSO)Cl2] and 9-methyladenine in presence of a strong Brønsted base led to the formation of a mixed valent Ru3 molecular triangle. 1H NMR, FAB and high-resolution mass spectra as well as elemental analysis established the formation of the tricationic Ru3 molecular triangles. The triangular complexes were found to be kinetically inert in all oxidation states.
Schmittel and coworkers have designed several heterometallic and heteroleptic triangles through a novel multicomponent approach.68 The metallacycles were assembled in a stepwise fashion. A kinetically locked copper(I) complex [Cu(phen)2]PF6 (40) was constructed as a hinge to which free phenanthroline units 41 were attached through Sonogashira coupling, resulting in hinge 42 (Scheme 13). Treatment of precursor 42 with linear ligand 43, containing two free phenanthroline units, in the presence of Cu(I) and Ag(I) ions resulted in the formation of trinuclear metallacycles [Cu3(43)(42)](PF6)3 (44, homometallic and heteroleptic) and [CuAg2(43)(42)](PF6)3 (45, heterometallic and heteroleptic). Using a similar strategy, it was possible to synthesize heterometallic isosceles molecular triangles.69
Scheme 13.

3.1.3. Molecular Squares
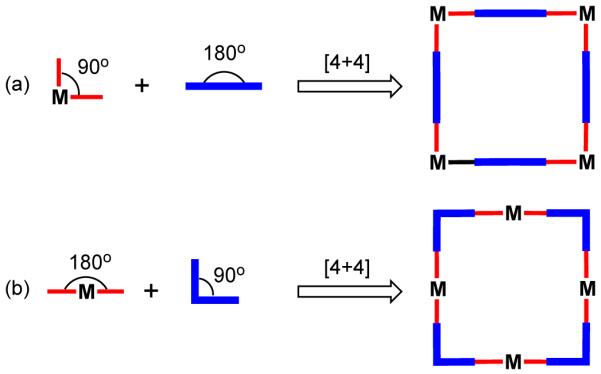
Among the metal mediated assembly of molecular polygons, molecular squares are the most extensively studied. 70 Interest in square metallacycles has been fueled by their ease of preparation, conformational stability and the distinctive properties they exhibit. According to the directional bonding approach, a molecular square can be formed by the combination of a 90° corner unit with a linear bridging ligand. Thus, there are two complementary ways to design a molecular square: a) by the combination of a 90° metal-containing acceptor having two accessible cis-coordination sites with linear donor units (Figure 13a or 13b) by using a 180° linear metal-containing acceptor and a ligand with 90° turn (Figure 13b). Almost any transition metal with a square planar, trigonal bipyramidal, or octahedral geometry can be used as a corner unit. However, transition metals with square planar geometries are most widely used because cis-protected 90° metal corner units can be easily derived by blocking adjacent coordination sites with strong chelating ligands, leaving the other two sites accessible for ligand substitution.
Figure 13.
General design strategy for the synthesis of M4L4 molecular squares.
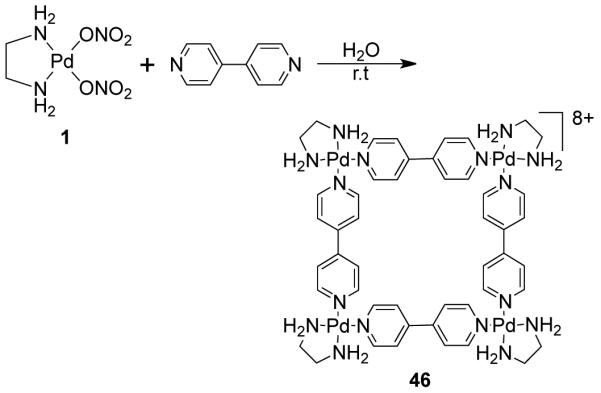
Fujita et al. described the first molecular square (46) via coordination-driven self-assembly of 90° cis-protected palladium(II) with 4,4′-bipyridine as the linear linker, under ambient conditions (Scheme 14).23,24 Since then, a vast number of molecular squares have been reported utilizing this strategy. Interestingly, the Pt(II) analogue of square 46 can be obtained only after heating the reaction mixture at 100 °C for four weeks. This is due to the kinetic inertness of the Pt-N bond relative to the Pd–N bond. Consequently, Pt(II) containing molecular squares are more stable than their palladium analogues. This exemplifies that kinetically stable macrocycles can be obtained under thermodynamic control.
Scheme 14.
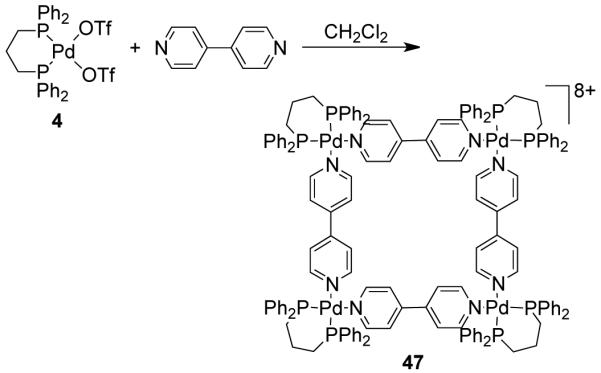
Stang and coworkers have used diphosphine-blocked cis-blocked Pd(II) and Pt(II) complexes extensively as 90° metallocorner units to synthesize a large library of molecular squares of varied size and functionality.8 For example, the phosphino-based counterpart (47) of Fujita’s square can be assembled from a 1:1 stoichiometric mixture of 90° cis-blocked [Pd(dppp)(OTf)2] (4) and 4,4′-bipyridine in CH2Cl2 (Scheme 15).25 The selection of phosphine complexes as corner units enables easy monitoring and reliable characterization of the self-assembled products through 31P NMR spectroscopy. However, the use of phosphine-based ligands makes the ensembles hydrophobic, in contrast to the ensembles derived from amine-based acceptor units.
Scheme 15.
A number of square assemblies containing various other rigid linking subunits, such as p-benzonitrile, 1,4-dicyanobenzene, 4,4′-dicyano-1,1′-biphenyl, diazapyrene and diazaperylene combined with both chelated and non-chelated bisphosphines have been reported by the same group.71,72 The versatility of this self-assembly strategy allows for the synthesis of a series of molecular squares that contain crown ethers or calixarenes as angular units73 or squares which contain porphyrins as linear linking components.74 All these molecular squares were isolated in high yields as robust, air-stable, microcrystalline solids and characterized by multinuclear NMR, mass spectrometry and X-ray crystallographic studies.
Several platinum-based, large metallosupramolecular squares with linear metal-containing acceptors on the edges and donor ligands with a 90° turn were also designed using a similar strategy.75,76 For example, molecular square 50 was made by the self-assembly of linear diplatinum ditopic acceptor 48 with alkynylpyridine-containing donor ligand 49 in CD2Cl2 at room temperature (Scheme 16). Use of bis(heteroaryliodonium) salts as corner units have also led to similar molecular squares with large dimensions. These novel supramolecular species were formed in quantitative yields as assessed by NMR via spontaneous self-assembly and their tetrameric natures were established by electrospray ionization–Fourier transform–ion cyclotron resonance ESI-FT-ICR-mass spectrometry, multinuclear NMR and physical data. Force field calculations and modeling established the size of these species as ranging from approximately 3.6 to 4.7 nm (diagonally) and 2.6 to 3.4 nm (side). Likewise, a series of molecular squares were recently assembled from 90° Pt(II)-based alkynylpyridine-containing donor ligands via coordination-driven self assembly.77 Photophysical studies of these metallacycles revealed high room temperature phosphorescence quantum yields and lifetimes attributed to the excited state becoming localized on the π-conjugated bridging-ligands following intramolecular charge transfer sensitization.
Scheme 16.

A series of terpyridine-based, cationic, novel heterometallic squares {(dppf)Pd[(pyterpy)2Ru]}4(PF6)8(OTf)8 and {fac-Br(CO)3Re[(pyterpy)2M]}4(PF6)8 (M = Fe, Ru, or Os, pyterpy = 4′-(4′″-pyridyl)-2,2′:6′,2′-terpyridine), were prepared by a self-assembly between Re(CO)5Br or [Pd(dppf)(OTf)2] (32) with (pyterpy)2M(PF6)2.78. Würthner et al. have constructed molecular squares of nanometer dimensions via the self-assembly of ditopic perylene bisimide bridging ligands with Pt(II) and Pd(II) phosphine corner units.79 Treatment of ditopic perylene ligand 51 with [M(dppp)(OTf)2] (M = Pd (4), Pt (52)) in CH2Cl2 leads to the exclusive formation of molecular squares 53 and 54 (Scheme 17). The squares were characterized by elemental analysis, 1H and 31P NMR spectroscopy and mass spectrometry. The optical and electrical properties of the ligands are conserved in the final assemblies with platinum-based molecular square 54 showing a fluorescence quantum yield of almost unity and multiple, fully reversible redox couples. Ferrocenyl80 and pyrene81 functional moieties were also incorporated in to the perylene bisimide bridging ligands. The assembly of these functionalized ligands with the Pt(II) phosphine corner unit gave functionalized molecular squares with interesting photophysical and electrochemical properties.
Scheme 17.

Several other molecular squares were also assembled using titanocene82,83 and fluxional thiacrown-based84 ligands as the corner units. The reactions of the low-valent titanocene sources [Cp*2Ti(η2-C2(TMS)2)] and [tBuCp2Ti(η2-C2(TMS)2)] (TMS = tetramethylsilane) with various ditopic bridging linkers such as pyrazine, tetrazine, 4,4′-bipyridine and trans-4,4′-azobispyridine resulted in the formation of titanium-based supramolecular squares.82 Similarly, treatment of a thiacrown-capped Pt(II) complex, [Pt([9]aneS3)Cl2] ([9]aneS3 = 1,4,7-trithiacyclononane) with 4,4′-bipyridine in presence of silver triflate led to a platinum(II) molecular square incorporating four fluxional thiacrown ligands at the corners.84 Hor et al. have described a cationic molecular square [(dppf)4Pt4(isonicotinate)4](OTf)4 obtained through a ligand displacement reaction in which a dinuclear Pt2Br2(dppf)2(C8H4S2) species exchanges with isonicotinic acid to release free bithiophene, giving the molecular square.85 Solid-state structural studies revealed that a highly symmetrical structure was formed with the four bridging isonicotinate ligands oriented in a head-to-tail fashion. In a separate study, the palladium analogue of the molecular square was crystallized from an equilibrating mixture of a triangle and square (vide infra).86
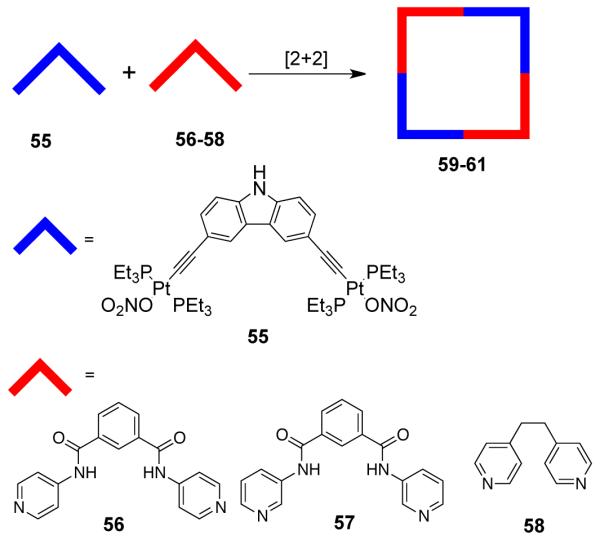
A series of [2 + 2] self-assembled molecular squares were recently reported utilizing a 90° platinum(II)-based carbazole ligand as the acceptor in conjunction with various flexible ditopic ligands as donors.87 The treatment of 3,6-bis[trans-Pt(PEt3)2(NO3)(ethynyl)]carbazole (55) with pyridyl donor ligands 1,3-bis(4-pyridyl)isophthalamide (56); 1,3- bis(3-pyridyl)isophthalamide (57), and 1,2-di(4-pyridyl)ethane (58) led to the formation of molecular squares 59-61 in good yields (Scheme 18). Multinuclear NMR and ESI-mass spectrometric studies established the formation of the squares. Fluorescence studies on the square 59 showed that it could selectively sense pyrophosphate anion. Enhancement of fluorescence intensity was observed upon titration with aqueous P2O74− anion in DMF solution. Interestingly, no such perturbation in fluorescence intensity was observed when the titration was carried out with other anions (F−, ClO4−, H2PO4−). A novel redox active [2 + 2] molecular square containing two vinylogous tetrathiafulvalenes and two molybdenum tetracarbonyl fragments was also reported via coordination-driven self-assembly of complementary angular units.88
Scheme 18.
Mirkin et al. have designed a tetranuclear heterobimetallic square through the cooperative ligand binding properties of square planar and tetrahedral metal centers.89 The treatment of a flexible phosphinothioether-based hemilabile ligand with a mononuclear Rh(I) source in a 2:1 ratio, followed by the addition of one equivalent of Zn(II) leads to the formation of heterobimetallic square 62 in high yield. X-ray structural studies have shown that the two square planar Rh(I) centers and the two distorted tetrahedral Zn(II) centers are bound by four hemilabile ligands. It was observed that the cis-phosphine/cis-thioether arrangement of the ligands around the Rh center is crucial in enforcing the directionality of the ligand.89 A homometallic Rh(I) tetranuclear metallacycle (63) was also isolated and structurally characterized (Figure 14).90
Figure 14.
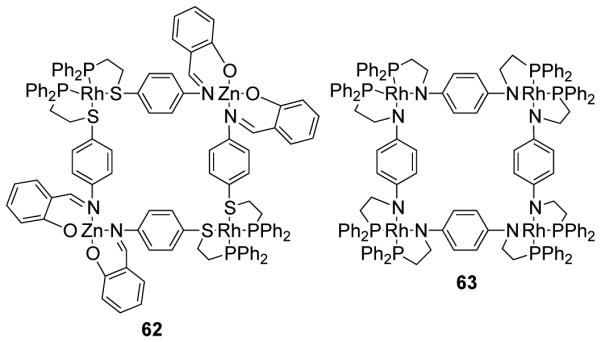
Rh(I) based squares through the cooperative ligand binding properties.
3.1.4. Triangle-Square Equilibrium
The composition of a self-assembly product depends upon the rapid chemical exchange between the complementary building blocks, reaction intermediates and the final ensembles. This enables the system to self-correct, giving the most stable thermodynamically-controlled product. However, thermodynamic control only affords the formation of a single product when there is a sufficient energetic advantage of one species over the other possible species. There are several metallosupramolecular systems where two or more macrocyclic species exist in dynamic equilibrium in solution, due to the lack of a clear thermodynamic preference for one species over the other.
According to the directional bonding approach the self-assembly of 90° metal corners with linear and relatively rigid bridging ligands, in principle, should result in molecular squares. However, several such systems have afforded instead an equilibrium mixture of supramolecular squares and triangles. These equilibria can be rationalized from a thermodynamic point of view; a delicate balance between entropy and enthalpy determines the ratio of squares and triangles. While a molecular square is enthalphically favored due to a smaller conformational strain as compared to triangles, entropy favors the formation of triangles since they are assembled from a smaller number of components. The equilibrium between triangle and square can be written as
Thus, according to Le Chatelier’s principle, an increase in the concentration of the components in the mixture shifts the equilibrium from triangles to squares, while an increase in temperature drives the equilibrium towards the triangles, since the transformation from squares to triangles is expected to be an endothermic process (Figure 15).
Figure 15.
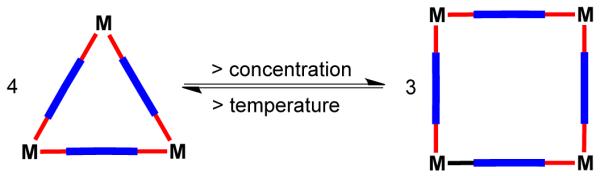
Equilibrium between a self-assembled molecular triangle and a square.
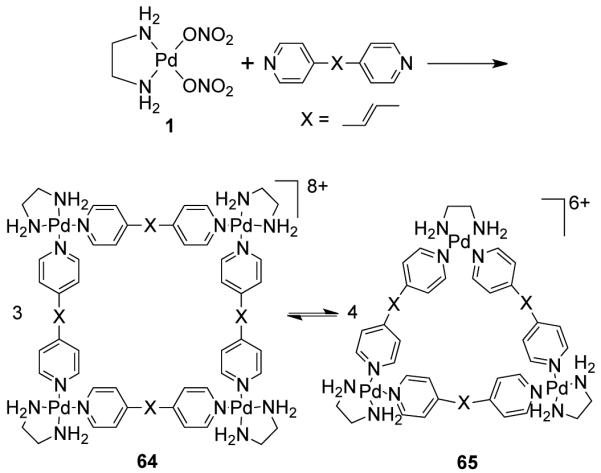
The nature of the linear ligands also plays a vital role in the triangle-square equilibrium. An increase in the flexibility of the ligands favors the formation of triangles by delocalizing strain through small deformations in the backbone. Sufficiently rigid linkers shift the equilibrium significantly in the direction of the molecular square since the enthalpic gain compensates for the entropic penalty of square formation. It was Fujita et al. who first observed that molecular triangles are favored over the corresponding squares when extended and constitutionally flexible bispyridyl linkers were employed.91 For example, the assembly of [Pd(en)(NO3)2] (1) with trans-1,2-bis(4-pyridyl)ethylene led to the formation of two different macrocycles: molecular square 64 and molecular triangle 65 (Scheme 19). The two macrocycles were in dynamic equilibrium with 64 being the major product. At higher concentration, the equilibrium shifted towards the square, producing three molecules of 64 from four molecules of 65.
Scheme 19.
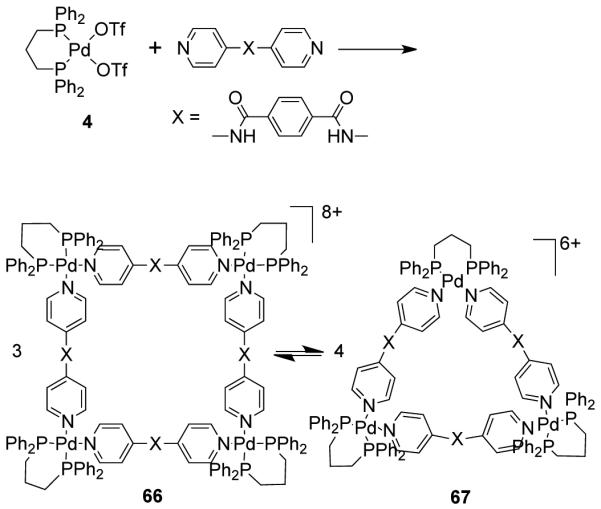
Schalley et al.92 have employed a series of extended and flexible bispyridyl linkers with cis-blocked [M(dppp)(OTf)2], (M = Pd (4), Pt (52)) complexes as the corner building units to demonstrate the triangle-square equilibrium in solution. Scheme 20 shows the equilibrium for Pd(II) square 66 and triangle 67 in the reaction of [Pd(dppp)(OTf)2] (4) and N,N’-di(4-pyridyl)terephthalamide. The complexes under study were characterized by a combination of multinuclear NMR, ESI-FT-ICR-mass spectrometry, and X-ray crystallography. Temperature, concentration and solvent type significantly influenced the equilibrium. At higher temperature, the equilibration of the squares and triangles is significantly faster as determined from variable temperature NMR spectroscopy. Due to the greater kinetic stability, the ligand exchange process in the Pt(II) complexes is slow compared to the Pd(II) complexes. Consequently, the triangle-square equilibration is significantly faster for Pd(II) complexes than their Pt(II) analogues. It was observed that the square/triangle ratio is influenced by the change in building block concentration. Increasing amounts of nonpolar solvents promotes the exchange processes resulting in an increased preference for triangle formation.92
Scheme 20.
The triangle/square equilibria also depend on the nature of the ancillary ligands on the metal fragments. It has been shown that even the rigid linker 4,4′-bipyridine can lead to a mixture of the two macrocycles: square and triangle when 2,2′-bipyridine replaces the ethylenediamine capping ligand.91 The effect of the ancillary ligands on the metal fragments on the equilibrium was suggested to be due to the steric repulsion between the 2,2′-bipyridine and 4,4′-bipyridine moieties present in the macrocycles, pushing the equilibrium to some extent towards the formation of the molecular triangle. A recent study on the influence of the steric properties of chelating diamines on the position of the trimeric/tetrameric equilibrium has also corroborated the above suggestion.93 In the self-assembly of [Pd(N-N)](NO3)2 with 4,4′-bipyridine as the bridging ligand, the effects of several diamine ancillary ligands (N-N) such as N,N,N’,N’-tetramethylethylenediamine, N,N,N’,N’-tetraethylethylenediamine, 1,3-diaminopropane, N,N’-dimethylpiperazine, homopiperazine and ethylenediamine on the final assembly were systematically investigated by 1H and DOSY NMR spectroscopy and X-ray diffraction. All the chelating diamine ligands exhibited dynamic triangle-square equilibria. Molecular triangles became the major components of the mixture when the diamine was bulky, while less bulky chelates favored molecular squares.
Ferrer and coworkers94 have also extensively investigated the effect of the ancillary ligands on the equilibria between molecular triangles and squares with different diphosphine chelating ligands. The reaction of cis-blocked [M(P-P)(OTf)2] precursors (M = Pt, Pd) obtained from different diphosphines - diphenylphosphinopropane (dppp), diphenylphosphinoferrocene (dppf) and 1,2-bis(diethylphosphino) ethane (depe) with the rigid linker, 1,4-bis(4-pyridyl)tetrafluorobenzene, led to the formation of a mixture of the corresponding molecular squares and triangles. The characterization of the dynamic triangle/square equilibria by multinuclear NMR in combination with mass spectrometry revealed a dependency of the square/triangle ratio on several factors, such as the nature of the metal corners, concentration and solvent. The use of sterically demanding diamines 4,4′-R2bpy (bpy = 2,2′-bipyridine; R = H, Me, t-Bu) as ancillary ligands in the cis-protected palladium(II) and platinum(II) complexes [M(4,4′-R2bpy)(OTf)2] (M = Pd, Pt; R = H, Me, t-Bu; bpy = bipyridine) with the same 1,4-bis(4-pyridyl)tetrafluorobenzene ligand also showed a dynamic equilibrium between molecular triangle and square.95 However, the analogous reactions where ethylenediamine was used as the ancillary ligand led to only molecular squares.94 Interestingly, it has also been shown that mononuclear species may also participate in square/triangle equilibria when a longer octafluoro containing rigid linkers are used in conjunction with [(tmen)Pd(NO3)2] (tmen = tetramethyl ethylenediamine).96
The nature of the linear linkers also has a profound impact on triangle-square equilibria. In their study of the self-assembly of 90° cis-protected [Pd(dppp)(OTf)2] (4) with phenoxy-substituted diazadibenzoperylene linear linker, 68, Würthner and coworkers have observed a complex dynamic equilibria between molecular triangle 69 and square 70 (Scheme 21).97 Linear linker 68 has a highly twisted and non-flexible aromatic backbone. Despite the possibility of π–π interactions between the two phenyl groups of the dppp ligand and the π-surface of the diazadibenzoperylene ligands, which should stabilize the molecular square, the steric requirement of the phenoxo groups on 70 drives the equilibrium towards the triangle, as more space is available. The additional space makes the triangle energetically favorable, despite the distorted geometry at the metal centers. However, the use of a perylene bisimide ligand that is significantly longer and thus can reduce the steric crowding of the phenoxo substituents results in the exclusive formation of the molecular square.98,99
Scheme 21.

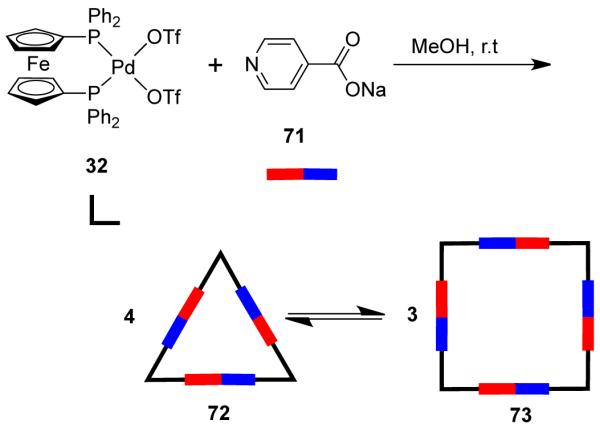
Unfortunately, selective isolation of both species involved in a square-triangle equilibrium is not always possible. In their investigation of the equilibrating mixture of a triangle and square obtained by the self-assembly of [Pt(PMe3)2(OTf)2] (38) with bispyridyl ethylene (bpe), Stang and coworkers accomplished the selective crystallization of either of the two species via the appropriate choice of solvents and nature of the anions present in the system.100 While the cationic molecular square was crystallized as the triflate salt, [cis-(PMe3)2Pt(bpe)4](OTF)8, the molecular triangle was crystallized as a mixed triflate/cobalticarborane (CoB18C4H22−) salt, [cis-(PMe3)2Pt(bpe)3](OTF)4(CoB18C4H22−)2. Similar results were observed in a study86 focused on the use of ambidentate linkers to construct supramolecular macrocycles with cis-blocked Pd(II) and Pt(II) complexes as corner pieces. The self-assembly reaction of 90° acceptor [cis-(dppf)Pt(OTf)2] (33) with an equimolar amount of the ambidentate ligand sodium isonicotinate (71) allows for the formation of a mixture of symmetrical triangles (72) and squares (73) in solution (Scheme 22). Selective crystallization of a rhomboid was achieved from a dynamic equilibrium between a supramolecular dimeric rhomboid and trimeric hexagon.101 However, the species isolated in the solid state might not be necessarily the predominant species in solution.
Scheme 22.
An analogous reaction using [(tmen)Pd(NO3)2] (tmen = tetramethyl ethylenediamine) also produced a mixture of symmetrical triangles and squares in solution. The assemblies were characterized by 1H, 31P and DOSY NMR spectroscopy and X-ray diffraction. Though only the molecular square crystallized in both cases, the solution composition was a mixture of square and triangle, with the latter as the major component in both cases. The square/triangle ratio depends on the temperature as well as the concentration of the building blocks.86
There are other systems where self-assembly reactions have led to the formation of mixture of triangles and squares without any equilibrium between them. For example, Yu et al. have reported the synthesis of bispyrazole-based molecular triangles and squares using Pd(II) and Pt(II) based dimetal clips.102 The self-assembly reaction of a series of tetramethyl substituted bispyrazolato ligands – 3,3′,5,5′-tetramethyl-4,4′-bipyrazolyl, 1,4-bis-4′-(3′,5′-dimethyl)-pyrazolylbenzene, 1,4-bis-4′-(3′,5′-dimethyl)-pyrazolylbiphenyl of different lengths with dimetal clips [M(2,2′-bpy)(NO3)2]2(NO3)2 (M = Pd or Pt, 2,2′-bpy = 2,2′-bipyridine) or [M(phen)(NO3)2]2(NO3)2 (M = Pd or Pt, phen = 1,10-phenanthroline) allowed for the formation of molecular triangles and squares through spontaneous deprotonation of the pyrazolato ligands. As determined by NMR, the macrocycles were stable and no dynamic equilibrium between the corresponding triangles and squares was observed. The treatment of Pd(II) dimetal clips with the longer bispyrazolate linkers led to the formation of a non-equilibrating mixture of the corresponding molecular triangles and squares. The triangle/square ratio is dependent on the length of connecting linkers. With longer linkers, the ratio of triangle/square is nearly unity. However, with shorter ligands, the triangle/square ratio significantly shifts in favor of the square. Interestingly, with the shortest bispyrazole ligand (3,3′,5,5′-tetramethyl-4,4′-bipyrazolyl), molecular triangles were obtained exclusively with both Pd(II) and Pt(II) dimetal clips. These tetrasubstituted bispyraole linkers have interplanar angles of about 50-90° due to the steric repulsion of the four methyl groups, resulting in the formation of distorted metallomacrocyclic assemblies. The synthesis of symmetrical triangular and square assemblies using an unsubstituted coplanar 4,4′-bipyrazolate ligand has also been reported.103
Sauvage et al.104,105 have observed the existence of a dynamic equilibrium between trinuclear and tetranuclear cyclic pseudorotaxanes. The Cu(I)-mediated assembly of trinuclear [Cu(74)]33+ and tetranuclear [Cu(74)]44+ pseudorotaxanes were attained by using a phenanthroline containing macrocycle which is rigidly attached to a filament bearing a second phenanthroline unit, 74 (Scheme 23).105 The coordination vectors of the two binding sites are orthogonal to each other, forcing the ligand to orient in an iso-fashion. A similar ditopic ligand having phenanthroline and terpyridine binding sites orthogonal to each other gave a non-equilibrating mixture of dinuclear and trinuclear macrocycles where the Cu(I) atom was pentacoordinated to a terpyridine unit and a phenanthroline unit from another ligand.106
Scheme 23.

3.1.5. Molecular Rectangles
The design and synthesis of molecular rectangles has motivated supramolecular chemists for some time, largely due to their interesting photophysical and molecular recognition properties.107 However despite the relative topological simplicity, the syntheses of molecular rectangles are not straightforward. Combination of a 90° metal precursor with two rigid linear linkers of differing lengths in the absence of any driving bias should result in a molecular rectangle. However, two molecular squares of different sizes are generally obtained instead of a rectangle due to the strong enthalpic driving force in favor of square formation (Figure 16) and the lack of selective recognition by the two different linear linkers. Only in case of Re(I) were a few rectangles obtained using a three-component assembly approach where two linear linkers of different lengths were utilized.108
Figure 16.
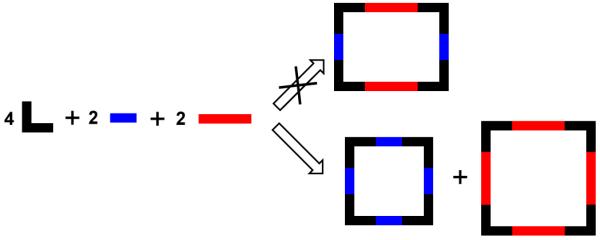
Formation of molecular squares of different sizes instead of a rectangle due to the strong enthalpic driving force.
As a result, various groups have developed several different approaches over the last few years for the preparation of supramolecular rectangles. Stang and coworkers have developed an approach wherein a predesigned platinum-based molecular clip containing two parallel donor sites are disposed in the same direction is treated with linear ditopic ligands to achieve self-assembled molecular rectangles.109 The [2 + 2] self-assembly of diplatinum(II) molecular acceptor 1,8-bis[trans-Pt(PEt3)2(NO3)]anthracene 75 with rigid bispyridyl bridging ligands – 4,4′-bipyridine, trans-1,2-bis(4-pyridyl)ethylene, 1,4-bis(4′-pyridylethynyl)benzene, 2,5-bis(4′-pyridylethynyl)furan and 3,8-bis-pyridin-4-ylethynyl-[1,10]-phenanthroline led to the formation of cationic molecular rectangles 76-80 (Scheme 24). Characterization of the rectangles was accomplished with multinuclear NMR and UV-vis spectroscopy, FAB-mass spectrometry, and X-ray crystallography. Spectroelectrochemical studies on 76 and 77 showed that the compounds could be reversibly reduced at the neutral bispyridyl acceptor ligands while oxidation occurs at the dianionic anthracene clips.110 The phenanthroline functionalized molecular rectangle 80 has been shown to act as an optical sensor for Ni(II), Cd(II) and Cr(III) ions.111
Scheme 24.
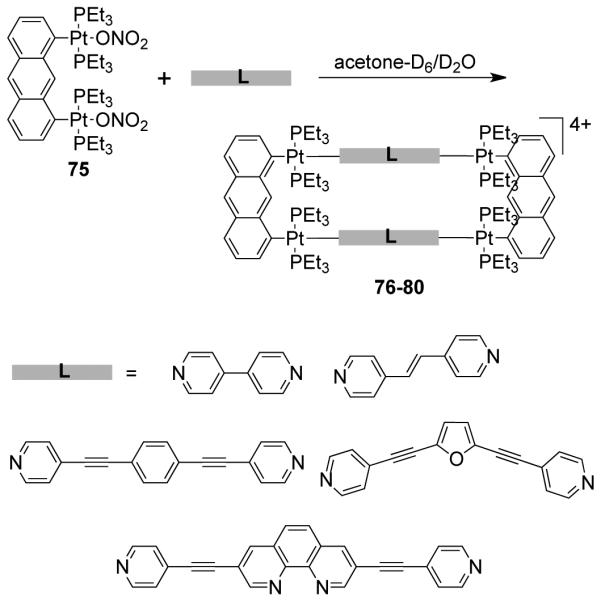
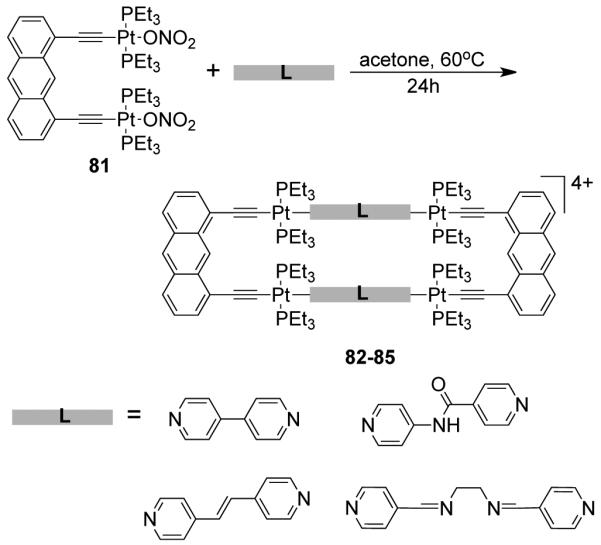
Similarly, the [2 + 2] self-assembly of a diplatinum(II) molecular clip (75) with a linear unsymmetrical bispyridyl ligand, N-(4-pyridinyl)isonicotinamide (30) led to the formation of a symmetrical rectangle.112 Of the two possible linkage isomers, only the most symmetrical one was observed based on multinuclear NMR, mass spectrometry and X-ray structural studies. The solid-state structure of the rectangle shows that the bispyridyl ligands occupy the long edge of the rectangle. A functionalized bis(pyridyl)-substituted perylene diimide ligand was also incorporated by treatment with the anthracene based clip (75) to form a functional molecular rectangle.113 Extending the same strategy further, a series of discrete cationic rectangles (82-85) were synthesized using a di-Pt(II) molecular clip containing ethynyl functionality 81 with bispyridyl linear linkers such as 4,4′-bipyridine, trans-1,2-bis(4-pyridyl)ethylene, N-(4-pyridyl)isonicotinamide and N,N’-bis(4-pyridylidene)ethylenediamine (Scheme 25). 114 The assemblies were characterized by multinuclear NMR and mass spectrometry. Rectangle 85 was designed with the aim of introducing a N4 pocket that could act as a receptor site for transition metal ions. The incorporation of ethynyl functionalities introduced fluorescent behavior into the discrete assemblies. Molecular rectangle 85 was fluorescent and showed quenching in solution upon the binding of hard transition metal ions (Fe3+, Cu2+, Ni2+, and Mn2+) into the N4 pocket. Ko et al. have reported a series of Pt4 rectangles from a diplatinum aromatic molecular clip bearing two symmetrically bound platinum moieties.115
Scheme 25.
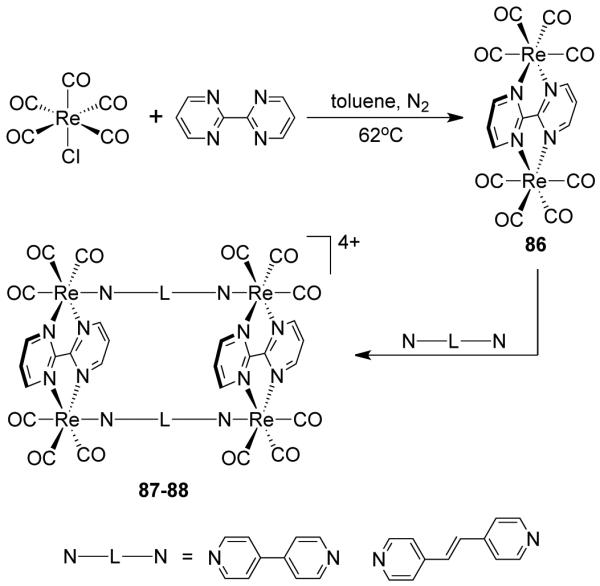
Bosnich et al. reported the synthesis of a molecular clip bearing two symmetrically bound terpyridyl chelators separated by aromatic spacers.116 Dipalladium(II) complexes of these spacer-chelators readily form large molecular rectangles with linear linkers such as 4,4′-bipyridine. Hupp and coworkers have designed rhenium based molecular rectangles via a step-wise process (Scheme 26).117 A stable bimetallic edge 86 was created first by treating fac-Re(CO)5Cl with a rigid 2,2-bipyrimidine ligand. Subsequent assembly of the edges with bispyridyl ditopic linker 4,4-bipyridine and trans-1,2-bis(4-pyridyl)ethylene resulted in the formation of cationic molecular rectangles 87 and 88.117 Cationic molecular rectangles containing cofacial porphyrin edges were assembled from the reaction of bis(4-ethynylpyridyl)porphyrin with bimetallic edge 86 in an equimolar ratio.118
Scheme 26.
Suss-Fink and coworkers119 have prepared the cationic molecular rectangle [Ru4(p-cymene)4(μ-bpy)2(μ-C2O4)2]4+ 91 by first creating a bimetallic edge 90, formed from the treatment of dimeric ruthenium arene precursors 89 with oxalate, and then assembling it with the ditopic linear linker 4,4′-bipyridine in MeOH (Scheme 27). Alternately, reactions of [Cp*MCl2]2 (M = Rh, Ir) with bidentate ligand (L = pyrazine; L’ = diisocyanide) gave the bimetallic edge [Cp*MCl2(L or L’)]2, which upon treatment with silver trifluoromethanesulfonate (AgOTf) led to tetranuclear cationic metallarectangular complexes [Cp*2M2Cl2(L)(L’)]2(OTf)4 containing different ligands.120
Scheme 27.

Building upon the same strategy, Therrien et al.121 prepared a series of cationic arene ruthenium-based molecular rectangles having large cavities. For example, metallarectangles [Ru4(η6-p-cymene)4(μ-N ∩ N)2(OO ∩ OO)2]4+ were synthesized from the dinuclear arene ruthenium complexes [Ru2(η6-p-cymene)2(μ-OO ∩ OO)2Cl2] (OO ∩ OO = 2,5-dihydroxy-1,4-benzoquinonato (dhbq), 122 2,5-dichloro-3,6-dihydroxy-1,4-benzoquinonato (dchq),122 5,8-dihydroxy-1,4-naphthoquinonato (dhnq), 123 9,10-dihydroxy-1,4-anthraquinonato (dhaq), 124 or 6,11-dihydroxy-naphthacene-5,12-dionato(dhtq)124) by reaction with pyrazine, bipyridine or 1,2-bis(4-pyridyl)ethylene linkers (N ∩ N = pyrazine, 4,4′- bipyridine, 1,2-bis(4-pyridyl)ethylene) in the presence of AgOTf in methanol. The complexes were characterized by spectroscopic methods, X-ray crystallography and cyclic voltammetry. Host-guest studies using aromatics as the guest molecules in these metallarectangles suggested that metallarectangles incorporating 1,2-bis(4-pyridyl)ethylene as the linkers were able to host an anthracene, pyrene, perylene, or coronene molecule in their cavity while rectangles having 4,4′- bipyridine as the linker can only encapsulate anthracene. However, out-of-cavity interactions were observed between 4,4′-bipyridine-containing rectangles and pyrene, perylene, or coronene. In contrast, the small pyrazine-containing metallarectangles show no interaction in solution. Jin and coworkers have also synthesized similar metallarectangles by the combination of the unsaturated dinuclear arene-ruthenium, iridium and rhodium clips and various linear bidentate pyridyl connectors, pyrazine, 4,4-bipyridine, 1,2-bis(4-pyridyl)ethylene and 4-[5-(4-pyridyl)-1,3,4-oxadiazol-2-yl]pyridine to generate the corresponding metallarectangles.125,126 The metallarectangles having 1,2-bis(4-pyridyl)ethylene as the connectors were also shown to undergo a [2 + 2] cycloaddition of the olefinic double bonds under UV radiation both in solution and in the solid-state.127,128 Recently, the same group reported the synthesis of half-sandwich iridium metallarectangles induced by C–H activation.129,130 Bimetallic edges, formed from half-sandwich iridium arene precursors and pyrazine or bispyridyl linkers, were converted to tetranuclear metallarectangles upon treatment with dicarboxylic acids such as fumaric acid, 1,3-benzenedicarboxylic acid, 5-amino-1,3-benzenedicarboxylic acid or azobenzene 4,4′-dicarboxylic acid. The Cp*IrIII fragment catalyzes the C–H activation of dicarboxylic acids to build the macrocyclic architectures.
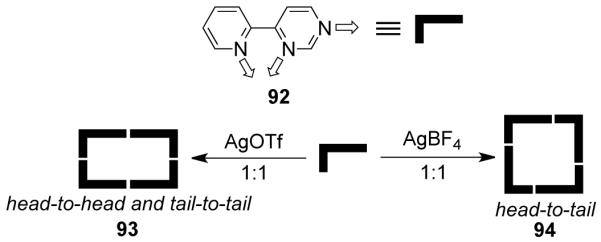
Silver-based cationic molecular metallacycles were designed using 4-(2-Pyridyl)-pyrimidine (92) as the ligand.131 Due to the presence of the two orthogonal metal binding sites, a head-to-tail aggregation would give a molecular square while the head-to-head or tail-to-tail aggregation would result in a rectangle. The reaction of 92 with AgOTf in a 1:1 stoichiometric ratio led to head-to-head and/or tail-to-tail aggregation, resulting in rectangular ensemble 93 (Scheme 28). However, the use of AgBF4 (instead of AgOTf) led to the formation of square assembly 94 with a head-to-tail aggregation. Treatment of a N-methylated bis(amidopyridine) ligand with silver salts AgNO3, AgO2CCF3, AgO3SCF3, AgBF4, and AgPF6 gave the corresponding cationic disilver(I) macrocycles.132 Heteronuclear cationic molecular rectangles having two redox active ferrocene subunits and a pair of transition metal atoms were reported by Lindner et al.133 A redox-active Pd-based rectangle [(η5-C5H4C2-3-py)2Fe]2Pd2Cl4 was prepared from PdCl2(COD) and (η5-C5H4C2-3-py)2Fe. The same donor clip yielded a Ag(I)-based molecular rectangle upon treatment with AgClO4. The Ni2 rectangle, [(η5-C5H4C2-4-py)2Fe]2Ni2(NO3)4, was obtained by treatment of [Ni(H2O)6](NO3)2 with (η-C5H4C2-4-py)2Fe.
Scheme 28.
3.1.6. Higher Polygons and Large Ring Systems
Several other higher order 2D polygons have been reported by using specifically designed organic acceptors with directional metal acceptors. Using the directional bonding approach, hexagonal assemblies can be realized in two complementary ways a) by the combination of six 120° units with six complementary linear units, or b) by the combination of three 120° units with three complementary 120° units. Similarly, discrete pentagonal ensembles can be assembled by the incorporation of five 108° building units with five complementary linear units. For example, Scheme 29 illustrates the assembly of a molecular hexagon via the combination of nitrogen containing corner units having 120° bond angles with complementary linear organometallic linking units. As the covalent angle of an sp2 hybrid carbon is 120°, in the case of bis(4-pyridyl) ketone (95), treatment with 96 in dichloromethane at room temperature afforded the desired hexamer, 97.134 In a complementary approach, the 120° diplatinum(II) acceptor forms the corner unit with 4,4′-bipyridine as the linear linker to also afford a [6 + 6] molecular hexagon. A [3 + 3] molecular hexagon was assembled from a 1:1 reaction of 95 with complementary 120° di-Pt(II) acceptor.135
Scheme 29.
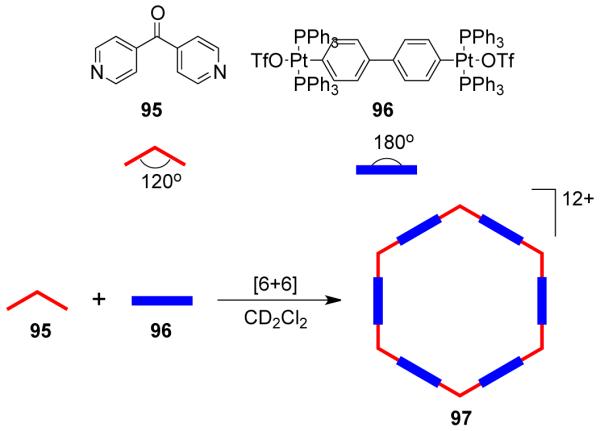
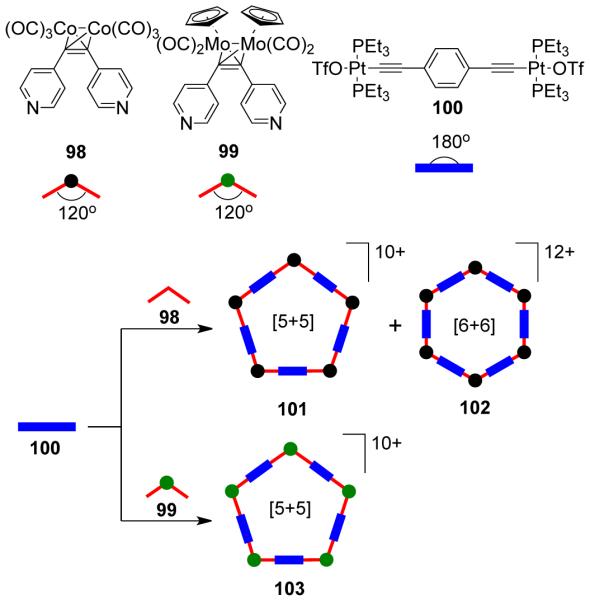
However, an equilibrium mixture of hexagonal and pentagonal metallacycles was often observed in such an assembly due to the small, 12° difference between the 120° internal turning angles needed for the hexagonal assembly and the 108° angle in analogous pentagonal assemblies. The relatively large size of the assemblies and the flexibility of the building units allow the distortion needed for shifting the equilibrium from one to the other. The use of two metal-carbonyl coordinated dipyridyl donors, 98 and 99, with linear diplatinum (II) acceptor 100 led to the interesting observation that the steric-bulk of the metal carbonyl corners controls the hexagonal/pentagonal equilibrium (Scheme 30).136 The use of a sterically less-imposing ligand (98) resulted in a mixture of a [5 + 5] pentagon (101) and a [6 + 6] hexagon (102), while the exclusive formation of a [5 + 5] pentagon (103) was achieved by the use of bulkier molybdenum donor ligand 99 with linear organoplatinum(II) acceptor 100. Multinuclear NMR and electrospray ionization–time of flight (ESI-TOF)-mass spectrometry established the formation of these assemblies.
Scheme 30.
Using a different approach developed by Lehn and coworkers, “naked” metal ions were utilized as the effectors to trigger the spontaneous ordering of individual components into higher order stereospecific circular helicates.137 Iron containing metallacyclic pentagons and hexagons were assembled by using a trisbipyridyl ligand, tris(2,2′-bipyridine). When the ligand was treated with FeCl2 in ethylene glycol at 170 °C, the formation of a pentanuclear assembly was observed, while replacement of FeCl2 with FeSO4 led to the generation of hexanuclear circular helicates. In the pentanuclear assembly, the chloride ion occupied the cavity of the macrocycle and is perhaps responsible for the preferred formation of the molecular pentagon in the first reaction. However, increasing the flexibility of the trisbipyridyl ligand by introducing ether linkages led only to molecular squares with different Fe(II) salts.138 These reactions demonstrate the remarkable influence of the ion-templating effect on the formation of polynuclear assemblies.
Dunbar et al. extensively investigated the role of anions in anion-templated self-assembly reactions between first-row transition metal ions and a divergent bis-bipyridine ligand.139,140 Anions can direct the course of an assembly process via noncovalent interactions due to their wide range of coordination geometries, pH sensitivity, and high free energy of solvation. In the reaction of 3,6-bis(2-pyridyl)-1,2,4,5-tetrazine (bptz) with Ni2+ ions, the larger anion [SbF6]− favored the formation of molecular pentagon [{Ni5(bptz)5(CH3CN)10}⊂SbF6]9+ while relatively smaller anions – ClO4− and BF4−, yielded molecular squares [{M4(bptz)4(CH3CN)8}⊂X]7+ (M = Ni(II), Zn(II); X = ClO4− and BF4−). The molecular pentagon can easily be converted to the square in the presence of excess of BF4−, ClO4−, or I− anions. However, the Ni(II) square can be partially converted to the less stable pentagon only under more forcing conditions in the presence of excess [SbF6]− ions. Thus, by proper choice of counterions, the assembly process can be tuned to the desired macrocyclic assembly. These circular helicates can have helical (interwoven) and parallel (grid-type) arrangements of the ligands, depending on the transoid or cisoid orientations of the coordinating sites in the ligand. This is in contrast to the assemblies generated through the directional paradigm, where all the ligands and the metal centers lie on the same plane. von Zelewsky and coworkers synthesized an interwoven molecular hexagon by employing a pinene-2,2′-bipyridine ligand.141 Treatment of the α,α’-bis(pinene-2,2′-bipyridyl)-p-xylene linker with AgPF6 in a mixture of acetonitrile and chloroform spontaneously led to the formation of circular single-stranded helicates, as observed from X-ray structural studies. NMR and CD spectroscopy in solution, along with ESI-mass spectrometry results, indicated that the assembly was stable in solution. An equilibrating mixture of a [2 × 2] grid-type tetranuclear assembly and a hexanuclear assembly was obtained when a ligand designed with a rigid 4,7-phenanthroline unit between the two binding sites was treated with copper(II) cations in a 1:1 ratio in acetonitrile.142 Interestingly, a solvent-dependent reversible switching between tetramer and hexamer was observed. While acetonitrile favored the formation of hexamer, nitromethane afforded the formation of the tetranuclear assembly only. Coronado et al. have reported the synthesis of a metallosupramolecular hexagon from the self-assembly of a mixture of Cu(II) ions and a rigid heteroditopic ligand containing phenanthroline and terpyridine binding units.143 The 1:1 reaction of a ditopic ligand containing a terpyridine tridentate ligand and a 1,10-phenanthroline bidentate moiety directly connected to one another (terpy-phen) with Cu(OAc)2 in methanol yielded the hexameric cation [Cu6(terpy-phen)6(PF6)6]6+ (Figure 17). Each copper ion is bound to two ligands through the phenanthroline and terpyridine binding sites and a weakly coordinated PF6− anion. As expected for grid-type assemblies, the coordinating ligands are disposed in a cisoid fashion above and below the mean plane of the hexamer.
Figure 17.
Molecular structure of [Cu6(terpy-phen)6(PF6)6]6+. Weakly coordinating PF6− were omitted for clarity. Color code: golden, Cu; red, O; blue, N; gray, C.
A number of higher ordered 2D multinuclear supramolecular assemblies were constructed using specifically designed mono- and bis-terpyridine ligands with various transition metals.144 Newcome and coworkers assembled both homonuclear145 and heteronuclear146,147 hexametallic macrocycles using a bis(terpyridyl) monomer possessing a 120° angle with respect to the two ligating moieties. The reaction of the bis(terpyridyl) monomer with two equivalents of RuCl3·nH2O or FeCl2·4H2O produced the corresponding bis-metallic adducts which, on further treatment with one equivalent of bis(terpyridyl) ligands under reducing conditions, yields the self-assembled diamagnetic hexameric ruthenium and iron-based macrocycle of benzenoid architecture. More recently, sterically congested, hexameric Pd(II)- and Cd(II)-tetrakispyridinyl-based macrocycles were reported using both mono- and tridentate coordination sites.148 In addition to 1H and 13C NMR, 2D COSY NMR studies, the formation of these hexagonal macrocycles was established using traveling wave ion mobility mass spectrometry (TWIM-MS) which allowed for the deconvolution of the isotope patterns of different charge states, thus avoiding the isomer superposition prevalent in regular ESI-MS and FT-MS. The same group also reported a novel family of metallocyclic pentagons constructed by the facile self-assembly of a bis(terpyridine)-carbazole (104) utilizing terpyridine-metal coordination as the driving force for the assembly process.149 The treatment of 104 with one equivalent of M2+ ion [M = Fe (105), Ru, Zn] in MeOH allowed for the formation of pentameric cationic macrocycles (Scheme 31). Multinuclear NMR, UV-vis and mass spectrometric established the structures of these pentagonal architectures. The photophysical studies on these pentagonal assemblies showed interesting light harvesting properties.
Scheme 31.
Recently, a series of anion-templated hexanuclear macrocycles has been reported, formed by treating half-sandwich Rh(III) metal corners with deprotonated 2,4-diacetyl-5-hydroxy-5-methyl-3-(3-pyridinyl)-cyclohexanone (dhmpc).150 The size of the counteranion significantly affects the nuclearity of the final self-assembled macrocycles. While larger counteranions OTf−, PF6− and SbF6− favor the formation of hexanuclear metallamacrocycles, BF4− favors the formation of a tetranuclear metallamacrocycle. Single crystal X-ray structural analysis for the macrocycles reveals that the two counteranions are encapsulated within the cavities of each of the host hexanuclear metallamacrocycles. Figure 18 shows the molecular structure of [Cp*Rh(dpmhc)6]6+. 1H NMR studies also indicated the retention of the counterions within the molecular cavities in solution. Interestingly, the tetranuclear macrocycle can be converted to hexanuclear macrocyclic assemblies by the addition of the appropriate anion as its [NBu4]+ salt with preferred selectivity for larger anions in the order of SbF6− > PF6− > OTf− > BF4− > Cl−.
Figure 18.
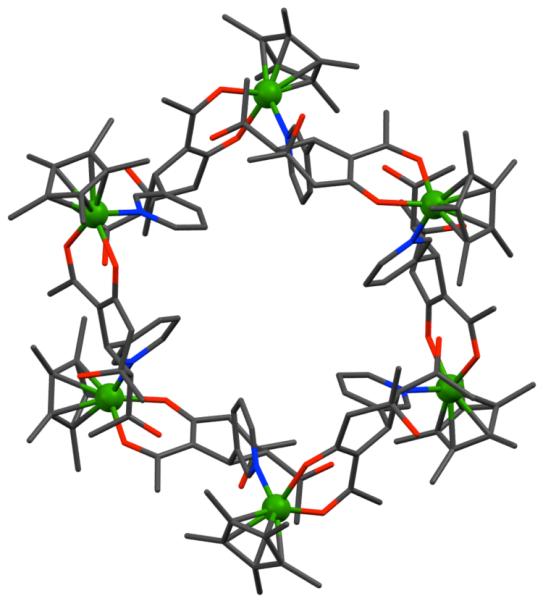
Molecular structure of [Cp*Rh(dpmhc)6]6+. Color code: green, Rh; red, O; blue, N; gray, C.
The weak link approach (WLA), a strikingly different design strategy, developed by Mirkin and coworkers has led to the realization of a vast plethora of flexible 2D homo- and heterodimetallic macrocyclic rings.11 These large ring systems have been prepared from specifically designed hemilabile ligands containing phosphanyl alkyl ether, thioether, and amine groups. The phosphorus group serves as the strong binding site for the late transition metals while ether, thioether, and amine groups serve as the weak binding sites. These ligands also provide a way to control the reactivity of the condensed structures and incorporate functionalities, such as fluorescent, redox-active, or catalytic moieties, into the final supramolecular architecture. In one early demonstration of this strategy, a Rh(I) containing homodimetallic cationic macrocycle was synthesized from phosphinoether-based hemilabile ligand 1,4-bis[2-(diphenylphosphanyl)ethoxy]-2,3,5,6-tetramethylbenzene (106). 151 A 1:1 stoichiometric reaction of 106 with a Rh(I) precursor formed by the reaction between [RhCl(COT)2]2 (COT = cyclooctene) and AgBF4 in CH2Cl2 resulted in a condensed macrocycle 107. The addition of strongly coordinating CO (1 atm) in CH2Cl2 resulted in displacement of the labile RhI-O bond leading to the formation of a large cationic macrocycle (108) in quantitative yield. Removal of CO from 108 and dissolution in MeCN resulted in the MeCN adduct of 109 (Scheme 32).
Scheme 32.
Subsequently, reports of the use of other weak linkers, such as thioether and amines to synthesize a number of bimetallic Rh(I) cationic macrocycles have emerged.152 For example, treatment of a Rh(I) source with the thioether-containing hemilabile ligand 1,4-bis[2-(diphenylphosphino)ethylthio]benzene led to the formation of a cationic fused macrocycle (110).153 Interestingly, due to the greater inertness of the metal-thioether bond, the RhI-S bonds of 110 are inert under conditions that result in the displacement of the ether moieties in complex 107 upon addition of CO, CH3CN, or both. However, altering the electronics of the aromatic core in the thioether containing ligand allows the opening up of the condensed structure by ancillary ligands. 154 Introduction of fluoride substituents on the aromatic core of the phosphinothioether-based ligand allows for the synthesis of condensed structure 111 that reacts similarly to the ether-based macrocycles to give expanded macrocycle 112 (Scheme 33). Also reported were several other Pd(II), Ir(I), Ru(II) and Cu(I) based cationic macrocycles. Treating a Pd(II) source with phosphinoalkyl ether-containing hemilabile ligands gave access to condensed cationic complexes (113) which could be opened up into larger metallacycles (114) by treatment with MeCN (Scheme 33).155 Similarly, the cationic bimetallic Cu(I) macrocycles, 116, has been synthesized by addition of small ancillary ligands such as acetonitrile, isonitriles, diimines, or pyridine to the condensed structures, 115 (Scheme 33).156 Heteorobimetallic macrocycles can also be synthesized by using dissymmetric hemilabile ligands that incorporate both ether and thioether weak links. Due to the differences in the labilities of metal-ether and metal-thioether bonds, different metals can be selectively placed on the macrocycles by changing the reaction conditions.157
Scheme 33.

3.2. Neutral Systems
As described above, the majority of synthetic strategies for metallamacrocycles involve the use of neutral polypyridyl ligands with multivalent metal ions that result in charged assemblies. In these charged species, counterions often block the cavities that are formed; however, in contrast, neutral assemblies can provide a cavity devoid of counterions that may serve as a host for the encapsulation of neutral organic guests. Neutral assemblies can be designed by (1) directly using anionic bridging ligands that can compensate for the cationic metal charge or (2) synthesizing a neutral precursor acceptor followed by treatment with a neutral donor.
Square planar divalent platinum and palladium complexes, in which two or three coordinating sites are occupied by strong M-P or M-C bonds and the remaining site(s) are coordinated by labile, weakly coordinating anion(s) such as nitrate, triflate, have been used as the acceptor units with dianionic bridging ligands as the donor linkers. The spontaneous self-assembly of neutral nanoscopic molecular rectangles 122-126 was achieved by a 1:1 stoichiometric combination of an acetone solution of a predesigned platinum-based molecular clip, 1,8-bis[trans-Pt(PEt3)2(NO3)]anthracene (75), with an aqueous solution of linear dicarboxylates 117-121 (Scheme 34).158,159 Multinuclear NMR, mass spectrometry and single crystal X-ray structural analysis established the formation of the macrocycles. The utilization of the Pt-O bonding interaction as the driving force for designing the discrete assemblies is an interesting feature of these macrocycles, as large supramolecules incorporating a Pt-O bond were believed to be unstable due to the unfavorable hard-soft combination. Platinum-based macrocycles incorporating cyclic oxocarbondianions, squarate and croconate and their acyclic analogue, oxalate, have also been synthesized in excellent yield via self-assembly. The combination of diplatinum molecular clip 75 with all three dianions afforded molecular rectangles.160 In all cases, multinuclear NMR spectra were consistent with the formation of single, highly symmetrical species. Single-crystal X-ray structural studies of these rectangles revealed that all the ligands coordinate to the molecular clip in bismonodentate mode. An analogous neutral Pd2 rectangle, 128, was reported which used a predesigned ethynyl containing Pd(II)-based molecular clip (127) in conjunction with disodium fumarate as the donor linker (Scheme 35).161 Rectangle 128 represents an example of a neutral Pd(II) macrocycle obtained using Pd-O coordination bonding as the driving force. While oxygen-based hard donors are expected to be unsuitable for the soft acid Pd(II), the electrostatic interaction between the anionic fumarate and Pd(II) plays an important role in obtaining this neutral molecular rectangle.
Scheme 34.
Scheme 35.
Building upon a similar strategy, neutral molecular rhomboids and triangles were assembled using dicarboxylate anions as the donor linkers.158,159 Both kinds of macrocycles use 60° Pt(II)-based building block 12 as an acceptor unit, and ditopic linear 118-120 (in the case of triangles 130-132) or angular 129 (in the case of rhomboid 133) dicarboxylate bridging ligands (Scheme 36). The formations of the macrocycles were unambiguously confirmed by single crystal X-ray structural analyses and NMR spectroscopy. The crystallographic studies revealed the presence of different types of channels within the crystal lattices that were occupied by solvent molecules. The diplatinum 60° acceptor unit 12 also produced a supramolecular rhomboid with croconate (C5O52−) ion and a molecular triangle with squarate ion.160 Multinuclear NMR, single-crystal X-ray crystallography and FAB-mass spectrometry established the formation of the assemblies. Heterobimetallic Pt4Fe2 neutral macrocycles were assembled using a flexible bridging 1,1′-ferrocenedicarboxylate as the donor linker.162 While the combination of the 1,1′-ferrocenedicarboxylate with 0° Pt-based molecular clip 75 as the acceptor unit leads to a neutral molecular rectangle, the use of 60° Pt-based molecular clip 12 leads to a neutral rhomboid. Spectroelectrochemical analysis of the molecular rectangle reveals a reversible two-electron transfer due to the ferrocenyl moieties.
Scheme 36.
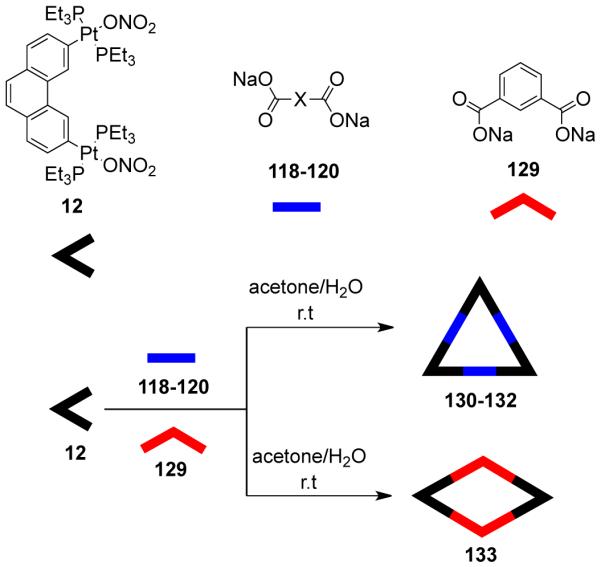
Severin and coworkers have demonstrated the formation of a series of neutral metallacycles from half-sandwich Ru(II), Rh(III) and Ir(III) metal corners and tridentate linkers.163 For example, treatment of [(p-cymene)RuCl2]2 (89) with 3-hydroxy-2-pyridone 134 in the presence of base (Cs2CO3 or K2CO3) allowed the formation of trinuclear metallacycle 135 in a diastereoslective manner in almost quantitative yield (Scheme 37).164 The macrocycle 135 showed high affinity and selectivity for Li+ and Na+ ions at its central receptor site. The formation of the macrocycle 135 was confirmed by mass spectrometric and single-crystal X-ray analysis. The dianion of 3-hydroxy-2-pyridone acts as a bridging, tridentate ligand with the half-sandwich ruthenium centers occupying the vertices of the triangle. A prerequisite for the formation of such trinuclear macrocycles is that the ligands must be relatively rigid and the two coordinate vectors must be orthogonal. Other di-tri- and tetranuclear metallacycles of Ru(II) and Rh(III) were also obtained using deprotonated heterocyclic compounds such as 2-hydroxy-nicotinic acid, 2-amino-nicotinic acid, imidazole-4-carboxylic acid, 2,3-dihydroxyquinoline, 2,3-dihydroxyquinoxaline, or 6-methyl-2,3-phenazinediol as linkers.165 Using the same strategy, hexanuclear coordination cages of the formula [(C5Me4R)M(C7H3NO4)]6 (M = Rh, Ir; R = Me, H) were obtained by the stepwise treatment of [(C5Me4R)MCl2]2 with AgOAc followed by pyridine-3,5-dicarboxylic acid.166
Scheme 37.
The same group has also demonstrated the formation of neutral macrocycles that contain four or ten palladium metal centers using 2,3-dihydroxypyridine and its derivative as the dianionic bridging ligands.167 The reaction of chloro-bridged palladium-based acceptor unit [(PEt3)PdCl2]2 (136) with 2,3-dihydroxypyridine 137 in methanol, using Cs2CO3 as the base, led to the formation of a neutral tetranuclear molecular square (138; Scheme 38). The use of 5-chloro-2,3-dihydroxypyridine under similar condition gave a decameric complex. Similar to 2,3-dihydroxypyridine, the use of 2-hydroxynicotinate also resulted in the formation of a tetranuclear molecular square. However, a potential drawback of these assembly reactions is that they do not give a single product and often need additional crystallization for purification.
Scheme 38.
Lindoy and coworkers utilized a range of aryl-linked bis-β-diketonato ligands incorporating both parallel and sixty-degree orientated coordination vectors to form a variety of neutral supramolecular architectures with appropriate transition metal ions. 168 While 1,3-phenylene-linked bis-β-diketonato ligands led to the formation of dinuclear metallacycles,169,170 1,4-phenylene-linked bis-β-diketonato ligands gave trinuclear assemblies.171,172 These planar ‘platform-like’ macrocycles, upon interaction with ditopic heterocyclic bases, led to the formation of both sandwich-like discrete and extended 3D supramolecular architectures.173,174 Recently, the same group reported one of the largest neutral triangular supramolecular assemblies.175 A biphenyl-spaced β-diketonate ligand with its donor sets oriented at sixty degrees was used to form the trinuclear metallacycle, Co3L3(pyridine)6 [H2L = 1,1′-(4,4′-Biphenylene)-bis-3,3-dimethylpentane-1,3-dione]. The single crystal X-ray structural analysis (Figure 19) showed the formation of a discrete triangular architecture in which three β-diketonate ligands bridge three Co(III) ions such that each ligand forms the side of a neutral, essentially equilateral triangle. Each Co(III) center is in a pseudooctahedral geometry wherein the axial coordination sites are occupied by pyridine ligands. The metallacycle contains a considerable triangular void area (~ 118 Å2), with a disordered pyridine molecule occupying this void in the solid state.
Figure 19.
Molecular structure of Co3L3(pyridine)6 [H2L = 1,1′-(4,4′-Biphenylene)-bis-3,3-dimethylpentane-1,3-dione]. Color code: green, Co; red, O; blue, N; gray, C.
Neutral molecular rectangles were also prepared using a step-wise synthetic route. Hupp et al.176,177 have synthesized rhenium based neutral molecular rectangles 141-146 in a step-wise fashion by first creating a stable bimetallic edge (140) using a rigid dianionic benzimidazole ligand (139) and then assembling the edges by adding bifunctional dipyridyl or diazine ligands, as shown in Scheme 39. The dimensions of the cavities, as determined from X-ray structural studies, range from 5.7 × 7.2 to 5.7 × 19.8 Å. Interestingly, electrochemical reduction of the rectangles yields unusual mixed-valent complexes in which the ligands themselves are the redox centers and interligand electronic communication is controlled by direct ligand orbital overlap rather than by superexchange through the metal ions. The overall charge neutrality and optical electron transfer properties of these rectangles make them attractive for application as sensory materials for neutral organic guests. Alkoxy bridged neutral molecular rectangles [{(CO)3Re(μ-OR)2Re(CO)3}2(μ-L)2] were obtained by treatment of Re2(CO)10 with bipyridyl ligands such as pyrazine (pz), trans-1,2-bis(4-pyridyl)ethylene (bpe), 1,4-bis-[2-(4-pyridyl)ethenyl]benzene (bpeb) and 4,4′-bipyridine (bpy) in the presence of higher aliphatic alcohols under solvothermal conditions. 178 Similarly, a furan-decorated neutral Re(I)-based 2D rectangle was recently obtained by the treatment of Re2(CO)10 with trans-1,2-bis(4-pyridyl)ethylene (bpe) in the presence of 2-furanmethanethiol in a one-pot synthesis.179 These works builds upon the strategy earlier developed independently by Hupp180 and Sullivan181 wherein rhenium complexes bridged by either thiolate or alkoxy ligands as the bimetallic edges were assembled using N-donor ligands.
Scheme 39.

A series of neutral luminescent molecular rectangles [{Re(CO)3(μ-bpy)Br}{Re(CO)3(μ-L)Br}]2 [147, L = pyrazine (pz); 148, L = 4,4′-dipyridylacetylene (dpa); 149, L = dipyridylbutadiyne (dpb); and 150, L = 1,4-bis(4′-pyridylethynyl)-benzene (bpeb)] having fac-Re(CO)3Br as corners and 4,4′-bipyridine (bpy) as the bridging ligand on one side and bipyridyl ligands of varying length (L) on the other side have been reported, as shown in Scheme 40.182 Similarly, neutral molecular rectangles [{Re(CO)3(μ-bpe)Br}{Re(CO)3(μ-L)Br}]2, (bpe = trans-1,2-bis(4-pyridyl)ethylene; L = pz, bpy), have also been shown to exhibit light emitting properties.183 Lees et al.184 also described a series of neutral self-assembled macrocycles featuring fac-Re(CO)3X (X = Cl, Br) as corners and linear bipyridyl bridging ligands as linkers. Depending on the flexibility, lengths and angles of the bridging bipyridyl ligands 4,4′-dipyridylbutadiyne (DPB), 4,4′-azopyridine (AZP), 1,4-bis(4′-pyridylethynyl)-2,5-dihexyloxybenzene (BPDB), 1,4-bis(4′-pyridylethynyl)-2,5-didodecyloxybenzene (BPDDB) and 2,5-bis(4-pyridylethynyl)thiophene (BPET), various macrocyclic complexes –squares {[ClRe(CO)3(DPB)]4 and [ClRe(CO)3(AZP)]4, triangles [BrRe(CO)3(BPDB)]3 and [BrRe(CO)3(BPDDB)]3, or a dimeric species [ClRe(CO)3(BPET)]2 were obtained.
Scheme 40.
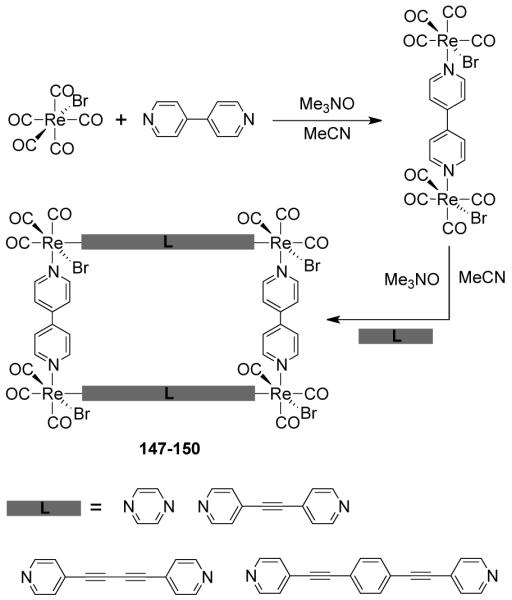
Through the dimetallic building block approach, Cotton and coworkers have described a large number of neutral metallamacrocycles such as loops, triangles and squares using dimetallic paddlewheel units as the corner pieces to link divalent anions, such as dicarboxylates.12 Because of the idealized orthogonality between the planes defined by the metal atoms and replaceable equatorial ligands, these dimetallic units, upon treatment with equatorial linkers, form square arrays. The first series of neutral molecular squares (159-165) derived from quadruple bonded dimolybdenum subunits were synthesized via the reaction of [Mo2(cis-DAniF)2(CH3CN)4]2+ (151; DAniF = N,N’-di-p-anisylformamidinate) and (Bun4N+)2(Carb2−) [Carb2− = oxalate (152), fumarate (153), acetylenedicarboxylate (154), tetrafluoroterephthalate (155), ferrocene dicarboxylate (156), 4,4′-biphenyldicarboxylate (157) and carborane dicarboxylate (158)] as shown in Scheme 41.185,186 The X-ray structural characterization of molecular squares 159-162 showed that each corner consisted of a quadruply bonded dimolybdenum unit, with two cisoid formamidinate paddles and two carboxylate paddles from the dicarboxylate linkers. The conformation of each dicarboxylate anion is such that both carboxylate groups are equatorially disposed. The dimensions of the squares, defined by the four Mo2 corners in each molecule are 7 × 7, 9 × 9, 10 × 10, and 15 × 15 Å2 for 159-162, respectively. Thus, tuning the shape and size of the linkers can modulate the dimensions of the channels created by the squares. 1H NMR studies on the squares showed that they are fairly stable in solution and are highly symmetrical. These compounds also display rich electrochemical behaviors that are affected by the nature of the carboxylate linkers. Extending this strategy to other metals, similar neutral molecular squares having Rh24+ units were reported by treating [Rh2(cis-DAniF)2(CH3CNeq)4(CH3CNax)2](BF4)2 with (Et4N+)2(Carb2−) where Carb2− represents the dicarboxylate anions bicyclo[1.1.1]pentane-1,3-dicarboxylate, tetrafluoroterephthalate, 1,4-cubanedicarboxylate, terephthalate, fumarate, and trans-1,4-cyclohexanedicarboxylate. 187 Neutral molecular squares having ruthenium corner pieces, Ru25+, using oxalate and terepthalate as the linkers were also reported by the same group.188 The groups of Bonar-Law189,190 and Schiavo191 have also reported a few other dirhodium neutral squares based on Rh24+ corner units and dicarboxylate anions as the linkers.
Scheme 41.

Several neutral molecular triangles were also synthesized using the dimetallic corner units such as [Mo24+],192 [Rh24+],193,194 and [Re25+]195 by varying the reaction conditions and length and flexibility of the dianionic linkers. Several neutral triangles designed using this strategy were obtained by treating dimolybdenum [Mo24+] subunits, [Mo2(cis-DAniF)2(CH3CN)4]2+ with flexible 1,4-cyclohexanedicarboxylate linkers.192 The molecular triangle [{cis-Mo2(DAniF)2(1,4-O2C-C6H10-CO2)}]3, as observed from crystal structure analysis, has Mo2(DAniF)2 units occupying the three vertices of the triangle while both carboxylate groups of the 1,4-cyclohexanedicarboxylate linker orient themselves in an equatorial (eq,eq) fashion. Figure 20 shows the molecular structure of the triangle.
Figure 20.
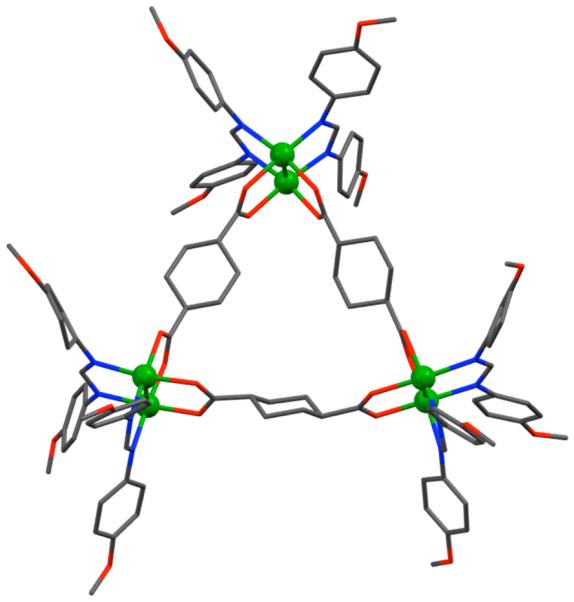
Single-crystal X-ray structure of [{cis-Mo2(DAniF)2(1,4-O2C-C6H10-CO2)}]3. Color code: green, Mo; red, O; blue, N; gray, C.
An analogous [Rh2(DAniF)2]2+ dimetallic corner unit also led to the formation of a neutral molecular triangle [Rh2(DAniF)2(μ4-C2O4)]3 (167) from a 1:1 equimolar mixture of [Rh2(cis-DAniF)2(CH3CN)4]2+ (166) and (C2O42−) (152), in near quantitative yield. Interestingly, using a 10-fold excess of oxalate linker, under the same reaction conditions, selectively gave the corresponding molecular square [Rh2(DAniF)2(μ4-C2O4)]4 (168; Scheme 42).193,194 In solution, a dynamic equilibrium between the triangle 167 and square 168 was found to exist. The dirhenium complex cis-Re2(μ-O2CCH3)2Cl2(μ-dppm)2 (dppm = Ph2PCH2PPh2), which contains two substitutionally labile acetate ligands, afforded the molecular triangle [cis-Re2Cl2(μ-dppm)2(μ-O2CC6H4CO2)]3 upon treatment with an excess of terephthalic acid in refluxing ethanol.195 By employing [cis-Mo2(DAniF)2]2+ as the vertex building block and terephthaloyldiamidate as the linker, dimolybdenum-containing neutral metallacycles of general formula [cis-Mo2(DAniF)2((ArNOC)2C6H4)2]n were synthesized and structurally characterized. 196 The geometries of the products are guided by the identities of the aromatic groups present in the linkers. When the aromatic group is phenyl, a molecular triangle is the exclusive product; changing the aromatic substitutents to p-trifluoromethylphenyl or m-trifluoromethylphenyl affords a molecular square selectively.
Scheme 42.

Cotton et al. have also investigated the dynamic equilibrium between a neutral molecular square, [cis-Mo2(DAniF)2]4(O2CC6F4CO2)4 (164), and its corresponding triangle, [cis-Mo2(DAniF)2]3(O2CC6F4CO2)3 in CDCl3 solution from a reaction mixture of [cis-Mo2(DAniF)2]2+ and the perfluoroterephthalate anion.197 The compounds were characterized by X-ray structural studies and solution NMR spectroscopy. Since the perfluoroterephthalate linkers are linear and relatively rigid, the formation of a molecular square was expected. However, multinuclear NMR studies show that a dynamic equilibrium exists between the square and triangle. As determined from NMR studies, the conversion of squares to triangles is entropically favored but enthalpically disfavored with an equilibrium constant of 1.98(7) × 10−4 at 23.7 °C. However, a recent study showed that use of sub-stoichiometric amounts of oxalate gave molecular triangle as the major product with no observable equilibrium with its molecular square counterpart.198
Mirkin and coworkers synthesized a number of neutral metallamacrocycles using a wide range of transition metal precursors with a variety of flexible hemilabile ligands by the weak link approach. In one of the early examples of this strategy, they reported the formation of a Rh(I)-containing, neutral macrocycle via the halide induced ring opening of a cationic condensed intermediate by breaking a weak RhI-S bond.153 A 1:1 equimolar mixture of [RhCl(COT)2]2 (COT = cyclooctene) and the phosphinothioether-based hemilabile ligand 1,4-bis[2-(diphenylphosphino)ethylthio]benzene were combined to form a cationic condensed intermediate (169; Scheme 43). The addition of [Me4N]Cl in the presence of CO (1 atm) resulted in the displacement of the labile RhI-S bonds leading to the formation of neutral macrocycle 170 in quantitative yield. Condensed macrocycles containing ether instead of thioether linkages can also be opened up by CO/Cl− to give neutral macrocycles. Using a bis(phosphinoalkyl-thioether)arene ligand with a fluorinated aryl group, [1,4-(Ph2PCH2CH2S)2C6F4], instead of benzene-based phosphinothioether-based hemilabile ligands, Rh(I) and Ir(I)-containing neutral macrocycles were reported.154 Treatment of [Rh(NBD)Cl]2 (NBD = norbornadiene) or [IrCl(COT)2]2 (COT = cyclooctene) with [1,4-(Ph2PCH2CH2S)2C6F4] generated condensed structures 171 and 172, respectively. Addition of CO/I− cleaved the Rh-S and Ir-S bonds to yield 173 and 174 respectively (Scheme 43). Expanding the paradigm to other d8 square planar transition metals, the same group reported Pd(II)-based neutral macrocycles by treating [Pd(MeCN)4][BF4]2 with phosphinoalkyl ether or a thioether-containing hemilabile ligand to give condensed cationic complexes 175, which can be opened up into neutral metallocycles 176 by introducing CN− ions (Scheme 43).155 Similarly, the d8 octahedral transition metal Ru(II) was used to synthesize neutral condensed and open macrocycles by addition of CO, pyridine and alkyl diamines.199
Scheme 43.

3.3. Chiral Systems
Due to their ability to mimic complex biological systems, chiral supramolecular assemblies have been extensively studied over the past two decades.200 The biological implications of chirality demand that we improve our ability to access chiral molecules as pure entities.201 Through properly designed and preprogrammed building units, supramolecular self-assembly can provide relatively easy, simple access to chiral supramolecules with controlled stereochemistry that may help in our understanding of nature’s intricate biochemical processes. Furthermore, these systems may also provide ways to study newer approaches to enantioselective molecular recognition, chiral sensing and asymmetric catalysis.
Chiral 2D metallacycles are among the most extensively studied due to their relative ease of preparation and their potential applications in enantioselective recognition, sensing and catalysis.200 The rational design of these discrete chiral ensembles by the directional bonding approach can be achieved by using (1) chiral metal auxiliaries as corner units with achiral multidentate ligands as the connectors; (2) achiral metal corner units with chiral multidentate ligands as the connectors; (3) inherently chiral metal centers having specific coordination geometries with achiral multidentate linkers; or (4) combinations of the above.
In the first strategy, appropriate metal corner units with optically active chiral auxiliaries202 are combined with achiral bridging ligands, resulting in chiral metallacycles. An early example of this strategy was provided by Stang et al. wherein optically active [M(R-BINAP)(OTf)2] (M = Pd (177) or Pt (178), BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthalene) were used as shape-defining corner units due to their high reactivity toward the coordination of nitrogen-containing ligands, as well as the significant degree of conformational rigidity of BINAP.203,204 When treated with 90° diaza ligands, bis(3-pyridyl)iodonium triflate (179) or bis(4-(4-pyridyl)phenyl)iodonium triflate (180), chiral supramolecular squares 181 and 182 were obtained (Scheme 44).203 The macrocyclic nature of these complexes was established by multinuclear NMR and confirmed by mass spectrometric analysis. Reactions of anthracene based C2h symmetrical diaza ligands 2,6-diazaanthracene and 2,6-diazaanthracene-9,10-dione with [M(R-BINAP)(OTf)2] (M = Pd (177) or Pt (178)), have also led to the formation of similar chiral molecular squares.203
Scheme 44.

Interestingly, as determined from 31P NMR studies, the use of 2,6-diazaanthracene in acetone at ambient temperature as the linker led to only a single diasteromer out of a possible six. However, the use of 2,6- diazaanthracene-9,10-dione under similar conditions led to a mixture of diastereomers. Chiral macrocyclic assemblies containing two or four porphyrin units were also readily synthesized by employing [cis-Pd(R-BINAP)(OTf)2] (177) as the chiral metal auxiliary with di- and tetra-pyridyl substituted porphyrins.205 The chiral macrocycles have a chirality-induced puckered geometry of D2 symmetry at ambient temperature. The high solubility and ease of synthesis of these self-assembled chiral porphyrin macrocycles provides excellent examples for the study of porphyrin exciton coupling interactions and induced circular dichroism.
A homochiral molecular triangle was reported by Mirkin et al., in which a chiral enantiopure (S)-binaphthyl diamine derivative was used as the corner linker.206 Each corner linker encloses a Cu(II) ion chelated by the N,N,O−,O− set of donor atoms which further connect to a [Cu(py)3(H2O)] fragment through the carboxylate groups present on the periphery of the salen precursors to form a molecular triangle (Figure 21). It is the inherent chirality of the Cu(II)-enclosed corner unit that imparts the chirality to the molecular triangle. Interestingly, an unusual solvent-induced, spontaneous and reversible transformation of the triangular macrocyclic complex into a homochiral helical polymer was also observed.
Figure 21.
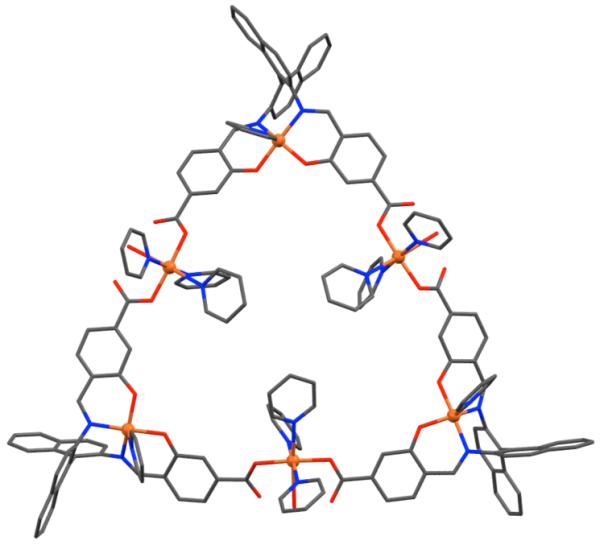
X-ray crystal structure of the homochiral molecular triangle. Color code: golden, Cu; red, O; blue, N; gray, C.
In the second approach, chiral metallacycles were assembled by interacting chiral organic multidentate ligands with achiral metal auxiliary units. An example of this approach was provided by Lin and coworkers wherein they used pyridyl functionalized-1,1′-binaphthyl ligands (183-185) derived from enantiopure 1,1′-binaphthyl-2,2′-diol (BINOL) with achiral fac-[ReCl(CO)5] metal corners to assemble a family of chiral molecular squares (186-188; Scheme 45).207 The formations of the chiral metallacycles were confirmed by NMR, mass spectrometry, UV-visible and CD spectroscopy. Using angular bipyridine bridging ligands derived from enantiopure 1,1′-binaphthyl-2,2′-diol (BINOL) and [M(dppe)]2+ (M = Pd or Pt and dppe = bis-(diphenylphosphino)ethane) metallocorners, a series of chiral molecular squares were readily assembled by the same group.208 The macrocycles were characterized by a variety of techniques including infrared, UV-vis, circular dichroism (CD), multinuclear NMR spectroscopy, and ESI-mass spectrometry. All of these chiral molecular squares were highly luminescent in solution at room temperature with good quantum efficiency.
Scheme 45.

A series of chiral molecular triangles, 193-195, was afforded by the equimolar treatment of bis(alkynyl) derivatives of BINOL ligands 189-191 with the cis-protected Pt(II) metallocorner, cis-[PtCl2(PEt3)2] (192), utilizing the facile insertion of Pt(II) into alkyne C-H bonds (Scheme 46).209 Although from a geometric consideration, molecular squares were expected, the kinetic preference for smaller metallacycles drives the assembly towards the exclusive formation of molecular triangles. Interestingly, replacement of cis-blocked ligand 159 with cis-[Pt(4,4-dtBuPy)Cl2], leads to both a molecular triangle and a square in 29% and 22% yields, respectively.210 However, using the same ligand systems, molecular squares can be exclusively formed via stepwise directed-assembly reactions in modest yields.211
Scheme 46.

A mixture of chiral organometallic molecular polygons, ranging from triangles to octagons, resulted when 1 equiv of trans-[PtCl2(PEt3)2] was treated with the atropoisomeric bridging ligand 2,2′-diacetyl-1,1′-binaphthyl-6,6′-bis(ethyne).212 The analytically pure molecular polygons were isolated by silica gel column chromatography and characterized by ESI-mass spectrometry, multinuclear NMR, and CD spectroscopy. By adopting a similar methodology, the robustness of the Pt-alkynyl bond was utilized to assemble chiral dinuclear metallocyclophanes with exo213 and endo214 functionalization. Lin et al.215 also reported a series of chiral mesoscopic metallacycles having as many as 47 metal centers using a stepwise assembly method. These molecular polygons were built from a similar binapthyl based ligand and trans-Pt(PEt3)2 as the metallocorner.
Another category of ditopic chiral linkers has side-chains connected by an ethylene-bridge with a rigid gauche conformation. Chiral, enantiopure metallosupramolecular rhomboids have been self-assembled in solution through the coordination of bis-pyridyl-substituted chiral ditopic ligands with cis-blocked Pd(II) and Pd(II) acceptors.216 For example, treatment of chiral ditopic ligand 196 with [(en)M(NO3)2] [M = Pd (1), Pd (37)] led to the assembly of enatiopure SSSS rhomboids, 197-198 (Scheme 47). The R-enantiomer also forms under the same conditions as the all-S enantiomer. The assemblies were characterized in detail by NMR, CD spectroscopy in solution and by ESI-FT-ICR-mass spectrometry in the gas phase and could be adsorbed and imaged at surfaces in an electrochemical environment. The chirality of these assemblies manifests itself on surfaces at both local and global levels.216 Jeong et al. have used a very similar carboxylate functionalized bis-pyridyl-substituted chiral ditopic ligand with Cu(II) ions in DMF to self-assemble a chiral triangular metallacycle.217 A chiral C3-symmetric hexanuclear triangular-prismatic copper(II) cluster was also assembled from a enantiomeric dipeptidic N,N’-terephthaloyl-bis(S-aminocarboxylato) ligand.218
Scheme 47.

Schalley and coworkers have also established the diastereoselective self-assembly of metallosupramolecular squares utilizing axially chiral 4,4′-bipyridine bridging ligands. Chiral molecular squares 199-201 were synthesized by the treatment of enantiopure chiral 4,4′-bipyridine ligands with cis-blocked Pd(II) building block, [Pd(dppp)(OTf)2] (4) as the corner units in a 1:1 molar ratio (Scheme 48).219 Corresponding Pt(II)-based chiral molecular squares were also prepared. Amino acids such as (S)-alanine incorporated into the axially chiral 4,4′-bipyridine ligands afforded amino-acid functionalized chiral metallosupramolecular squares.220 As established from multinuclear NMR and X-ray structural studies, only one diastereomer was formed in which the side chains were positioned outwards and the pair-wise, opposite bipyridines had the same axial chirality. Although the chiral centers of the alanines favor one of the axially chiral biaryl configurations in the free ligand, the ratio of both diastereomers is strictly 1:1 in the squares. This diastereoselectivity is governed by the stereoelectronic effects of the dppp ligand, which lead to the exclusive formation of heterochiral assemblies.
Scheme 48.
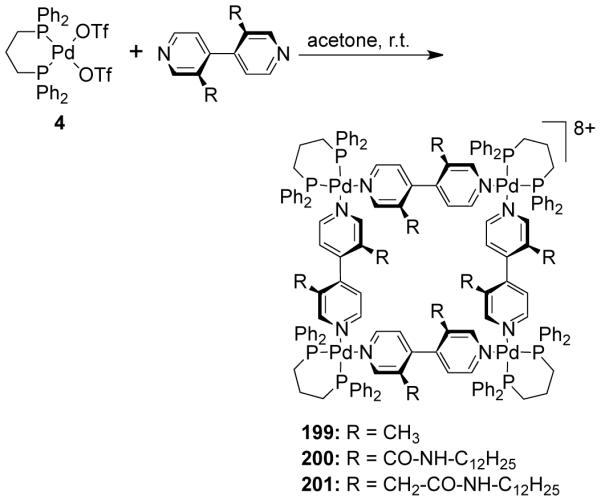
The synthesis of enantiopure molecular rectangles was achieved using the platinum-based molecular clip, 1,8-bis[trans-Pt(PEt3)2(NO3)]anthracene (75) with (2R,3R)-(+)-tartarate and (2S,3S)-(−)-tartarate. 221 These enantiopure rectangles showed the phenomenon of induced circular dichroism (ICD). Although (2R,3R)-(+)-tartaric acid is CD-inactive in the long wavelength and shows only one negative Cotton effect at 214 nm, the corresponding molecular rectangle showed bands which corresponded to anthracene moiety above 350 nm. This indicated the presence of ICD in the anthracene moiety and, as such, in the chiral supramolecule upon association with chiral inducers, (2R,3R)-(+)-tartarate dianions.
In an interesting study, Raymond et al. observed supramolecular chiral induction in dinuclear triple-stranded helicates composed entirely of achiral components.222 The anionic helicates show strong intermolecular assymetric induction in the presence of chiral organic cationic molecules such as N-methyl-s-nicotinium (s-nic), N-methyl cinchoninium (cinc), and N-methylquininium (quin). These chiral cations can transmit chirality to the metal centers of the helicates (by associating in the cleft between the ligand planes) leading to the determination of the chirality of the entire helicate. The van der Waals contact and cation-π interaction plays an important role in communicating chirality to these metallo-helicates. In fact, due to the precise nature of these interactions, minor variations in the structures of the chiral cation as well as the central ligand scaffold of the helicate significantly affect the level of intermolecular chiral induction.222
In the third strategy, inherently chiral octahedral (tris-chelated) metal complexes were used to assemble chiral metallacycles with achiral multidentate ligands as the building blocks. Chiral molecular triangles and squares of Co(III) were reported by Yamanari et al.223 through the self-assembly of inherently chiral mononuclear octahedral complexes based on the multidentate bridging ability of the achiral ligands. The treatment of fac-[CoCl3(tacn)] (202; tacn = 1,4,7-triazacyclononane) with purine-6-thiones (203) in the presence of an equimolar amount of NaOH initially produced a chiral species, 204. Subsequent deprotonation resulted in the formation of chiral molecular square 205 via self-assembly of the mononuclear complex, 204 (Scheme 49). Along the same line, reaction of octahedral Co(II) ions with multidentate achiral ligands which have a ~90° twist (D2h symmetry), such as the tetraacetylethylene dianion, facilitated the formation of chiral molecular squares.224 The reaction between [Mo(CO)6] and 3,6-di-tert-butyl-1,2-benzoquinone in the presence of trace quantities of dioxygen led to the initial formation of molybdenum complexes of 3,6-di-tert-butylcatechol. Condensation of this monomer through bridging oxo ligands gives cyclic, chiral [{Mo-(μ-O)(3,6-DBCat)2}4] square with four Mo centers of the same chirality.225
Scheme 49.
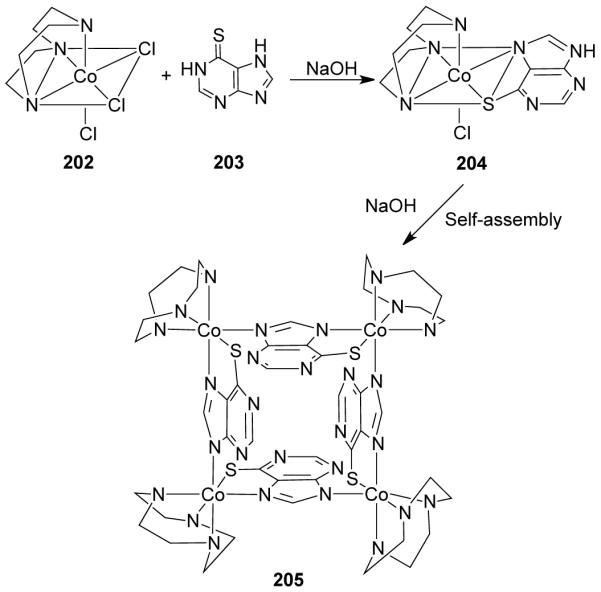
Using the dimetallic building block approach, Cotton and coworkers described a series of chiral molecular triangles containing Rh24+ dimetallic corner units.226 The reaction of cis-Rh2(C6H4PPh2)22+ units with the tetraethylammonium salts of the linear dicarboxylates, in organic solvents, produced racemic crystals of the corresponding chiral molecular triangles [{Rh2(C6H4PPh2)}3(μ-L)3X3] (L = oxalate, terephthalate, and 4,4-biphenyldicarboxylate; X = DMF, py).227 Due to the head-to-tail arrangement of the two orthometalated bridging ligands, the cis-Rh2(C6H4PPh2)22+ unit is inherently chiral, and the molecular triangles based on this unit are thus chiral. However, the chirality induced by the conformational arrangement of the ligands in these supramolecules is only secondary when compared to the chirality (R or S) that arises from the configurational arrangement of the molecules. Interestingly, as determined from single crystal structural analysis, multinuclear NMR, mass spectrometry and electrochemical studies, molecular triangles were exclusively formed both in solution and in the solid state. The reason for this well-defined preference was due to the preferred twist (ca. 23°) of the cis-Rh2(C6H4PPh2)2 corners which induces very little strain on the triangular structures as compared to the corresponding square structures. Thus, the entropic preference for the smaller ring becomes the controlling thermodynamic factor. A resolved enantiomer of the same starting dimetallic corner unit cis-Rh2(C6H4PPh2)2+ resulted in enantiopure molecular triangles (RRR and SSS).228 As ascertained from the crystal structural analysis, the enantiomers are isostructural. Figure 22 shows the single-crystal X-ray structure of the SSS-[cis-Rh2(C6H4PPh2)2(O2CC6H4CO2)(py)3(MeOH)3]3. Cotton et al. have also reported enantiopure chiral molecular loops RR-[cis-Rh2(C6H4PPh2)2(py)2(O2C(CF2)nCO2)]2 (n = 2, 3) using the same Rh24+ dimetallic unit cis-Rh2(C6H4PPh2)22+ in organic solvents.229
Figure 22.
Single-crystal X-ray structure of SSS-[cis-Rh2(C6H4PPh2)2(O2CC6H4CO2)(py)2]3. Axial py and MeOH ligands were omitted for clarity. Color code: green, Rh; pink, P; red, O; gray, C.
3.4. Functional Systems and their Properties
The rational design and synthesis of functionalized metallosupramolecular architectures through coordination-driven self-assembly has generated a great deal of interest in recent times and has been extensively reviewed.230 Several of these functional assemblies can be employed as precursors of electronic, catalytic and photophysical materials and used for molecular recognition and encapsulation. Assemblies containing transition metals are generally more sensitive and responsive to electro- and photo-chemical stimuli as compared to metal-free organic structures. The rigid architecture of the supramolecular ensembles offers well-defined scaffolds for the incorporation of functional moieties where the stoichiometry and position of the individual functional groups can be precisely controlled. Moreover, upon formation, these functional moieties can interact further to give rise to a higher level of functionality. The directional bonding approach, due to its highly modular nature, allows the incorporation of various functionalities into the edge and corner of a building block. Likewise, tethering of functional moieties through a covalent attachment to the building blocks allows the positioning of the functionality into the cavity or on the periphery of a resulting assembly (Figure 23).
Figure 23.
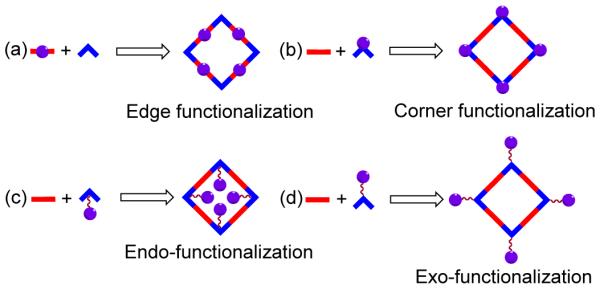
Incorporation of functional moieties into assemblies through the use of functionalized edge or corner building blocks or covalent attachment of functional groups endo- or exo- to the metallacycles.
A variety of functional moieties such as crown ethers, carboranes, cavitands, dendrimers, ferrocenyl units, lipophilic and lipophobic groups onto the edges, corners and on the periphery of a number of discrete supramolecular assemblies.231 The combination of functionalized diaza-crown ethers with a 0°, 60°, or 90° di-Pt(II) acceptors led to the facile formation of a series of geometrically distinct edge and corner functionalized supramolecules. Incorporation of a flexible pyridine-functionalized 18-member diaza crown ring was achieved by treatment with varying Pt(II) acceptors, for example, the [2 + 2] self-assembly of 60° di-Pt(II) acceptor 12 with a diaza crown ring, 206, led to the formation of cationic metallacycle 207 (Scheme 50).232 Neutral, Pt-based discrete assemblies containing flexible 32-member dibenzo crown ether were also synthesized and characterized by NMR and X-ray structural studies.233
Scheme 50.
If properly functionalized, cavitands can be utilized as building units for supramolecular ensembles to give unique structures. The introduction of diametrically opposed pyridyl donor groups at the bridging positions on the rim of a cavitand bowl can lead to the formation of corner-functionalized metallacycles.234 For example, the tethering of two tolyl pyridyl moieties as bridging ligands on a cavitand bowl and subsequent self-assembly with fac-[Re(CO)5Br] in chloroform gave a self-assembled rhombic metallacycle after 24 h, which is neutral and kinetically stable at room temperature. 235 1H NMR and ESI-mass spectrosmetric studies established the formation of the metallacycle. Similarly, diametrically opposed pyridyl groups substituted on the rim of a cavitand bowl which projecting outward at an angle of 60° relative to one another, generated corner-functionalized metallosupramolecular assemblies when assembled with linear ditopic Pt(II) acceptors.236 The [2 + 2] and [3 + 3] self-assembly of a pyridyl-containing cavitand corner donor units with 60° and 180° diplatinum acceptors led to cavitand-functionalized rhombic and triangular supramolecular assemblies, respectively. The assemblies and building blocks were characterized with multinuclear NMR spectroscopy, ESI-mass spectrometry, and elemental analysis. Similarly, a number of metallacycles were assembled by incorporating carborane-functionalized linear donors as well as linear and 120° carborane functionalized di-Pt(II) acceptors.237,238 Treating the linear carborane-containing bis-pyridyl donor with 0°, 60° and 90° Pt(II) acceptors led to the formation of [2 + 2] rectangular, [3 + 3] triangular, and [4 + 4] square metallacycles, respectively.237 Likewise, the treatment of an edge-functionalized carborane-containing linear diplatinum(II) acceptor units, upon combination with a 90° bis-pyridyl donor unit generated a large metallosupramolecular square. The assemblies and building blocks were characterized with multinuclear NMR spectroscopy, ESI-mass spectrometry, and elemental analysis. In addition to these examples, neutral molecular rectangles and rhomboids were designed using platinum-oxygen bonds as the driving force for assembly.238 The combination of m- and p-carborane dicarboxylate units with a 0° diplatinum acceptor yielded a neutral molecular rectangle and a rhomboid, respectively. Multinuclear NMR and X-ray structural studies established the formations of the assemblies.
Edge-functionalized hydrophobic and hydrophilic self-assembled supramolecular rectangles were synthesized from functionalized 180° 1,4-bis-(4-ethynylpyridyl)-2,3-substituted benzenes with 0° diplatinum(II) acceptors via coordination-driven self-assembly to give supramolecular rectangles.239 The hydrophobic rectangles were functionalized with C6H13, C12H25, and C18H37 alkyl chains while the hydrophilic rectangles were decorated with methyl terminated di-, tetra-, and hexaethylene glycol units. Multinuclear NMR (1H and 31P) and ESI-mass spectrometry were used to characterize the assemblies. Molecular force-field modeling suggested that the hydrophobic and hydrophilic chains of each exohedrally-functionalized rectangle prefer to wrap around and aggregate in solution. Similar edge-functionalized alkoxy-bridged Re(I) supramolecular rectangles decorated with long alkyl chains (C4H9, C8H17, and C12H25) have also been reported.240 In the presence of increasing water concentrations these Re(I)-rectangles aggregated in solution. Such self-aggregation of led to an enhancement luminescence efficiency presumbably from the restriction of torsional molecular motion in the aggregates. Furthermore, intermolecular quenching experiments of Re(I)-aggregates with various amines and quinones showed aggregation-facilitated electron transfer between the rectangles and electron donors and acceptors via both static and dynamic quenching processes.
Considerable research effort has been expended on the functionalization of metallosupramolecular squares by employing functional ligands or/and metal corners in the assembly processes. Upon square formation, these functions may interact, leading to a higher level of functionality. Moreover, the cavities of the molecular square may accommodate guest molecules. Notably, Würthner et al. have designed light-harvesting metallosupramolecular squares having of perylene bisimide walls and fluorescent antenna dyes.241 The fluorescent dye 4-dimethylamino-1,8-naphthalimide was incorporated at the bay area of N,N’-bispyridyl perylene bisimide ligand. Subsequent assembly of this ditopic ligand with [Pd(dppp)(OTf)2] gave a self-assembled molecular square. Optical studies of the macrocycle by UV/Vis and steady-state and time-resolved fluorescence spectroscopy revealed that the light captured by the dye antennas gets transported to the perylene-bisimide core by a fluorescence resonance mechanism with high quantum efficiency. Thus, this multichromophore square assembly with aminonaphthalimide antenna dyes is an artificial model for the cyclic light-harvesting complexes in purple bacteria. Earlier a molecular square having sixteen pyrene chromophores attached to four ditopic bay-functionalized perylene bisimide chromophores exhibited extremely fast electron-transfer processes that are normally only observed in solid materials.81
Multiple crown ethers were incorporated into the metallosupramolecular assemblies by exo-functionalization of the building blocks with crown ethers to yield 2D macrocycles with well-defined cavities with varied geometries.242,243 Dibenzo[24]crown-8 was appended to both the 120° donor and the 120° acceptor units through covalent attachment. The combination of the predesigned 120° bispyridyl donor unit, 208, containing Dibenzo[24]crown-8 pendent with the complementary 120° di-Pt(II) acceptor, 209, in a 1 : 1 stoichiometric ratio allowed the formation of a [3 + 3] hexagonal metallocyclic assembly, 210. A [6 + 6] hexagonal assembly, 211, was afforded when 208 was treated with the 180° di-Pt(II) acceptor, 48 (Scheme 51).
Scheme 51.

Likewise, by using a similarly designed dibenzo[24]crown-8 pendent containing 120° diplatinum(II) acceptor unit, different types of [3 + 3] and [6 + 6] hexagonal assemblies can be generated through combination with di-2-pyridyl ketone (60° donor) and 4,4′-bipyridine (180° donor), respectively. Rhomboidal assemblies containing multi-crown ethers can also be synthesized via the [2 + 2] self-assembly of 120° di-Pt(II) acceptor and 120° bis-pyridyl donor unit (208) with their complementary 60° building blocks. These multi-crown architectures have been investigated as the hosts for the multivalent self-assembly of poly[2]pseudorotaxanes. Dialkylammonium ions can form a 1:1 host-guest complex with dibenzo[24]crown-8 ether in nonprotic solvents held together by H-bonding, π-π stacking interactions and electrostatic forces. Hence, dibenzylammonium ions were used to form a new class of poly[2]pseudorotaxanes from the above discrete molecular rhomboids and hexagons having pendant multi-crown ether moieties in their periphery.242,243 As determined by multinuclear 1D and 2D NMR as well as mass spectrometry, the hexagonal assemblies such as 210 and 211 can complex three and six dibenzylammonium ions, respectively. The rhomboidal assemblies can complex two such ions. In all cases, the underlying rigid nature of the 2D polygonal cavity is retained while the flexibility of each crown-ether is reduced as a result of host-guest complexation. The orthogonal, non-interacting nature of the coordination-driven self-assembly of the underlying polygons allows for rhomboidal and hexagonal poly[2]pseudorotaxanes to be prepared.
Dendritic moieties were also incorporated into metallosupramolecular assemblies via the directional bonding approach by functionalizing the building blocks with predesigned dendrons. Dendrimers are highly branched, monodisperse macromolecules emanating from a central core. Incorporation of these dendritic moieties as functional units in supramolecular assemblies is of interest due their potential application in biology, 244 catalysis 245 and photo- and electrochemistry.246 A number of metallodendrimers of varying size and shape were assembled by the combination of predesigned bis-pyridyl donor building blocks 212-215 on which Fréchet-type dendrons were tethered through covalent attachment. The self-assembly of rhomboidal metallodendrimers, 217-220, was achieved by the combination of 120° Fréchet-type dendritic donor units 212-215 and 60° di-Pt(II) acceptor unit 12 in a 1 : 1 stoichiometric ratio (Scheme 52).247,248 Single crystal X-ray structural studies of rhomboidal metallodendrimers 217 and 218, showed well-defined rhombic structure with approximately 2.3 × 1.3 nm cavities. Treatment of 120° donors 212-215 with 120° diplatinum(II) acceptor units 216, and 180° diplatinum(II) acceptor 48 units led to the formation of [3 + 3] hexagons 221-224, and [6 + 6] hexagons 225-228, respectively (Scheme 52). In a complementary approach, [3 + 3] coordination-driven self-assembly between 120° diplatinum acceptor subunits containing tethered dendrons with the bis-pyridyl donor building blocks afforded six-component hexagonal metallodendrimers. 249 Likewise, neutral dendritic metallacycles were achieved through the platinum-oxygen coordination bonds. The combination of rigid 120° dicarboxylate donor linkers funtionalized with Fréchet-type dendrons with complementary 60° and 120° di-Pt(II) acceptor subunits 12 and 216 yielded neutral rhomboidal metallodendrimers and hexagonal metallodendrimers, respectively. 250 All these assemblies were characterized with multinuclear NMR, mass spectrometry and elemental analysis. Molecular modeling studies revealed that these exo-functionalized cavity-cored metallacycles have well-defined cavities and may thus function as transport vehicles for small, biologically active molecules. Similar Fréchet-type dendrons were also incorporated on the edges of Pd(II)- and Pt(II)-based metallosupramolecular squares.251 Functionalization of 4,4′-bipyridines in their 3,3′-positions with different generations of Fréchet-type dendrons and subsequent self-assembly with [M(dppp)(OTf)2] (M = Pd, Pt) led to nanometer-sized metallosupramolecular squares. ESI-FT-ICR-mass spectrometry and NMR experiments established the formation of these dendritic molecular squares, which showed interesting dynamic behaviour. Variable-temperature NMR studies revealed the coexistence of different conformers in solution, which interconvert slowly due to high barriers for both ligand rotation, caused by the dppp phenyl groups and for the racemization of the bipyridine ligands. The ligand exchange process was much slower for the Pt(II) squares as compared to their Pd(II) analogs.
Scheme 52.


The design and synthesis of macromolecules that contain multiple redox centers is of interest due to their potential application in electro-and photochemical devices.246 However, traditional covalent synthetic strategies to prepare functionalized dendritic and polymeric architectures have often led to relatively unsatisfactory yields and demand considerable synthetic effort. Exo-functionalization of the building blocks at the vertices with ferrocene moieties and their subsequent self-assembly with complementary subunits provides an efficient synthetic pathway with precise control over the location and number of the ferrocenyl moieties introduced into the system. Following a similar synthetic strategy as above, the combination of a 1:1 stoichiometric ratio of ferrocene appended bispyridyl 120° donor unit 229 with 60°, 120° and 180° diplatinum acceptors 12, 209 and 48 allows the self-assembly of [2 + 2] rhomboidal 230, [3 + 3] hexagonal 231, and [6 + 6] hexagonal 232 metallacycles, respectively (Scheme 53).252 In a complementary approach, ferrocenyl tethered 120° di-Pt(II) acceptor units were assembled into a [6 + 6] hexagon by treatment with the linker 4,4′-bipyridine.253 A [3 + 3] self-assembly between the ferrocenyl appended bispyridyl 120° donor unit 229 with complementary ferrocenyl tethered 120° di-Pt(II) acceptor units yields a hexagonal metallacycle with six ferrocenyl units. Multinuclear NMR and mass spectrometric analysis confirmed the formation of these assemblies. Electrochemical investigations on the multiferrocenyl assemblies 230-232 revealed that the redox sites are well isolated from each other electrochemically and are active. The sizes of the assemblies have an effect on the observed diffusion coefficients and half-wave potentials. Larger hexagonal assemblies led to a decrease in diffusion coefficient and an increase in half-wave potential. Heterofunctional hexagons containing orthogonal functionalities: redox-active ferrocenyl - host-guest crown ether,254 ferrocenyl - Fréchet-type dendrons,255 and host-guest crown ether - Fréchet-type dendrons256 were also assembled through a coordination-driven self-assembly approach. The precise positioning of the functional groups present at the vertex of the hexagons leads to supramolecules that are capable of carrying out a variety of functions both simultaneously and independently. Recently, Newcombe et al.257 reported the formation of pentameric and hexameric metallamacrocycles functionalized with sugar moities at the corners from the Fe(II) mediated complexation of 120° 3,5-bis(terpyridinyl)arene ligands. Treatment of 120° 3,5-bis(terpyridinyl)arene donors with FeCl2· 4H2O in MeOH for 24 h gave a mixture of pentameric (4.3%) and hexameric (11.9%) metallamacrocycles which were separated by gradient column chromatography. Although the hexamer was the expected product, the formation of pentamer was a result of an initially formed kinetic product. 1H NMR, 13C NMR, UV-vis spectroscopy, cyclic voltammetry and mass spectrometry established the structures of the macrocycles. These macrocycles generated nanofibres having diameters ranging from 10-80 nm.
Scheme 53.

Endo-functionalized two-dimensional supramolecular architectures have also been designed via coordination-driven self-assembly.258 The construction of endo-functionalized metallacycles including two [2 + 2] rhomboids (235 and 236) and a [3 + 3] hexagon (237) were achieved by the self-assembly of a 120° dipyridyl donor ligand (233) with three different di-Pt(II) acceptors 12, 234 and 209, respectively (Scheme 54). The metallacycles, containing several nitrobenzyl moieties at their interior surfaces, were characterized by multinuclear NMR (31P and 1H) and ESI-mass spectrometry. Significant C–H⋯O hydrogen bonding between the nitrobenzyl acceptor and the edge molecules of the supramolecular architecture was observed in the [2 + 2] rhomboid 235 and this interaction gradually decreased upon the enlargement of the resulting polygonal structures from 235, through a rhomboid 236 to a hexagon 237.
Scheme 54.

The incorporation of porphyrin moieties into supramolecular assemblies has generated much interest in recent years due to their potential application in varied fields such as photo-driven molecular switches, artificial photosynthetic systems, fluorescence-based chemical sensors, photonic wires, reaction catalysis and anticancer pharmaceutical behavior.259 Ligands derived from porphyrin units have the ability to serve as either linear or angular building units depending on the position of the donor functional groups, leading to both edge and corner functionalized supramolecular matallacycles. One of the earliest examples where porphyrin units were used to assemble metallacycles was provided by the work of Lehn et al. in 1994.260 The formation of edge-functionalized neutral molecular squares was achieved by treating 5,15-(4-pyridyl)-10,20-phenyl-porphyrin (4′-trans-DPyP) or its Zn complex with one equivalent of cis-Pt(NCPh)2Cl2. Similar treatment of 5,10-derivatized isomers (4′-cis-DPyP or its Zn complex) with trans-Pt(NCPh)2Cl2 gave corner-functionalized assemblies. Studies performed by Stang et al.205 also involved the use of derivatized porphyrin units not only as the corners but also as the edges of self-assembled metallacyclic squares using bisphosphine coordinated Pd(II) and Pt(II) angular and linear modules as the metallic precursor. Heterobimetallic [2 + 2] metallacycles of porphyrins have been assembled through a two-step modular approach using octahedral Ru(II) units as the building blocks.261 Treatment of [trans,cis,cis-[RuCl2(dmso)2(CO)2] (238) with 5,10-(4-pyridyl)-10,20-phenyl-porphyrin (4′-cis-DPyP; 239) in a 1:4 molar ratio allowed for the formation of bis-porphyrin ruthenium intermediate 240 which can be quantitatively assembled by reaction with [Pd(dppp)(OTf)2] (4) into a [2 + 2] rhomboidal metallacycle, 241 (Scheme 55). X-ray structural studies revealed a butterfly geometry due to stacking interactions between a phenyl ring of the dppp and the pyridyl ring of the porphyrin. The structural features compare well with the palladium [2 + 2] metallacycle [Pd(dppp)(cis-DPyP)2](CF3SO3)4 reported earlier by Stang et al.36 Homometallic neutral metallacycles of porphyrins [RuCl2(dmso-S)2(cis-DPyP)]2 (242) can be obtained by treatment of the bis-porphyrin ruthenium intermediate, 240, with one equivalent of 238 (Scheme 55).262 Interestingly, treatment of 238 with 5,10-(3-pyridyl)-10,20-phenyl porphyrin (3′-cis-DPyP) rather than of 4′-cis-DPyP yielded the corresponding [2 + 2] metallacycle with a staggered geometry in which two porphyrin units were held in a slipped cofacial arrangement by the ruthenium metal corners.263 This staggered geometry, as determined by X-ray structural studies, is reminiscent of those of the special pair of bacteriophylls in the reaction centers and of adjacent B850 units in the LH2 light-harvesting antenna systems of photosynthetic bacteria.264 Metalation of 242 with zinc acetate led to incorporation of Zn in to the porphyrin cavities. These Zn metalated metallacycles can further be assembled through axial coordination with linear bridging ligands to form molecular sandwiches and wires. As determined from NMR studies, the titration of Zn metalated macrocycles with one equivalent of 4,4′-bipyridine leads rapidly to the quantitative assembling of sandwich-like 2:2 supramolecular adducts where the porphyrin moieties adopt cofacial geometry, as evidenced by X-ray crystallographic studies.262
Scheme 55.

Hupp et al. have studied in some detail the synthetic and functional chemistry of molecular squares having porphyrins as edges and Re(CO)3Cl as corners.265 The nanoscale cavities of these prophyrin assemblies were chemically tailored for incorporation of receptor groups with high affinities for specific ions or molecules.266,267 For example, the edge-funtionalized supramolecular square, 268 244, derived from 2,8,12,18-tetrabutyl-3,7,13,17-tetramethyl-5,15-bis(4-pyridyl)-porphyrin (243) and Re(CO)5Cl (Scheme 56) renders lattices of 244 responsive to aqueous Zn(II) ions upon cavity functionalization with tris(2-aminoethyl)amine, whereas functionalization with 1,6-hexanedithiol enhanced the lattice response to molecular iodine by a mechanism involving formation of a charge-transfer complex.266,267 Fabrication of these porphyrinic squares as thin membrane films led to size-selective transport and thus they act as a nanofilters for various probe molecules. 269 Photoelectrochemical cells based on these cavity-containing porphyrinic molecular squares were able to sensitize flat indium-tin oxide (ITO) electrodes to photocurrent production with visible light.270,271 Recently, it has been demonstrated that large porhyrinic squares derived from meso-triazolyl appended Zn(II) porphyrins can show efficient energy trasfer between the meso – meso-linked Zn(II) porphyrin units thus have interesting implications towards development of artificial light-harvesting apparatus.272 Covalently linked cyclic porphyrin arrays have been synthesized to mimic natural light-harvesting systems and to investigate the highly efficient energy migration processes occurring in these systems for future applications in molecular photonics.273,274 Molecular rectangles containing cofacial porphyrin edges were synthesized by the same group through the reaction of bis(4-ethynylpyridyl)porphyrin with [Re2(CO)6Cl2(μ-bipyrimidine)] or a [Re2(CO)6(μ-bis(benzimidazolate)] in an equimolar ratio.118 The collapsed structure of the macrocycles brings the two porphyrin faces in close proximity, thereby substantially reducing the cavity for the incorporation of guest molecules. However, the distortion of the nominally rigid ethynylpyridyl linkages in the porphyrin ligand edges imparts significant charge-transfer characteristics to these rectangles. A series of neutral rhenium-based symmetric porphyrin dimers synthesized by the reaction of the methoxyphenyl derivatives of 4′-cis-DPyP with octahedral Re(CO)5Cl in THF, display efficient intramolecular porphyrin-porphyrin energy transfer. 275 A cyclic self-assembled tetramer of an asymmetric meso-ethynylpyridyl-functionalized Zn(II)-porphyrin was also structurally characterized.276 Femtosecond transient absorption and anisotropy spectroscopies have established fast energy transfer within a self-assembled cyclic porphyrin tetramer between porphyrin subunits.
Scheme 56.

Metallosalen complexes have long been known for their ubiquitous use in a variety of catalytic transformations.277 Salen scaffolds have been incorporated into metallosupramolecular assemblies via the coordination-driven directional bonding approach.278 The availability of a large and diverse library of salen scaffolds and the relative ease with which salen moities can be derivatized makes them ideal building blocks for the construction of functional, supramolecular assemblies for their potential application in various areas of research such as catalysis,277 materials279 and biology.280 Hupp et al.281 have assembled a series of metallacycles having salen scaffolds at the edges. For example, the reaction of bis(4-pyridyl)-functionalized salen ligand 245 with cis-(PEt3)2Pt(OTf)2 8 gave access to a [2 + 2] molecular loop 246 and a [4 + 4] molecular square 247 (Scheme 57). When free base semiflexible ligand 245 was used, preferential formation of a molecular loop was observed in nearly quantitative yield. The curvature that allows for loop closure, as determined from X-ray structural studies, was achieved via backbone distortion of the bridging salen ligand and likely entropic effects predominate in the assembly process. Alternatively, molecular square 247 was obtained directly by combination of cis-(PEt3)2Pt(OTf)2 8 with Zn(II)-245 ligand. Using a similar approach, the same group has introduced pyridyl containing salen scaffolds into molecular squares using Re(CO)5Cl as corner units. The use of the bimetallic edge [{(CO)4Re}2BisBzIm] 86 instead of Re(CO)5Cl resulted in the formation of molecular rectangles.282 Interestingly, post-metalation of the salen cavities of the molecular loop 246 with ZnEt2, which reduces the flexibility of the salen segments, leads to quantitative supramolecule-to-supramolecule transformation to the analogous molecular square 247.281 Similar structural conversion from tetranuclear to dinuclear squares induced by light was also observed earlier.283 Introduction of donor and acceptor centers in the same molecule can induce self-complementarity, leading to self-coordination and formation of supramolecular structures without participation of other ligands.
Scheme 57.

The templated approach to higher order supramolecular assemblies using bis-Zn(II)-salen building blocks and various ditopic bipyridine donors through coordinative Zn(II)-Npy interactions have been demonstrated.284 The use of bipyridine ligands of various sizes such as 4,4′-bipyridine, 1,4-di(4-pyridyl)benzene and 1,2-di(4-pyridyl)ethane enabled the modulation of the dimensions of the resulting assemblies. For example, [2 + 2] assembly of bis-Zn(II)-(salphen) 248 with 4,4′-bipyridine generated dimeric assembly 249 (Scheme 58). Binding and NMR studies showed that macrocyclic assemblies were formed under dilute conditions. The solid-state structure of the bipyridine congener shows the presence of porous channels that could be potentially exploited to make porous materials and in the construction of supramolecular transition metal catalysts. Similarly, a [2 + 2] assembly of bis-Zn(II)-(salphen) with a bispyridyl Ni(salphen) derivative led to the formation of a rectangular metallacycle through Zn(II)-N coordination.285 Salen-based molecular scaffold that contains both pyridyl donor and Lewis acidic Zn(II) acceptor sites in the same molecule have been designed and as a consequence, the molecular scaffolds assemble into a tetrameric vase structure through self-coordination.286 Although building block was achiral, the tetrameric assembly was chiral and both enantiomeric forms were present in the solid-state structure. A metal-templated approach recently afforded a tetra-Zn(salphen) supramolecular assembly from the combination of a diimine based on 3,3′-diaminobenzidine scaffold and a dialdehyde precursor, 4,6-dihydroxy-1,3-benzene-dicarboxaldehyde, in the presence of Zn(OAc)2·2H2O as a templating agent.287 The macrocycle showed strong cooperative self-assembly mediated by the formation of Zn(salphen) dimer units held together via μ2-phenoxo interactions.
Scheme 58.
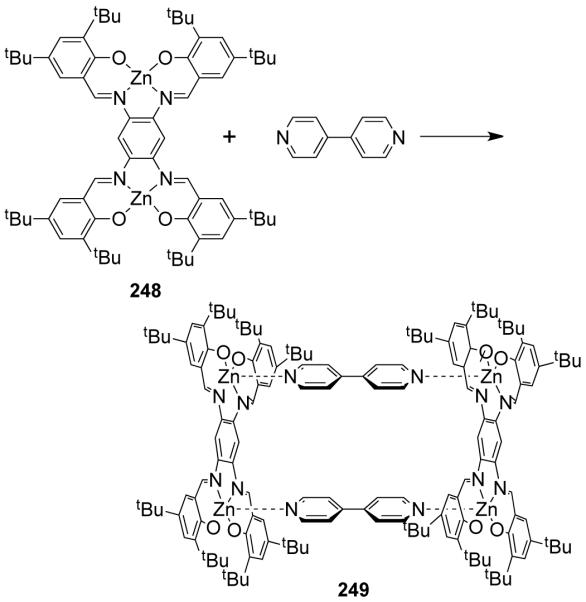
3.5. Applications and Uses of 2D Systems
Over the past few decades, metallosupramolecular architectures have found applications in various fields such as molecular recognition, photo- and electrochemical sensing, cavity controlled catalysis and molecular biology. These applications exploit the presence of the cavity in the assemblies.
3.5.1. Molecular Recognition
The design of discrete molecular entities that can selectively recognize molecules and signal the presence of a specific analyte is one of the main achievements of supramolecular chemistry.230 The use of supramolecular assemblies as hosts for molecular recognition is governed by various non-covalent interactions such as electrostatic, hydrogen bonding and van der Waals interactions. Metallosupramolecules have generated a great deal of interest due the relative ease with which they can be designed from simple complementary metal precursors and organic ligands. The necessary components to detect analytes and generate analytically useful and observable signals can be incorporated into the ensembles and the stoichiometry and position of the individual functional groups can be precisely controlled. From a biological point of view, studies on molecular recognition using supramolecular ensembles allow for the development and design of small discrete molecules that can interact with biological systems for therapeutic use, e.g. as anti-cancer drugs. One of the early examples of the application of metallosupramolecular assembly towards molecular recognition was provided by the investigations carried out by Maverick et al. in 1986,288,289 in which they studied the binding properties of various guest molecules such pyridine, quinuclidine, pyrazine and diazabicyclo-[2,2,2]octane (DABCO) in chloroform solutions by a cofacial copper(II) dinuclear metallacycle. X-ray crystallographic and solution studies have shown that DABCO preferentially binds at the cavity with a binding constant of 220 L/mol.
Molecular squares, with their tunable and easily accessible cavities, can lead to the selective inclusion of a wide range of organic molecules. Some of the early pioneering work of Fujita and Stang has shown that molecular squares can selectively recognize wide variety aromatic guests through various non-covalent interactions. For example, the first molecular square, [{Pd(en)(4,4′-dipyridyl)}4]8+ 46 synthesized by Fujita and his coworkers23,24 showed the ability to recognize neutral aromatic guests such as benzene, naphthalene etc. through charge-transfer interactions as well as hydrophobic interactions between the electron-deficient pyridine nuclei and the electron-rich aromatic guests. While the binding of small molecules such as dimethoxybenzenes showed small binding constants,290 the larger 1,3,5-trimethoxybenzene formed 1:1 host-guest complexes in D2O (Figure 24). However, Incorporation of a highly electron-deficient perfluorinated phenylene moiety in molecular rhomboid 250 led to high size and shape-specific molecular recognition of various polymethoxybenzenes such as 1,3,5-trimethoxybenzene, p-dimethoxybenzene, m- dimethoxybenzene, and o-dimethoxybenzene with association constants Ka = 2500, 2680, 1560, and 1300 M−1, respectively.291 Other large substrates were poorly or hardly recognized.
Figure 24.
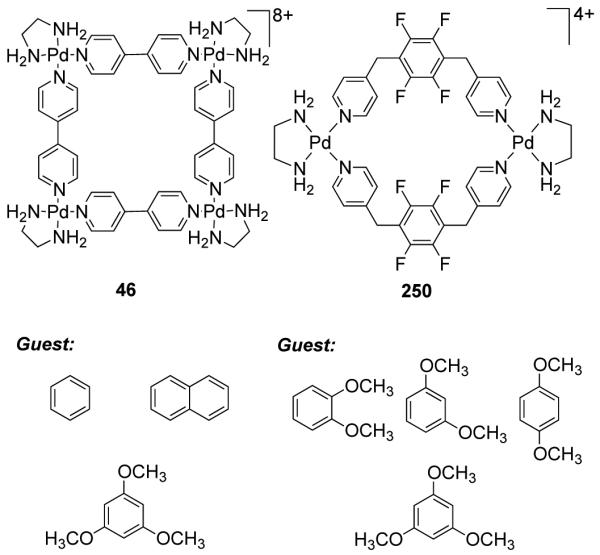
Molecular square, [{Pd(en)(4,4′-dipyridyl)}4]8+ (46) and molecular rhomboid 250 for selective binding of aromatic guest molecules.
Stang et al. successfully encapsulated dihydroxynaphthalenes in a related molecular square [{Pd(dppp)(4,4′-dipyridyl)}4]8+ (47) in CD3OD.292 Later, they were able to introduce Lewis base recognition sites into the squares by encapsulating AgOTf molecules through interactions with the ethynyl bonds.293 This host-guest precursor, 251, that contain silver atoms coordinated via the “π-tweezer effect” between each respective set of acetylene moieties, serves as the receptor for bidentate coordinating Lewis bases such as pyridine, pyrazine, phenazine, or 4,4′-dipyridyl ketone (Figure 25).292 Single crystal X-ray structure determination of the phenazine encapsulated molecular square revealed a 1:1 stoichiometry of the Lewis acid-base receptor host-guest complex. The overall geometry of the perimeter for this complex is almost planar with the guest phenazine oriented nearly orthogonal to the Pt–Pt2+–Pt plane.
Figure 25.
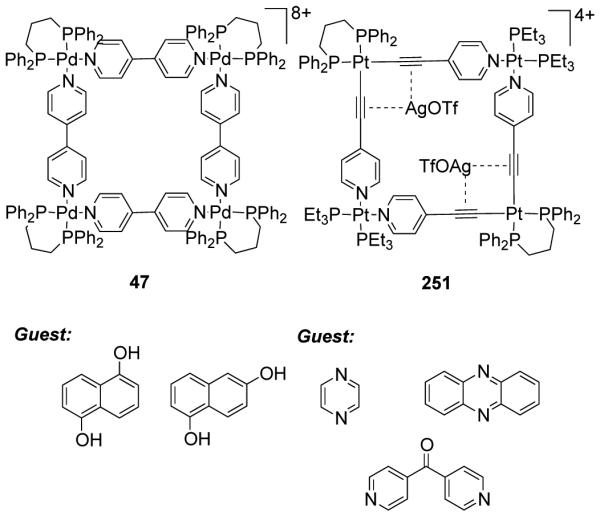
Molecular square, [{Pd(dppp)(4,4′-dipyridyl)}4]8+ (47) and AgOTf encapsulated host-guest precursor 251 for dihydroxynaphthalenes and lewis bases, respectively.
Jeong at al.294 reported a palladium-based metallomacrocycle 252 with two topologically discrete binding subcavities that showed homotropic cooperative binding behavior towards N,N,N’,N’-tetramethylterephthalamide through hydrogen bonding (Figure 26). It was proposed that the binding of the guest molecule in one subcavity allosterically activates the other subcavity to form stronger hydrogen bonds with the second guest molecule. The same group also designed and explored a series of osmium(IV) molecular squares and rectangles for the molecular recognition of various diamide guests.295 While the osmium-based molecular square selectively binds adipamide and terephthalamide, osmium(IV) rectangle was found to preferentially bind larger amide guest, such as biphenyldicarboxamide. The strong binding of the diamides was due to the presence of hydrogen-bonding pockets of the hosts. Hydrogen-bonding sites have also been utilized for the size selective molecular recognition of a number of mono and disaccharides in neutral platinum(II) based matallocycles.296 Fluorescence and 1H NMR studies established a 1:1 binding stoichiometry in chloroform through intermolecular hydrogen bonding interactions.
Figure 26.
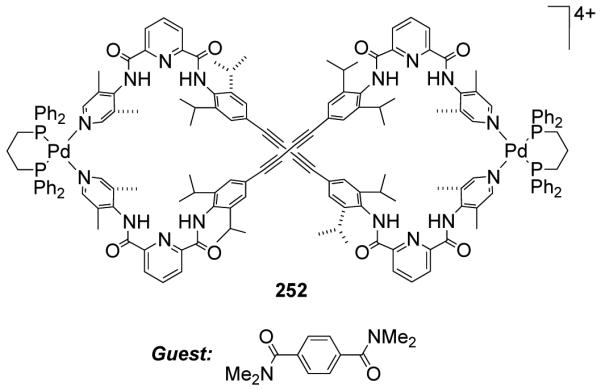
Metallomacrocycle 252 containing two topologically discrete binding subcavities for homotropic cooperative binding of guest molecules.
Heterotropic cooperative binding of fullerene and a diamine (4,4′-bipyridine or N,N,N’,N’-tetramethylhexane-1,6-diamine) was achieved in a macrocycle having cofacial, fused Zn-porphyrin arrays (Figure 27).297 The guest molecules, with complementary electronic effects on the binding properties of 253 communicate with one another through the π-electronic conjugation. The binding of diamine increases the electron density in the porphyrin ring, which disfavors the binding of a second diamine molecule but favors the binding of an electron acceptor such as fullerene. Similarly, binding of fullerene into the host activates the subsequent binding of the diamine into the other cavity.
Figure 27.
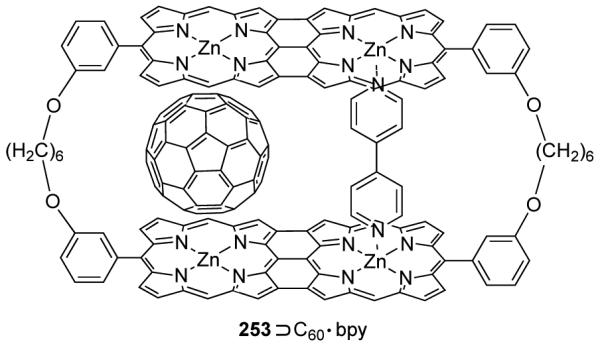
Heterotropic cooperative binding in macrocycle 253 having cofacial, fused Zn-porphyrin arrays.
3.5.2. Chemosensing
The essential components of a fluorescent chemosensor are a molecular recognition unit for selective interaction with an analyte and a signaling unit, generally a chromophore or a fluorophore. These are designed in such a way that the binding unit and the event reporter (chromophore or fluorophore) are structurally integrated so as to maximize the communication between the units. Since luminescence is very sensitive to subtle changes in geometry upon binding of a guest, fluorescent chemosensors are attractive for sensing of various analytes. Recently, electron-rich and photoluminescent heterometallic self-assembled molecular squares were shown to exhibit high quenching selectivities and sensitivities towards nitroaromatics, particularly picric acid.298 No significant quenching was noticed with electron-rich molecules such as benzene, xylene and 2,4,6-TMP, even at relatively higher quencher concentrations. A similar quenching pattern was observed with polar aromatics such as benzoic acid and phloroglucinol. The highly selective quenching effect with electron-poor molecules was ascribed to the induced π–π interactions between the π electron system of the metallic donor of the molecular square and the analyte.
Chiral, luminescence molecular squares have been shown to exhibit interesting enantioselective fluorescence quenching behavior by chiral amino alcohols in THF.207 The emission was assigned to both ligand-centered and MLCT excited states. The luminescence signal of the enantiopure molecular square can be quenched by both enantiomers of 2-amino-1-propanol but at significantly different rates. The quenching of the luminescence is proposed to be due to the inclusion of the chiral amino alcohols in the cavity of the squares through hydrogen bonding interactions. Enantioselective molecular sensing of chiral mandelic acid was investigated by using specifically designed tweezer complexes.299 The enantiometrically pure monomeric four coordinate Cu(I) metallacycle (S)-254 upon chelation of 2,2′-bipyridine (bpy) to the Cu(I) center breaks the weak Cu-S links and opens up the condensed structure, (S)-254, into a 27-membered macrocycle (S)-255 which can recognize mandelic acid (Scheme 59). Thus, the binding of the (R)- or (S)-mandelic acid was pseudo-allosterically regulated by binding of the anciliary ligand bpy, resulting in an increased cavity size. The condensed structure (S)-254 shows no binding of the guest molecules due to the inaccessibility of the binding pocket, as determined from fluorescence intensities.
Scheme 59.
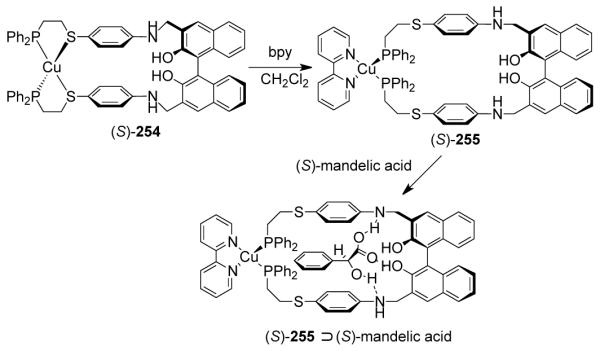
Severin and coworkers have designed a number of half-sandwich complexes of Ru(II), Rh(III) and Ir(III) with arene or cyclopentadienyl ligands as sensors for small cations and anions.163 Many of these metallacycles were found to act as highly selective receptors for lithium and sodium ions, the binding constants being dependent upon the nature of the half-sandwich complex, the ligand, and the pH.300,301 While the trinuclear Ru(III) metallomacrocycles (256, Figure 28) showed high affinity and selectivity for Li+ and Na+ ion,164,302 the Ir(III) macrocycles act as a fluoride ion receptor in which a pre-coordinated lithium ion serves as the binding site.303 The quantification of the host-guest complexes were done by NMR, electrochemical and colorimetric methods. Extending the same strategy further, they have also designed and synthesized a Ru-based metallomacrocycle for the selective detection of lithium ions in water and in serum by fluorescence spectroscopy.304 An interesting feature of these macrocycles is the incorporation of different pendent fluorophores – pyrene, dansyl and methoxycoumarin into the trinuclear metallacycles. The cavities in the assemblies serve as a Li+ ion recognition unit. Addition of LiCl to an aqueous solution of metallomacrocycle led to an increase in the fluorescence signal intensity due to a ‘turn on’ response of the receptor upon binding of Li+. Fluorescent titration with NaCl instead of LiCl showed a negligible response, indicating its highly specific and selective nature. The application of methoxycoumarin-appended sensor 256 in a complex matrix such as human serum also revealed its high specificity and selectivity towards biologically important lithium ion.
Figure 28.
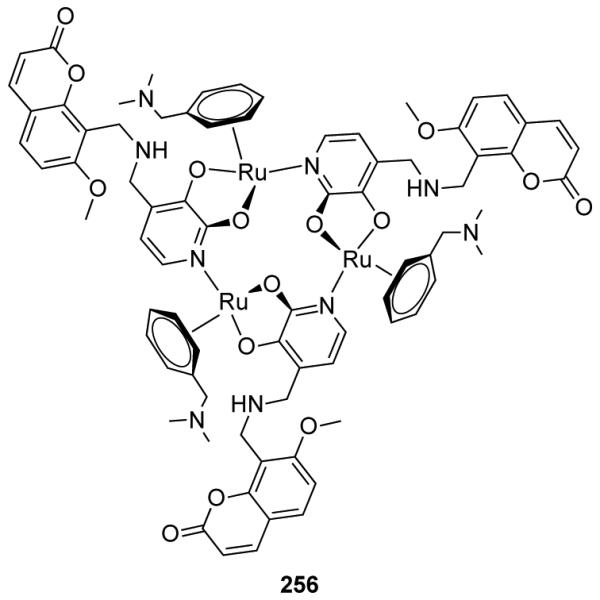
Trinuclear Ru(III) metallomacrocycles (256) for high affinity and selective binding of Li+ and Na+ ion.
Puddephatt and coworkers305 have synthesized several Pd(II) based molecular triangles using a 4(3H)-pyrimidone ligand which act as efficient anion sensors. Binding of the anions was determined by X-ray structural and NMR studies in solution. The 1H NMR titration data demonstrated that the molecular triangles are an effective host for planar nitrate and tetrahedral oxoanions. X-ray structures determined for triangles showed that binding of BF4−, ClO4− and CF3SO3− follow the 1:1 binding model. The oxoanions, including ClO4−, NO3−, HSO4−, and CF3SO3−, are bound more strongly than BF4− while PF6− is bound more weakly. A series of dinuclear cationic Ag(I) macrocycles synthesized from N-methylated bis(amidopyridine) ligands showed easy anion exchange properties.132 As determined from structural studies, the counterions occupy the cavity of the macrocycles. Anion exchange studies carried out with ESI-mass spectrometry demonstrated that these counterions could be replaced by other anions of a similar nature. Similarly, Pd(II) “lantern complexes” synthesized from bispyridyl amide ligands were shown to act as hosts for either cations or anions by adjusting the conformation of the amide substituents to accommodate the guests.306,307
A molecular rectangle 85 that can selectively sense 3d transition metal ions has also been designed and synthesized.114 The anthracene-based clip acts as the fluorophore, while the edge functionalized imine-based donor that contains a N4 pocket for binding moderately hard transition metal ions acts as a molecular recognition site (Figure 29). Due to the presence of a metal ion receptor site, the solution fluorescence intensity was quenched efficiently upon titration with 3d transition metal ions like Mn2+, Fe3+, Ni2+, and Cu2+. Interestingly, no such quenching was observed when the titration was performed with soft metal ions (Zn2+ or Cd2+) with a d10 electronic configuration. The metal-containing design of these rectangles and the presence of a specific binding core for metal ions make them suitable as fluorescent sensors for transition metal ions. Stang et al. have also designed a molecular rectangle (80) containing the phenanthroline moiety on the longer edge and have shown that it is an efficient chemosensor for Ni2+, Cd2+, and Cr3+ up to a 1:1 host-guest ratio (Figure 29).111 Binding of the metal ions in the N4 pocket was determined by UV-visible spectroscopy.
Figure 29.
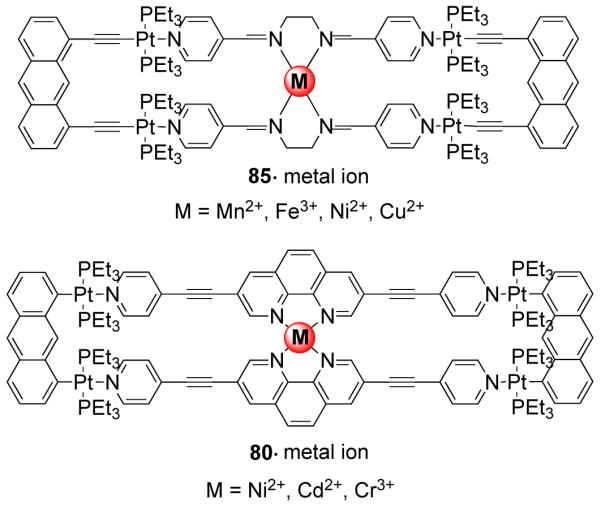
Molecular rectangles having N4 pockets for selective Chemosensing of 3d transition metal ions.
Lu et al. have assembled two gondola-shaped tetrarhenium metallocyclophanes, which were capable of selectively recognizing metal ions and planar aromatic molecules.308 The metallacycles were formed via a reaction of Re(CO)3, a ditopic heterocyclic clip, and a bischelating-bridging unit using an orthogonal-bonding approach. These highly luminescent cyclophanes selectively bind mercury cations and anthracene molecules. With Hg2+ ions, quenching of the emission was observed with formation of a 1:1 host-guest complex, as determined by binding studies. With other metal ions such as Li+, Sr2+, Co2+, Ni2+, Cu2+, Zn2+, Pb2+ and Ag+ these metallocyclophanes showed no significant fluorescent quenching activity.
3.5.3. Cavity Controlled Catalysis
One of the major applications of supramolecular chemistry is the design and understanding of catalysts and catalytic processes. Supramolecular architectures provide a unique opportunity for the study of non-covalent interactions that govern the binding of the reactants into conformations suitable for lowering the energy of the transition state of a given transformation. The cavity of an assembly can also stabilize reactive species or lead to enhanced reactivity and selectivity by isolating encapsulated guests from the bulk environment and can catalyze reactions effectively due to guest discrimination.17 Supramolecular catalysts can also activate otherwise unreactive substrates without the use of distinct covalent interactions between the catalyst and substrate, promoting reactions which cannot occur under normal conditions. With the vast number of metallosupramolecular assemblies known and the ease with which new molecules of predictable geometries can be generated, the study of their catalytic activity is an attractive proposition for supramolecular chemists.
One of the earliest examples of the application of metallosupramolecular architecture in a catalytic organic transformation was provided by Sanders and coworkers where they used a covalently bound trimeric porphyrin (257) as a host for the acceleration of the intramolecular Diels-Alder reaction of a furan-based diene and a maleimide-based dienophile (Scheme 60).309 In the absence of any catalyst, both endo- and exo-adduct were observed. However, with one equivalent of 257, the reaction rate was enhanced 200-fold with the exo-adduct as the sole product observed. The metallosupramolecular assembly was also found to be an active catalyst for the acyl transfer reaction. The reaction proceeded through a tightly-bound intermediate, in which both the pyridyl ring and the imidazole group were coordinated to two of the Zn(II) centers within 257, bringing the reactive sites into close proximity.310
Scheme 60.

Lin et al.213 investigated enantioselective catalysis using chiral metallosupramolecular cyclophane 258 derived from the enatiopure BINOL ligand and cis-Pt(PEt3)2Cl2. A combination of 258 with Ti(OiPr)4 forms an active catalyst for the enantioselective diethylzinc additions to aromatic aldehydes to afford the corresponding chiral secondary alcohols (Scheme 61). With 1-naphthalehyde as the substrate, a 95% conversion with 94% enantiomeric exess (ee) was observed. Interestingly, enantioselectivity drops when smaller aromatic aldehydes were used as substrates and was postulated to be due to the rigid architecture of the metallocyclophane 258. However, the disposition of chiral dihydroxy groups in an endo fashion renders the catalyst less enantioselective due to the steric congestion preventing the reaction with Ti(OiPr)4 to form an active catalyst.214 Ti(IV) complex of metallosupramolecular triangle 193 was also found to efficiently catalyze the enantioselective (ee upto 92%) additions of diethylzinc to aromatic aldehydes.209
Scheme 61.
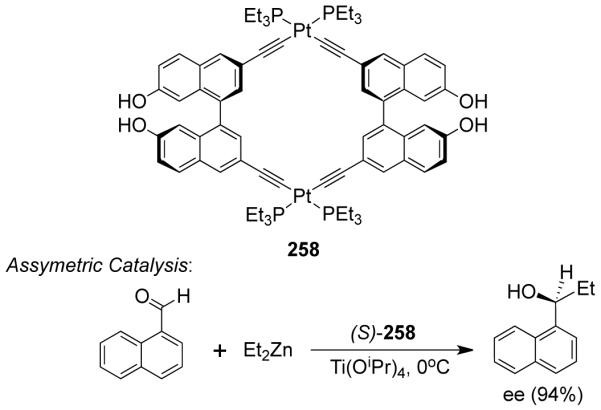
Hupp et al.311 have used a Zn porphyrin-lined metallacyclic square to encapsulate a simple Mn porphyrin unit, a known catalyst for epoxidation reactions. Mn porphyrin catalysts suffer deactivation under aerobic conditions. The protective encapsulation provided by the supramolecular square to the dipyridyl and tetrapyridyl form of the Mn porphyrin catalyst units prevents this deactivation and affords a 10 to 100-fold increase in turnover number and a substantial increase in the lifetime of the Mn catalyst. With the dipyridyl catalyst, the square also induced substrate size-selectivity by preventing access to the active site by large substrate molecules.311
Allosteric regulation is a ubiquitous form of control for biological molecules to regulate substrate binding but is a rarity for synthetic catalysts.312,313 Mirkin et al. have developed metallosupramolecular architectures containing two structural domains that can mimic an abiotic assay similar to the enzyme-linked immunosorbent assay (ELISA).314 An important design feature of these allosteric catalysts is the structural Rh(I) metal centers which act as allosteric regulators while the chiral Cr(III) or Zn(II)-salen moities act as the catalytic active sites. The reversible binding of CO and Cl− at the Rh(I) centers in 259 (M = Cr(III)) and 260 (M = Zn(II)) results in the opening up of the structure to 261 and 262, respectively (Scheme 62). This opening up allowed substrate molecules to enter so that they could undergo a fast intramolecular reaction. The investigations of the acyl transfer reaction between acetic anhydride and pyridyl carbinol showed that the reaction rate increased significantly upon binding of CO and Cl− at the Rh(I) centers, which opened up the structure.315 Since metallosalens can catalyze the reaction in a bimetallic fashion, the substrate molecules can access the cavity and can undergo a faster intramolecular reaction. Coupling of the catalytic amplification step to a pH-sensitive fluorophore (diethylaminomethylanthracene) that interacts with the reaction byproduct, acetic acid, provides a straightforward method for visually and spectrophotometrically monitoring the reaction. Recently, the same group has developed a supramolecular triple-layer allosteric catalyst for the ring-opening polymerization of ε-caprolactone.316 The allosteric supramolecular structure contains a monometallic catalytic site buried in the middle layer of a triple-layer of the complex, which can reversibly open and close upon addition of small molecules and elemental anions thus reversibly exposing and concealing the catalytic center. The ring-opening polymerization of ε-caprolactone can be turned on by the in-situ opening of the triple-layer complex and then completely turned off by reforming it through the abstraction of Cl−, the allosteric effector agent, without appreciable loss of catalytic activity.
Scheme 62.

Supramolecular allosteric cofacial porphyrin assemblies can also function as catalysts for acyl transfer reactions.317,318 The cavities of these macrocycles are tunable via introduction of simple ancillary ligands that bond to the structure control domains. This enables the cofacial porphyrin structures to act as allosteric catalysts capable of discriminating different substrate combinations and selectively transforming them into the desired products. The change in cavity size that occurs allows the acyl transfer reaction between pyridylcarbinol and 1-acetylimidazole. A tetrametallic macrocycle having two Zn catalytic centers assembled via the weak link approach was shown to be an efficient allosteric catalyst for the hydrolysis of 2-(hydroxypropyl)-p-nitrophenyl phosphate (HPNP), a model substrate for RNA.319 The binding of CO and Cl-induces a transition of the catalytically inactive condensed structure to the highly active open structure. The chromium and zinc containing catalytic molecular tweezer complexes 263 and 264 were found to be active enantioselective catalysts for the asymmetric ring opening reaction of cyclohexane oxide by azidotrimethylsilane (TMSN) (Scheme 63).320
Scheme 63.
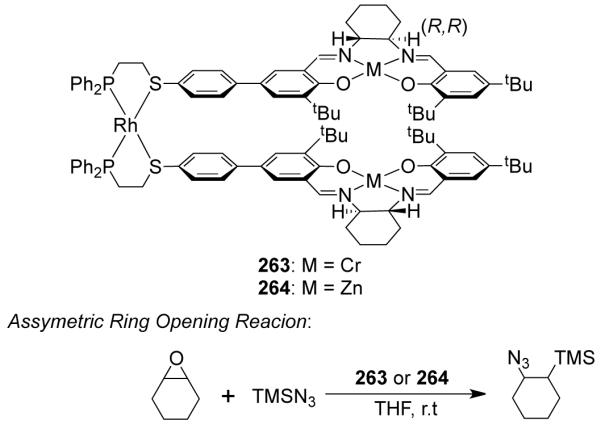
Cotton et al.228 investigated a metal carbine transformation using the model intermolecular cyclopropanation of styrene with thyl diazoacetate in both homogeneous and heterogeneous phases by using enantiopure chiral molecular triangles SSS-[cis-Rh2(C6H4PPh2)2(C8H4O4)(py)2]3 (265), SSS-[cis-Rh6(C6H4PPh2)6(C2O4)3(py)5(CH2Cl2)] (266), and SSS-[cis-Rh6(C6H4PPh2)6(C14H8O4)3(py)4(CH2Cl2)2] (267) obtained from the dimetallic corner unit cis-Rh2(C6H4PPh2)22+ (Scheme 64). These studies showed that the metallosupramolecular ensembles were very active and had remarkable enationselectivity.228 The enatiocontrol achieved was superior to that of known dirhodium(II) catalysts with chiral carboxamide ligands.321
Scheme 64.

3.5.4. Biological Applications
Recently, 2D metallosupramolecular assemblies have been investigated as potential candidates for selective anti-tumor therapies. G-quadruplexes are the most recent secondary DNA structures that are being considered as targets for anti-cancer drug design.322 Guanine rich stretches of DNA can self-assemble into a cyclic arrangement of four guanines (G-quartet) held together by eight Hoogsteen hydrogen bonds. Further association of these G-quartets leads to the stacking of tetramers on top of each other to form four-stranded structures called a G-quadruplex. These G-quadruplexes appear at the ends of chromosomes in the telomeric and transcriptional regulatory regions in several important oncogenes. Because telomere maintenance mechanisms and the transcriptional regulation of oncogene expression are important targets for drug design, G-quadruplexes represent potentially important targets in drug development. Sleiman et al.323 have shown that Fujita’s platinum based metallosupramolecular square23 [Pt(en)(4,4′-dipyridyl)]4 (46) could efficiently bind to G-quadruplex DNA and effectively inhibit telomerase activity. Thermal denaturation studies with duplex and quadruplex FRET probes and enzymatic assays demonstrated that platinum square 46 strongly binds to G-quadruplexes and can act as an inhibitor of telomerase. The highly electropositive nature of 46, as well as hydrogen bonding interactions between the ethylenediamine ligands and phosphates of the DNA backbone, contributed to the observed strong binding affinity to the G-quadruplex. The interaction of DNA with supramolecular squares 46 and [Pt(en)(1,4-bis(4-pyridyl)tetrafluorobenzene)]4 showed modification of the secondary and tertiary structure of DNA due to intercalation between base pairs.324 Assays against HL-60 tumor cell lines displayed discrete antiproliferative behavior for both molecular squares as compared to cisplatin. Appending a long lipophilic chain on one of the carbon atoms of the ethylenediamine moiety of 268, followed by self-assembly with 4,4′-bipyridine (bpy) formed molecular square 269, and led to the interesting observation of channel formation in planar bilayer lipid membranes (Scheme 65).325 It was hypothesized that the membrane-bound lipophilic metallosupramolecular square led to the formation of long-lived and highly conducting ion channels due to extended aggregation of the complexes and lipids surrounding a toroidal pore in the membrane.
Scheme 65.

Therrien et al. have investigated a series of cationic arene ruthenium122 and osmium326 based tetranuclear molecular rectangles for their anticancer activity against ovarian A2780 cancer cells lines. The large rectangles incorporating 1,2-bis(4-pyridyl)ethylene linkers are more cytotoxic than the 4,4′-bipyridine-containing smaller cationic rectangles. However, the observed cytotoxicity may also arise due to fragmentation of the rectangles within the cell, leading to cytotoxic mononuclear complexes. Similar ruthenium-based molecular rectangles having dipyridyl amides as linkers also showed potent anticancer activity against various human cancer cell lines.327 Dyson et al., in their study on the cytotoxic activity of a series of trinuclear p-cymene ruthenium metallacycles on human cancer and fibroblast cells, observed the interconversion between trimeric metallacycles and monomeric complexes as a function of pH.328
4. THREE DIMENSIONAL (3D) NANOSCOPIC CAGES
Self-assembly processes to form complex 3D structures, such as the icosahedral or dodecahedral shapes of the viral capsids, are abundant in nature. However, abiological preparation of such complex structures has proved challenging. Thus, not surprisingly, many of the first examples of complex 3D topologies were the result of serendipity rather than design. However, the development of different design principles over the last couple of decades (Section 2) for the preparation of high-symmetry coordination cages via self-assembly has led to an incredible variety of structures ranging from simple ‘Platonic’ solids (tetrahedron, octahedron, cube, dodecahedron and icosahedron) which have faces consisting of only single regular polygons, to more complex ‘Archimedean’ solids, which have faces consisting of two or more regular polygons, with precise control over the geometry. Coordination-driven self-assembly provides synthetic control on a supramolecular scale to carefully design and understand the geometric requirements of a particular ligand vis-à-vis the stereoelectronic preferences of a specific metal center. In this section, we will discuss the synthetic and topological aspects of the self-assembled coordination cages, their functional properties and various applications and uses.
4.1. Archimedean and Platonic Systems
4.1.1. Tetrahedra
A tetrahedron, the simplest of the platonic solids, can be assembled utilizing a few different metal-ligand stoichiometries: a) M4L6 tetrahedra, where the four metal ions occupy the vertices and the six ligands act as the edges; b) M4L4 tetrahedra, where the metal ions act as the four vertices, and the four faces of the tetrahedra are covered by ligands with threefold symmetry and c) M6L4 tetrahedra, or truncated tetrahedra, where the ligands occupying each of the four faces of the tetrahedron are connected by metal centers on the middle of the edges (Figure 30).
Figure 30.
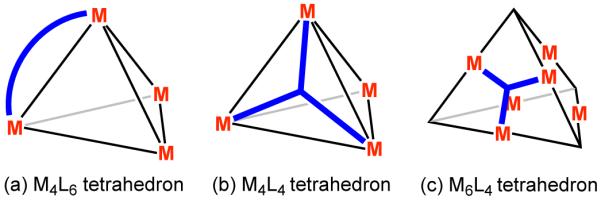
Schematic representation of the different ways of assembling a tetrahedral topology. Only one of the ligands is shown for clarity.
In 1988, Saalfrank and coworkers serendipitously stumbled upon the first coordination-driven tetrahedral assembly.329 In their pursuit of tetradonor-substituted allenes, the double deprotonation of malonic ester with methyl Grignard to generate its dianion led to the in-situ formation of a ditopic, bisbidentate ligand (L) via C-C coupling of two dialkyl malonate monoanions with oxalyl chloride and spontaneous deprotonation of the bis(enol) intermediates (Scheme 66). The tetraanionic assembly [(NH4)4∩{Mg4(L)6}] (270) consists of a distorted tetrahedron composed of four magnesium(II) ions occupying the apices, which are linked along each of the six edges by the bisbidentate ligands such that each of the four magnesium(II) ions are octahedrally coordinated. The ammonium counterions are hydrogen bonded to the oxygen donors at the triangular faces of the tetrahedron.330 Temperature-dependent 1H NMR studies showed the presence of homochiral, racemic complexes that were kinetically stable on the NMR timescale. Replacement of magnesium(II) by divalent transition metals allowed the syntheses of the corresponding tetranuclear complexes [(NH4)4∩{M4(L)6}] (M=Mn(II), Co(II), Ni(II), Zn(II)).331 Homovalent and mixed-valent tetranuclear iron complexes [M⊂{FeIInFeIII4-n(L)6}]n± (M = NH4+, Na+, K+, Cs+ K+, H2O) can also be accessed by using FeCl2 or FeCl3.332 The use of 4,4′-phenylene and 4,4′-biphenylene as spacers in the bisbidentate ligands led to tetrahedra with enlarged cavities. For instance, deprotonation of the stable tetramethyl 2,2′-terephthaloyldimalonate compound with sodium hydride yielded a negatively charged ditopic tetradentate ligand, which reacted with iron(III) chloride to form a tetrahedral tetrairon core bridged by six chelate ligands with a much larger cavity.333
Scheme 66.
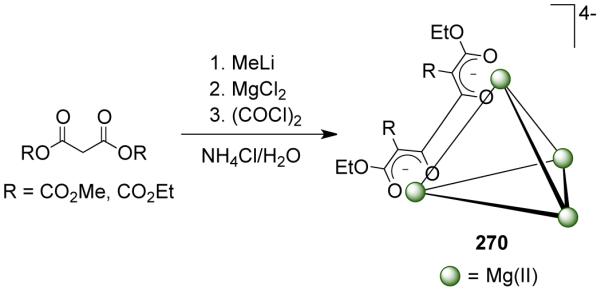
Raymond and coworkers assembled early examples of anionic M4L6 and M4L4 tetrahedral clusters with trivalent and tetravalent metal ions such as Ga(III), Al(III), In(III), Fe(III), Ti(IV) and Sn(IV) using the symmetry interaction approach and delineated the design principles in order to predict and construct the formation of self-assembled systems, as discussed in section 2.2.9 Though composed of achiral building units, many of these tetrahedral assemblies are intrinsically chiral (vide infra, section 4.3). The use of bis-hydroxamate334,335 and bis-catecholate336 based ligands having C2-symmetry and octahedral metal ions have led to the assembly of a vast number of tetrahedral clusters of varying sizes. For example, deprotonation of six equivalents of 271 with KOH, followed by the addition of Et4NCl and four equivalents of Ga(acac)3 led to the isolation tetrahedral assemblies [NEt ⊂ Ga4(271)6]11− (272; Scheme 67).336 The naphthalene spacer between the two pryocatechol units provides rigidity and predisposes the ligand for interaction with the trivalent metals to exclusively form tetrahedral assemblies over the entropically more favorable M2L3 helicates.336 These anionic, tetrahedral assemblies can selectively encapsulate a variety of monocationic guests (vide infra, section 4.5.1),337 and can form without thermodynamically-templating cationic guest molecules. However, the formation of M4L6 tetrahedra with larger cavities, using anthracene in the ligand backbone, can only be achieved in presence of suitable cationic guest molecules. In the absence of such a guest, entropically favored M2L3 triple helicates are formed. Although the use of a bulkier spacer such as pyrene338 prevents the formation of helicates, tetrahedra are formed only in the presence of a guest molecule. Larger M4L6 cages with 1,1′-binaphthyl as the spacer have also been designed with an aim to accommodate larger guest molecules.339
Scheme 67.
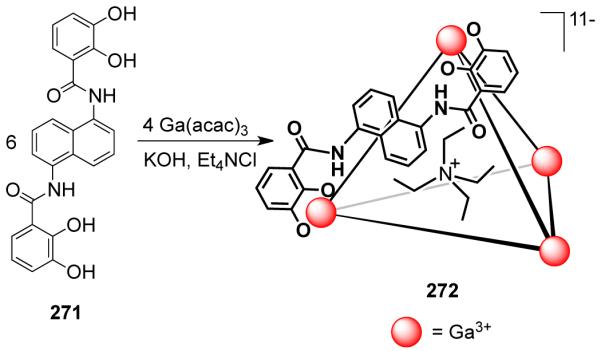
McCleverty, Ward and coworkers have reported a number of cationic M4L6 tetrahedra ustilizing symmetry interaction approach.340 These tetrahedra were assembled from flexible bispyrazolylpyridine ligands with 1,2-phenyl,341,342 2,3-napthyl,343,344 9,10-anthracenyl,344 and biphenyl345,346 spacers between the binding sites. Treatment of bispyrazolylpyridine ligands having a 1,2-phenyl central spacer (273) with a divalent metal Co(II) as their fluoroborate or perchlorate salt afforded racemic tetrahedral cages [Co4(273)6X]X7 [X = BF4−, ClO4−] (274; Scheme 68).341,342 X-ray structural studies showed that the ligands in 274 bridges along each of the six edges, spanning two metal ions. Each pseudo-octahedral metal ion is coordinated by a bidentate unit from each of three different ligands with one of the tetrahedral counterions trapped in the cage cavity. In fact, the formation of these intrinsically chiral assemblies was templated by the trapped anions. The encapsulated BF4− anion is complementary in both charge and shape to the cavity with respect to the Co4 tetrahedron. Each fluorine atom of the trapped anion is directed toward the center of a triangular face of the Co4 tetrahedron. Interestingly, variable-temperature 11B and 19F NMR studies revealed that the BF4− anion is effectively trapped within the cavity and does not exchange with the seven outer-sphere BF4− counterions.342 Similar flexible bispyrazolylpyridine ligands having 1,8-napthyl spacers led to a M12L18 assembly having a topology consistent with a truncated tetrahedron with T symmetry upon reaction with Co(II) or Zn(II) in the presence of NaBF4 or NaClO4.347 However, in a study using a related tetrahedral [Fe4L6]8+ (L = 5,5′″-dimethyl-2,2′:5′,5″:2″,2′″-quaterpyridine) complex it has been shown that templating by polyatomic anions is not always necessary for the assembly of cage structures as demonstrated by the isolation of the anion-free cage.348
Scheme 68.
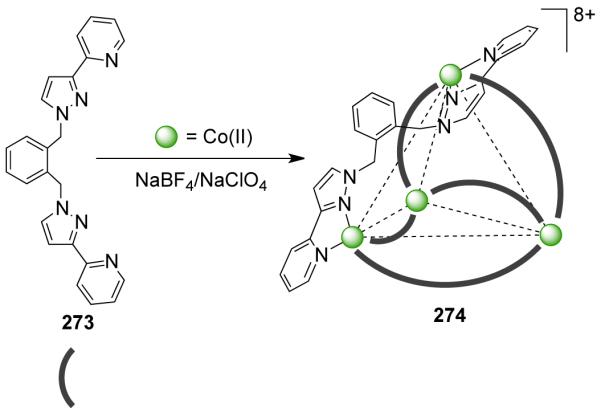
In an elegant approach, Nitschke and coworkers349 designed a M4L6 tetrahedral cage utilizing dynamic covalent and coordinative bonds in tandem from a multicomponent system. The synthesis of the tetrahedral cage, 277, was achieved by treatment of 2-formylpyridine (275) and 4,4′-diaminobiphenyl-2,2′-disulfonic acid (276) subcomponents with iron(II) and base in aqueous media (Scheme 69). The single crystal X-ray structure analysis of the tetramethylammonium salt of 277 revealed that the dynamic covalent bonds formed in-situ between a 4,4′-diaminobiphenyl-2,2′-disulfonic acid and two 2-formylpyridine moities give rise to a bis-bidentate ligand which cheleates the Fe(II) atoms occupying the vertices of the tetrahedron in a hexaimine ligand environment. Strong binding interactions between the Fe(II) and imine ligands confer structural stability to cage 277. The sulphonate groups of 4,4′-diaminobiphenyl-2,2′-disulfonic acid are symmetrically exposed to the exterior of the cage thereby contributing towards its solubility in aqueous solutions. Tetrahedral cage 277 was also assembled from a multicomponent system via a sequential self-assembly approach.350 Each of the imine ligands formed from three different amines have flexible, bent and linear rigid motifs, respectively. When treated with 2-formylpyridine they form entropically-favored, self-assembled architectures through a self-sorting process. The cage has an internal void volume of 141Å.
Scheme 69.

Recently, the same group reported a new class of M4L6 tetrahedral cages, which have reconfigurable exteriors in solution.351 The Fe(II)-templated reaction of 3,3′-bipyridine-6,6′-dicarboxaldehyde with para-substituted anilines led to the formation of these M4L6 tetrahedral cages decorated with externally-directed amine residues. For example, treatment of 3,3′-bipyridine-6,6′-dicarboxaldehyde with p-chloroaniline in the presence of an appropriate Fe(II) salt gave a homochiral tetrahedral cage having twelve p-chloroaniline residues on the periphery. The C2-symmetric bis-bidentate pyridylimine ligands define the edges of the tetrahedron, bridging the four Fe(II) ions at the vertex. Addition of p-toluidine or p-methoxyaniline to a solution of p-chloroaniline-containing cage resulted in the quantitative displacement of p-chloroaniline residues to form p-toluidine-containing and p-methoxyaniline-containing cages, respectively. Electronic effects drive these subcomponent substitutions wherein electron-rich anilines effectively substitute for the electron-poor aniline residue. Each of these cages encapsulates an anion within their cavities. The peripheral substitution does not impact the anion binding preferences in these cages and follows the order of BF4− < OTf− < PF6−. Interestingly, a large library of 91 distinct heteroleptic M4L6 cages, assembled from a reaction of 3,3′-bipyridine-6,6′-dicarboxaldehyde with p-chloroaniline, p-bromoaniline and p-iodoaniline in presence of Fe(II) ions, collapsed into a single homoleptic p-methoxyaniline-containing M4L6 cage upon addition of more electron-rich p-methoxyaniline to the mixture.
Lindoy and coworkers assembled neutral M4L6 tetrahedral cages by utilizing a β-diketonate ligand having phenylene spacers with its donor sets oriented at sixty degrees. Treatment of iron(III) chloride with 1,4-phenylene-bridged bis-β-diketonato ligands in THF in a 2 : 3 molar ratio led to the formation of a series of M4L6 tetrahderal cages of the type [Fe4L6] · (solvent)n.171 Single crystal X-ray structural studies of these cages revealed a tetrahedral topology with four iron(III) ions situated at the vertices of the tetrahedron and the six bis-β-diketonato ligands defining the edges. Increasing the size of the bis-β-diketonato ligand by extending the bridge between the coordination domains from phenylene to biphenylene led to the assembly of a large neutral M4L6 tetrahedral cage.352 Treatment of biphenylene-spaced bis-β-diketone ligand, 1,1′-(4,4′-Biphenylene)-bis-3,3-dimethylpentane-1,3-dione, with iron(III) chloride led to the formation of the cage with four encapsulated tetrahydrofuran guest molecules. A corresponding tetrahedral cage was assembled from a phenylene-spaced bis-β-diketone ligand, 1,1′-(1,4-phenylene)bishexane-1,3-dione, and could encapsulate only one tetrahydrofuran molecule as a guest.
A M4L4 tetrahedron can be assembled by utilizing ligands that have three bidentate metal binding sites with a C3-axis of symmetry in their idealized structure in conjugation with metal ions that prefer octahedral coordination geometries.9,353 Raymond et al.354 assembled Ti(IV), and Sn(IV)-containing M4L4 tetrahedrons, 279 and 280, using a catecholamide trigonal trisbidentate ligand (278) under basic conditions (Scheme 70). 1H NMR and mass spectrometric studies indicated the formation of M4L4 clusters. The X-ray structural analysis of (Et3NH)8[Ti4(278)4] (279) revealed that the cluster lies on a crystallographic threefold axis but has the molecular symmetry of the rotation group T, such that all the metal ions have the same chirality (all D or L). Similarly, tetranuclear Fe(III) cages generated in a one-pot reaction from benzene-1,3,5-tricarboxylic acid trichloride, bis-t-butyl malonate, methyllithium, and iron(II) dichloride under aerobic conditions,355 and tris-pyrazolyl borate based M4L4 tetrahedral assemblies were also reported.356,357 The internal cavities of these tetrahedral cages are too small to encapsulate guest molecules. Utilizing the same design strategy, Raymond et al. designed larger M4L4 tetrahedra based on a C3-symmetric ligand, 1,3,5-tris(4′-(2″,3″-dihydroxybenzamido)-phenyl)benzene.358 Tetrahedral assemblies with Al(III), Ga(III), In(III) and Ti(IV) as the pseudo-octahedral metal centers were synthesized with cavity sizes of around 450 Å3. In contrast to tetrahedral clusters 279 and 280, where no evidence of guest encapsulation was observed in the small cavity, these cages were able to encapsulate charge-, size-, and shape-selective guest molecules within their cavities. Along the same line, the assembly of triscatechol imine ligands with titanium(IV) ions in the presence of alkali metal carbonate as a base (Li, Na, K) in DMF led to the formation of metallosupramolecular tetrahedra with huge internal cavities.359,360 The X-ray crystal structure of the Ti(IV) catecholate octaanionic cluster showed the presence of an internal cavity with four potassium cations bound to the internal catecholate oxygen atoms. Additionally, three DMF molecules were bound to each of the potassium ions.
Scheme 70.
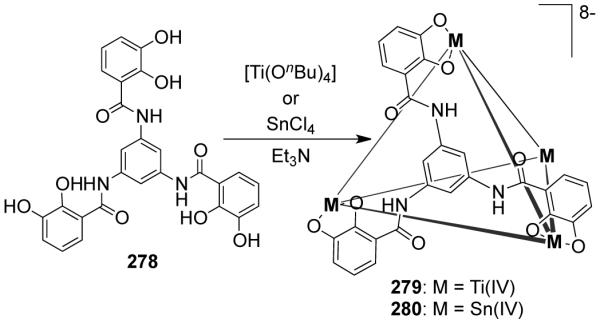
N-centered tripodal heptadentate ligands have been used to assemble M4L4 tetranuclear clusters.361 Treatment of 281 with Cs2CO3 followed by the addition of In(ClO4)3 in methanol resulted in a tetrahedral cluster [Cs⊂{In4(281)4}]ClO4 (282) with a cesium ion as the encapsulated guest molecule (Scheme 71). The same reaction in the absence of base gave N-protonated tetranuclear In(III) complex [In4(H-281)4](ClO4)4 (283) without the cesium guest molecule. Furthermore, treatment of In(ClO4)3 with 281 and subsequent addition of triethylamine afforded the neutral tetranuclear complex [In4(281)4] (284). The tetrahedral cluster, 282, was also accessible from 283 by deprotonation with Cs2CO3. Cluster 284 can be formed by the deprotonation of 283 with triethylamine (Scheme 71). All these complexes were characterized by 1H NMR spectroscopy, mass spectrometry and single crystal X-ray structural studies. While 282 and 283 crystallize as racemic mixtures of homochiral tetrahedra with ΔΔΔΔ or ΛΛΛΛ-fac stereoisomers with T molecular symmetry, complex 284 crystallizes in an idealized S4 symmetry with a ΔΔΛΛ configuration due to the loss of C3-symmetry of ligand 281 during deprotonation.
Scheme 71.

M6L4 tetrahedra or truncated tetrahedra contain tripodal ligands at each of their four faces connected by metal centers at the middle of the edges. They can also be envisioned as derived from a tetrahedron whose corners have been capped. In 1995, Fujita et al.26 reported the first synthesis of truncated tetrahedra (Fujita and coworkers refer to them as octahedral cages, depicting them as octahedral molecules with missing faces) using the molecular panelling approach. M6L4 tetrahedral cages 286-290 were quantitatively assembled by the treatment of cis-capped metal ion hinges with 2,4,6-tri(pyridyl)-1,3,5-triazine 285 (Scheme 72). Each panel spans a face of the tetrahedron and is linked to another at the edges by six metal ions. For example, (en)PdII cornered cage 286 was readily prepared by treating [(en)Pd(NO3)2] (1) with 285 in a 6:4 ratio in aqueous medium. Replacement of the end-capping ligand with (N,N,N′,N′-tetramethylethylenediamine)PdII, (2,2′-bipyridine)PdII or (en)PtII resulted in cages 287, 288 and 289, respectively, with altered bulk properties.362 Use of [Ru([12]aneS4)(H2O)(dmso)](NO3)2 under similar reaction conditions led to the formation of Ru(II)-cornered coordination cage 290 having interesting sensing properties.363 NMR, cold-spray ionization mass spectrometry (CSI-MS) and X-ray crystal structural analysis established the formation of these assemblies.
Scheme 72.
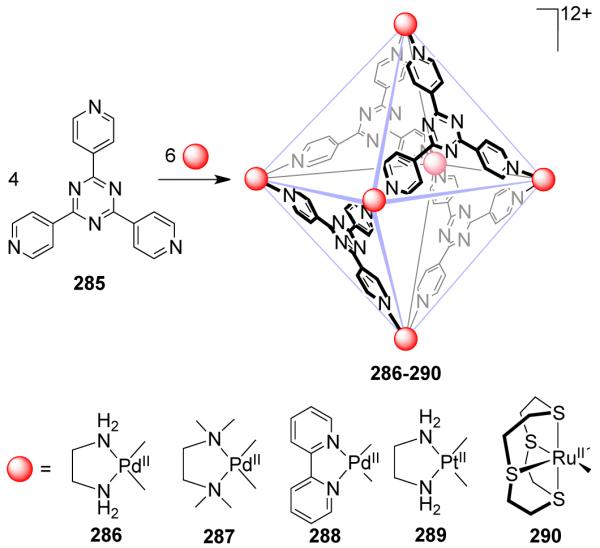
Using the directional bonding approach, Stang et al. have designed truncated tetrahedra via the face-directed self-assembly of planar tritopic 120° subunits with angular ditopic 90° corner units. The ratio of faces to corners must be 2:3 with a total number of 10 subunits to obtain a closed structure. The [6 + 4] self-assembly reaction of 1,3,5-tris-(4-pyridylethynyl)benzene (291) with [cis-(dppf)M(OTf)2] (M = Pd (32), Pt (33)) resulted in the formation of cationic M6L4 truncated tetrahedra 292 and 293 (Scheme 73).364 The use of optically active, chiral 90° angular units, [M(R-BINAP)(OTf)2]365 (M = Pd (177) or Pt (178), BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthalene) or [cis-(PMe3)2Pt(OTf)2]366 38 in conjugation with 291 has led to the formation of similar truncated tetrahedrons. The structures of these tetrahedra were established by multinuclear (31P and 1H) NMR spectroscopy, ESI-mass spectrometry and X-ray crystallography. Recently, the same group reported the coordination-driven self-assembly of a truncated tetrahedron (Figure 31) using 90° platinum acceptors and a hexapyridyl ligand with six-fold symmetry, under mild conditions.367 The tetrahedron was characterized by multinuclear (31P and 1H) NMR spectroscopy, ESI-mass spectrometry, X-ray crystallography, and pulsed-field-gradient spin-echo (PGSE) NMR, along with computational simulations. X-ray diffraction studies showed that the highly symmetrical hexapyridyl ligand acts as the faces and the 90° platinum acceptors are connectors at the edges. These truncated tetrahedra show a unique 3D nanoscale pore suitable for encapsulation of guest molecules and preliminary studies indicated that the nanocavity is able to encapsulate upto three molecules of 1,3,5-triphenylbenzene.
Scheme 73.

Figure 31.
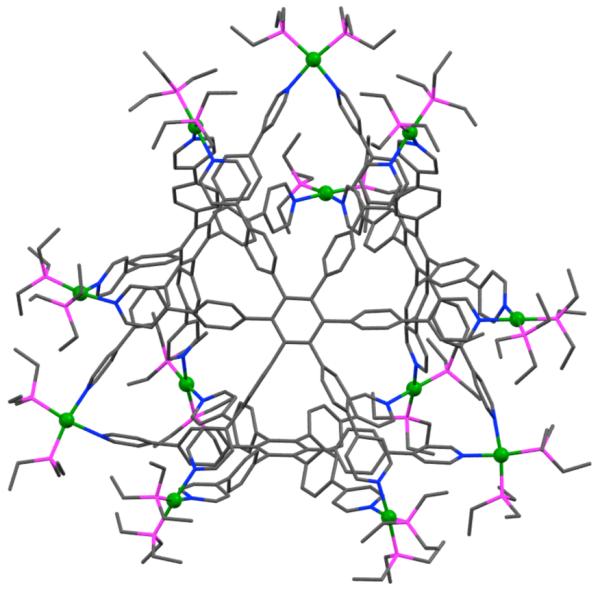
Single crystal X-ray structure of a truncated tetrahedron formed using a hexapyridyl ligand. Protons, solvent molecules and PF6− are omitted for clarity. Color code: green, Pt; pink, P; blue, N; gray, C.
In an elegent approach, Severin and coworkers have recently assembled molecular cages with tetrahedral geometries by combining metallasupramolecular chemistry with dynamic covalent chemistry in an orthogonal fashion.368, 369 Polycondensation of triamines with metallamacrocyclic building blocks, containing pendent aldehyde groups, has enabled the facile synthesis of these nanoscopic cages. Reactions of [(arene)RuCl2]2 [ arene = p-cymene, 1,3,5-C6H3Me3, 1,3,5-C6H3(i-Pr)3] with formyl-substituted 3-hydroxy-2-pyridone ligands gave trinuclear metallamacrocycles with pendant aldehyde groups. Subsequent condensation with C3-symmetric triamines led to the formation of tetrahedral cages. For example, treatment of [(p-cymene)RuCl2]2 (89) with 4-Formyl-3-hydroxy-2(1H)-pyridone (294) in the presence of Cs2CO3 provided trinuclear metallamacrocycle 295.369 As observed from X-ray crystallographic studies, three pendent aldehyde groups in the metallomacrocyclic trimer were oriented towards the same face of the macrocyclic framework. A subsequent [4 + 4] condensation reation of the macrocyclic trialdehyde, 295, with C3-symmetrical triamine 1,1,1-tris(4-aminophenyl)ethane (296) in a mixture of CH2Cl2 and MeOH led to the formation of tetrahedral cage 297 after several days. The formation of the molecular cage was supported by NMR and mass spectrometric studies. The X-ray crystal structure of the supramolecular tetrahedron 297 revealed that the trinuclear metallamacrocycles occupy four vertices of a tetrahedron, while the triphenylamine units span each of the four faces. This paradigm can also be used to design even larger cages. Indeed, the use of an expanded triamine with the trialdehydic metallamacrocycles gave [4 + 4] condensation products with larger dimensions.
4.1.2. Cubic Systems
Supramolecular cubic systems can be achieved by employing two distinct, yet related, design paradigms: (a) edge- and (b) face-directed self-assembly. In the edge-directed self-assembly method, the precursor subunits define the edges of the cube. Thus, an edge-directed cube can be assembled via the reaction of 8 tritopic 90° corner units with 12 ditopic linear linkers (Figure 32a). In the face-directed self-assembly paradigm, some or all of the faces of the target assemblies are spanned by the linkers themselves, which hold together the overall architecture. Thus, in this paradigm, a supramolecular cube results from the combination of 6 tetratopic, planar, 90° faces with 12 ditopic, 90° subunits (Figure 32b-c). Here, the corners of the cube are missing, while the surfaces are covered.
Figure 32.

General design strategy for the synthesis of: a) M8L12; b) M6L12 and c) M12L6 molecular cubes.
Thomas et al.370 have reported an edge-directed M8L12 supramolecular cube from 8 tritopic 90° corner units and 12 linear linkers following the directional bonding approach. The treatment of [([9]aneS3)Ru(DMSO)Cl2] (298) with 4,4′-bipyridine in a 8:12 molar ratio in the presence of silver triflate in nitromethane resulted in the cationic supramolecular cube, 299, over a period of four weeks (Scheme 75). NMR and ESI-mass spectrometry confirmed the structure of the cage compound. The kinetic stability of cube 299 in coordinating solvents such as acetonitrile also indicated the formation of an unstrained cubic structure. Electrochemical studies on 299 showed three irreversible oxidation processes.
Scheme 75.
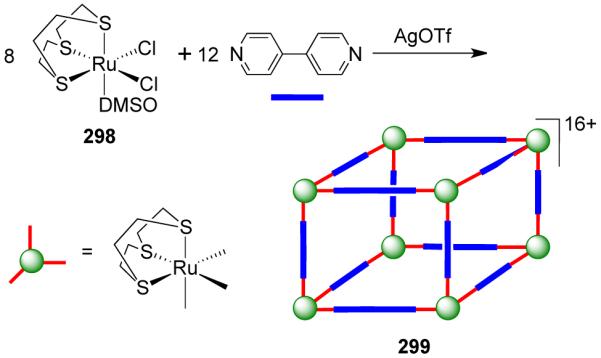
Fujita and coworkers371 have assembled a face-directed M6L12 supramolecular cube by combining 90° ditopic linkers with palladium ions, which act as tetratopic acceptors. The reaction of ditopic ligand 300 with Pd(NO3)2 in DMSO-d6 led to the quantitative self-assembly of a face-directed supramolecular cube (301; Scheme 76). In the molecular cube, Pd(II) ions are located in each face and ligand 300 acts as corner pieces. 1H NMR spectroscopy was in agreement with the formation of a single, highly symmetric product. After anion exchange from NO3 to OTf ions, CSI-mass spectrometric studies confirmed the formation of a M6L12 cube. Single crystal X-ray structural studies revealed the dimensions of nanoscale cube 301 to be approximately 3 × 3 × 3 nm (27 nm3). Similar M6L12 cubes were also realized using a bimesityl-based tetrapyridyl ligand.372 Slow evaporation of a solution of a 1:1 molar equivalent of Cu(OAc)2 ·H2O or Ni(NO3)2 ·6H2O and the tetrapyridyl ligand in MeOH over a period of 7-10 days yielded the molecular cubes. Although NMR and mass spectrometric studies were inconclusive, the single crystal X-ray studies showed the compounds to be supramolecular cubes with Oh symmetry. The nature of the counterions plays a vital role in the assembly of these cages.
Scheme 76.
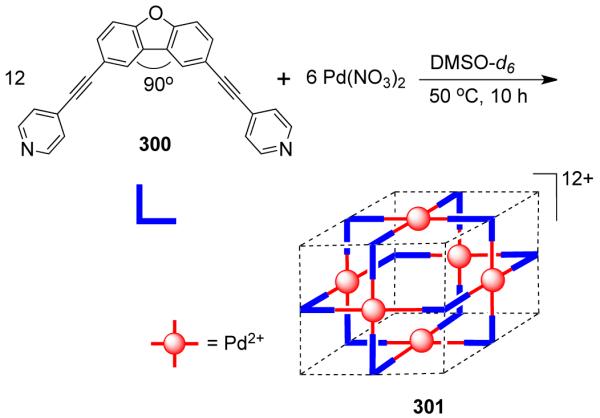
Brisbois et al.373 designed a M12L6 supramolecular cube by assembling 12 ditopic 90° metal corner units and 6 tritopic linkers through the directional bonding paradigm. The treatment of 12 equiv of [Pd(en)(NO3)2] (1) with 6 equiv of tetrapyridyl ligand scaffold 302 in a methanol-water mixture led to the quantitative assembly of metallosupramolecular cube 303 (Scheme 77). NMR and mass spectrometric studies confirmed the formation of the cube. The tetrapyridyl ligand, 302, spanned the faces of the cube while the (en)PdII hinges cliped the edges.
Scheme 77.
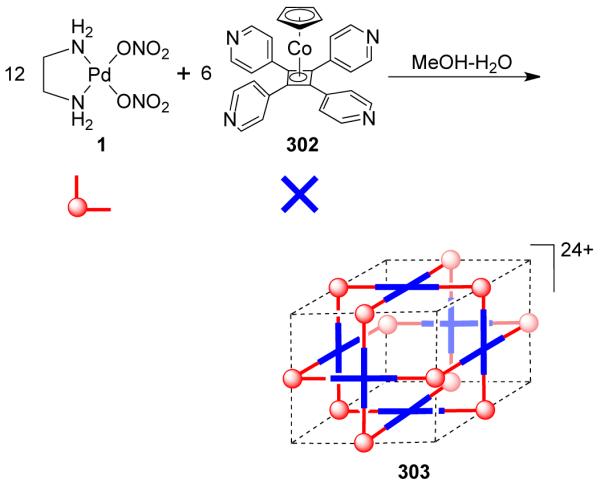
In other strategies, cyanides have been utilized as effective ditopic linkers in a design approach wherein dimerization of two molecular squares in a complementary orientation results in a molecular cube. Rauchfuss and coworkers374 successfully applied this approach to design novel organometallic cubes where metal centers occupy all eight corners. For example, in the first step in this synthesis, the tricyanometalate Et4N[Cp*Rh(CN)3] (Cp* = C5Me5) was employed to prepare a Rh4 molecular square by treatment with the dimeric rhodium chloride complex [Cp*RhCl2]2. The synthesis of the final molecular cube was achieved by removing the chloride ligand from the Rh4 molecular square with AgPF6 in acetonitrile. The same group also reported the formation of molecular cubes {M[Cp*Rh(CN)3]4[Mo(CO)3]4}2− (M = K+, Cs+) isolated from reactions of [Cp*Rh(CN)3]− with equimolar [(η6-C6H3)Mo(CO)3] in the presence of K+ or Cs+.375 The alkali metal ions act as templates in the formation of the organometallic cubes. X-ray crystallographic studies on the complexes revealed box-like M8(μ-CN)12 cages containing alkali metal cations at the center of the cages. The cages feature 12 external CO and 4 external C5Me5 ligands. The directionality of the metal-cyanide bond was also used to assemble a supramolecular cube, [(Cp*WS3Cu3)8Cl8(CN)12Li4] from a precursor, the incomplete cubane-like compound [PPh4][Cp*WS3(CuCN)3].376
Long et al. prepared several molecular cubic clusters having analogous structures inherent to Prussian blue by utilizing ditopic cyanides as linkers. Reactions between [M(H2O)3(tacn)]3+ (M = Cr, Co; tacn = 1,4,7-triazacyclononane) and [Co(CN)3(tacn)] gave molecular box clusters [Cr4Co4(CN)12(tacn)8]12+ and [Co8(CN)12(tacn)8]12+ with core structures consisting of single cubic units.377 As determined from single crystal X-ray crystallography, the structure of the latter species is shown to consist of a cube of eight CoIII ions occupying the corners capped by tacn ligands with each edge spanned by a linear cyanide bridge. The same group was also able to generate larger cubic metal-cyanide clusters by using a blocking ligand on one of the mononuclear reaction components.378 This design paradigm was demonstrated by the synthesis of a fourteen-metal [(Me3tacn)8Cr8Ni6(CN)24]12+ cluster. The geometry of this cluster consists of a cube of eight Me3tacn-ligated Cr(III) ions connected via bridging cyanide ligands to six square-planar Ni(II) ions situated just above the center of each cube face. A still larger tetracapped edge-bridged cubic [(Me3tacn)12Cr12Ni12(CN)48]12+ cluster was synthesized from the direct assembly reaction between [(Me3tacn)Cr(CN)3], NiI2 and KCN in aqueous solution.379 The structure of this cluster features a cube of eight Cr(III) centers linked along the edges by 12 trans-coordinated [Ni(CN)4]2− units, and capped on four faces by [(Me3tacn)Cr]3+ moieties.
Ward and coworkers have reported a number of supramolecular M8L12 cubes using flexible bispyrazolylpyridine ligands and octahedral metal ions via the symmetry interaction approach.340 For example, the bispyrazolylpyridine ligand, 304, having a pyridyl spacer, reacts with Co(II) salts to afford [Co8(304)12]16+ cubic cage 305 (Scheme 78).380 The metal ions occupy each vertex of the cube and the ligands coordinate in a bis-bidentate bridging manner via the two bidentate pyrazolyl-pyridine units spanning each edge. Although ligand 304 is potentially pentadentate, the central pyridyl unit does not participate in the coordination and thus behaves like a bidentate bridging ligand having a meta-phenylene spacer.381 In contrast to the M4L6 tetrahedral clusters synthesized using the same flexible bispyrazolylpyridine ligands, these assemblies are achiral due to the fact that the tris-chelate metal centers in these ‘cubes’ do not have the same optical configuration and possesses crystallographic inversion centers. There is a C3 axis in each case along the long diagonal, with the two M(II) centers on this axis having a facial tris-chelate coordination, and the other six all having a meridional geometry. The combination of a C3 axis and an inversion center means that these cages actually have non-crystallographic S6 symmetry. There is extensive π–π stacking between the overlapping portions of the ligand moieties. Similar octanuclear cubic assemblies with 1,5-napthyl (Lnapth) and 9,10-anthracenyl (Lanth) spacers afforded [M8L12]16+ cages (M = Cu, Zn, Co, Ni and Cd).382 While the cages based on Lnapth have S6 symmetry, with diagonally opposite pairs of corners having facial tris-chelate configuration and the other six being meridional, all metal centers in these cages based on Lanth have meridional tris-chelate configurations. The cubic cages based on Lnapth show extensive interligand aromatic π-stacking interactions at their peripheries. In contrast, cubes containing Lanth do not show significant interligand aromatic stacking interactions.
Scheme 78.
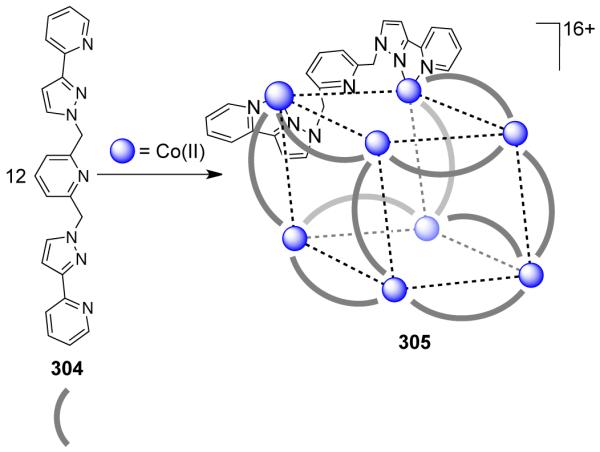
Recently, Nitschke and coworkers self-assembled a M4L6 cubic cage utilizing dynamic covalent and coordinative bonds in tandem via a reaction between tetrakis(4-aminophenyl)porphyrin, 2-formylpyridine, and iron(II) triflate in DMF as evidenced by 1H NMR and ESI-mass spectrometric studies. 383 Replacing tetrakis(4-aminophenyl)porphyrin with nickel(II)- or zinc(II)-tetrakis(4-aminophenyl)porphyrin under identical conditions yielded the nickel-containing and zinc-containing congeners. Single crystal X-ray structure determined for the nickel-containing M4L6 cubic cage showed that each face of the cube was spanned by the porphyrin ligands with each corner of the cube defined by a six-coordinate low-spin Fe(II) ion. All of the Fe(II) centers within each cage adopted the same Λ or Δ configuration. In the crystal lattice, both the enantiomers were present. The cubic cage has a large internal void volume (>1300 Å3), which enabled it to selectively encapsulate higher fullerenes from fullerene soot.
4.1.3. Octahedra
Formation of supramolecular octahedral architectures using a coordination-driven self-assembly is quite rare in the literature. Similar to cubic systems, supramolecular octahedral assemblies have been achieved through edge- and face-directed self-assembly paradigms. Shionoya et al. have reported the construction of M6L8 octahedral metallosupramolecular assemblies using a series of divalent d5-d10 transition metal ions M2+ (M = Mn, Fe, Co, Ni, Pd, Pt, Cu, Zn, Cd, and Hg) and tritopic pyridyl ligand 306.384 A [6 + 8] assembly led to the formation of cationic [M6(306)8]12+ (307) octahedral-shaped cages in which six metal ions occupy the vertices and the eight faces of the octahdron are spanned by eight ligands (Scheme 79). Single crystal X-ray structural studies on [Hg6(306)8]12+ reaveled a 3 nm sized octahedron-shaped capsule with sides of 1.8 nm. It was also demonstrated that the [Hg6(306)8]12+ capsule is reversibly interconvertible with a [Hg6(306)4]12+ cage by changing the ratio of ligand to metal. 385 The structural interconversion between the two distinct complexes was accompanied by reversible changes in the coordination mode of the Hg2+ ion between octahedral and linear coordination geometries. A similar [Pd6L8]12+ cage has been reported with the cyclotriveratrylene-type ligand - tris(isonicotinoyl)cyclotriguaiacylene and a “naked” palladium ion.386 Lah et al. assembled similar a face-directed molecular octahedron from a combination of 3,3′,3″-[1,3,5-benzenetriyltris(carbonylimino)]trisbenzoate as a C3-symmetric tritopic ligand and a paddlewheel Cu2(COO)4 moiety as a corner unit.387 Here, eight ligand molecules span the faces of the octahedron connected by six Cu2(COO)4 paddle-wheel units at the corners.
Scheme 79.
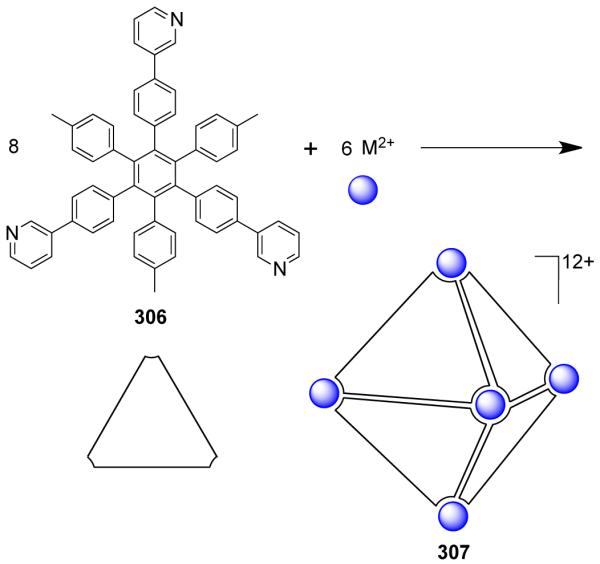
Müller et al. utilized a threefold chelating ligand tris(2-hydroxybenzylidene)triaminoguanidinium chloride [H6L]Cl (308) to form the triangular faces of an octahedral metallosupramolecular assembly.388 The one pot reaction of PdCl2, Et4NCl, [H6L]Cl (308), sodium 5,5-diethylbarbiturate (NaHbar, 309), and Et3N in acetonitrile at room temperature leads to the formation of a anionic octahedral molecular assembly [{Pd3L}8{μ-(bar)}12]16− (310; Scheme 80). Octahedral complex 310 was characterized by 13C MAS-NMR spectroscopy and X-ray crystallography. The solid-state structure showed that ligand 308 forms triangular molecular panels through N, N, O chelation with paladium leaving a free coordination site for the bridging ligand. The ditopic sodium 5,5-diethylbarbiturate (NaHbar, 309) ligands connect the faces of the octahedron through the middle of the edges.
Scheme 80.

Zhou and coworkers389 have assembled an edge-directed supramolecular octahedron from an angular (90°) dicarboxylate ligand and a paddlewheel Cu2(COO)4 cluster. A solvothermal reaction between 9H-carbazole-3,6-dicarboxylic acid (H2CDC; 311) and Cu(NO3)2·2.5H2O in a 1:1 DMA/EtOH solution (DMA = N,N-dimethylacetamide) afforded molecular octahedron [Cu2(CDC)2(DMA)(EtOH)]6 · xS (S = noncoordinated solvent molecule; 312; Scheme 81). Single crystal X-ray structural studies showed that the molecular octahedron is composed of six axially coordinated Cu2(COO)4 paddlewheel clusters occupying the vertices joined by 12 CDC linkage units along the edges. Interestingly, molecular octahedron 312 can be reversibly assembled into a 3-dimensional metal organic framework by interlinking the octahedra through ditopic 4,4′-bipyridine ligands.
Scheme 81.
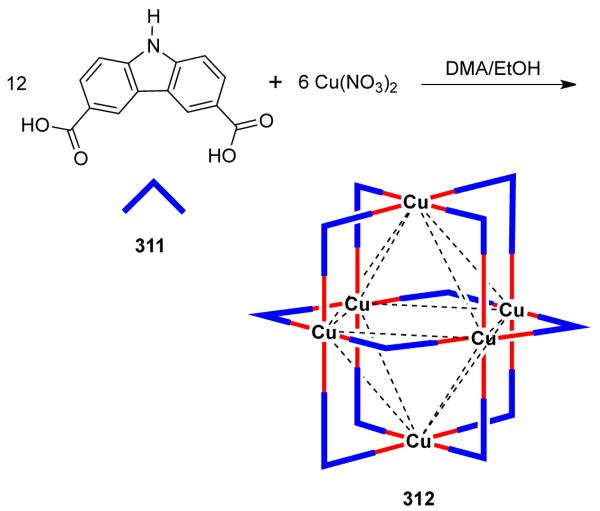
4.1.4. Dodecahedra
In principle, the most complicated member of the five Platonic polyhedra is a dodecahedron. It contains 12 fused five-membered rings that comprise the highest symmetry group Ih. The 12 pentagonal faces are formed from 20 vertexes and 30 edges. Hence, a dodecahedron can be prepared via edge-directed assembly of 20 tripodal subunits with approximately 108° directing angles in combination with 30 bidentate linear subunits. Only a few examples of Pt(II) based molecular dodecahedra are known, where di-platinum based linear linkers (48, 96) were assembled with tripodal ligand tris(4-pyridyl)methanol 313 to obtain dodecahedra 314 and 315 of nano-dimensions (Scheme 82).390,391 Though no direct structural evidence was provided for their exact shape, NMR along with mass spectrometric results and TEM images demonstrated the formation of the expected dodecahedra. These dodecahedra are significantly larger than the hydrocarbon C20H20 of the same symmetry, synthesized by classical covalent synthesis in 23 steps.392,393 The cavities of the self-assembled dodecahedra 314 and 315 are expected to be capable of encapsulating large globular oligomers and molecules with solvation shells.
Scheme 82.
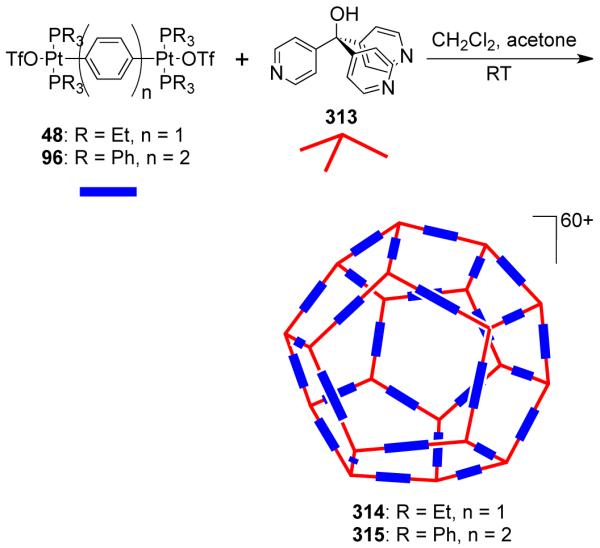
4.1.5. Cuboctahedra
The cuboctahedron is a rare archimedean semiregular polyhedron that combines square and triangular faces. Overall it has 12 vertices, 24 edges, and a dihedral angle of 125° between the triangular and the square surfaces. Using the directional bonding approach, a cuboctahedron can be constructed by combining planar tridentate faces with 108° turning angles via bidentate angular components. As a 109.5° angle is close to 108°, the bidentate angular linking subunit may have a tetrahedral atom connected to a rigid linear donor or acceptor site. Stang and coworkers have reported the formation of nanoscopic molecular cuboctahedra from the combination of 12 tritopic planar units and 8 ditopic angular units.394 The tridentate Pt3 acceptor, 316, was treated with bidentate 4,4′-bipyridylacetal 317 in 2:3 molar ratio to obtain the first Pt-based molecular cuboctahedron 318 (Scheme 83). Despite its large molecular mass (26592 Da) and high charge (24+), cuboctahedron 318 is remarkably soluble in organic solvents, such as dichloromethane or acetone. In a complementary approach, 1,3,5-tris-(4-pyridylethynyl)benzene 291 was assembled with bis(4-[trans-Pt(PPh3)3OTf]phenyl)ketone to yield similar cuboctahedra.394 The self-diffusion coefficient at 25°C was 1.92 × 10−2 cm−2 s−1, which gave an experimental hydrodynamic diameter of 5.0 nm, was in good agreement with predictions based on force-field simulations as well as the size of the component building units. Finally, ESI-mass spectrometry was used to establish the exact composition.
Scheme 83.

Ward and coworkers have also reported the formation of cuboctahedral cages from the combination of ligands having different binding modes with octahedral metal ions.395,396 A 3:1:3 reaction of tris-bidentate ligand 319, which can cap one triangular face of a metal polyhedron, a bis-bidentate ligand 320, which can span one edge of a metal polyhedron, and a range of M2+ ions results in the assembly of mixed-ligand cuboctahedral cages [M12(μ3-319)4(μ2-320)2]24+ [M = Co (321), Cu, Cd] in MeNO2 (Scheme 84).
Scheme 84.

Single crystal X-ray structural studies of [M12(μ3-319)4(μ2-320)]24+ (M = Cu, Cd) show that they are isostructural, having a dodecanuclear cuboctahedral metal framework containing eight triangular and six square faces. Four of the eight triangular faces were capped by triply bridging ligands 319. Twelve doubly bridging ligands 320 span the remaining edges. Counterions and solvent molecules occupy the central cavity. A cooperative interaction between the two types of ligand renders the formation of the mixed ligand cuboctahedral cage more favorable than the homoleptic cages. The ESI-mass spectra on solutions of redissolved crystals showed a clear sequence of peaks corresponding to the intact mixed-ligand cage with no peaks for the homoleptic cages. All 12 tris-chelate metal centers have meridional geometry, and again, all have the same chirality, indicating that the same chiral configuration at each metal center is necessary for the closed cage to form.
4.2. Other Coordination Cages
4.2.1. Trigonal Bipyramids and Double Squares
A large number of molecular trigonal bipyramids synthesized via coordination-driven self-assembly have been reported in the literature. In principle, a M3L2 trigonal bipyramid can be designed by assembling two tritopic, tetrahedral donors with three ditopic 90° subunits through the edge-directed self-assembly. Fujita and coworkers prepared one of the earliest trigonal bipyramidal cages in 1995.397 Treatment of 2 equivalents of the tritopic ligand 1,3,5-tris(4-pyridylmethyl)benzene, 322, with 3 equivalents of ditopic 90° [Pd(en)(NO3)2] (1), in the presence of sodium 4-methoxyphenylacetate led to the exclusive formation of trigonal bipyramidal cage 323 (Scheme 85). However, in the absence of a specific guest, self-assembly resulted in the formation of oligomeric products. Monocarboxylate guests with bulky hydrophobic moieties induce the assembly of 323 more effectively as compared to less hydrophobic substrates.
Scheme 85.
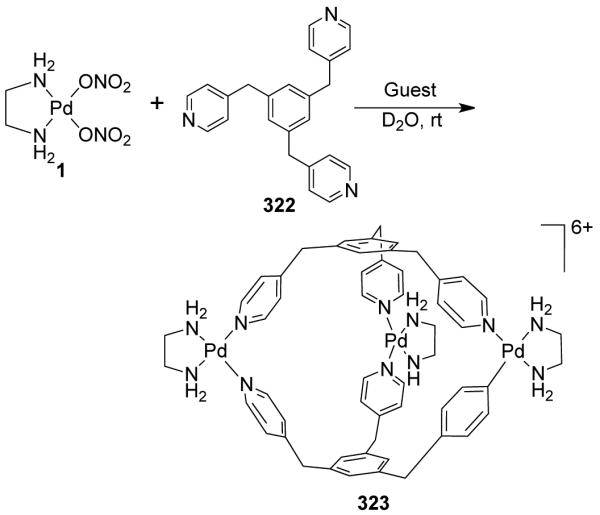
Stang et al. reported the synthesis of a number of M3L2 trigonal bipyramidal cages using tritopic 109° linkers.398,399 A [2 + 3] self-assembly reaction between ~ 109° tripodal angular unit 324 and generic 90° angular units such as cis-Pt(PMe3)2(OTf)2 (38), cis-Pt(dppp)(OTf)2 (52) and cis-Pt(dppe)(OTf)2 (325) resulted in trigonal bipyramidal cages 326-328 (Scheme 86).398 Multinuclear NMR studies and mass spectrometric analysis confirmed the formations of these cages. Similarly, the use of flexible tritopic tectons containing amide399 and ester 400 functionalities resulted in the exclusive formation of trigonal bipyramidal cages. Such a reaction, however, with less flexible ligands resulted in the formation of double square architectures.401,402 For instance, treatment of [Pd(en)(NO3)2] (1) with the tripyridylmethane acetate (329) resulted in self-assembly of a double square cage (330) in which four ligand molecules are held together by six metal ions (Scheme 87).401 Similarly use of preorganized tripodal metalloligand incorporating octahedral metal centers, aluminium or gallium, in which the orientation of the pyridyl coordination sites for self-assembly were controlled by the metal’s octahedral coordination environment, afforded novel heterometallic trigonal bipyramidal cages when assembled with appropriately angled (60° or 90°) platinum acceptors.403
Scheme 86.
Scheme 87.

The reaction of a rigid tripodal ligand having ester functionalities with a 90° Pt(II) acceptor also gave an assembly of similar architecture.402 Thus, if enough flexibility was incorporated into the tritopic units, the thermodynamics of the reaction would favor the formation of trigonal bipyramids over the double square architectures. The double square represents a balance between entropy, where the smallest structure is favored, and enthalpy, where the structure with the least strain is prevalent. A flexible tripodal ligand, 331, containing pyrazolyl functionality led to a 2:3 self-assembled double square cage 333 upon treatment with cis-(tmen)Pd(NO3)2 (332; tmen = N,N,N′,N′-tetramethylethane-1,2-diamine) in a 2:3 molar ratio (Scheme 88).404 A similar reaction using CuCl2·3H2O instead of the cis 90° acceptor, 332, yielded a Cu3 trigonal bipyramidal cage (334). For the M3L2 trigonal bipyramidal cage, 334, the tripodal ligand adopts a symmetrical cis-, cis-, cis-conformation while in the double-square cage 333 the ligand adopts a nonsymmetrical cis-, trans-, trans-conformation.
Scheme 88.
Youngs et al.405 reported the synthesis of a trigonal bipyramidal supramolecular cage based upon rhodium and platinum metal centers in a step-wise process. The treatment of 4-ethynyl-pyridine with tert-butyllithium, followed by its addition to (Me3tacn)RhCl3 afforded the formation of facial octahedral complex (Me3tacn)Rh(CCPy)3 335. Subsequent assembly of the tritopic linker with the square planar complex cis-(DCPE)Pt(NO3)2 336 [DCPE = 1,2-Bis(dicyclohexylphosphino)ethane] resulted in a self-assembled hexacationic trigonal bipyramidal cage (337; Scheme 89) with Rh(III) and Pt(II) atoms occupying the vertices. Multinuclear NMR and crystal structural studies were used to characterize the assembly.
Scheme 89.

4.2.2. Adamantanoids
Adamantanoids have a shape similar to a tetrahedron that has its edges kinked into 109.5 angles at their points of bisection. While these distortions cause the sides and angles to deviate from those of a tetrahedron, adamantane retains its overall symmetry that of a point group Td. Supramolecular adamantanoids can be designed according to the directional bonding approach by adopting an edge-directed strategy. The shape itself consists of four fused, chair-conformed cyclohexane-like rings, which requires that all of the pertinent angles should be 109.5°, matching the ideal tetrahedral geometry of sp3-hybridized carbon atoms. Thus, a combination of six 120° ditopic units with four 109.5° tritopic units can lead to a self-assembled molecular adamantanoid. For example, a [6 + 4] self-assembly reaction between ditopic units (338 or 339) with tritopic units (340) in CD2Cl2 led to the formation of adamantanoids 341 and 342 in quantitative yields (Scheme 90).406 1H and 31P NMR as well as ESI-mass spectrometric studies confirmed the formation of these assemblies. As a result of the stereogenic centers present in the backbone of the ester moiety of ligand 340, the resulting assemblies were chiral in nature. Fréchet-type dendron-decorated adamantanoids, assembled utilizing similar building units, have also been reported recently.407
Scheme 90.

Lin et al.408 reported chiral M4L6 adamantanoids from lanthanide salts and C2 symmetric racemic mixtures of binaphthalene-based ligands. The treatment of lanthanide nitrate or perchlorate with 2,2′-hydroxy-1,1′-binaphthalene-6,6′-dicarboxylic acid (6,6′-H2BDA) forms distereoselective porous molecular adamantaniods [Ln4(6,6′-H2BDA)6(H2O)12]·12DMF (Ln = Gd(III), La(III)). X-ray structural studies on [Gd4(6,6′-H2BDA)6(H2O)12]·12DMF (Figure 33) revealed the formation of a porous nanosized assembly having an internal cavity with a volume of ca. 1700 Å. Within the adamantanoid, Gd atoms reside on the crystallographic 3-fold axis, adopting idealized tricapped trigonal prismatic coordination geometries by ligating three chelating carboxylate groups from three different 6,6′-BDA ligands and three water molecules. In the solid-state, six DMF molecules occupy the internal cavity.
Figure 33.
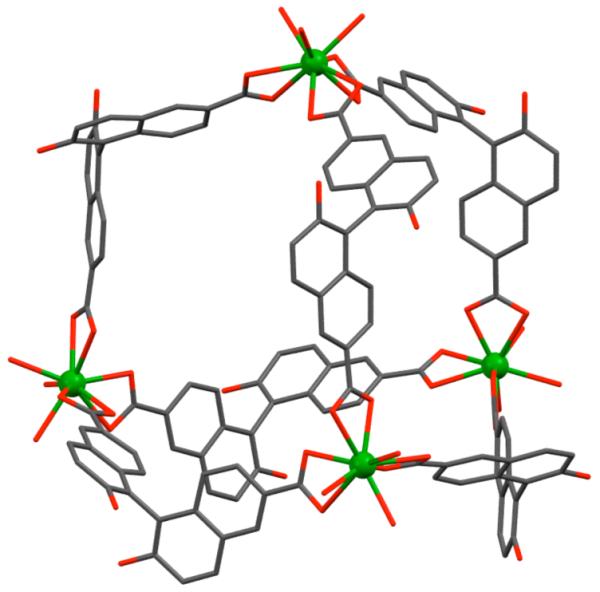
X-ray crystal structure of [Gd4(6,6′-H2BDA)6(H2O)12]·12DMF. Color code: green, Gd; red, O; gray, C.
4.2.3. Trigonal Prisms
The simplest way of designing a trigonal prism is the combination of an end-capped acceptor, a tritopic planar donor and a bidentate linear donor in a 6:2:3 molar ratio. Fujita and coworkers have reported a few trigonal prisms using this multi-component self-assembly approach. For example, the self-assembly of the 90° cis-blocked [Pd(en)(NO3)2] (1), tritopic electron poor triazine panel 2,4,6-tri(pyridyl)-1,3,5-triazine (285) and pyrazine, which acts as bidentate pillar, led to the formation of trigonal prism 344 (Scheme 91).409 The charged metal ion hinges endows the cages with water solubility.
Scheme 91.

In this three-component self-assembly approach there is the possibility of the formation of smaller analogues, like molecular tetrahedra and squares, from the combination of individual donors with the metal acceptors. However, large aromatic molecules such as coronene (343) effectively template the selective formation of the trigonal prism and can be removed after assembly by simple extraction with organic solvents. Prism 344 remains stable after removal of the template and allows the intercalation of various organic molecules. In the presence of 1,3-diketone the ketone is found to exist only in its enol form, suggesting a stabilization effect of prism 344, as confirmed by NMR spectroscopy.409 The pyrazine pillar can be replaced by tetramethyl-4,4′-bipyridine or longer 1,4-bis(2,6-dimethylpyridin-4-yl)benzene which results in cages 345 and 346 with larger cavities for stacking of two and three equivalents of aromatic guests, coronene,410 pyerene410 and porphine411 and mononuclear M(II) Complexes412 (M = Pt, Pd, and Cu). Use of an elongated 4,4′-bipyridine extends the cavity of the cage to an ideal size for the accomodation of three guest molecules.411 The stacking of these aromatic molecules within the cavity is driven and controlled solely by non-covalent interactions.
Using a two-component approach, Stang and coworkers have designed trigonal prisms 348 and 349 using a 0° Pt-based molecular clip (75) as the acceptor unit in combination with planar tripodal linkers 291 and 347 by a 3:2 self-assembly reaction (Scheme 92).413 The structures of these macrobicyclic species were established with multinuclear NMR and ESI-mass spectrometry. Although several M3L2 prismatic cages using flexible ligands have been reported,414 they often require the use of templates to assemble in solution or assemble only in the solid state. Use of these rigid donors and acceptors has made it possible to synthesize prismatic cages in the absence of templates. Buliding upon this strategy, Ko and coworkers115 have reported the formation of trigonal prisms using a bulkier Pt2 molecular clip in a 3:2 self-assembly reaction with several tritopic planar donors. Multinuclear NMR and mass spectral data confirmed the formation of trigonal prisms. The use of isocyanide as the linker also led to the facile formation of molecular trigonal prisms. In a complementary approach, the use of tritopic planar Pt3 acceptors in combination with 0° organic clips has also led to the formation of several trigonal prisms in 2:3 self-assembly processes.415,416 These π-electron rich fluorescent cages showed efficient quenching of fluorescence intensity both in solution and solid state in the presence of planar oxidizing nitroaromatics, which are the chemical signatures of many commercially available explosives. The cages have open spaces to accommodate electron-deficient small nitroaromatics and the bulky PEt3 groups help to avoid intermolecular stacking, thus preventing self-quenching of fluorescence. The solution fluorescence was quenched efficiently by adding nitroaromatics such as DNT and TNT.415,416
Scheme 92.

Distorted trigonal prisms 351-353 were also designed by using trigonal pyramidal acceptors 313, 324 and 350 in conjunction with the 0° Pt-based molecular clip (75) in 2:3 molar ratio (Scheme 93).417 NMR and mass spectrometric results, along with single crystal X-ray analysis, established that the sizes of the propeller-type 3D structures range from 1 × 2 nm to 1 × 4 nm. The formation of the smallest of these prisms, 351, was accompanied by the entrapment of a nitrate ion inside its molecular cavity. The PF6− salt of the largest prism of this series, 353, however, crystallizes with no counteranion inside the cage. A complementary approach using a 0° donor organic molecular clip also led to the assembly of similar molecular prisms.418 Tetrahedral carbon, silicon, and phosphorus were used as structure-defining elements in these coordination based cages. Multinuclear NMR, elemental analysis and ESI-FT-ICR-mass spectrometry established the formations of these structures.
Scheme 93.

Bosnich et al. have constructed trigonal prisms from a palladium-based cleft, 354, that contains cofacially disposed terpyridyl-Pd units. The 1:1 reaction of planar tritopic linkers, 285 and 291, with 354 led to the quantitative formation of trigonal prisms 355 and 356 (Scheme 94). Multinuclear NMR and ESI-mass spectrometric data confirmed the formations of the prisms. These supramolecular trigonal prisms, each bearing three molecular clefts, are shown to form 1:6 and 1:7 host–guest complexes with 9-methylanthracene and one of the prisms also forms a 1:2 host–guest complex with a tritopic tri-anthracene guest that registers with the recognition sites of the host.
Scheme 94.

A number of Re(I)-, Ru(II)-, Ir(I)-, and Rh(I)-based metalloprisms with tridentate triazine ligand, 2,4,6-tris(4-pyridyl)-1,3,5-triazine (285) possessing N,N′- or O,O′-donor spacers acting as pillars have also been reported. Hupp et al.419 reported the synthesis of trigonal prism 359 by using the bimetallic edge 86 prepared from fac-Re(CO)5Cl and 2,2-bipyrimidine ligands and subsequently reacting these edges with triazine ligand 285 (Scheme 95). Alternately, the trigonal prism, 359, can be assembled in a one-step process by treating Re(CO)5(OTf), 2,2-bipyrimidine with 285 in a 6:3:2 ratio in THF.420 The use of 2,2′-bisbenzimidazolate (357) or a pair of benzylthiols (358) based bimetallic edges led to the formation of trigonal prisms 360 and 361, respectively (Scheme 95).421 The prisms 360 and 361, in their singly reduced form, comprise of highly coupled mixed valence systems with orbitally degenerate organic redox centers. The orbital degeneracy of the redox sites of 285 has pronounced effects on light-induced (IT) charge redistribution within the molecules. The self-assembly of Re2(CO)10 and triazine panel, 285, in ROH (R = C4H9, C8H17, C12H25), using solvothermal methods at elevated temperatures, led to the formation of thermodynamically stable trigonal cages in excellent yields.422 In an extension of the methodology, indigo was used as the organic pillar to assemble a trigonal prism in good yield.423 The structural features of this metalloprism showed very strong π-π interactions between the π-clouds of 285 to pull the inner-core of the prism inwards with a distance one-half of that of the outer core. In THF, the metalloprism displayed intense bands in the near UV region (200-300 nm) due to π-π* transitions of both 285 and indigo ligands.
Scheme 95.
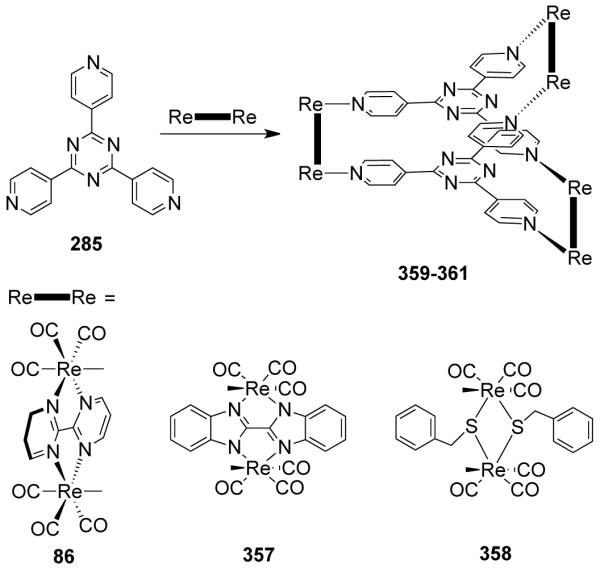
Several trigonal-prismatic cationic cages in which six (η6-arene)ruthenium(II), (η5-pentamethylcyclopentadienyl)rhodium(I) or (η5-pentamethylcyclopentadienyl)iridium(I) units are held together by two trigonal 2,4,6-tris(4-pyridyl)-1,3,5-triazine panels (285) and three dichloro424,425 or oxalate426,427 bridges were also reported. An extension of this principle led to the construction of larger cationic hexanuclear metalloprisms from p-cymene or hexamethylbenzene (C6Me6) ruthenium building blocks and 2,5-dihydroxy-1,4-benzoquinonato (dhbq) or 2,5-dihchloro-3,6-dihydroxy-1,4-benzoquinonato (dchq) or 5,8-dihydroxy-1,4-naphthoquinonato (dhnq) or 9,10-dihydroxy-1,4-anthraquinonato (dhaq), or 6,11-dihydroxy-naphthacene-5,12-dionato(dhtq) bridges.428,429 For example, treatment of diruthenium bridges 362-366 with 285 in a 3:2 molar ratio led to the formation of cationic trigonal prisms [Ru6(p-cymene)6(285)2(dhbq)3]6+ (367), [Ru6(p-cymene)6(285)2(dchq)3]6+ (368), [Ru6(p-cymene)6(285)2(dhnq)3]6+ (369), [Ru6(p-cymene)6(285)2(dhaq)3]6+ (370), [Ru6(p-cymene)6(285)2(dhtq)3]6+ (371) (Scheme 96). Similarly, use of 2,4,6-tris(3-pyridyl)-1,3,5-triazine as the trigonal panel instead of 285 in conjunction with dhnq or dhtq bridges have also led to the formation of hexanuclear arene ruthenium complexes with trigonal-prismatic architecture.430 These prisms were capable of encapsulating various aromatic molecules to form the corresponding inclusion systems as established from one-dimensional ROESY 1H NMR experiments, mass spectrometry, and single-crystal structure analysis. The self-assembly of these cationic prisms was also achieved in the presence of Pt(acac)2 or Pd(acac)2 [acac = acetylacetone] to yield Pd(acac)2 or Pt(acac)2 encapsulated prisms in almost identical isolated yields.431 The formation of the Pd(acac)2/Pt(acac)2 encapsulated cages was monitored by NMR and finally by X-ray structure determination. Interestingly, the use of an oxygen-donor tritopic panel, 1,3,5-benzenetricarboxylate, instead of N-donor panels in combination with oxalate bridged di-ruthenium unit has led to the assembly of an incomplete RuII8 trigonal prism, which exhibits a remarkable shape-selective binding affinity for neutral phenolic compounds via hydrogen-bonding interactions.432
Scheme 96.

Along the same line, Severin et al.433 designed a trigonal prism having an adaptable cavity size. Using a carboxylate-based di-ruthenium unit in combination with tritopic ligand 285, the formation of a trigonal prismatic cage was achieved. The cage can adopt two distinct conformations: a compressed anti-prismatic structure without an internal cavity and an elongated structure having a trigonal prismatic geometry that is able to accommodate two corronene molecules as guests. The modulation of the cavity space from nearly zero to more than 500 Å was achieved by a conformational change in the flexible ligand rather than the more commonly observed guest-induced constitutional rearrangement.
Hupp et al. have designed supramolecular porphyrinic prisms featuring three, six, or nine porphyrin panels via coordination-driven self-assembly. These molecular prisms were formed by reversible coordination of planar tritopic panels to the Zn(II) sites of the porphyrinic dimers and trimers.434,435 However, due to the weaker Zn-ligand bonds, these assemblies dissociated in highly polar solvents or at high dilution. The use of templated ring-clousure metathesis (tRCM) to permenently “set” the structure resulted in the formation of both metallated and metal-free torsionally rigid, irreversible porphyrin prisms.436 A 3:2 molar ratio reaction of a Zn-diporphyrin conjugated through a butadiyne linker and 2,4,6-tris(4-pyridyl)-1,3,5-triazine (285) or triethynylpyridylbenzene (291) panels preorganizes these panels into assemblies that are ready for ring-clousure metathesis. The tRCM using a first generation Grubb’s catalyst afforded the trigonal prisms in high yield. The tridentate panels are important for the formation of the corresponding prisms. Würthner and coworkers described the characterization of a porphyrin-based assembly based on Zn porphyrins as planar units induced by coordination to a bispyridyl functionalized perylene bisimide.437,438 The perylene bisimide ligands act as pillars via two axial coordination bonds with the porphyrinic Zn(II) ions, fixing the planes of the porphyrin units in a nearly cofacial orientation and inducing the formation of molecular rectangles and trigonal prism-like structures.
A novel approach towards the construction of heteroleptic trigonal prisms was recently demonstrated from a three-component reaction of carboxylate and pyridyl donor ligands with platinum acceptors. 439 Such multicomponent, selective self-assembly represents a unique assembly process in which multiple, varying components can selectively recognize and combine to generate only one discrete structure within a mixture. Self-assembly of cis-[(PEt3)2Pt(OTf)2] (8) with linear dicarboxylate 119 and triethynylpyridylbenzene (291) in 6:3:2 molar ratio led to supramolecular trigonal prism 372 as the predominant species after equilibration (Scheme 97). Multinuclear (31P and 1H) NMR spectroscopy, ESI-mass spectrometry, and PGSE NMR measurements, along with the computational simulations, established the selective self-assembly of heteroleptic trigonal prism 372. The self-assembly process is directed, possibly, by a charge separation effect, whereby the anionic carboxylate ligand and a neutral pyridyl donor favor a heteroligated motif upon coordination with Pt(II) centers rather than relying only on the stoichiometry and directionality of their molecular components. Furthermore, this selective self-assembly can be achieved via supramolecule-to-supramolecule transformations wherein a homoleptic truncated tetrahedron 373 can be transformed into th heteroleptic trigonal prism 372 by addition of a solution of homoleptic neutral triangle having carboxylate ligands as the edges (Scheme 98).439 Similar supramolecule-to-supramolecule transformations in self-assembled polygons,440,441 helicates442 and cavitand cages443 via coordination-driven assembly based on thermodynamic or kinetic control have also been observed. In a complementary approach, heteroleptic Pd(II)-based trigonal prisms were assembled using tritopic carboxylates and linear ditopic pyridyl ligands in conjunction with 90° Pd(II) as an acceptor.444 Isolation of the guest-free cage as well as guest-induced selective formation of the encapsulated cage was achieved using this paradigm.
Scheme 97.

Scheme 98.
Using a three-component self-assembly approach, Schmittel et al.445 designed molecular trigonal prisms from tritopic trisphenanthroline (374) and various bisterpyridine ligands 375-377. The reaction of 374 with [Cu(MeCN)4]PF6 in dichloromethane, followed by the addition of bisterpyridine ligands 375-377, led to the formation of heteroleptic trigonal prisms[Cu6(374)2(X)3]6+ (X = 375-377) 378-380 (Scheme 99). These prisms exist in a dynamic equilibrium with smaller oligomers as determined from NMR and mass spectrometric studies. However, the use of tripyridines as templates drives the equilibrium completely towards the trigonal prisms. In a complementary approach, use of a trispyridine as the tritopic panel with bisphenanthroline as the pillar also led to the formatiom of molecular nanoprisms.446
Scheme 99.

4.2.4. Tetragonal Prisms
A number of tetragonal prisms have been assembled in recent years using various design paradigms. One of the earliest M2L4 cages having a tetragonal prismatic architecture was reported by Atwood and coworkers447 wherein two octahedral Cu(II) ions were bridged by four bispyridyl bidentate building units. While all four equatorial positions of each Cu(II) were coordinated by the pyridyl nitrogens of the ligands, the axial positions were occupied by water molecules. The use of semirigid ligands in conjunction with Cu(II) ions has also led to the assembly of tretragonal prisms. A 1:2 reaction of Cu(II) ion with semirigid bisbenzimidazole ligand 381 led to the formation cationic tetragonal prism [Cu2(381)4 ⊃ ClO4−]3+ 382 having S4 symmetry (Scheme 100).448 Single crystal X-ray structural analysis showed that the ligand 381 adopts a syn conformation in order to bridge two Cu(II) ions. Each of the Cu(II) ions are held by the benzimidazole arms of the four different ligands via Cu–N coordination to metal ions in a square planar geometry. The ClO4− counterion resides in the cavity of the cage. Under similar conditions, the use of NiCl2 led to the formation of a neutral tetragonal prism [Ni2(381)4Cl2 ⊃ H2O].449 The use of ditopic, semirigid ligands that contain long central bases and two pyridyl coordinating rings linked by methylene groups, 2,6-bis(3-pyridylmethyl)hexahydro-4,8-ethenopyrrolo[3,4-f]isoindole-1,3,5,7-tetrone also led to a M2L4 tetragonal prism.450 Twisted dinuclear Cu(II) cages of the M2L4 type with an encapsulated chloride between the metal centers were also reported.451 Use of other metal ions with preferred square planar coordination geometries, such as Pd(II), have also led to the formation of tetragonal prismatic cage structures. Fujita and coworkers452 have reported a Pd(II) based M2L4 cage using a rigid organic clip-type donor with “naked” square planar Pd2+ as tetratopic acceptors. Pd(NO3)2 yielded the tetragonal cage 384 upon 2:4 self-assembly with a bidentate pyridyl ligand, 383 (Scheme 101), which was characterized by NMR and single crystal X-ray structural analysis. An analogous dinuclear Pd(II) M2L4 tetragonal cage encapsulating both anionic453 and cationic454 guest molecules inside the cavity was also investigated.
Scheme 100.
Scheme 101.
Bruno et al.455 have reported a series of cationic tetragonal prisms incorporating a 2,5-dihydroxy-1,4-benzoquinonato (dhbq) ligand instead of an oxalato bridge. The tetragonal prisms, having general formula [Ru8(η6-arene)8(μ-tppH2)2(dhbq)4]8+ (389, arene = C6H5Me; 390, arene = p-cymeme; 391, arene = C6Me6), were assembled by a 1:2 reaction between 5,10,15,20-tetra(4-pyridyl)porphyrin (tppH2, 385) and ruthenium building blocks 386-388 bridged by a dhbq ligand (Scheme 102). Similarly, toluene or p-cymene ruthenium building blocks bridged by the dhbq ligand and connected by a zinc porphyrin 5,10,15,20-tetra(4-pyridyl)porphyrin-Zn(II) (tppZn) tetradentate ligand led to the formation of tetragonal prisms: [Ru8(C6H5Me)8(μ-tppZn)2(dhbq)4]8+ and [Ru8(p-cymene)8(μ-tppZn)2(dhbq)4]8+.456 These supramolecular tetragonal prisms interact strongly with duplex and human telomeric quadruplex DNA.
Scheme 102.

Using the same design principle, Jin and coworkers457 have reported tetragonal prisms incorporating tetrakis(4-pyridyl)porphyrin as a tetratopic donor in combination with oxalato bridged half-sandwich Ir(III), Rh(III) and Ru(II) connectors. Single crystal X-ray structure analysis of the Ru8 tetragonal prism [Ru8(p-cymene)8(μ-tppH2)2(μ-C2O4)4]8+ (Figure 34; tppH2 = 5,10,15,20-tetra(4-pyridyl)porphyrin, 385) showed the complex possessed helical chirality with a porphyrin-porphyrin separation of 4 Å. The open space volume inside the prism was approximately 800 Å3. The cavity of the octanuclear prism was too small to accommodate a guest molecule, and strong π-stacking interactions between two porphyrin panels were observed. As a consequence of the twist in the oxalato clips, two porphyrin panels, although being approximately parallel, are twisted along their normal vector and thus give rise to two helical isomers. Chiral discrimination of these enantiomers can be achieved by using anionic chiral Λ-BINPHAT as NMR solvating agent.458
Figure 34.
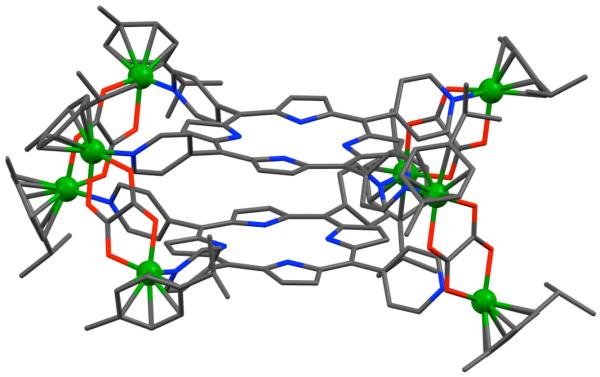
The single crystal X-ray structure of Ru8 tetragonal prism [Ru8(p-cymene)8(μ-tppH2)2(μ-C2O4)4]8+. Color code: green, Ru; red, O; blue, N; gray, C.
Lu and coworkers459 have reported a series of octametallic tetragonal prismatic cages, [{(CO)3Re(μ2-OR)2Re(CO)3}4(μ4-tpeb)2] (393, R = −C8H17; 394, R = −C12H25; 395, R = −C7H7) obtained in excellent yields at elevated temperatures under solvothermal conditions by mixing Re2(CO)10 and the tetradentate ligand 1,2,4,5-tetraethynyl(4-pyridyl)benzene (tpeb, 392), in a 2:1 ratio in high boiling alcohols (Scheme 103). These prisms 393-395 were the first examples of neutral luminescent molecular tetragonal prisms possessing eight octahedral Re(I) centers and two different kinds of ligands. Single crystal X-ray structural studies on 393 established that two tpeb ligands were coordinated to four [(CO)3Re(μ2-OCH2C6H5)2Re(CO)3] edge moieties, thereby forming a tetragonal prism. Molecular recognition studies revealed that these prisms engage in host-guest interactions with pyrene molecule.
Scheme 103.
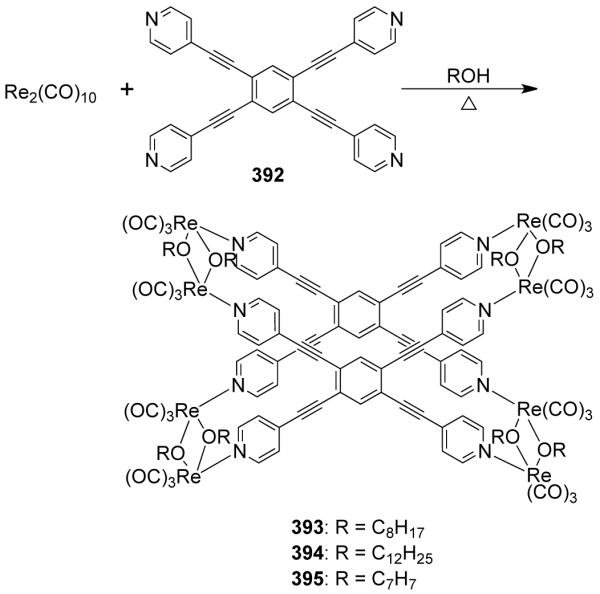
Discrete tetragonal prisms were constructed via a multicomponent coordination-driven self-assembly approach from a combination of a tetraphenylethylene-based tetratopic donor 396, a linear dipyridine donor and a 90° platinum metal complex in appropriate stoichiometric ratios in the absence of any template.460 The 1:2:4 reaction of 396, 4,4′-bipyridine or (E)-1,2-di(4-pyridyl)ethane and cis-[(PEt3)2Pt(OTf)2] (8) in CDCl2/CD3NO2 led to the formation of tetragonal prisms 397 and 398 (Scheme 104). Heteroleptic tetragonal prisms were assembled from tetratopic donor 396 and 5,10,15,20-tetra(4-pyridyl)porphyrin (tppH2) ligand using sodiumterepthalate as the pillar with 8 as the acceptor.439 These heteroleptic prisms can be achieved through supramolecule-to-supramolecule conversion from their homoleptic counterparts. Using this methodology, the self-assembly of supramolecular hexagonal prisms has also been achieved upon mixing a hexakis[4-(4-pyridyl)-phenyl]benzene donor ligands and carboxylate donor ligands such as sodium terephthalate, sodium (1,10-biphenyl)-4,4′-dicarboxylate and sodium 4,4′-(diazene-1,2-diyl)dibenzoate with 8 in a 1:3:6 ratio.461 Recently,the combination of tetraphenylethylene-based tetratopic donor 396 and ditopic bipyridine or carboxylate ligands functionalized with hydroxyl or amine groups, hydrophobic alkyl chains, or electrochemically active ferrocene was reported to yield a suite of self-assembled tetragonal prisms under mild conditions.462 These metallacages were characterized by multinuclear NMR (31P and 1H) and ESI-mass spectrometry. Their shapes and sizes were established using molecular force field simulations and pulsed-field-gradient spin-echo (PGSE) NMR experiments.
Scheme 104.
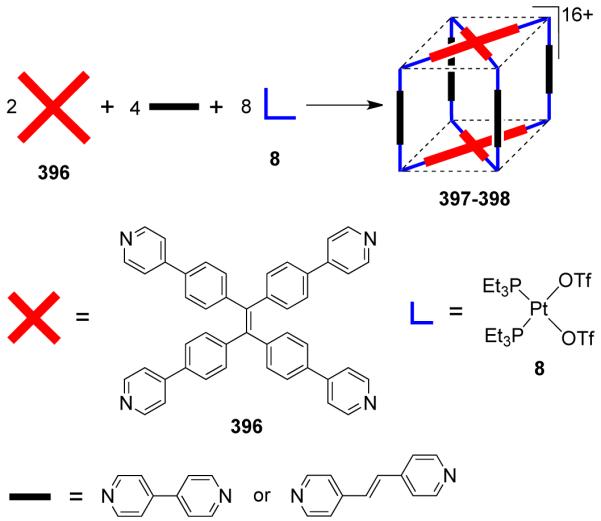
4.2.5. Molecular Boxes
Tetratopic buiding units, in combination with a 90° acceptor, may generate several discrete molecular arctictures ranging from a trifacial box to higher analogues or even molecular cubes (Figure 35). Though the smallest homologue (trifacial box) is the entropically most preferred isomer, higher analogues like cubes, tetrafacial boxes and even hexagonal barrels have been reported.
Figure 35.
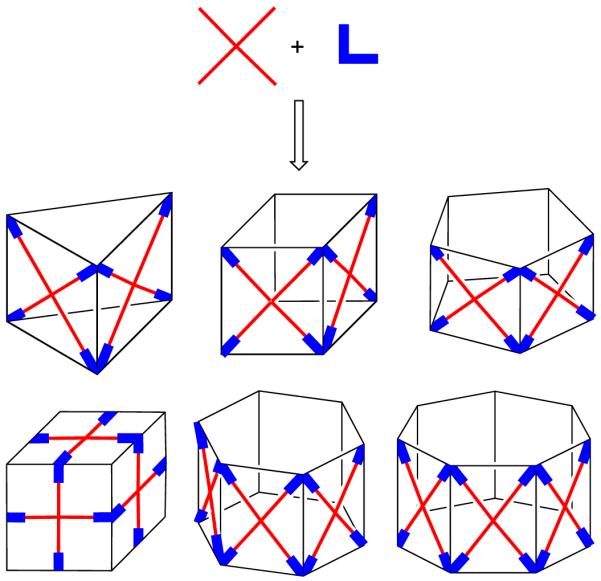
A few possible discrete architectures from 1:2 self-assembly of tetratopic donor with 90° acceptor.
The coordination-driven self-assembly of several trifacial boxes was achieved using rationally designed tetrapyridyl star connectors with 90° platinum linkers. 463 The 1:2 combination of tetratopic planar donors 302, 392, 399 and 400 with [cis-(PMe3)2Pt(OTf)2] (38) in nitromethane led to the formation of trigonal prisms 401-404 (Scheme 105). This face-directed approach gives high yields without template assistance. Each trifacial box consists of three tetrapyridyl star connectors that span the faces with Pt(II) hinges clipping the vertices. The prisms have been characterized by multinuclear (1H and 31P) and DOSY NMR and dual electrospray ionization–Fourier transform–ion cyclotron resonance dualESI-FT-ICR-mass spectrometry. The use of a conformationally chiral star connector, 302, leads to a face-based conformationally chiral prism (401) when the connector arm ends attached to a vertex have a strongly correlated twist sense and chirality is communicated across polyhedral faces, edges, and vertices. Molecular mechanics studies suggested that in the smallest trifacial box 401, collective effects dominate and the all-P and all-M conformers are strongly favored. 1D EXCHSY NMR studies revealed pyridine edge interconversion processes in 401-404 compatible with pyridine rotation around the Pt-N bond. Theoretical studies showed that largest trifacial box, 404, displayed a highly unusual kinetic behavior during the pyridine ligand rotation.464
Scheme 105.

Fujita and coworkers have employed zinc tetrakis(3-pyridyl) porphyrin (405) in conjunction with [(en)Pd(NO3)2] (1) to assemble a M6L3 trifacial box, (406; Scheme 106), utilizing the coordination-driven face-directed self-assembly paradigm.28 1H NMR, ESI-mass spectrometric and X-ray analyses confirmed the formation of the prismatic structure. The crystal structure of 406 showed that the three porphyrin-based ligands span the faces of the prism with six palladium atoms occupying the apical positions. Addition of a pyrene guest molecule for encapsulation triggered a conformational change in host 406 from D3h to C2 symmetry.
Scheme 106.
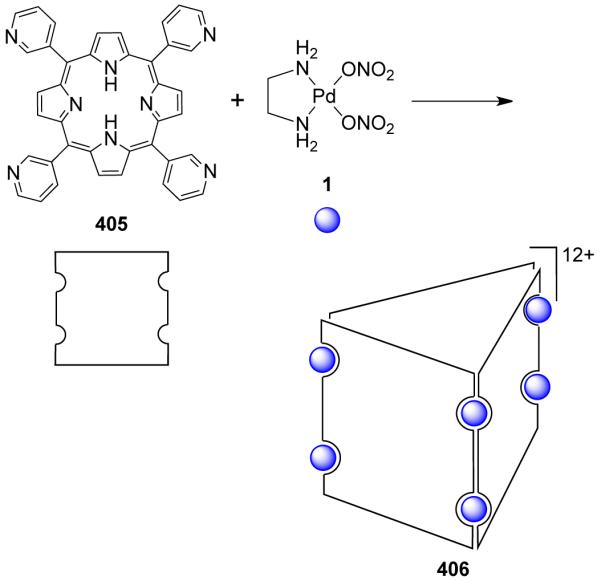
The treatment of tetrapyridyl rectangular molecular panel, 407, with [(en)Pd(NO3)2] (1) at 50°C for 4 d in D2O/CD3OD led to the formation of tetrafacial box 408 as the major product (Scheme 107).465 As detemined from NMR and CSI-mass spectrometric studies, tetrafacial box 408 exists in a dynamic equilibrium with several other products ranging from a trifacial box to a pentafacial box. However, this product equilibrium can be tweaked to favor the quantitative formation of trifacial box 409 through use of biphenyl as a template in the assembly reaction. The use of a 60° donor, 1,2-bis(4-ethynylpyridine)benzene, in combination with Pd(NO3)2 yielded the solvato-controlled assembly of trifacial M3L6 and tetrafacial M4L8 open boxes. Owing to the lability of Pd(II)-N bonds, a facile interconversion between M3L6 and M4L8 boxes was observed by removal or addition of solvent.466 Addition of CD3CN to a solution of M4L8 in DMSO-D6 promoted the smooth conversion of complex M4L8 into a M3L6 box, as revealed by 1H NMR spectroscopy.
Scheme 107.

A hexagonal barrel was reported using 5,10,15,20-tetra(4-pyridyl)porphyrin (tppH2; 385) in combination with a 90° Pt(II) acceptor.29 The molecular box [{cis-(dppf)Pt}12(tppH2)6]24+ (410; dppf = 1,1-bis(diphenylphosphino)ferrocene) was obtained in quantitative yield by treatment of a solution of tppH2 in dichloromethane with two equivalents of cis-[(dppf)Pt(OTf)2] (33) in nitromethane (Scheme 108). NMR and single crystal X-ray diffraction studies established the exclusive formation of this hexagonal Fe-Pt heterometallic barrel. Structural analysis revealed the formation of a hollow barrel of 2.7 nm × 2.7 nm × 1.9 nm dimensions with an internal void volume of around 13,550 Å3. In contrast to the self-assembly reactions in which similar tetratopic donors in conjunction with 90° Pt(II) acceptors give trifacial boxes,463 the use of bulky dppf ligands to cap the 90° Pt(II) acceptor led to the assembly of a wider hexagonal box.
Scheme 108.
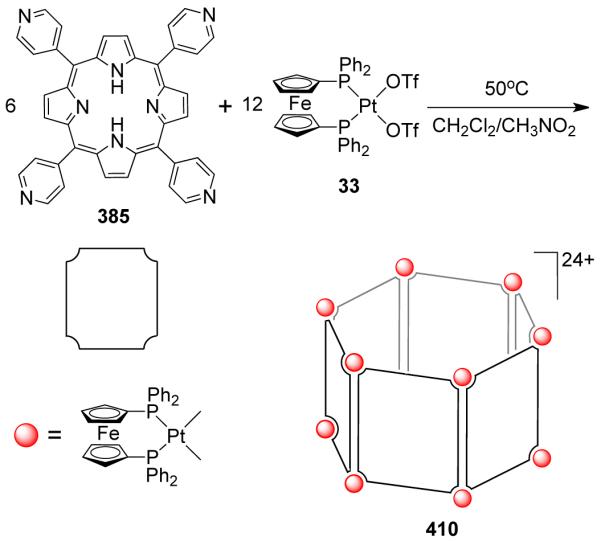
4.2.6. Molecular Spheres
A molecular sphere/ball can be formulated as eight trigonal curved surfaces and six square planar centers. Proper selection of tripodal ligands which can fit on these trigonal faces, in conjunction with square planar metal centers, may generate molecular balls by an 8:6 face-directed self-assembly. An early example of a coordinatively saturated Pd6 molecular ball was reported by Liu et al.467 A flexible tripodal imidazole donor ligand (titmb = 1,3,5-bis(imidazol-1-ylmethyl)-2,4,6-trimethylbenzene) produced a nanometer sized M6L8 molecular sphere [Pd6(titmb)8]12+ upon treatment with PdCl2 (Figure 36). The inner cavity of the ball was determined to be 1000 Å3, which was enough to accommodate eight Cl− ions inside the ball. Despite the possibility of several conformational isomeric forms due to the flexibility of the ligand, only one configuration was adopted in the structure. An analogous molecular sphere [Pd6(322)8]12+ was reported by Fujita and coworkers using a flexible tripodal pyridyl ligand 1,3,5-tris(4-pyridylmethyl)benzene (322) in combination with Pd(NO3)2 (Figure 36).468 The average distance between axially located Pd(II) centers was 15.2 Å and that between equatorial centers was 10.7 Å. The distance between the central benzene rings of the ligands situated at the terminus of the three-fold axis was 19.2 Å. The same group also prepared a neutral, nanometer-sized metallosupramolecular sphere [Ni6(tpst)8Cl12] (tpst = 2,4,6-tri[(4-pyridyl)sulfanylmethyl]-1,3,5-triazine) from an assembly reaction of NiCl2 and the tpst ligand in DMF.469
Figure 36.

Molecular structure of M6L8 molecular spheres [Pd6(titmb)8]12+ and [Pd6(322)8]12+. Color code: pink, Pd; blue, N; gray, C.
Using a similar approach, a nanosized molecular sphere [Pd6(tpbc)8]12+ 412 (tpbc = N,N’,N”-tris(3-pyridinyl)-1,3,5-benzenetricarboxamide) was reported independently by Lah470 and Mukherjee471 using suitably designed C3-symmetric triangular ligands as facial components and C4-symmetric Pd(II) metal ions as corner linkers. Treatment of 4 equiv of tpbc (411) with 3 equiv of Pd(NO3)2 in DMSO-d6 led to the quantitative self-assembly of a single product (412; Scheme 109) as determined by 1H NMR spectroscopic studies. X-ray crystallographic studies470 revealed that the nitrogen donor atom at the meta position of the carboxamido pyridinyl group and the tilted pyridyl versus the facial plane of the ligand both provide the needed curvature for the formation of a molecular sphere. Along similar lines, a Pd(II)-based molecular sphere, [Pd6(tpte)8]12+, incorporating ester functionalities, was assembled using a C3 symmetric tripodal linker, N,N’,N”-tris(4-pyridylmethyl) trimesic ester (tpte), with Pd(NO3)2 in a 4:3 molar ratio. NMR, ESI-mass spectrometry, TEM, and MM2 force-field calculations support the formation of the nanoball with eight trigonal faces occupied by C3 symmetric donor linkers. Host-guest studies on molecular sphere [Pd6(tpte)8]12+ showed efficient encapsulation of Et4N+ cations despite the sphere’s high positive charge.471
Scheme 109.
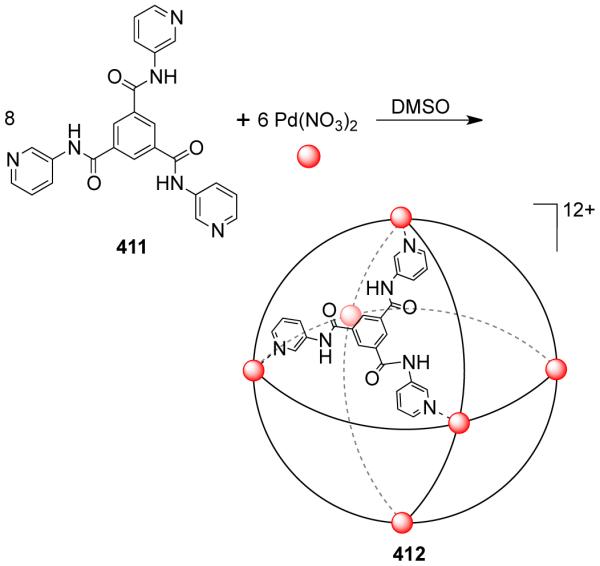
Batten and coworkers472 have reported a new approach for preparing bimetallic molecular balls using tripodal metallo-ligands in combination with metal centers of appropriate coordination environments. This efficient, one-pot assembly process involves two distinct steps. First, the central tris(pyrazolyl)hydroborate core of the bifunctional ligand, [tris-{3-(4-pyridyl)-1-pyrazolyl}hydroborate] (413) is utilized in an in situ, preorganizational step to form the neutral metalloligand [CuI(413)(CH3CN)] (Scheme 110). This building block conformationally locks an otherwise flexible organic ligand, thereby providing directionality to the peripheral 4-pyridyl donor groups for further reaction with an octahedral metal ion. In the second step, crystallization of the discrete nanoball species [{CuI(413)(CH3CN)}8{FeII(NCS)2}10/3{FeII(NCS)(CH3CN)}8/3]·(ClO4)8/3·(CH3CN)n (414) occurs by self-assembly upon addition of FeII ions and a thiocyanate salt. Single crystal X-ray structural studies revealed that nanoball 414 possesses a rigid [FeII{CuI(413)}] shell in which the octahedral arrangement of FeII ions and cubic arrangement of CuI ions forms a distorted rhombic dodecahedron. The fact that FeII ions exist in two different magnetic environments helps the nanoball reversibly switch between HS and LS states triggered by thermal, light and guest perturbation. Treatment of metallo-ligand [CuI(413)(CH3CN)] with a range of divalent metal salts (MX2) (MII = Cu, Mn, Zn, Cd, Fe; X = ClO4−, NO3−, BF4−) gave a series of nanoballs of the general formula [(413)CuI(MeCN)8MII6(X)10(MeCN)2](X)2·xMeCN, producing isostructural crystalline samples in a matter of hours.473 These porous nanoballs showed hydrogen gas adsorption as well as solvent adsorption behavior in the solid state.
Scheme 110.
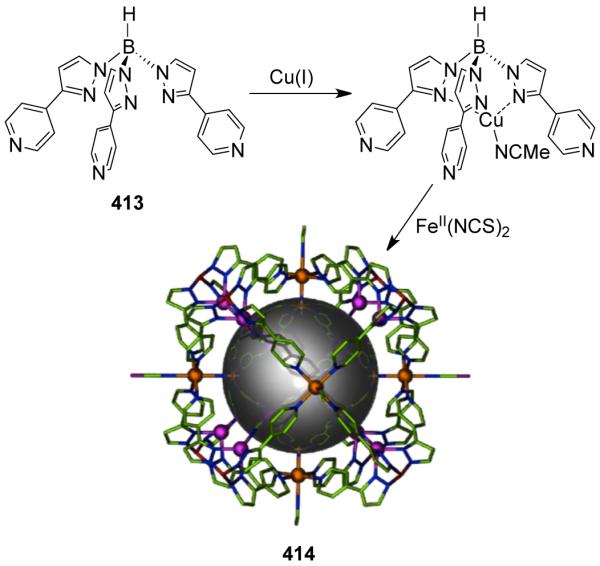
Unlike the approach used in previous examples of face-directed molecular balls, Fujita and coworkers have revealed a new strategy using edge-directed self-assembly. In this approach, 12 Pd(II) square-planar metal ions were assembled with 24 units of rigid bent dipyridyl linkers 415-417 to obtain spherical hollow structures 418-420 (Scheme 111).474 1H NMR and CSI-MS spectroscopic studies confirmed the formation of M12L24 molecular spheres. X-ray structural studies on 441 revealed that the spheres have cuboctahedron symmetry in which each of the 12 Pd(II) atoms occupy the 12 vertices, while the dipyridyl bridging ligands span the 24 edges. Related molecular spheres having cuboctahedral symmetry with copper(II) dinuclear tetracarboxylate junctions have also been reported.475
Scheme 111.

Interestingly, when a ligand analogous to 416, dipyridylthiophene, was used in conjunction with Pd(II)-square planar metal ions, it resulted in the formation of 72-component self-assembled giant Pd24L48 coordination spheres using 24 palladium ions (M) and 48 curved bridging ligands (L).476 X-ray crystallographic analysis showed that the Pd24L48 coordination sphere (Figure 37) forms a rhombicuboctahedron, which is an Archimedean solid with eight triangular and 18 rectangular faces (26 in total), with one triangle and three rectangles meeting at each vertex. Each of the 48 curved bridging ligands span the edges while 24 metal ions occupy the vertices. Although the ligand dipyridylthiophene differs very little from 416, the difference in ligand bend angle from 127° in 416 to 149° in dipyridylthiophene, stemming from longer C–S bonds (1.80 Å) in thiophene as compared with the C–O bonds (1.39 Å) in furan, accounts for the difference in the final polyhedral geometry. Furthermore, a self-organization study using a mixture of both the bent ligands with Pd(II) ions, where the ligand ratio was varied to reflect the continous modulation of mean ligand angle from 149° to 127°, revealed that a slight change in the mean ligand bend angle critically switches the final structure between M24L48 and M12L24 coordination spheres.
Figure 37.
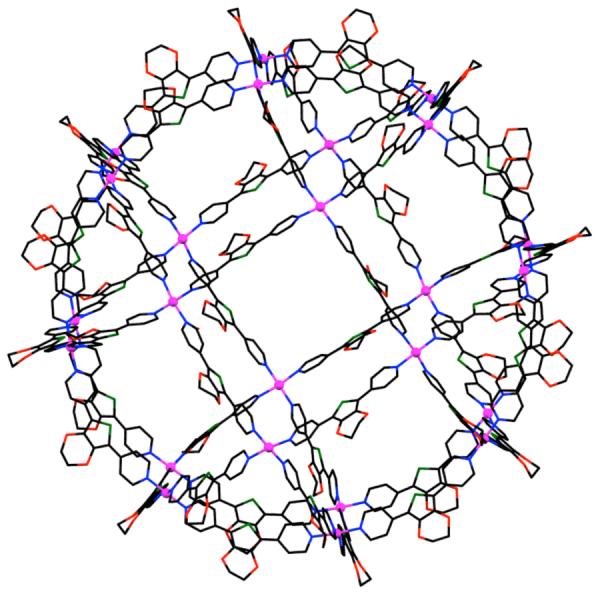
The X-ray crystal structure of Pd24L48. PF6− counter ions and protons are omitted for clarity. Color code: pink, Pd; red, O; blue, N; green, S; gray, C.
4.3. Chiral Systems
Chirality is an inherent property of almost all biomolecules since they are made up of building blocks that are chiral in nature. Almost all biochemical reactions that occur in nature are stereospecific and extremely efficient. Thus, the creation of chiral, 3D spaces for carrying out enantioselective reactions, molecular recognition and catalysis in confined cavities using natural systems for motivation is a much sought after goal. In recent years, discrete, chiral 3D supramolecules of finite sizes have been assembled by means of weak intermolecular non-covalent interactions477 as well as metal-ligand coordination.478 These assemblies have proved themselves as powerful tools to aid in the understanding and exploration of chirality and its effects in host-guest interactions, chemosensing and enantioselective catalysis.
All Platonic and Archimedean solids, along with prisms and anti-prisms, with the exception of snub cube and snub dodecahedra, are inherently achiral. However, chirality can be introduced in these architectures through coordination-driven self-assembly by removing symmetry elements: centers of inversions and mirror planes. This can be achieved through different design strategies: a) through the use of achiral building blocks where chirality arises at the metal center due to strong mechanical coupling between metal sites through the ligands used, resulting in exclusive homochiral assemblies; b) using achiral components where chirality is introduced as a consequence of the twisting of the ligands; c) using enantiopure or racemic scaffolds to assemble chiral supramolecules. The stereogenic center may be incorporated into the organic ligand bridging the metal centers or on an auxiliary ligand; d) through the encapsulation of a guest molecule within the cavity which perturbs the architecture to lose symmetry elements, thereby making it chiral.
Among the chiral supramolecular architectures, tetrahedral assemblies are perhaps the most extensively studied. Saalfrank et al. reported the earliest example of a chiral supramolecular tetrahedron as described above (section 4.1.1).329,330 The teranuclear magnesium(II) assembly, [(NH4)4∩{Mg4(L)6}] 270 was formed as a racemic mixture of homochiral ΔΔΔΔ and ΛΛΛΛ ensembles as determined by time-dependent 1H NMR spectroscopy. The overall T symmetry of the ensembles is due to the restricted rotation around the C–C single bonds in the tetracarboxylate ligands, which make the ligands twist in the same sense when coordinating to the four metal centers.329 The replacement of the R group in the tetracarboxylate ligand (Scheme 77) with hydrogen results in isosturctural homochiral ΔΔΔΔ and ΛΛΛΛ assemblies. 479 Variable-temperature 1H NMR studies revealed that the assembly undergoes nondissociative enantiomerization from ΔΔΔΔ to ΛΛΛΛ symmetry without the formation of diastereomeric intermediates.480 The interconversion involves four simultaneous Bailar twists associated with (Δ)/(Λ) isomerization at the four octahedral metal centers that are synchronized with the atropenantiomerization process on the four sterically unhindered ligands. When bulky R group such as CO2Et in the tetracarboxylate ligand are present, a restriction of the rotation around the central C-C bond in L2− occurs, thereby preventing enantiomerization.
Raymond and coworkers designed a series of chiral M4L6 tetrahedra based on bis-hydroxamate334,335 and bis-catecholate336 ligands through the symmetry interaction approach. As described above (section 4.1.1), these tetrahedral anionic cages were assembled from four metal ions such as Ga(III), Al(III), In(III), Fe(III), Ti(IV) Ge(IV) and Sn(IV), which are coordinated by six bifunctional bis-chelating ligands spanning along the edges of the tetrahedra. The chiral environment of the cavities were probed using a monocationic ruthenium(II) half-sandwich complex [CpRu(p-cymeme)]+ as a guest molecule.481 The two enantiotropic methyl groups of the isopropyl substituent of p-cymene become diastereotopic upon binding with the chiral host and are thus distinguishable, as evidenced by NMR spectroscopy. This implies that the guest is increasingly able to sense the chiral environment of the host molecule.
Though the bis-chelating ligands are achiral in nature, the chirality in these tetrahedral assemblies results from the trisbidentate coordination at each metal center, leading to either a Δ or Λ configuration. Due to strong mechanical coupling between the metal centers through the ligands, the chirality is communicated from one vertex to the next, resulting exclusively in homochiral tetrahedra (with either ΔΔΔΔ or ΛΛΛΛ configuration). Depending on the chiralities at the metal centers, these clusters can have idealized C3 (ΔΛΛΛ/ΛΔΔΔ), S4 (ΔΔΛΛ), or T (ΔΔΔΔ/ΛΛΛΛ) symmetry. However, the use of achiral components almost always leads to racemic chiral supramolecules due to the lability of the metal-ligand bonds, which facilitate interconversion of the enantiomers. These racemates can be enantiomerically resolved through use of a chiral auxiliary molecule. For example, as shown in Scheme 112, the tetrahedral assembly [Ga4(271)6]12− preferentially precipitates the diastereomeric ion pair of (ΔΔΔΔ)-KH3(S-nic)7[(S-nic) ⊂ Ga4(271)6] when treated with chiral auxiliary (S)-N-methylnicotinium (S-nic), leaving the soluble ΛΛΛΛ-K6(S-nic)5[(S-nic) ⊂ Ga4(271)6] species in solution.482,483 The chiral auxiliary S-nic can then be replaced through ion exchange by an achiral guest cation such as NMe4+ or NEt4+. The mechanism for chiral resolution of the M4L6 tetrahedron was shown to be proton dependent and relies on interactions of the chiral S-nic+ counterion with the exterior of the anionic host. Interestingly, the M4L6 tetrahedron [NEt4 ⊂ Ga4(271)6]11− (272) retains the “chiral structural memory”.484 In a solution of (ΔΔΔΔ)-[NEt4 ⊂ Ga4(271)6]11−, as many as three of the six-edge bridging ligands can be replaced by phenyl derivatives while maintaining the original chirality.
Scheme 112.

McCleverty, Ward and coworkers have assembled chiral M4L6 tetrahedral clusters using flexible bispyrazolylpyridine ligands having a central aromatic spacer between the binding sites.340 These cationic cages are chiral, having non-crystallographic T symmetry with all four metal centers possessing the same tris-chelate optical configuration with a C3 axis running through each vertex but no mirror planes.345 X-ray structural studies have shown that the assemblies crystallize as a racemic homochiral mixture with equal numbers of ΔΔΔΔ/ΛΛΛΛ enantiomers. The chiropticity of the internal cavities of the tetrahedral cages of the type [Co4(274)6(BF4)]7+ can be probed by using an enentiopure chiral solvating agent [tris-(tetrachlorocatecholato)phosphate(V)] (TRISPHAT).485 Addition of 2 TRISPHAT Δ-1, to a solution of racemic mixtures of ΔΔΔΔ and ΛΛΛΛ enantiomers of [Co4(274)6(BF4)]7+ in 5% CD3NO2 in CDCl3 led to the enantiodifferentiation of the protons of the ligands. The 19F NMR signal (a singlet) for the encapsulated achiral BF4− anion also gets split into two peaks with a separation of 2 ppm between the components arising from the two diastereo-isomers. The chiral environment can thus be detected through the enantiodifferentiation of an achiral guest in the chiral cavity. In an alternative approach, diastereoselective formation of a chiral tetrahedral cage has been achieved using a ligand in which a chiral auxiliary, α-pinene, was appeneded to a similar flexible bispyrazolylpyridine ligand, having a central 1,2-phenyl aromatic spacer.486 As evidenced from its 1H NMR spectrum, the cage exists as a single diastereoisomer in solution and crystallizes as a single diastereoisomer in the acentric space group C2.
A number of chiral M4L4 tetrahedral cages have been assembled from tris-bidentate chelating ligands and pseudo-octahedral metal ions by the groups of Raymond,354,358 Saalfrank,355 Ward356,357 and Albrecht.359,360 The C3-symmetric ligands in these cages span the faces of M4L4 tetrahedra with octahedral metal ions occupying the vertices. The chirality in these tetrahedral assemblies originates from the tris-bidentate metal chelates, where each metal center can adopt a Δ or Λ configuration within each tetranuclear structure. As a result, a M4L4 tetrahedron can adopt T (ΔΔΔΔ or ΛΛΛΛ), C3 (ΔΔΔΛ or ΛΛΛΔ), or S4 (ΔΔΛΛ) symmetry. The chiral induction studies carried out on [Ga4(278)4]12− showed that the chiral auxiliary (S)-N-methylnicotinium (S-nic) can induce an enantiomeric excess in a racemic population of [Ga4(278)4]12− tetrahedra.358 Larger M4L4 clusters from analogous tris-bidentate chelating ligands having phenylene spacers fail to show enatiomeric enrichment due to the rotational freedom of the phenylene spacers in the ligand.
Achiral tetratopic ligands derived from hydrogen bond-directed solid-state synthesis have also been used to assemble chiral polyhedral cages. A linear template based on resorcinol was used to direct a regiocontrolled synthesis of tertrapyridyl ligand 421 from trans-1-(2-pyridyl)-2-(2-pyridyl)ethylene).487 Addition of an acetonitrile solution of 421 to an acetonitrile solution of Cu(NO3)2 ·2.5 H2O led to the formation of a chiral tetrahedral assembly [Cu4(421)4(H2O)4]8+ (422; Scheme 113). X-ray structural studies revealed that each of the four Cu(II) ions occupies a corner of the tetrahedron. Similar tetratopic ligands derived from trans-1-(2-pyridyl)-2-(4-pyridyl)ethylene) have earlier been shown to assemble with Cu(II) ions to form a hexanuclear cage structure that conforms to a trigonal antiprism.488
Scheme 113.

Chiral molecular assemblies have also been achieved using achiral components in which chirality arises as a consequence of the way the organic ligands are twisted in space. For example, Robson 489 and Müller 490 reported chiral truncated tetrahedra having M6L4 stoichiometries utilizing guanidine-based ligands. The chirality in these assemblies originated from the screw-like orientation of the ligands upon coordination to the metal centers at each face of the tetrahedra. The reaction of CdCl2 with tris[(5-bromo-2-hydroxybenzylidene)amino]guanidinium chloride, [H6Br3L]Cl in methanol in the presence of Et4NCl and Et3N led to the formation of chiral M6L4 tetrahedra.490 X-ray structural studies revealed that each cadmium atom lies in a square pyramidal center with three of the basal coordination sites occupied by a ligand and one Cl−. The fifth site was occupied by a phenolate oxygen atom from the neighboring [(CdCl)3(H6Br3L)] triangular panel, formed upon the binding of three CdCl units. This led to to the formatiom of the chiral cage (Et4N)5(Et3NH)3[{(CdCl)3(H6Br3L)}4]. Of the eight counterions, one [Et4N]+ gets trapped in the cavity of the terahedron. These guanidine-based ligands have also been used to assemble a chiral trigonal bipyramid491 and a molecular octahedron [{Pd3L}8{μ-(bar)}12]16− (310).388
Hannon and coworkers492 have reported the assembly of a M12L12 chiral ball (424) from bis-bidentate ligand 423 and Cu(I) ions. X-ray structural studies revealed that three bis-bidentate ligands (423) assembled to form a chiral trinuclear circular helicate {Cu3(423)3}3+ with a Cu(I) ion occupying the vertices of the triangle in a four coordinate pseudo-tetrahedral environment. Four of these bowl-shaped circular helicates {Cu3(423)3}3+ aggregrate to form a tetrameric ball-shaped molecular assembly [{Cu3(423)3}4]12+ (424) held together by CH-π interactions (Scheme 114). The chirality of each triangular circular helicate makes the overall ball-shaped structure chiral. The chirality of 424 is due to the propeller-type twist arrngement of the pyridyl rings of each triangle.
Scheme 114.
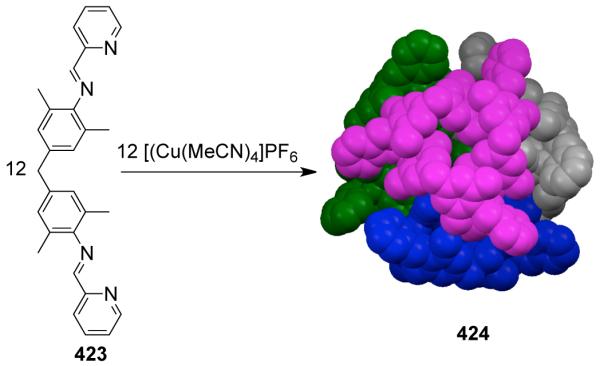
Molecular scaffolds that are intrinsically chiral have been used as both enantiopure and racemic mixtures to assemble chiral supramolecular assemblies. The chiral center may be introduced into the bridging ligand and/or on the auxiliaries attached to the metal center. Using the directional bonding approach, M6L4 adamantanoids have been assembled using enatiopure chiral building blocks having stereogenic centers in their carbon backbones. For example, the presence of a chiral center on the ligand backbone makes adamantanoids 341 and 324 chiral.406 Their stereogenic centers reduce the overall symmetry of the adamantanoids from Td symmetry, as expected for achiral species, to D2 symmetry. A racemic mixture of binaphthalene-based ligands with lanthanide salts led to chiral adamantanoids having perfect T symmetry, as a result of the C2 symmetric nature of the ligand and C3-symmetric lanthanide centers.408 Cui et al.493 reported the synthesis of distereoselective tetrahedral cages using the enantiopure atropisomeric biphenyl bridging ligand 5,5′,6,6′-tetramethyl-3,3′-diketone-2,2′-bis(methoxy-methoxy)-biphenyl, and C3-symmetric metal ions. A 3:2 molar reaction of the bridging ligand with M(III) chloride (M = Fe, Ga) in DMF, followed by layering the solution with methanol, afforded the homochiral (ΔΔΔΔ) or (ΛΛΛΛΛ) M4L6 tetrahedral cages in moderate yields. These cages demonstrated a high enantioselective ability to resolve small racemic alcohols by crystallization inclusion.
Jeong et al.494 reported a chiral nanoball encapsulating lanthanum ions in a ferritin-like assembly. The enatiopure carboxylate ligand (S,S)-425, synthesized from 4,4′-dibromostilbene through Sharpless asymmetric dihydroxylation, assembled into a molecular cluster having the formula [La18{(S,S)-425}24(CO3)2(H2O)32]2+ (426; Scheme 115) in the presence of LaCl3· 6H2O. X-ray structural studies showed that 24 ligands encompass 18 metal ions in the assembly. The ball consists of six La3[(S,S)-425]4 units which are related by pseudo-octahedral symmetry. Each unit is further interconnected with another four equatorial units by four organic ligands, completing the assembly. These ferritin-like chiral assemblies form double helices in the solid-state with single-handedness.
Scheme 115.
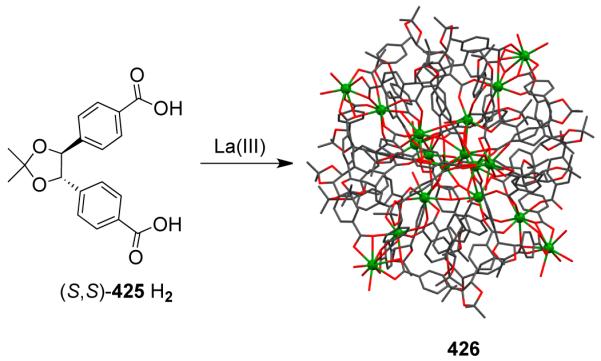
Stang and coworkers365 were able to assemble chiral M6L4 truncated tetrahedra by introducing stereogenic centers on the auxiliary ligands attached to metal donor units. A 3:2 combination of [M(R-BINAP)(OTf)2] (M = Pd (177) or Pt (178), BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthalene) and tridentate ligand 1,3,5-tris[(4-pyridyl)ethynyl]benzene (291) led to the formation of an assembly having a truncated tetrahedral geometry with T symmetry. Both NMR and ESI-FT-ICR-mass spectrometric studies established the formation of these assemblies. Similarly, utilizing the [(R)-(+)-BINAP]Pd(II) bistriflate as a chiral donor unit, Ikeda et al.495 assembled Zn-based porphyrins bearing four pyridyl groups into a chiral M4L2 square prismic supramolecule possessing D4h-symmetry. Chirality in these assemblies arises due to the chiral auxiliaries present on the periphery of each cage. Fujita et al.496 have used chiral (R,R)-diaminocyclohexane as an auxiliary attached to a Pd(II) center to assemble C2 chiral M6L4 truncated tetrahedra. A chiral M6L4 cage, 429, was assembled from the D2h-symmetric pyridyl ligand, 427, with 90° cis-[(R,R)-diaminocyclohexane]Pd(NO3)2 (428) in the presence of 1,4-dichlorobenzene (Scheme 116). The self-assembly process was driven by the template-effect of the aromatic guest molecule. 1H NMR studies revealed the formation of two diastereomers in a 1.3:1 ratio. With (S,S)-diaminocyclohexane auxiliary ligands, the resulting assembly showed a negative Cotton effect.
Scheme 116.
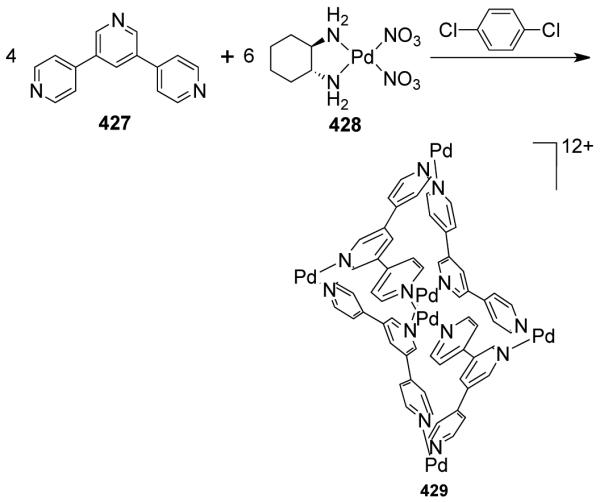
Chirality in a supramolecular assembly can also be induced through encapsulation of guest molecules into the achiral assembly, triggering a conformational change from achiral to chiral. Fujita and coworkers observed that the inclusion of flat, achiral 1,3,5-benzenetricarboxylic acids as guest molecules distorts trigonal bipyramidal cage 323 (Scheme 85) into a chiral cage.497 The chirality of this assymetric cage was shown by complexation of R-mandelic acid, which induces diastereomeric organization resulting in P- and M-cages. Furthermore, the observation of two diastereomers also indicated that no racemization takes place on the NMR time scale. Notably, diastereomers were not observed when racemic mandelic acid was used. Encapsulation of a spherical guest such as CBrCl3 or CBr4 led to the formation of an achiral trigonal bipyramidal cage under similar conditions. This was explained through molecular modeling studies wherein it was observed that the asymmetric cage possesses flat internal cavities while the symmetric cage possesses spherical cavities. The same group also observed a conformational change from D3h-symmetry to C2-symmetry in a porphyrin-based M6L3 trifacial box (406; Scheme 106) upon encapsulation of pyrene as a guest molecule.28
4.4. Functionalized Systems
The functionalization of 3D supramolecular assemblies has been intensively investigated over the last few years with an aim to develop nanoscale ensembles that can emulate biological systems and can find applications in various fields such as host-guest chemistry, cavity directed synthesis, catalysis, photonics, redox activity, magnetic behavior, self-organization and sensing.230 Although various functionalized nanoscopic systems have been developed through conventional covalent synthesis, when issues such as the control of functional groups and structural precision, the ability to perform selective encapsulation, and synthetic ease and building-block versatility come into question, coordination-driven self-assembly emerges as a powerful tool to assemble 3D functional supramolecules with relative ease. The conformational rigidity of supramolecular ensembles allows for greater structural control and positioning of functional groups on the tectons to form novel, multifunctional structures. Precise control over the number, location, orientation and relative distribution of functional moieties is possible in these systems. Using the directional bonding approach, functional groups have been introduced both on the exterior and interior of supramolecules.
Exo-functionalization of metallacages can be achieved by the attachment of a functional moiety on the convex side of a building block. This positions the groups on the periphery of the resulting self-assembled suprastructure. Fujita and coworkers have designed a number of Pd12L24 exo-functionalized molecular spheres (Scheme 117) decorated with many different functional groups on the surface by simply attaching the desired moiety to the convex dipyridyl ligands (430-435). In one early example, metal porphyrins and fullerene nanonballs were introduced on the periphery of a Pd12L24 sphere (436 and 437).474
Scheme 117.

Similarly, attachment of saccaride groups at the periphery led to saccharide-coated Pd12L24 molecular spheres that form aggregates upon interaction with proteins.498 Studies with concanavalin A, which is a well-known lectin that selectively recognizes α-mannopyranoside and α-glucopyranoside at its four binding sites, showed the formation of aggregates with α-mannopyranoside and α-glucopyranoside coated Pd12L24 spheres 439 and 440, respectively, but not with α-galactopyranoside or β-glucopyranoside coated ones, indicating that concanavalin A can recognize the terminal saccharide units of Pd12L24 molecular spheres. Recently, the same group also reported the facile preparation of self-assembled Pd12L24 spherical complexes 438 and 441 with their surface modified with oligonucleotide chains499 and hexapeptide aptamers (Arg–Lys–Leu–Pro–Asp–Ala) respectively. 500 The oligonucleotide-coated spheres can recognize complementary nucleobases through Watson-Crick base pair formation. These well-defined DNA nanoparticles templated by self-assembled Pd12L24 molecular spheres may lead to systems that can recognize natural DNA in aqueous media.
Stang et al. reported the construction of multifunctional cuboctahedra having crown ether and ferrocene as functional groups on the periphery (Scheme 118).501 Treatment of 120° exo-functionalized acceptors 442 and 443 with the planar tritopic donor 1,3,5-tris-(4-pyridylethynyl)benzene (314) in 3:2 resulted in the formation of cuboctahedral complexes 444 and 445 with 12 pendant crown ether or ferrocene groups, respectively, at the vertexes. Multinuclear NMR (1H and 31P), ESI-mass spectrometry and cyclic voltammetric studies established the structures of the complexes. Both functionalized supramolecular systems were obtained in high yield and all of the functional units retained their functional fidelity while surrounding a nanoscopic hollow “core” environment ideal for guest encapsulation. The multiferrocenyl cuboctahedron molecule shows a single redox couple indicating that the redox species react more or less independently of one another. These assemblies may find uses as multielectron catalysts and sensors. Also, the complementary recognition properties of the peripheral crown ether hosts and the interior cavity of the crown ether-functionalized cuboctahedron may be used to develop both an exo and endo receptor.
Scheme 118.

Endohedral functionalization of supramolecular architectures can be achieved by covalently attaching a functional moiety on the concave side of a directional building block, precisely orienting the group inward. Fujita et al. assembled a variety of discrete, endo-functionalized Pd12L24 molecular spheres (452-457) by combining 12 “naked” Pd(II) ions with 24 bis-4-pyridyl ligands (446-451) involving two acetylene spacers (Scheme 119). The acetylene spacers play an important role in the overall architecture of the resulting ensembles. In addition to providing a larger cavity space as compared to that in exohedral Pd12L24 cages assembled using dipyridyl ligands 430-435, the acetylene spacers prevent the ligands from adopting unfavorable nonplanar conformations, which are caused by steric repulsion between the pyridyl groups and the core benzene ring in the absence of the spacer. The large cavities of the Pd12L24 molecular spheres allow for the introduction of a variety of pendant functional moities such as oligo(ethylene oxide) chain (452),502 perfluoroalkyl chain (453),503 extended aromatic system (454), 504 polymerizable methyl methacrylate 505 (455), sodium p-styrenesulphonate units, 506 peptide (456),507 and sugar508 (457) lining the interior surface. The well-defined nanoscale cavities of these functionalized assemblies have helped in the understanding and illustration of interesting properties otherwise not observable under standard conditions. For example, the attachment of perfluoroalkyl chains generated a fluorous nanophase within the cavity, allowing it to solubilize up to ~ eight perfluoroalkane molecules per spherical complex. Their solubility can be tuned by varying the length of the pendant perfluoroalkyl chains.503 Lining the inner surface with extended aromatic systems such as corronene created an aromatic nanophase resulting in increased solubility of fullerenes, which are sparingly soluble in most solvents.504 Fujita and coworkers also prepared Pd12L24 molecular spheres (455) that can confine 24 methyl methacrylate (MMA) units at their internal cavities.505 These monomer units can be polymerized to PMMA through radical polymerization using 2,2′-azobis(isobutyronitrile) (AIBN) as a radical initiator. The sphere with tri(ethylene oxide) linkers afforded the best efficiency, with 73% conversion. These nanomolecular materials may find applications as high-density data storage materials.
Scheme 119.

One of the key challenges in materials science is to gain a high degree of control over the size and shape of nanoparticles. Recently, Fujita and coworkers508 succeded in restricting the size and shape of growing silica nanoparticles by using a Pd12L24 molecular cage (457) whose inner surface was lined with sugar residues, creating an environment for the sol-gel condesation of tetramethoxysilane. The hydrophilic cavity of the cage served as an endo-template for the condensation process to give monodispersed silica nanoparticles with diameters of 2-4 nm relative to the dimensions of the Pd12L24 spheres (2-4 nm). As monitored by NMR spectroscopy, signals corresponding to free siloxane disappeared during the reaction, whereas a signal arising from methanol appeared and increased in intensity, indicating the formation of condensation products. The sol-gel condesation process took place within the endo-template rather than on the bulk solvent, as evidenced by the homogeneity of the solution. Remarkably, the polydispersities of the silica nanoparticles achieved with this method approached unity, with Mw/Mn < 1.01. Recently, the Pd12L24 molecular cage 457, whose inner surface was lined with sugar residues, was also utilized to generate core-shell SiO2/TiO2 and SiO2/ZrO2 nanoparticles with high monodisperse molecular weight distributions (Mw/Mn <1.01) as characterized by TEM and MS analyses.509
Similarly, tethering of azo-benzene units 510 created a confined and well-defined hydrophobic phase inside the cavity of a Pd12L24 self-assembled spherical complex. The hydrophobicity of the cavity can be switched through the photoisomerization of appended azobenzene chromophores. Azobenzenes are well known to undergo cis-trans isomerization upon irradiation with UV light resulting in large changes in size and polarity. The use of a hydrophobic guest, 1-pyrenecarboxaldehyde, as a probe showed that the hydrophobic environment of the confined cavity becomes less hydrophobic due to the reversible isomerization of tethered azobenzene. Likewise, lining the inner surface of a Pd12L24 cage with alkyl chains511 allowed the solubilization of hydrophobic dyes such as Nile red in polar solvents.
Recently, endo-functionalized Pd12L24 (L = 2,6-bis(4-pyridylethynyl)toluene) molecular spheres were shown to assemble into large, monolayered, hollow, vesicle-like blackberry structures in polar solvents.512 As established from laser-light scattering studies, these Pd12L24 cationic cages self-assemble into larger structures in DMSO-D6 mediated by both electrostatic and hydrophobic interactions similar to viral-capsid formation in biological systems. A continuous increase in total scattered intensity from the solution in static light scattering (SLS) studies indicated the formation of large self-assembled structures. Dynamic light scattering (DLS) studies revealed that the large assemblies have a narrow size distribution. The very similar values of average radius of gyration (Rg) and average hydrodynamic radius (Rh) obtained fron light-scattering studies indicated the formation of large hollow spherical structures. Like viral-capsids, these assemblies form monolayer spheres with individual nanocages evenly distributed in the wall of the vesicle-like structures. The vesicle-like structures formed by the nanocages also responded to external stimuli (i.e. ionic strength and solvent polarity) and subsequently changed their sizes. Similar spontaneous self-assembly of M4L6 truncated tetrahedral cages [(en)Pd6L4](NO3)12 (286, L = 2,4,6-tris(4-pyridyl)-triazine) into large monodisperse hollow vesicle-like structures was also observed in dilute solutions.513
Self-assembly of trimeric Zn(II)-porphyrin units into trigonal prismatic architectures was achieved by using linear ditopic N-donor ligands such as 1,4-diazabicyclo[2.2.2]octane (DABCO), 4,4′-bipyridine and 5,15-(4-pyridyl)-10,20-phenyl-porphyrin (4′-trans-DPyP). These ligands act as pillars via two axial coordination bonds with the porphyrinic Zn(II) ions, locking the planes of the porphyrin units in an almost cofacial orientation.514,515 For example, treatment of Zn(II)-trisporphyrin (458) with DABCO in a 2:3 ratio led to the formation coordination cage (459; Scheme 120). Measurement of stability constants using UV-vis and 1H NMR spectroscopic studies revealed that in presence of an excess of the ditopic N-donor ligand, these coordinations cages fall apart to give open species. At micromolar concentrations, the formation of a fully assembled coordination cage was highly favored over the formation of intermediate species stabilized by fewer interactions. In contrast, at millimolar concentrations, the relative stability of intermediate species increased, leading to a stepwise self-assembly process. The presence of a 2:2 intermediate was identified using 1H NMR spectroscopy. Cage 459 was found to effectively recognize benzene-1,3,5-tricarboxamide derivatives through hydrogen bonding and aromatic interactions.
Scheme 120.

Reek and coworkers also reported a similar DABCO-induced porphyrinic architecture using a Zn(II)-trisporphyrin phosphite ligand. Self-assembly of two Zn(II)-trisporphyrin phosphite ligands with three ditopic DABCO ligands led to the formation of a trigonal prismatic cage.516 The cage can encapsulate an active Rh(I) catalytic center through coordination to the phosphorus atoms of the Zn(II)-trisporphyrin phosphite panels. Remarkably, the cage showed high selectivity for linear aldehydes in the rhodium-catalyzed hydroformylation of 1-octene. Similar porphyrin cages were assembled by the same group through a ligand templated approach wherein bifunctional phosphine ligands possess groups to form cages through coordination to porphyrinic Zn(II) ions while also complexing an active transition metal center for regioselective catalytic applications (vide infra).18,19
Hupp and coworkers demonstrated an elegant way to assemble cavity-tailored porphyrinic boxes through coordination-driven self-assembly. Treatment of porphyrinic trimer 460 with pyridine-derivatized porphyrin dimer 461 in a 2:1 ratio led to the generation of a symmetrical 16-porphyrin box, 462 (Scheme 121).517 Chemically orthogonal metallation of 460, with Zn(II) and 461 with Sn(IV) ensured that self-recognition between trimers or dimers was avoided. The steric demand of the axial tert-butyl benzoate groups on 461 ensured that four of them were directed inwards so as to force the dimeric units 461 to link selectively with the first and third porphyrins of 460, leaving the central Zn sites unoccupied. NMR (1H and PGSE) and UV-vis spectroscopy established the exclusive formation of the porphyrinic box 462. Solution-phase small-angle X-ray scattering (SAXS) measurements confirmed the formation of monodisperse assemblies of precisely the size expected from model box structures. The large cavity (22 × 14 × 10 Å) of the assembly was exploited for both size-selective and enantioselective catalysis. Introduction of bulky R groups such as 3′,5′-di(4-tert-butylphenyl)-biphenyl-4-carboxylates on prophyrin dimer 461 produced an unusual twisted box. The steric crowding makes the dimer incapable of combining with trimeric unit, 460, thus forming a porphyrinic box having the architecture of 462.
Scheme 121.

Osuka and Kim have extensively investigated the formation of a number of supramolecular porphyrin boxes via enantiomeric self-sorting of racemic 4-pyridyl appended meso-meso linked Zn(II) diporphyrins. 518 As established by 1H NMR and CSI-mass spectrometric studies, meso-meso linked Zn(II) diporphyrin units (463) quantitatively self-assembled into supramolecular porphyrinic box 464 in CDCl3 (Scheme 122).519 Due to the presence of different meso-aryl substituents, the diporphyrin unit 463 is intrinsically chiral, and hence two enantiomers (R and S) are present in equal abundance in solution. Accordingly, both (R)-464 and (S)-464 porphyrin boxes were formed in solution through homochiral self-sorting from their respective (R)-463 and (S)-463 isomers. The optical resolution of these chiral boxes were achieved with a chiral HPLC setup. Nanoscale porphyrin box, 464, has a cavity dimension of 22 × 14 × 10 Å. Extension of the pyridyl arms of the Zn(II) diporphyrin units led to the formation boxes with larger dimensions. These porphyrin boxes were demonstrated to serve as a well-defined light-harvesting models, exhibiting excitation energy hopping rates that depend on the vertical distance between the meso-meso linked Zn(II) diporphyrin subunits.520 The 90° dihedral angle between the diporphyrin and the 4-pyridyl groups played a key role toward the formation of the box-shaped assemblies.
Scheme 122.

Cavitands, which have half-bowl shaped structures, can be used as building blocks for supramolecular cages. Early ground-breaking works of Cram and coworkers demonstrated that covalent linking of two cavitand bowls in a “rim-to-rim” manner resulted in the generation of the closed-shell host molecules, named carcerands and hemicarcerands, that could accommodate complementary guest molecules or ions.521 More recently, attention has been turned toward the development of larger covalent cavitand cages, which can lead to higher order supramolecules that incorporate a greater number of cavitand hosts.522 Incorporation of donor groups at the apical positions on the rim of cavitands makes them interesting molecular building blocks for the self-assembly of coordination-driven metallacages and the majority of these cavitand-based assemblies involve tetrasubstituted cavitands connected by metal centers.234 In 1997, Dalcanale and coworkers reported the first metal directed, dimeric cavitand-based coordination cages (466 and 467) from a 1:2 reaction of tetracyano cavitands 465 with [M(dppp)(OTf)2] (M = Pd (4), Pt (52)) at room temperature (Scheme 123).523 The X-ray crystal structure of 466 showed the presence of an encapsulated triflate ion within the cage. A further comprehensive study on a series of similar cavitand-based coordination cages showed that the self-assembly process depends on a P-M-P angle close to 90° between the chelating ligand and the metal precursor (Pd and Pt), weakly coordinated counterions, and preorganization of the tetradentate cavitand ligand.524,525 Calorimetric measurements on the formation of these cages together with dynamic NMR (1H, 19F, PGSE, NOE, and EXSY) experiments helped determine the thermodynamics, interionic sructure, kinetic stability, and degree of anion encapsulation in these assemblies. The introduction of phenyl spacers to tetracyano cavitand 465 led to the formation of cavitand cages with larger internal cavities of nanoscopic dimensions.526 In this case, as monitored by 19F NMR spectroscopy, no permanent inclusion of triflate ion was observed due to the rapid exchange with the external counterions because of the larger dimensions of the lateral portals. Heteronuclear cavitand-based coordination cages were also assembled from a tetrasubstituted cavitand having one 4-pyridylethynyl and three 4-cyanophenyl donor groups at the apical positions with Pd(II) and Pt(II) as the metal centers.527
Scheme 123.

Using a different synthetic route Kobayashi et al. were able to introduce pyridyl, pyridylethynyl and cyanophenyl groups on the apical positions of the cavitands to assemble both dimeric homo- and hetero- cavitand cages via metal coordination based on thermodynamic and kinetic control.443,528 A 1:1:4 mixture of tetra(4-pyridyl)-cavitand (468), tetrakis(4-cyanophenyl)-cavitand (469) and Pd(dppp)(OTf)2 (4) in CDCl3 selectively self-assembled into thermodynamically stable hetero cavitand cage 470 (Scheme 124). Interestingly, a 1:1:4 mixture of tetrakis(4-cyanophenyl)-cavitand (469), tetrakis(4-pyridylethynyl)-cavitand and Pd(dppp)(OTf)2 (4) gave rise to only homo cavitands in CDCl3 indicating that a combination of factors such as coordination ability and steric demand of cavitand ligands can control the selective self-assembly of homo- or hetero-cavitand coordination cage. The fact tetra(4-pyridyl)-cavitand, 468, failed to form any discrete assembly as compared to tetrakis(4-pyridylethynyl)-cavitand, which formed homo cavitand assembly indicated that tetra(4-pyridyl)-cavitand, 468, is sterically more hinderd than tetrakis(4-pyridylethynyl)-cavitand. Similarly, a 1:1:4 mixture of cyanophenyl-cavitand, pyridylethynyl-cavitand and Pd(dppp)(OTf)2 self-assembled into the kinetically as well as thermodynamically most stable homo pyridylethynyl cavitand cage and most labile homo cyanophenyl cavitand cage in a 1:1 ratio as determined by 1H NMR spectroscopy. However, heating the reaction mixture at 50 °C for 2 h shifts the thermodynamic equilibrium towards hetero cavitand assembly.
Scheme 124.

Other examples of coordination-driven self-assembly of cavitand-based Pd(II) and Pt(II) dimeric cages were shown by Hong and coworkers using tetrasubstitued cavitands having fexible pyridyl donors at the apical positions of the cavitands.529,530 Formation of these cavitand cages was affected by stuble changes in the solvent system. While interclipped homo cavitand cages were readily formed in organic solvents (acetone, CH2Cl2 or CHCl3), intraclipped supramolecular bowls were formed in aqueous solutions. 531 Subsequest studies using nitromethane as a solvent showed the existence of a dynamic equilibrium between the interclipped homo cavitand cage and the intraclipped supramolecular bowl.532 1H NMR and CSI-mass spectrometric studies confirmed the coexistence of two ensembles in nitromethane. Variable temperature 1H NMR studies have shown that the conversion from the interclipped homo cavitand cage to the intraclipped bowl is entropically favored and enthalpically disfavored. Haino et al. introduced phenyl-bipyridine donors at the apical positions of cavitands to form stable dimeric cages via coordination of four Ag(I) metal centers.533 Each metal center coordinates with two bipyridine units in a tetrahedral fashion. The large internal cavity of the cavitand cage allowed for the selective encapsulation of large aromatics as well as aliphatic mono and diacetates as guest molecules. The detailed thermodynamic studies on guest encapsulation revealed that both CH–π interactions between the methyl groups on the guest termini and the aromatic cavity and desolvation of the inner cavity play a key role in guest encapsulation.534 Attachment of iminodiacetate moieties at the apical position of resorcinarene-based cavitands followed by treatment with CoCl2 or FeCl2 under basic conditions led to the formation of dimeric coordination cages consisting of four octahedrally-coordinated metal ions holding together two cavitand scaffolds.535,536 X-ray structural studies on both cages revealed that each metal ion is coordinated to two doubly deprotonated iminodiacetate moieties, resulting in an octaanionic complexes. These pH-dependent, water soluble cages were found to selectively encapsulate a wide variety of guests including aromatic molecules, alkanes, alkenes, haloalkanes and alcohols.537 For example, in the case of the Co(II) cavitand cage, the encapsulation preference for p-xylene was 26700 times more favorable as compared to that of o-xylene or m-xylene. Calorimetric studies have shown that guest molecules having optimum sizes, shapes, and polarities occupy the greatest percentage of cavity space and have the greatest number of favorable bonding interactions with the internal cavity of the cage.538
In an alternative design strategy, pendant donor groups were introduced as bridges between the phenolic OH groups of resocinarene scaffolds. Treatment of tetrasubstituted cavitand 471 with [M(dppp)(OTf)2] (M = Pd (4), Pt (52)) in a 1:2 molar ratio at room temperature led to the formatiom of coordination cages 472 and 473, respectively (Scheme 125).539,540 The formation of the cages was established by multinuclear NMR (1H and 31P) and mass (ESI and MALDI-TOF) spectrometry and by single crystal X-ray structural studies in case of cavtiand cage 472. Cage 472 has a large internal cavity of ca. 840 Å3, ideal for the encapsulation of guest molecules such as fullerenes. Cavitand cages with larger cavity dimensions were similarly assembled using a tetrasubstituted cavitand ligand having a phenyl spacer between the methylene bridge and pyridine donor unit.541 Host-guest studies using cage 472 as host gave a 1:1 host-guest complex (472 ⊃ Guest) with methano[60]fullerene derivatives as evidenced 1H NMR and ESI-mass spectrometric studies.540
Scheme 125.

Since the early studies by Shinkai and coworkers542 on metal mediated self-assmebly of calix[4]arene-based dimeric cages, a number of novel coordination cages using tetrasubstituted calix[4]arenes in conjunction with various metal ions have been reported. Hunter and coworkers assembled DABCO-induced calixarene-porphyrinn cages utilizing calixarenes having two and four pendant Zn(II)-porphyrin units.543 While the calix-bisporphyrin formed a 2:2 complex with DABCO, calix-tetraporphyrin formed four different complexes depending on the stoichiometry and concentration of DABCO. Careful titration experiments, coupled with a combination of UV-visible and 1H NMR spectroscopy, revealed that during the course of a titration, all four intramolecular and intermolecular calix-tetraporphyrin-DABCO sandwich complexes were populated, and only at a precise calix-tetraporphyrin: DABCO ratio of 2:4 was the dimeric cage assembly was observed.
Cotton et al. reported a dimeric calixarene coordination cage using a calixarene scaffold, calix[4]arene(CO2)4, functionalized with four carboxylic acid groups.544 Two tetrasubstituted calixarene subunits, calix[4]arene(CO2)4, were assembled via four [Rh2(DAniF)2]2+ (DAniF = N,N’-di-p-anisylformamidinate) dimetallic corner units to give dimeric cage {NEt4 ⊂ [cis-Rh2(DAniF)2(H2O)1/2(MeCN)1/2]4[calix[4]arene(CO2)4]2}BF4, with a tetraethylammonium cation trapped inside the cavity. 1H NMR, IR, mass spectrometry and X-ray crystallographic studies established the formation of the dimeric cagehaving NEt4+ as an entrapped cation. Figure 38 presents the single crystal X-ray structure of dimeric cage {NEt4 ⊂ [cis-Rh2(DAniF)2(H2O)1/2(MeCN)1/2]4[calix[4]arene(CO2)4]2}+.
Figure 38.

Single-crystal X-ray structure of {NEt4 ⊂ [cis-Rh2(DAniF)2(H2O)1/2(MeCN)1/2]4[calix[4]arene(CO2)4]2}+. The anisyl groups of all formamidinate ligands and the axial ligands (two H2O and two MeCN molecules which occupy one axial position of each of the four Rh24+ units) have been removed for clarity. Color code: green, Rh; red, O; blue, N; gray, C.
Cavitand-based trimeric and tetrtameric cages via coordination-driven self-assembly were first reported by Beer and coworkers. Tetrasubstitued dithiocarbamate-functionalized cavitands were assembled into trimeric and tetrameric cavitand cages with late transition metals (Ni, Pd, Cu, Au, Zn, and Cd).545,546 The coordination geometry of the transition metal used dictated the architecture that was adopted. X-ray crystallographic studies showed that square planar coordination geometries result in tetrameric cages, whereas square-based pyramidal metal geometries favor trimeric architectures. Thus, while Zn(II) and Cd(II) formed trimeric cages, with Cu(II), a tetrameric cage was observed. Ni(II), Pd(II) and Au(II) also formed tetranuclear cages. These cavitand cages possess large internal cavity dimensions suitable for encapsulation of C60 and C70 fullerenes. Host-guest studies using these cages as hosts have showed the formation of strong 1:1 host-guest complexes with fullerenes, with the Cd(II) trimeric cage being the best host system.547 Electrochemical, NMR and molecular modeling studies suggested that the electronic character of dithiocarbamate sulfur donor atom, which was affected by the nature of the metal ions, dictated the binding efficiency of the electron-deficient fullerene guests. A hexameric spheroidal cavitand cage based on cavitand-terpyridine subunits was assembled from a reaction between a tetrasubstituted terpyridyl-phenyl cavitand with [Zn(NCMe6)][TFPB]2 (TFPB = tetrakis-(3,5-bis-(trifluormethyl)-phenyl)-borate) containing lipophilic anions, which enhanced the solubility of the highly charged assembly in non-polar organic solvents.548 The hexameric assembly was characterized by ESI-mass spectrometry, elemental analysis, small angle X-ray scattering (SAXS), and diffusion NMR spectroscopy.
4.5. Applications and Uses of 3D Cages
The last decade has seen remarkable progress in the application and use of 3D cages, designed primarily through metal-ligand coordination8-19 and hydrogen bonding,549 in diverse fields such as host-guest chemistry and molecular recognition, reactivity modulation, catalysis and biology. 3D cages engineered via the coordination-driven self-assemly paradigms have a distinct advantage over conventional covalent container molecules due to the synthetic ease with which the coordination cages can be realized, the availability of a large library of building blocks, and the ability to exhibit selective guest encapsulations. The strong and highly directional nature of metal-ligand interactions results in the formation of stable and rigid coordination cages with well-defined internal cavities. The confined nanospaces within the enclosed cavities in these coordination cages create a unique environments and geometric constraints different from the bulk media. It is this localized microenvironment that allows access to hitherto inaccessible molecules, stabilization of short-lived intermediates and leads to unusual reactions.
4.5.1. Host-Guest Chemistry
Host-guest studies using discrete 3D supramolecular cages having well-defined cavities as hosts for the selective binding of guest molecules play a crucial role in understanding the underlying mechanism, thermodynamics and specific interactions involved in guest binding. The fundamental knowledge thus gained forms the basis for molecular recognition and sensor applications, allowing the chemistry of supramolecular ensembles to be expanded into the field of catalysis and drug delivery. A variety of 3D supramolecular cages have been utilized to encapsulate multiple numbers of guests driven both by specific interactions between the guest and host, as well as nonspecific, weak, supramolecular interactions such as Coulombic, van der Waals, hydrogen-bonding, ion-association forces, and steric interactions. Furthermore, a synergistic combination of charge, dielectric, shape, size and solvation also dictate the guest binding preferences in host molecules. Mechanistic studies have shown that in these systems the guest encapsulation is entropically driven and operates either through an associative mechanism, where the guest replaces the non-covalently bound molecules/solvents from the cavity in a concerted fashion, or through a dissociative mechanism, where guest egress yields an ‘empty’ assembly which is trapped by an incoming guest.550
4.5.1.1. Molecular Encapsulation and Recognition
In the context of host-guest studies, Raymond and coworkers utilized a series of anionic tetrahedral M4L6 (M = GaIII, AlIII, InIII, FeIII, TiIV and SnIV) cages wherein the metal atoms occupying the corners of a tetrahedron were bridged by six bis-catecholate based ligands having C2-symmetry.9,336 Although composed of achiral components, these tetrahedral assemblies are intrinsically chiral due to the strong mechanical coupling between the metal centers, resulting in homochiral assemblies of ΔΔΔΔ or ΛΛΛΛ-configuration. The tetrahedral assembly [Ga4(271)6]12−, derived from naphthalene-based bis-catecholate ligand 271, was used extensively as host due to its favorable properties, including its solubility in water, structural integrity during guest exchange, and non-interconvertible and resolvable enatiomers. The anionic tetrahedral assembly [Ga4(271)6]12− was soluble in various polar solvents and contains a hydrophobic cavity volume of 450 Å. The assembly could selectively encapsulate a variety of monocationic guests such as NMe4+, NEt4+, Me2Pr2N+, Pr4N+, and PEt4+ with remarkable discrimination to form host-guest complexes [Guest ⊂ Ga4(271)6]11−. The affinity of the guest molecules for the cavity depends on their size, hydrophobicity, enthalpy of desolvation, and charge.337 X-ray crystallographic analyses to probe the host-guest interactions in [M4L6]n− cages of different metals (M = GaIII, FeIII, TiIV) and different encapsulated guests (NEt4+, BnNMe3+, Cp2Co+, Cp*2Co+) showed that the nature of the metal ions have little impact on the assemblies. However, the encapsulated guests significantly distort the sizes and shapes of the interior cavities of the assemblies.551 Studies to understand the guest encapsulation into [Ga4(271)6]12− through van’t Hoff thermodynamic analysis showed that the encapsulation of cationic guests is an endothermic process with positive ΔH and ΔS values. Spontaneous, entropically driven processes are a consequence of the desolvation of bound waters from the solvation shell of the guest and release of water from the cavity host into the bulk solvent, thus paying an enthalpic penalty through entropic gains.337 Further studies using a series of guests ranging from simple alkylammonium cations to complex organometallic Iridium species have confirmed that guest encapsulation in polar protic solvents involved initial desolvation of the guest with concomitant entropy-enthalpy compensation effects due to the rearrangement of the hydrogen bond network in solution.552 A recent study using a combination of NMR, UV-vis, and isothermal titration calorimetry has shown remarkable differences between the internal and external binding of simple alkylammonium cations, NEt4+ and NMe4+ into [Ga4(271)6]12−.553 The encapsulation process was entropically driven while external binding was enthalpically driven and involved ion association554 and the loss of degrees of freedom as a consequence of the high charge and hydrophilic outer space of the cage. Mechanistic studies to elucidate the guest exchage process in these tetranuclear assemblies using [Ga4(271)6]12− as the host for a series of tetraalkylammonium, phosphonium, and organometallic cations have shown that, unlike hydrogen-bonded cages,549 guest exchange in these tetrahedral cages occurs by the expansion of the cage apertures without metal-ligand bond rupture. 1H NMR kinetic studies have shown that the rate of guest exchange is significantly retarded with sterically demanding Cp*2Co+ as the anionic guest compared to smaller guests such as NMe4+, NEt4+, and CoCp2+.555 This indicated that the steric repulsions that must be overcome by the guest in order to escape increases with guest size and rigidity. Furthermore, guest exchange in hosts composed of more inert metal-ligand bonds was as facile as guest exchange in more labile analogues, suggested that the exchange was independent of metal-ligand interations. Computational studies revealed that guest exchage takes place through one of the four C3 apertures at the center of each face of the tetrahedron. Further kinetic studies of the host-guest dynamics of [Ga4(271)6]12− suggested a non-dissociative mechanism.556 The fact that larger guests have much more negative ∆S‡ values than their smaller counterparts substantiate the squeezing of the guests through host apertures rather than bond rupture.
Nitschke and coworkers designed an anionic tetrahedral cage, 277, through a combination of dynamic covalent and metal ligand coordination bonds, which could reversibly disassemble and reassemble upon application of an external stimulus.349 A change in pH of the aqueous solution of 277 having cyclohexane as a guest induced a reversible opening of the cage (Scheme 126). Addition of p-toluenesulphonic acid made the cage to come apart and liberate the cyclohexane guest. Changing the solution pH to a basic regime by addition of sodium bicarbonate induced the reassembly of the cage. Tetrahedral cage 277 was effectively used to store the potent greenhouse gas SF6 in aqueous solution.557 The stored payload could be released in a controlled fashion through different physical and chemical stimuli such as temperature, pH and imine exchange reactions. However, in contrast to tetrahedral host cage [Ga4(271)6]12−, it failed to encapsulate NMe4+ and other small alkylamonium cations despite being anionic and having an ideal shape and size. Recently, the same group designed a M8L6 porphyrin-based cubic cage that can efficiently encapsulate large aromatic guest molecules such as coronene and fullerenes.383 Addition of excess of coronene to the M8L6 cube led to the formation host-guest complexes in which exactly three equivalents of this flat, aromatic guest were encapsulated per host, as confirmed by NMR spectroscopy, ESI-Mass spectrometry, and microanalysis. The cube also showed selective binding affinity for fullerenes. From a commercially available fullerene soot, the cube selectively encapsulated C70, C76, C78 and C84 to form 1:1 host-guest complexes.
Scheme 126.
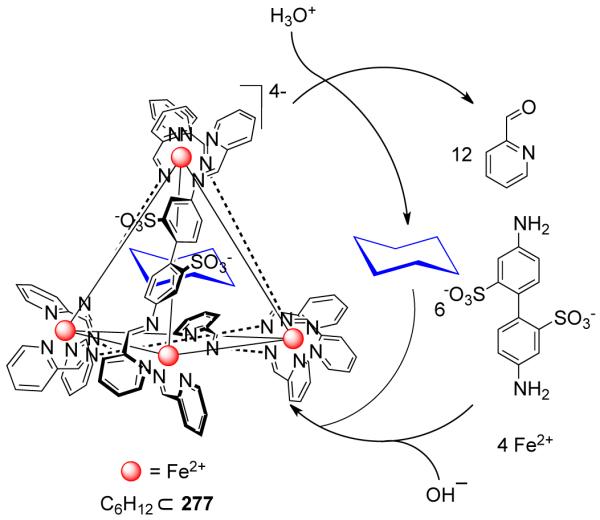
Raymond, Bergman and coworkers were also able to encapsulate neutral, hydrophobic guests including n-alkanes, cycloalkanes, arenes and enantiopure terpenoids into a tetrahedral host, [Ga4(271)6]12−, in aqueous solution.558,559 The driving force for the binding of these neutral guests was attributed to a hydrophobic effect. The molecular host assembly selectively encapsulated different substitutional isomers of disubstituted benzenes with ortho- substitution leading to the encapsulation of two guests, while meta- or para- substitution lead to the encapsulation of only one guest. Therrien et al. were able to encapsulate different planar aromatic molecules such as phenanthrene, pyrene, fluoranthene, benzo[e]pyrene, triphenylene, and coronene into the hydrophobic cavity of a series of cationic (arene)ruthenium-based trigonal prisms [Ru6(η6-p-cymene)6(285)2(OO ∩ OO)3]6+ 363-367 [OO ∩ OO = 2,5-dihydroxy-1,4-benzoquinonato (dhbq), 2,5-dichloro-3,6-dihydroxy-1,4-benzoquinonato (dchq), 5,8-dihydroxy-1,4-naphthoquinonato (dhnq), 9,10-dihydroxy-1,4-anthraquinonato (dhaq), or 6,11-dihydroxy-naphthacene-5,12-dionato(dhtq)] by using them as templating guests.428,429 Empty trigonal prismatic cages 363-367 have been used for host-guest studies and have shown that small aromatic molecules (phenanthrene and pyrene) show rapid inclusion into the hydrophobic cavities of the cages upon gradual addition of guests. However, molecules that are too large to exit the portal of the cages get permanently incarcerated, thus giving rise to stable carceplex systems.
Lindoy et al. have recently reported the selective encapsulation of a [FeIIICl4]− anion over a [FeIICl4]2− anion within the central cavity of a tetrahedral cage of the type [FeII4L6]8+ (L = 5,5′″-dimethyl-2,2′:5′,5″:2″,2′″-quaterpyridine).560 The treatment of quaterpyridine ligand L with FeCl2 · 5H2O in acetonitrile at reflux for 24 h followed by the addition of KPF6 led to the isolation of an [FeIIICl4]− encapsulated tetranuclear cage [FeII4L6 ⊃ FeIIICl4](PF6)7 as established from 1H NMR, microanalysis and X-ray crystallography. The crystal structure of [FeII4L6 ⊃ FeIIICl4](PF6)7 showed that [FeIIICl4]− occupies the central cavity and has a perfect tetrahedral symmetry with Cl–Fe–Cl bond angles of 109.5°. It is noteworthy that the tetrahedral cage [FeII4L6]8+ selectively extracts a [FeIIICl4]− anion from a mix of FeII and/or FeIII chloro species undoubtedly present in the solution. Such an observation is consistent with the earlier observation by Raymond and coworkers337 where they found that larger enthalpy of desolvation associated with doubly charged species significantly hampers their ecapsulation within a confined cavity, making it less favourable. The same system was earlier shown to encapsulate both BF4− and PF6− counteranions within the central cavity with marked selectivity for PF6− over BF4−.348
Fujita and coworkers have extensively investigated self-assembled trigonal prismatic cages constructed from a combination of end-capped Pd(II) ions as hinges, two tridentate planar panels and N-donor bidentate pillars for the intercacaltion of flat aromatic molecules (scheme 102).561 For example, the trigonal prism 344, pillered by pyrazine, could encapsulate one coronene molecule, stabilized through π–π interactions with the host assembly in aqueous media.409 The aromatic molecule, coronene, acts as templating agent in the selective formation of the trigonal prism architecture and can be removed after assembly through extraction with CHCl3. The efficient intercalation of planar guest molecules within the cavity of 344 was applied to control the equilibration between keto and enol forms of 1,3-bis(4-methoxyphenyl)propane-1,3-dione. As cofirmed by by 1H, 13C, and COSY NMR spectra, exclusive enolization was observed due to the selective encapsulation of the planar enol form over the nonplanar keto form. The cavity space of these trigonal prisms can be altered by changing the length of the pillars, thus facilitating encapsulation of more than one flat, aromatic molecule inside the cavity.410 The extended cavity afforded through the use of longer pillars, 2,2′,6,6′-tetramethyl-4,4′-bipyridine and 1,4-bis(2,6-dimethyl-4-pyridyl)benzene, results in cages 345 and 346, repectively, ideal for for stacking of two or three equivalents of an aromatic templated guest. Thus, two equivalents of eletron rich aromatic guests such as pyrene, coronene or porphine form acceptor–donor–donor–acceptor (A–D–D–A) quadruple stacks through charge transfer interactions with donor guests sandwiched between electron-poor triazine panels in cage 345. Recently, a dissymmetrical hetero A–D–A–A quadruple stack was also obtained with two different guest molecules, triphenylene and naphthalenediimide, using trigonal prism 345 as a host.562 Cage 346 showed quintuple (A–D–A–D–A) stacking with an intercalated triazine panel and two coronene or porphine molecules.410,411 The use of a less electron-rich, planar red flurorescent dye, tetraazaporphine, led to templated encapsulation of three equivalents of tetraazaporphines.563 The host-guest complex 346 ⊃ (azaporphine)3 was highly soluble in water as compared to free tetraazaporphine, which was sparingly soluble in aqueous solutions. Fluorescent studies with the 344 ⊃ (tetraazaporphine) system showed that tetraazaporphine retains its fluorescent properties upon encapsulation, in contrast to those in aqueous solution, which quench due to aggregation.564 The fact that tetraazaporphine retained its fluorescent properties indicated there is a minimal charge transfer interaction between the host and the guest molecule. Interestingly, reversible quenching of the fluorescence was achieved in aqueous solution through the addition of NEt3, leading to the abstraction of an acidic proton from the tetraazaporphine, forming a non-emissive 344 ⊃ (tetraazaporphine) species, which then can be treated with acid to reestablish emission. The selective encapsulation of tetraazaporphine/porphine stacks allowed for the generation of metal-containing homo- and hetero-arrays Cu(II)–M–Cu(II), (M = Cu(II) or Pd(II)).563 The inclusion complex 345 ⊃ [Cu(II)-azaporphine]3 showed a ferromagnetically coupled spin-spin interaction between the three Cu(II)-azaporphine units, as determined from ESR studies, which revealed a quartet state of the Cu(II) centers despite the absence of any covalent or non-covalet bonds between the stacked azaporphine nuclei. The hetero metal ion arrays also showed strong metal-metal interactions as indicated by broadening of their ESR spectra.
Fujita and coworkers have also demonstrated the formation of columns of aromatic molecules consisting of seven to nine discretely stacked aromatic molecules. The reaction of organic pillars of appropriate length, planar panel ligands, aromatic planar templating guests, and end capped Pd(II) metal hinges in a suitable ratio led to the quantitative formation of two-fold interpenetrated self-assembled trigonal prismatic cages.565 For example, combination of trans-1,2-bis(4-pyridyl)ethene (bpe) as a pillar, triazine panel 285, triphenylene as a aromatic templating guest and [(en)Pd(NO3)2] (1) in a 6:4:3:12 molar ratio in aqueous solution gave a two-fold interpenetrated self-assembled trigonal prismatic cage (474; Scheme 127) containing three triphenylene guest molecules sandwiched between four triazine panels. The 1H NMR spectrum, which showed two sets of signals in a 2:1 ratio for the intercalated triphenylene guest, and CSI-mass spectrometric studies, indicated the formation of a septet aromatic stacked motif, 474. X-ray crystallographic studies using a (tmen)Pd(II) hinge (tmen = tetramethylethylenediamine) and pyrene as guest confirmed the formation of septet (A–D–A–D–A–D–A) aromatic stacked architectures. Adjusting the pillar lengths facilitated the assembly of taller octuplet (A–D–A–D–D–A–D–A) and nonuplet (A–D–A–D–D–D–A–D–A) stacks. These discrete, interpenetrating, self-assembled hosts were utilized as “magnetic aligners” to orient a small organic molecule in a magnetic field.566 Septet columnar stacks with solubilizing side chains showed aggregation at higher concentrations in aqueous solution via intermolecular hydrophobic and π–π interactions.567
Scheme 127.

The entrapped molecules within the cavities of the trigonal prisms are unrestrained and have rotational freedom within the cages. This enabled encapsulated guests to dispay unique stacking and orientation preferences, depending upon the number of stacked guests. Three highly polarized aromatic guest molecules, pyrene-4,5-dione, formed a triple-layered host-guest complex when mixed with triazine panels, end-capped Pt(II) as hinges and trans-1,2-bis(4-pyridyl)ethylene (bpe) as pillars.568 The pyrene-4,5-dione molecules rotated freely within the cage, as indicated by the 1H NMR resonances which suggest highly symmetry. However, in the solid-state, three pyrene-4,5-dione molecules stack in a staggered fashion by 120° with respect to each other (Figure 39). By adopting such an orientation, the dione triple stack achives a negligible overall dipole moment. By systematically altering the pillar heights, the number of stacking molecules can be controlled without restricting the orientation of the guests.569 When the number of stacks was even, the dione molecules adopted a head to tail orientation to cancel the local and net dipole moments. In odd numbered stacks the net dipole is canceled at the expense of local dipole interactions.
Figure 39.
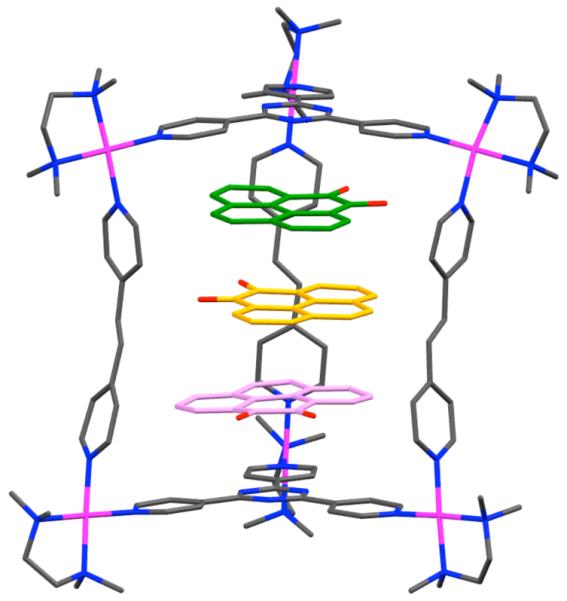
X-ray crystal structure of three pyrene-4,5-dione molecules stacked in a 120° twisted fashion in a trigonal prism cage. Solvents (H2O) and counter anions (NO3) are omitted for clarity. Color code: pink, Pd; red, O; blue, N; gray, C.
Using the same approach as described above, Fujita et al. engineered discrete, stacked arrays of planar transition metal complexes via d-d interactions between the metal centers for studying interesting chemical phenomena otherwise unobservable in infinite assemblies, in crystalline or liquid crystalline phases.561 For example, the suspension of two equivalents of planar M(acac)2 (M = Pt2+, Pd2+, or Cu2+, acac = acetylacetonate) in an aqueous solution of trigonal prismatic cage 345 led to the formation of host-guest complexes 345 ⊃ [M(acac)2]412. X-ray crystallographic analysis and UV-vis measurements of host-guest complex 345 ⊃ [Pt(acac)2]2 showed strong Pt(II)-Pt(II) d-d interactions. The inclusion complex 345 ⊃ [Cu(acac)2]2 showed spin-spin interactions as evidenced by a broadening of ESR signals at low temperature and Cu(II)-Cu(II) interactions involving nonbonding stacking of two dx2-y2 orbitals. Recently, 3-dimensional m × n stacked arrays of discrete, planar polynuclear metal complexes were assembled within the cavity of triginal prismatic cages. 570 Cyclic trinuclear Au(I) complexes [3 × 2] and [3 × 3] Au(I) clusters were encapsulated along the C3 axis of trigonal prismatic cages to form 3D columnar metal arrays. Therrien et al. have also achieved the encapsulation of M(acac)2 (M = Pt or Pd) as guests into the hydrophobic cavity of an (arene)ruthenium-based trigonal prism [Ru6(p-cymene)6(285)2(dhbq)3]6+ (367) using M(acac)2 as a templating guest.431 Studies of their cytotoxicity towards cancer cells revealed that these inclusion complexes 367 ⊃ [M(acac)2] were more active as compared to the free cage or Pd/Pt(acac)2 complexes.
The host-guest complexes containing square planar Ni(II) and Co(II) complexes as guest in trigonal prismatic hosts 344-346 showed interesting spin-crossover phenomena without changing the coordination number or geometry of the metal complexes.571 The encapsulation of the square planar low-spin NiII(acen) (acen = N,N”-ethylenebis(acetylacetoneiminato)) complex in cage 344 and 345 gave inclusion complexes 344 ⊃ [NiII(acen)] and 345 ⊃ [NiII(acen)]2 respectively. 1H NMR and CSI-mass spectrometry comfirmed their stoichiometry. Upon encapsulation, the spin state of NiII(acen) changed from low-spin to high-spin, as indicated by magnetic measurements. SQUID measurements showed paramagnetic behavior, confirming the high-spin state of the complex within the host assembly. X-ray structural studies on the 1:2 host-guest complex 345 ⊃ [NiII(acen)]2 revealed that the square planar geometries of the two NiII(acen) guests were perfectly preserved. The two planar molecules were twisted by 150° due to steric repulsion of the methyl groups of the acen moities. A spin-crossover phenomenon was also observed when CoII(tetetraazaporphine) was intercalated along with two coronenes into 346 to give inclusion complex 346 ⊃ [coronene · NiII(acen) · coronene]. The anisotropic and temparature dependent nature of the ESR signal is indicative of the involvement of a high-spin state. The observed spin-crossover of the Ni(II) and Co(II) complexes, upon entrapment in the host cages, was ascribed to interactions between the metal dz2 orbital and the π orbitals of aromatic cage panels or coguests.
To study the noncovalent spin-spin interaction between the host and the guest, Fujita et al. designed spin cages by replacing the typical triazine panels 285 with verdazyl panels 475.572,573 The trigonal prismatic-shaped cage assembled efficiently from a mixture of verdazyl panels, (en)Pd(II) hinges and pyrazine pillars.572 The encapsulation of triphenylene by this host showed spin exchange interactions between the two parallel panels with verdazyl cores, as evidenced by the broadening of the ESR signal at room temperature. At low temperature, the ESR spectrum showed characteristics of magnetic dipole interactions. Non-covalent host-guest spin-spin interactions were also observed when Cu(acac)2 was encapsulated as a guest. The ESR studies of the resulting inclusion complex clearly indicated that the Cu(II) center magnetically interacts with the radical panel of the cage. Similarly, a M6L4 tetrahedral spin cage (476) was assembled by using a 2,2′-bipyridine capped PdII complex with a verdazyl radical ligand, 475, in place of triazine panels (Scheme 128). The cage demonstrated host-guest spin-spin interactions.573 The four spin centers of cage 476 showed intramolecular magnetic interactions indicated by the broadening of the ESR signal as compared with free ligand 475, which showed hyperfine splitting. Upon the encapsulation of a stable nitrosyl radical, 477, the 1:1 host-guest inclusion complex 476 ⊃ 477 showed antiferromagnetic spin-spin host-guest interactions. Similar treatment of 478 with spin cage 476 formed a 1:2 host-guest complex 476 ⊃ (478)2 which showed enhanced magnetic interactions through host-guest and guest-guest interactions.
Scheme 128.
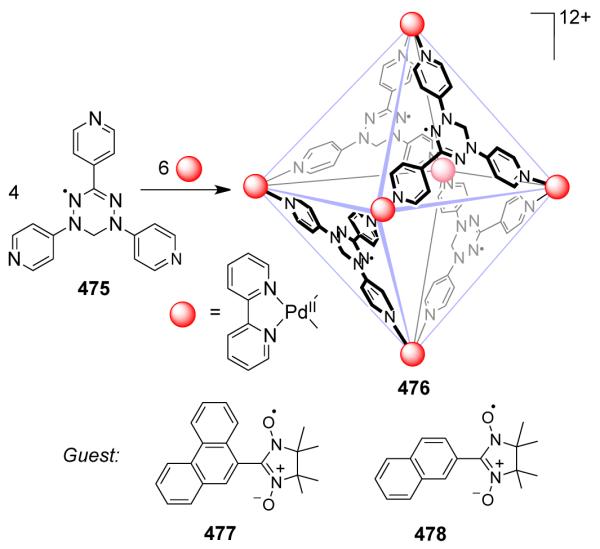
Fujita and coworkers also demonstrated that the confined cavities of the M6L4 coordination cages could induce through-space spin-spin interactions between encapsulated stable organic radicals in solution. When stable organic nitrosyl radical 478 was encapsulated in self-assembled cage 288, a 1:2 host-guest complex 288 ⊃ (478)2 was formed.574 Due to the restriction imposed by the confined cavity of the cage, two organic radicals were forced to be close to each other within the cavity. Such close encapsulation induced through-space spin-spin interactions between the two radicals, as observed by ESR spectroscopy. The ESR spectrum of 288 ⊃ (478)2, both in solution and the solid-state, showed a broadened signal assignable to a dimeric aggregate of radical 478 in a triplet state, stemming from an intermolecular spin-spin interaction. The quantity of the triplet state generated by encapsulation also increases with an increase in the affinity of the radical to the cage.575 Since these organic radicals are held together only through weak hydrophobic forces, the through-space spin-spin intercations are sensitive to external stimuli such as temperature and pH. The use of an organic radical having an amine group (479), which can be protonated, reduced the affinity of the radical to the cage due to cationic repulsions (Scheme 129).576 As a result, the encapsulated organic radicals were released from the 288 ⊃ (479)2 1:2 inclusion complex into an acidic solution, as indicated by the disappearance of the signal for the triplet state in the ESR spectrum of the host-guest complex in solution. Upon treatment with K2CO3, the triplet signal reappeared, signaling the reencapsulation of the radicals into the host.
Scheme 129.
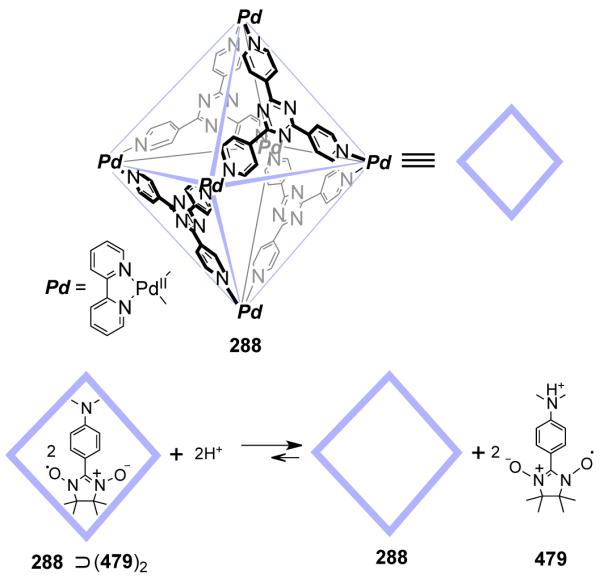
Enantioselective recognition and resolution of chiral guest molecules was also exploited using self-assembled coordination cages having well-defined cavities. Raymond et al. utilized the homochiral tetrahedral assembly [Ga4(271)6]12− as a host for the chiral recognition of half-sandwich organometallic species of a general formula [Cp*Ru(η4-diene)(H2O)]Cl (Cp* = C5(CH3)5).577. The combination of an aqueous solution of racemic [Ga4(271)6]12− with an ethereal layer of racemic Cp*Ru(η4-diene)Cl led to the formation of a 1:1 host-guest complex, [Ga4(271)6] ⊃ Cp*Ru(η4-diene)(H2O)]11−, with two diastereomeric pairs of enantiomeric host-guest complexes (Δ/R, Λ/S and Δ/S, Λ/R). When Cp*Ru(2-ethylbutadiene)Cl was used as the encapsulated guest, the ensemble showed a diastereomeric excess of 70%. Studying the scope of enantioselective recognition with a series of 1- and 2-substituted diene containing complexes revealed that the encapsulation process was controlled not only by the guest’s size, but also by its shape. Using enatiopure host cage ΛΛΛΛ-[Ga4(271)6]12− with Cp*Ru(2-ethylbutadiene)Cl as a guest in a 1:2 ratio led to the quantitative formation of ΛΛΛΛ-[Ga4(271)6] ⊃ Cp*Ru(η4-diene)(H2O)]11−.
Fujita and coworkers utilized a bowl-shaped host 480,27 assembled from the reaction of planar triazine panels tris-(3-pyridyl)triazine and (en)PdII hinges in a 6:3 ratio, to study the preferential encapsulation of a homochiral pair of 1,1′-binaphthyl-2,2′-diol (BINOL). 578 Treatment of an aqueous solution of the bowl-shaped host, 480 with an organic solution of (S)-BINOL at a 50% enatiomeric excess led to the formation of a 1:2 host-guest complex with one equivalent each of (S)-BINOL and (R)-BINOL selectively encapsulated within the cavity of 480 (Scheme 130). The efficient heterorecognition of enantiomers afforded a significant enantiomeric enrichment of BINOL in the organic phase from 50% to 81%.
Scheme 130.

4.5.1.2. Stabilization of Reactive Molecules and Intermediates
Ever since the early pioneering work of Cram and coworkers, where a covalently constructed carcerand derived from calix[4]arene was able to stabilize cyclobutadinene,579 there has been a sustained interest in designing molecular containers that can incarcerate unstable molecules and reactive intermediates within their cavities.521,522 The unique microenvironment that exists within a cavity protects otherwise unrealizable molecules and intermediates from the bulk phase, making them amenable to detection and characterization. Raymod and coworkers have utilized a homochiral tetrahedral assembly, [Ga4(271)6]12−, to stabilize phosphonium,580,581 diazonium,582 tropylium,582 iminium 583 ions and organometallic reactive intermediates. 584 Reactive phosphonium ions [R1MeC(OH)PR3]+ generated by the reaction of phosphines and ketones in acidic medium can be stabilized by encapsulation as guests within the tetrahedral assembly, [Ga4(271)6]12−, in aqueous solution.580 Although these phosphonium ions have a transient existence in an aqueous medium, entrapment within the hydrophobic cavity of the cage increases their lifetimes considerably. Encapsulation of a range of phosphonium ions showed that the sizes and shapes of the guest cations play a vital role in the stability of the inclusion complexes. The stability of the phosphonium cations also increases with decreasing pH of the solutions. Similarly, tetrahedral assembly [Ga4(271)6]12− effectively encapsulated diazonium and tropylium cations within its hydrophobic cavity.582 The combination of a tetrafluoroborate diazonium salt and [Ga4(271)6]12− in aqueous solution resulted in the formation of a 1:1 host-guest complex. Subsequent addition of 2,4-pentanedione showed no reaction, indicating stabilization of the diazonium ion within the cage.
Iminium ions generated in-situ from amines and ketones have a transient stability in aqueous solutions at neutral or basic pH.583 However, stabilization of such ions in water can be achieved via encapsulation within the hydrophobic cavity of the tetrahedral assembly, [Ga4(271)6]12−. For example, reaction of pyrrolidine with a wide variety of ketones in the presence of tetrahedral cage [Ga4(271)6]12− in aqueous solutions led to the formation of an encapsulated iminium ion complex (Scheme 131). Once encapsulated, these iminium ions were stable for months at room temperature. In the absence of host [Ga4(271)6]12−, no iminium ion could be observed by 1H NMR spectroscopy. The binding efficiency of the iminium cations depended on theie size, shape, charge and hydrophobicity. Encapsulation of proton-bound amine homodimers in self-assembled host cage [Ga4(271)6]12− was achieved in water. These homodimers are otherwise not obersevable in aqueous medium.585 A wide range of cyclic amines showed a proton-bound homodimer for N-alkyl aziridines, azetidines, pyrrolidines, and piperidines. Cyclic amines were chosen as guests due to their reduced degrees of rotational freedom, which attenuates the entropic price paid for encapsulation of multiple guests. Theoretical calculations suggest that formation of proton-boung homodimers is highly enthalpically favorable.
Scheme 131.
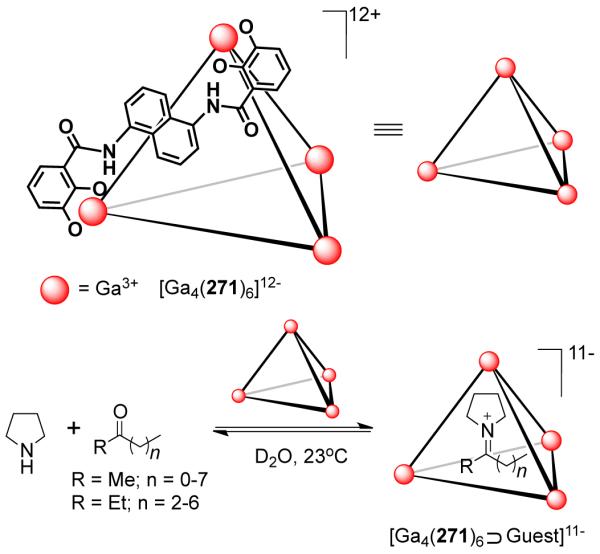
Expanding on the incarceration of water-sensitive guests, Raymond, Bergman and coworkers investigated the stabilization of reactive organometallic intermediates inside the self-assembled host [Ga4(271)6]12−.584 [CpRuCl(cod)] (cod = 1,5-cyclooctadiene), a catalyst for C-C bond formation that undergoes halide dissociation in polar media to form a cationic solvated species [CpRu(cod)(H2O)]+. Surprisingly, suspending [CpRuCl(cod)] in an aqueous solution containing tetrahedral host [Ga4(271)6]12−, resulted in the formation of an unusual 1:1 host-guest complex [Ga4(271)6 ⊃ CpRu(1,3,5-octatriene)]11− containing a highly unstable 16-electron ruthenium complex [CpRu(1,3,5-octatriene)]+ within the cavity. The encapsulation provided enough binding energy to prevent the decomposition of the reactive species for several weeks. The incarcerated complex, [CpRu(1,3,5-octatriene)]+, still retained its reactivity and reacted stoichiometrically with CO to form [CpRu(cod)(CO)]+. The addition of CO was belived to happen inside the cavity of the cage.
Fujita and coworkers were able to generate and characterize an elusive organometallic species within the confined cavity of self-asembled coordination cage 287.586 Treatment of an aqueous solution of coordination cage 287 with an excess amount of stable organometallic complex Cp’Mn(CO)3 (Cp’ = methylcyclopentadienyl) gave a 1:4 host guest complex 287 ⊃ [Cp’Mn(CO)3]4. Photoirradiation of crystals of 287 ⊃ [Cp’Mn(CO)3]4 at 100K generated a 16-electron coordinatively unsaturated species Cp’Mn(CO)2 in-situ due to the dissociation of a single CO from one of the four guest molecules. The unsaturated manganese species Cp’Mn(CO)2 retains its pyramidal geometry within the cage. Earlier, the same group was able to isolate and characterize reactive intermediates of a polycondensation reaction of trialkoxysilanes using tetrahedral coordination cage 286.587,588 Unstable cyclic silanol trimers formed during the polycondensation reaction were stabilized within the cavity of the coordination cage. The stability of the reactive species was attributed to its large size and rigidity, making it unable to escape the cavity of the cage. Modulating the cavity space through the use of a variety of coordination cages led to the observation of other short-lived species generated in the polycondensation of trialkoxysilanes.
In aqueous medium, short nucleotide fragments such as mono- and dinucleotides do not form stable hydrogen-bonded base pairs at molecularities less than four, due to competition from water. However, within the hydrophobic confines of biomolecules, even very short nucleotides can selectively form base pairs during translation, replication and transcription. Fujita et al. have recently achieved the selective formation of stable mono- and dinucleotides in water using self-assembled trigonal prismatic cages.589,590 In the presence of coordination cage 344, 5′-adenosine monophosphate (AMP) and 5′-uridine monophosphate (UMP) formed a 1:2 host-guest complex 344 ⊃ (AMP · UMP) in aqueous solution (Scheme 132), as indicated by an upfield shift of the nucleobase proton resonances in the 1H NMR spectrum. The encapsulation of AMP–UMP base pairs was driven by a combination of hydrophobic, electrostatic and π–π interactions.
Scheme 132.

X-ray crystal structural studies of the encapsulated base pair revealed that it is not linked through the usual Watson-Crick hydrogen bonding observed in duplexes, but rather through an anti-Hoogsteen hydrogen-bonding pattern usually observed in triplexes. 15N NMR spectroscopic studies also supported the formation of anti-Hoogsteen nucleobase pairs within the cavity of the cage. Similarly, dinucleoside monophosphate duplexes were formed when an aqueous solution of thymidylyl-(3′-5′)-2′-deoxyadenosine (TA) was stirred with the 4,4′-bypyridine pillared cage having an extended cavity to form a 1:2 host-guest complex. NMR (1D and NOESY) spectroscopic studies suggested the formation of a discrete base paired (TA)2 dimer through anti-Hoogsteen hydrogen-bonding within the cavity. The X-ray crystal structure of the host-guest complex, however, showed an infinite network, whereas dinucleotides formed hydrogen-bonded chains threaded through adjacent host cages. The authors suggested that the two structures (disctere and polymeric) were in equilibrium with discrete species being dominant in solution, while the polymeric structure was trapped in the solid-state.
Nitschke et al. utilized a self-assembled, water-soluble tetrahedral cage (277) to encapsulate highly porophoric white phosphorus P4, thus making it inert and air-stable.591 Uptake of P4 within the hydrophobic cavity of 277 of the anionic cage was observed in aqueous solution. 1H NMR and X-ray crystallographic studies indicated the incorporation of P4 through the formation of a 1:1 host-guest complex 277 ⊃ P4 (Scheme 133). The host-guest complex remained stable upon exposure to air for four months. Despite oxygen molecules’ smaller size than P4 molecules, suggesting that they could gain access to the interior of the cage, O2 did not react with P4 confined within the cavity. The reason for such an unusual behavior was attributed to the unavailability space to form the reaction intermediates inside the cavity during the steps of phosphorus oxidation. The incarcerated P4 molecule could be easily released by layering an aqueous solution of host-guest complex 277 ⊃ P4 with benzene, causing benzene molecules to replace P4 as the guests.
Scheme 133.

4.5.2. Cavity Controlled Reactions
Self-assembled coordination cages have been successfully applied as nanovassels for carrying out various stoichiometric and catalytic reactions. A wide variety of reactions such as thermal, photochemical and encapsulated transition metal catalysed reactions have been shown to occur within the nanospaces of coordination cages. The unique microenvironment created within the nanometer-sized cavities of these assemblies, due to their specific sizes, shapes and hydrophobicities, can force substrates to adopt unusual conformations often unobservable in bulk phases through π–stacking and hydrophobic effects. Furthermore, the stabilization of transition states and reactive intermediates within the cavities can also change the course of the reaction by manipulating the reaction energetics. Protection of these species from the bulk phase, similar to enzyme pockets, blocks the bulk reaction pathway, leading to new, unsual regio-, chemo- and stereoselective products.
4.5.2.1. Cavity Controlled Stoichiometric Reactions
Fujita and coworkers utilized steric constriction within the nanospaces of coordination cages to achieve unusual regio- and stereoselective Diels-Alder reactions between anthracenes and maleimides.592 For example, suspending a mixture of 9-hydroxymethylanthracene 481 and N-cyclohexylmaleimide 482 in an aqueous solution of a stoichiometric amount of coordination cage 287 resulted in the formation of 1:2 host-guest complex 287 ⊃ (481 · 482). Upon heating the reaction mixture to 80 °C, unusual stereo- and regioselective formation of a [2 + 2] cycloaddition product, 483, was observed in more than 98% yield (Scheme 134). NMR and X-ray crystallographic studies of 287 ⊃ 482 revealed the formation of a syn-1,4-Diels-Alder adduct via a reaction at the terminal anthracene ring. The observation of such an unusual Diels-Alder reaction was due to coordination cage 287 enforcing the two substrates to adopt specific orientations, thus preventing interactions at the expected 9,10 position of the anthracene.
Scheme 134.

Similarly, otherwise inert arenes also underwent unusual [2 + 2] and [2 + 4] thermal cycloaddition reactions within the cavities of analogous palladium and platinum tetrahedral cages. 593 Highly stable aromatic molecules, such as triphenylene, perylene, substituted naphthalenes, aceanthrylene and 1H-cyclopenta[l]phenanthrene afforded the corresponding [2 + 2] and [2 + 4] endo Diels-Alder adducts with N-cyclohexylmaleimide (482) in good yields.594,595 The steric demand of the N-cyclohexyl group on 482 is important as it directs the preorganization of the substrates within the hydrophobic cavity thus promoting energetically unfavorable stereo- and regioselective reactions.
Fujita and coworkers investigated cavity-directed, highly stereoselective photodimerization of olefins mediated by self-assembled coordination cages. For example, when excess amounts of the bulky olefin, acenapthylene (484) were suspended in an aqueous solution of coordination cage 286, formation of a 1:2 host-guest complex 286 ⊃ (484)2 was observed, as indicated by the upfield shift of the 1H NMR signals corresponding to the encapsulated guest molecules (Scheme 135a).596 Photoirradiation of this host-guest complex led to the exclusive formation of the syn dimer 485 in quantitative yield. Similarly, encapsulation of two molecules of 1-methylacenapthylene followed by irradiation gave exclusively the head-to-tail syn dimer. Exclusive formation of syn dimer 485 was also observed, even in the presence of sensitizers such as xanthine dyes, although the sensitized reactions generally go through triplet states.597 X-ray structural analyses of 286 ⊃ (484)2 and 286 ⊃ 485 both before and after [2 + 2] photodimerization allowed in-situ crystallographic tracking of the photochemical transformation.598 Before photoirradiation, the two encapsulated substrate molecules interacted with the cage through π–interactions and were disordered over three positions at 240K, providing a fluid-like environment even in the crystalline state. This allowed the guest molecule to thermally tumble within the host cavity. Nevertheless, quantitative formation of syn dimer 485 was observed upon irradiation at 240K, as revealed by the single crystal X-ray structure of 287 ⊃ 485. Bowl-shaped host 480 also showed the exclusive formation of the syn dimer in more than 98% yield in the [2 + 2] photodimerization of napthoquinones.596 X-ray structural studies of the dimer-encapsulated cage revealed a syn conformation for the dimer, which was held through π–π and CH–π interactions. Ramamurthy et al., working with coordination cage 286, also investigated the [2 + 2] photodimerization of trans-cinnamic acid esters in water.599 Irradiation of a series of trans-cinnamic acid methyl esters encapsulated host-guest complexes showed mostly the formation of syn head-to-head dimers in a range of 21-63% yields along with the corresponding monomeric isomerization products. In two cases, however, anti head-to-tail dimers were also observed, indicating that substituents can also play a role in the packing of the substrates and thus the photochemical outcome of the reaction. A series of derivatives of coumarin also led to the exclusive formation of syn head-to-head dimers with coordination cage 286 as the nanovassel for the [2 + 2] photodimerization reaction.600 Selective [2 + 2] cross photodimerization was also investigated using self-assembled coordination cage 286 as a host in aqueous medium.601 Such reactions are challenging, as the host cage must selectively and pairwise recognize two different olefins in a proper orientation for the cross photodimerization to occur. Suspension of acenaphthylene 484 and 5-ethoxynaphthoquinone 486 in an aqueous solution at 80°C in the presence of coordination cage host 286 led to the formation a 1:2 host guest complex, 287 ⊃ (484 · 486), as determined by the upfield shift of the signals due to the encapsulated guests in the 1H NMR spectra. Upon photoirradiation with a mercury lamp, a hetero syn dimer 487 was obtained in 92% yield (Scheme 135b). However, olefins having smaller or no substituents gave both hetero and homodimers. The steric bulk of the substrate is essential for preorganization of the substrate within the host cavity, which prevents the formation of other possible products. Thus, the use of sterically demanding N-cyclohexylmaleimide and its derivatives afforded a [2 + 2] photodimerization with planar arenes such as acenapthylene, pyrene, phenanthrene and fluoranthene.594,601 Chiral induction of an assymertic [2 + 2] cross-photoaddition was also observed with chiral cages analogous to 286 by replacing the ethylenediamine end-cap on each Pd center with chiral diamines.602 The [2 + 2] cross-photoaddition reaction of N-cyclohexylmaleimide and fluoranthene led to the formation of a [2 + 2] product with an enantiomeric excess of up to 50%. The remote chiral auxiliaries induce chiral deformation of the central cavity through deformation of the triazine panels.
Scheme 135.

The confined cavity of a coordination cage was also utilized to suppress the photocleavage of α-diketones to give cyclization products through kinetically unfavorable pathways.603 Typically, α-diketones undergo homolytic cleavage followed by degradation upon photoirradiation. However, encapsulation of benzil, a α-diketone, within the cavity of coordination cage 287 gave a 1:2 host-guest complex which, upon irradiation, gave unusual rearranged products with an overall yield of 52%, indicating that homolytic clevage is completely suppressed and a kinetically unfavorable pathway becomes dominant. The photomediated 1,4-radical addition of o-quinones to a bulky toluene derivative was also observed within the cavity of self-assembled host 286.604 The photoexcited quinone abstracts a proton from the neighbouring methyl group of the toluene derivative to generate a benzylic radical and semiquinone radicals, which subsequently recombine to selectively produce the 1,4-adduct in 70% yield.
Fujita and coworkers have also the achieved oxidation of alkanes through photochemical excitations of self-assembled coordination cage 287.605 It is important to note here that the host cage actively participates by influencing the reaction pathway rather than remaining an inert and passive component. To explore the photoresponsible nature of the cage 287, an inert alkane guest, adamantane was encapsulated to give a 1:4 host-guest complex 287 ⊃ (adamantane)4. Upon irradiation under aerobic conditions, regioselective oxidation of adamantane afforded 1-adamantylhydroperoxide and 1-adamantanol in 24% yield (or 96%, assuming that only one guest is oxidized per cavity). Detailed spectroscopic, electrochemical and theoretical studies revealed that host-guest complex 287 ⊃ (adamantane)4 was well suited for photoinduced electron transfer. 606 The electron poor triazine panels accept an electron from the encapsulated adamantane to give rise to an adamantyl radical. Subsequent quenching of the adamantyl radical by oxygen or water resulted in the formation of oxidation products 1-adamantylhydroperoxide and 1-adamantanol. Tight host-guest contact brings triazine panels close to the adamantane molecules, thus promoting unusual photoinduced electron transfer.
Encapsulation of half-sandwich iridium complexes within the cavity of supramolecular host cage [Ga4(271)6]12− showed selective C–H bond activation of aldehydes.607 The addition of monocationic iridium complex [Cp*(PMe3)Ir(Me)(C2H4)4]+ and tetrahedral cage [Ga4(271)6]12− in aqueous medium led to quantitative encapsulation resulting in a 1:1 host guest complex 272 ⊃ [Cp*(PMe3)Ir(Me)(C2H4)], as indicated by the upfield NMR shifts of the resonances of guest molecule. Heating an aqueous solution of host-guest complex [Ga4(271)6]12− ⊃ [Cp*(PMe3)Ir(Me)(C2H4)] and acetaldehyde at 75 °C led to the formation of a new encapsulated product, [Cp*(PMe3)Ir(Me)(CO)], which resulted from the C–H bond activation of the acetaldehyde. Substrate scope and mechanistic studies showed that size and shape selectivities play vital roles in the encapsulated C–H bond activation reactions.608 The fact that the dissociation of the guest was slower than the rate of C–H bond activation indicated that the reactions occur within the cavity of the host-guest assembly. Kinetic studies revealed that the iridium guest dissociation proceeds through a strongly bound ion pair intermediate through a two step process in which the guest exits the cavity but remains bound to the outside of the host cage before fully dissociating from the host.
4.5.2.2. Cavity Controlled Catalytic Reactions
Self-assembled coordination cages can be efficient reaction nanovassels for various catalytic reactions. The unique microenvironments of the hydrophobic cavities coupled with the size and shape-defined nanospaces drive reactions often unobservable in bulk solution. However, in many such cavity-mediated reactions, strong complexation and/or the large size of the reaction product traps it within the cavity, resulting in a low catalytic turnover. Judicious choice of the host and substrate can prevent such product inhibition, leading to truly catalytic systems. Raymond, Bergman and coworkers demonstrated that the problem of product inhibition might be circumvented through prudent choice of compatible hosts and guests. The anionic self-assembled cage [Ga4(271)6]12− was shown to act as a true catalytic host in the unimolecular aza-Cope rearrangement of enamonium cations in aqueous medium.609 Enamonium cation 488, the reaction substrate for the cationic aza-Cope rearrangement reaction, are analogous to the known337 strong binding ammonium cationic guest NMe2Pr2+ and thus effectively bind within the cavity of anionic self-assembled cage [Ga4(271)6]12− to form 1:1 inclusion complex [Ga4(271)6 ⊃ 488]11− (Scheme 136). Subsequest [3,3] sigmatropic rearrangement led to the formation of iminium cation 489, which escape from the interior of the cavity to an exterior binding site where they undergo hydrolysis to give the corresponding γ,δ-unsaturated aldehyde (490) with turn over numbers (TON) of ca. 8. Since the neutral molecules are bound weakly within the cavity of the host assembly [Ga4(271)6]12−, neutral aldehydes were rapidly displaced from the cavity by the cationic substrate molecules, preventing product inhibition.
Scheme 136.

When enamonium ions were substituted unsymmetrically (R2 ≠ R3), the rearrangement generated a chiral center, resulting in the formation of chiral product aldehydes. A series of prochiral enamonium tosylates, upon treatment with catalytic amounts of (NMe4)12[ΔΔΔΔ-Ga4(271)6], resulted in the asymmetric induction of a catalyzed aza-Cope rearrangement.610 The enantioselectivities achieved for the products were highly dependent on the substrate size and charge. Detailed substrate scope and mechanistic studies revealed that upon encapsulation, the confined cavity of the host assembly enforces the substrates to adopt reactive conformations, as evidenced by kinetic, activation parameter and quantitative NOE studies, thus accelerating the rate of rearrangement by factors up to 3 orders of magnitude.611 Extention of the study to the rearrangement of less reactive propargyl enamonium cationic substrates showed rate accelerations by factors of up to 184 as compared with the background reaction rate.612 Detemination of the kinetic parameters revealed that the catalytic reactions follow the Michaelis-Menten model of enzyme kinetics. Activation parameters indicated the reasons for the rate acceleration were due to a more positive value of ΔS‡.
Raymond, Bergman and coworkers recently achieved a reaction rate accleration by a factor of 106, comparable in size to those observed in the domain of enzymatic catalysis. The tetrahedral self-assembled cage [Ga4(271)6]12− was shown to acclerate the Nazarov cyclization of pentendiols.613 Suspension of 10 equiv. of 3,4,5-trimethylhepta-2,5-dien-4-ol in an aqueous solution of coordination cage [Ga4(271)6]12− at 50 °C resulted in the quantitative formation of the Nazarov cyclized product, pentamethylcyclopentadiene. The pseudo first order reaction rate was, however, impeded due the competitive binding of the cyclized product within the host cavity, leading to product inhibition. This problem was avoided by using maleimide as a trapping agent for the cyclized product. The resulting Diels-Alder adducts have less affinity for the host as compared to the substrate, accordingly restoring the pseudo first order kinetic behavior. The rate-determining step for the reation was found to be the rearrangement of the encapsulated substrate to adopt a bent conformation prior to or at the transition state. Thus, the constricted environment within the cavity of [Ga4(271)6]12− facililates a million-fold rate enhancement due to the combination of an increase in the basicity of the alcohol functionality upon encapsulation, preorganization of the bound substrate, and stabilization of the transition state of the electrocyclic reaction.
In previous studies, the same group demonstrated that tetrahedral cage [Ga4(271)6]12− could mediate acid-catalyzed reactions within its cavity in basic solutions. Due to the high binding affinity of cationic guest molecules over neutral species, addition of neutral guests leads to thermodynamically driven encapsulation and stabilization of the corresponding protonated species within the cavity of the anionic cage. Since hydrolysis of orthoformates goes through a protonated reactive intermediate, orthoformates HC(OR)3 undergo rapid hydrolysis to the corresponding formates in the presence of catalytic amount of coordination cage [Ga4(271)6]12− in a basic aqueous solution (pH 11).614 The catalytic cycle for the process is shown in Scheme 137. Initially, orthoformate gets encapsulated within the hydrophobic cavity of the cage to give a 1:1 inclusion complex, [Ga4(271)6]12− ⊃ [HC(OR)3]. The protonated ester HC(O)OR, generated through protonation from water, is subsequently hydrolysed by OH− to the fomate ion. Mechanistic studies revealed that electrostatic and hydrophobic effects drive the initial encapsulation of the neutral guest. Protonation of the substrate within the cavity through proton transfer from water is the rate-limiting step. Determination of the kinetic and activation parameters revealed that the hydrolysis follows Michaelis-Menten kinetics in parallel to enzymatic pathways. The observed entropy of activation and solvent isotope effect suggested that the initial step of the hydrolysis proceeded through an A-SE2 mechanism in contrast to an A-1 mechanism for orthoformate hydrolysis in bulk solution. A kinetic analysis of the rate constants by comparing with the uncatalyzed reaction showed a 103-fold rate acceleration. For tri-n-orthoformate, a 3900-fold accleration was observed. 615 Similarly, the acid-catalyzed hydrolysis of acetals was also investigated using tetrahedral self-assembled coordination cage [Ga4(271)6]12−. Acetals are stable in neutral and basic media and require Brønsted or Lewis acid catalysts for their hydrolysis. This makes tetrahedral cage [Ga4(271)6]12− an ideal catalytic host for the reaction. For example, addition of 2,2-dimethoxypropane as a substrate in a basic aqueous solution containing a catalytic amount of [Ga4(271)6]12− at pH 10 showed the expected hydrolyzed products.616,617 Mechanistic studies revealed that the mechanism of hydrolysis takes place inside the assembly and proceeds through an A-2 mechanism, in contrast to the A-1 mechanism operating in the uncatalyzed reaction. The rate-limiting step of the reaction involves negative entropy of activation and an inverse solvent isotope effect.
Scheme 137.

As discussed above, the Diels-Alder cycloaddtion reaction between substituted anthracenes with N-cyclohexylmaleimide in aqueous medium requires use of a stoichiometric amount of coordination cage to mediate the reaction, due to the strong complexation of the product to the host molecule.592 However, the same reaction proceeded catalytically upon use of bowl-shaped cage 480 as the host. For example, in the presence of a catalytic amount of coordination cage 480 (10 mol%), 9-hydroxymethylanthracene (481) with N-phenylmaleimide efficiently produced the Diels-Alder adduct with good TON (ca. 10).592 In this reaction, however, a conventional 9,10-adduct was formed in contrast to the unusual 1,4-adduct observed when coordination cage 287 was used. The open hydrophobic cavity of bowl 480 ensured facile encapsulation of the substrate molecules through hydrophobic, π–stacking and/or charge transfer interactions. The bent shape of the anthracene moiety in the resulting adducts hampers π–stacking within the host cage, thus reducing the affinity to the host as compared to the substrates. This enables substrate molecules to displace adducts and maintain the catalytic cycle.
Self-assembled coordination cages having encapsulated active organometallic catalysts have led to new and hithero unobserved reactions. Encapsulation of a catalyst within the cavity of self-assembled host cage may alter its activity through changes in its structure, giving rise to different properties. The rate-determining step in a catalytic cycle of a known organometallic catalyst may also be altered due to encapsulation, leading to new selectivities. Raymond, Bergman and coworkers utilized self-assembled tetrahedral cage [Ga4(271)6]12− to encapsulate an active rhodium complex to investigate the catalytic isomerization of allylic alcohols.618 A combination of the tetrahedral cage [Ga4(271)6]12− and small bisphosphine rhodium complexes of the general formula [(P-P)Rh(diene)]BF4 (P-P = PMe3 or 1,2-bis(dimethylphosphino)ethane (dmpe), diene = 1,5- cyclooctadiene (COD) or norbornadiene (NBD)) in aqueous solution led to the formation of discrete 1:1 inclusion complexes [Ga4(271)6 ⊃ {(P-P)Rh(diene)}]11−. Upfield shifting of the NMR resonances of the encapsulated guest molecules indicated the formation of a host-guest assembly. The encapsulated catalyst precursor was hydrogenated to generate a reactive catalyst, [Ga4(271)6 ⊃ {(P-P)Rh(OD)2}2]11−. Upon addition of allylic alcohol to this activated, catalytic host-guest assembly, the corresponding isomerized products were formed within 30 minutes at room temperature. While the shape and size selection of the catalytic host-guest assembly allowed encapsulation of small and unsubstituted allylic alcohols, it prevented encapsulation of large and branched substrates by denying access to the active site of the catalyst. Apertures of the host-guest assembly regulate the encapsulations of substrate molecules. Recently, gold(I)-phosphine complexes encapsulated by a self-assembled tetrahedral cage [Ga4(271)6]12− were utilized to investigate the intramolecular hydroalkoxylation of allenes.619 The combination of Me3PAuBr with the tetrahedral cage [Ga4(271)6]12− in D2O or MeOD led to the formation of a discrete 1:1 inclusion complex [Ga4(271) ⊃ (Me3PAu+)6]11− as observed by 1H NMR spectroscopy. Addition of 6-methylhepta-4,5-dien-1-ol to an aqueous solution of [Ga4(271)6 ⊃ (Me3PAu+)]11− showed 48% conversion of the allene to the desired cyclized product after 18 h at room temperature. In contrast, Me3PAuBr under similar reaction conditions gives only an 11% yield of cyclized product after 18 h. This reaction constitutes an elegant example of the enhancement of catalytic activity of an organometallic complex in which the reactivity and lifetime of the catalyst are both enhanced by supramolecular encapsulation.
Fujita et al. utilized coordination cage 286 as a phase transfer catalyst because of its amphiphilic nature in the Wacker-type oxidation of styrene and its derivatives. For example, the addition of styrene to an aqueous solution containing a catalytic amount of 286 led to the formation of host-guest system 286 ⊃ (styrene)n (n = ca. 3) as indicated by its 1H NMR spectrum. Subsequent addition of a catalytic amount of [(en)Pd(NO3)2] followed by heating at 80°C for 24 h gave acetophenone in an 82% yield with a TON of 8. The oxidation proceeds through a biphasic reaction wherein styrene is encapsulated by the host cage and then transferred into the aqueous phase where it is oxidized to acetophenone by the Pd(II) reagent. The reaction product, acetophenone, being less hydrophobic, was displaced from the host cavity by the substrate molecules.620 Similarly, coordination cage 287 catalyzed the Wacker-type oxidation of linear alkenols.621
Hupp, Nguyen and coworkers utilized a supramolecular porphyrinic box as catalyst for selective olefin epoxidation and enantioselective sulfoxidation. 622 The cavity-tailored supramolecular catalytic box consists of 18 porphyrin units – four Zn(II)-porphyrin trimers, two Sn(IV)-porphyrin dimer and one Mn(III)-porphyrin dimer. The catalytically active Mn(III)-porphyrin dimer unit was encapsulated within the rigid porphyrin box cavity that was tailored with carboxylate ligands via a self-sorting process. A competitive study of size-selective epoxidation between cis-stilbene and cis-3,3′,5,5′-tetra(tert-butyl)stilbene showed that cis-stilbene was 5.5-fold more reactive than sterically bulky cis-3,3′,5,5′-tetra(tert-butyl)stilbene. The steric bulk of the carboxylate ligands present on the adjacent Sn(IV)-porphyrin dimer inhibits the access of the larger olefin to the Mn(III) catalytic center resulting in size-selective catalysis. When the cavity was modified with a chiral amino acid ligand, N-acetyl-(D)-phenylalanine, on the Sn(IV) dimer, an enantioselective sulfoxidation of methyl p-tolyl sulfide with an enantiomeric excess of 12% ee was observed. Remarkably, the sense of chiral excess was reversed when N-acetyl-(L)-phenylalanine was used. No enantiomeric excess was observed when the free Mn(III)-porphyrin dimer was used in the presence of free Sn(IV)-porphyrin dimer.
Recently, a trigonal bipyramidal cage was utilized as a catalyst for Suzuki-Miyaura C–C cross-coupling reactions. 623 The molecular cage [{(Me4en)Pd}3L2](BF4)6 (L = 1,3,5-tris(isonicotinoyloxyethyl)cyanurate), assembled from a reaction of (Me4en)Pd(BF4)2 and 1,3,5-tris(isonicotinoyloxyethyl)cyanurate, has a truncated trigonal bipyramidal architecture with a C3h symmetry point group. Each L occupies a pyramid, and three Pd(II) ions lie in equatorial positions with a single, disordered water molecule nestled into the cavity. Both [{(Me4en)Pd}3L2 ⊃ H2O]6+ and desolvated [{(Me4en)Pd}3L2]6+ efficiently catalyzed C–C cross-coupling reactions in N,N-dimethylformamide with K2CO3 as a base without phosphine or any other additives. The cage [{(Me4en)Pd}3L2 ⊃ H2O]6+ gave higher yield of the cross-coupled product compared to [{(Me4en)Pd}3L2]6+ due to the water-assisted effects of the Suzuki-Miyaura C–C cross-coupling reaction.
4.5.3. Biological Applications
Biological applications of self-assembled supramolecular ensembles are still in their infancy. Only in the last few years have researchers begun to apply coordination-driven self-assembled supramolecular ensembles towards biological applications by building functional structures that mimic the complex recognition, regulatory, and other properties of biological systems. Incorporation of biological functionalities into these supramolecular structures and control of their relative arrangements has led to the observation of interesting phenomena. Furthermore, these self-assembled supramolecular metallacages are increasingly being applied to hitherto underexplored biochemical and biomedical applications.624
The binding of proteins regulates the expression of genetic information encoded in DNA in a reversible and noncovalent manner through weak interactions. Thus, the study of noncovalent interactions of DNA with synthetic, small molecules may provide insight into DNA structure control by proteins. Supramolecular chemistry allows the design of DNA-binding agents of similar dimensions to protein DNA-recognition motifs.624 Furthermore, such studies are also of great importance towards effective drug-design and development. Hannon and coworkers have extensively studied supramolecular DNA recognition by coordination-driven metallo-helicates. The supramolecular triple helicates [M2L2]2n+ (n = charge on each metal) were assembled through the interaction of metal ions with bis(pyridylimine) ligands (L) having diphenylmethane spacer.625 As determined from induced circular dichroism, linear dichroism, AFM, NMR and computational studies, racemic [FeII2L3]4+ was found to interact with DNA in a single binding mode consistent with major groove binding, an often preffered site for recognition by protein, and induces DNA bending and intramolecular coiling.626,627 The enantiomers of [FeII2L3]4+ bind differently with DNA. The M enantiomers bind the major groove with partial insertion of one chelate between DNA bases. P isomers bind in the minor groove, spanning the two phosphate backbones.628 However, an extension of the structure of the triple helicate through ligand modulation affects the DNA binding ability significantly. The extended helicates, having two ketimine spacers, do not bind to DNA, as established from induced circular dichroism studies.629 This is due to the loss of CH–π interactions among the ligand strands resulting in the disruption of the relay of chiral information between the two metal centers, leading to the formation of a mixture of rac and meso isomers, with the meso isomer being dominant. A dicationic Cu(I) double helicate [CuI2L2+]2+ binds less strongly to DNA and in a slightly different orientation as compared to tetracationic helicate [FeII2L3]4+.630 This indicates that the charge, size and shape of the helicates serve as important parametrs for effective DNA binding. The same group also observed a rare mode of DNA recognition wherein the tetracationic helicate [FeII2L3]4+ interacts with a palindromic DNA hexanucleotide. X-ray structural studies revealed that the helicate binds into the central trigonal hydrophobic cavity of a 3-way DNA junction.631
G-quadruplex DNA structures based on guanine quartets are found in the telomeric and transcriptional regulatory regions of chromosomes. In cancer cells, the telomeres are elongated by the enzyme telomerase, thereby conferring immortality to the cancerous cell line. Thus, molecules that can bind specifically to quadruplex DNA may lead to potential anti-cancer drugs.322 Therrien et al. studied the interaction of arene ruthenium based tetragonal coordination cages [Ru8(η6-arene)8(μ-tppH2)2(dhbq)4]8+ (389, arene = C6H5Me; 390, arene = p-cymeme; dhbq = 2,5-dihydroxy-1,4-benzoquinonato; tppH2 = 5,10,15,20-tetra(4-pyridyl)porphyrin) and [Ru8(η6-arene)8(μ-tppZn)2(dhbq)4]8+ with both duplex and quadruplex DNA.456 A fluoroscence intercalation displacement assay (FID) and surface Plasmon resonance (SPR) study showed that the metallacages bind strongly to telomeric and c-myc DNA. Although SPR results indicated that these cages bind more strongly with quadruplex DNA than duplex DNA, selectivity for quadruplex over duplex DNA was modest, supposedly due to the high cationic charge of the tetragonal cages, which increases non-specific binding of DNA via eletrostatic interaction.
In the context of recognition and folding of peptides, Fujita et al. utilized a variety of self-assembled molecular cages as hosts. Using tetrahedral cage 287 as a supramolecular host, sequence selective recognition of oligopeptides within the hydrophobic cavity of was observed in aqueous media.632 Coordination cage 287 showed a strong and specific binding affinity towards Ac–Trp–Trp–Ala–NH2 over other sequences of tripeptides. A tetrapeptide and hexapeptide possessing the Trp–Trp–Ala sequence also bind strongly to tetrahedral cage 287. The specific binding affinity for the Trp–Trp–Ala sequence was attributed to efficient π–π interactions between the indole rings of Trp residues and triazine panels of the host cage. Likewise, the bowl-shaped host, 480, encapsulated a nine-residue peptide (Ac-Trp1-Ala2-Glu3-Ala4-Ala5-Ala6-Glu7-Ala8-Trp9-NH2) inside the hydrophobic pocket in aqueous solution (Scheme 138).633,634 The binding of the peptide within the cavity induced and stabilized a α-helix conformation. 1H NMR titration studies revealed that the two terminal tryptophan residues (Trp1 and Trp9) bind to the bowl leading to a 1:1 host-guest complex. TOCSY and NOESY measurements, coupled with molecular modeling, revealed that the 9-mer peptide adopts an α-helical conformation with two Trp residues deeply buried into the hydrophobic pocket. However, the 2:1 host-guest complex, wherein two bowls wrapped the whole 9-mer residue, was predominantly formed in the presence of an excess amount of cage. Varying the hydrophobic residues in the short oligopeptides revealed that the aromatic-aromatic interactions are the key driving force for enclathration.635 Further studies using a trifacial cage by the same group have showed that it has a remarkable ability to fold Ala-Ala-Ala sequences into β-turns via encapsulation in the hydrophobic cavity.636,637 This result demonstrated that peptide recognition within the large cavity of self-assembled cages is a powerful method to produce the secondary structures of peptides, even if the peptide fragment is considerably short.
Scheme 138.

Recently, Therrien and coworkers have utilized a trigonal prism, [Ru6(p-cymene)6(285)2(dhbq)3]6+ (367), as a prototype for a drug delivery system. Cage 367 can act as a carrier for cisplatin and its high charge potentially facilitates uptake in cancer cells, although it leaches rapidly from the hydrophobic pocket in water.431 However, more hydrophobic complexes [M(acac)2] (M=Pd, Pt) are strongly immobilized within 367, while being almost insoluble in water in their free form under ambient conditions. The cage 367 was moderately cytotoxic towards human ovarian A2780 cancer cells, while the encapsulated cages were more active. The platinum containing species was about twice as active as the empty cage and the palladium entrapped species was more than one order of magnitude more cytotoxic. Recently, a series of biologically relevant, planar hydrophobic pyrenyl guest (Pyrene-R) molecules (491-498) were encapsulated within the hydrophobic cavity of cage 367 to form 1:1 cationic host-guest complexes [367 ⊃ Pyrene-R]6+, as evidenced by 1H NMR and ESI-mass spectrometric studies (Scheme 139).638 The in vitro anticancer activity of these water soluble systems showed that these inclusion complexes were more cytotoxic than the empty cage itself. Cytotoxicity studies of empty cage 367 on human ovarian A2780 cancer cells using an MTT assay, which measures mitochondrial dehydrogenase activity as an indication of cell viability, showed it to be moderately active. However, encapsulation of pyrenyl derivatives within the cavity of the cage 367 either increases or has negligible effect on the cytotoxicity. The host-guest complex [367 ⊃ 492]6+ and [367 ⊃ 493]6+ have IC50 values of 2 ± 0.6 μM and 3 ± 1.1 μM respectively that are comparable to IC50 value of cisplatin (1.6 μM). Similar antiproliferative activity studies on both A2780 (cisplatin sensitive) and A2780cisR (cisplatin resistant) human ovarian cancer cells using porphyrin based open arene ruthenium assemblies showed that both charge and length of the spacer between the porphyrin panels significantly affect the cytotoxic acitivity.639 The ability of the trigonal prism 367 to deliver guest molecules into the A2780 cells was studied using the host-guest complex [367 ⊃ 491]6+.640 The uptake and release of guest molecule 491 was monitored by means of flow cytometry. Once incarcerated within the cavity of the cage, the natural fluoroscence of the guest molecule was quenched. The freed molecules were monitored by fluorescence spectroscopy following incubation of [367 ⊃ 491]6+ with A2780 cell lines. Efficient uptake and release of the incarcerated payload was observed from the cages. The rate of uptake and release was found to be dependent on the incubation time and concentration of the host-guest complex. Recently, the same group studied the effect of sizes of the guest molecules on cytotoxicity by encapsulating a series of large pyrenyl-containing dendrimers of different generations within hexanuclear arene-ruthenium cage [Ru6(p-cymene)6(285)2(dhnq)3]6+ (370; dnhq = 5,8-dihydroxy-1,4-naphthoquinonato).641 The cytotoxicity of the host-guest systems, with the pyrenyl moiety being encapsulated in the hydrophobic cavity of the cage and the dendritic functional group dangling outward, was evaluated on human ovarian A2780 and A2780cisR cancer cell lines. The host-guest systems show similar cytotoxicity to the empty metalloprism 370 and remain intact before internalization by the cancer cells. This demonstrated that metallacage host system were able to deliver hydrophobic payload with large appandages into cancer cells.
Scheme 139.

A similar hexanuclear arene ruthenium complex [Ru6(p-cymene)6(291)2(dhnq)3]6+ having a trigonal prismatic architecture assembled from dinuclear arene ruthenium complexes [Ru2(η6-p-cymene)2(μ-OO ∩ OO)2(OTf)2] (OO ∩ OO = 5,8-dihydroxy-1,4-naphthoquinonato (dhnq)) and 1,3,5-tris-(4-pyridylethynyl)benzene (291) also showed effective growth inhibition activity in SK-hep-1 (liver cancer), HeLa (ovary cancer), HCT-15 (colon cancer), A549 (lung cancer), and MDA-MB-231 (breast cancer) tumor cell assays.642 The cage showed low micromolar inhibition against all cancer cell lines. In particular, the hexanuclear cage [Ru6(p-cymene)6(291)2(dhnq)3]6+ was found to inhibit the proliferation of A549 cells with an IC50 of 3.4 μM, which is comparable to the effect seen with cisplatin (IC50 = 2.4 μM). Mechanistic studies to understand the antiproliferative effect of the nanocage suggested that the anticancer activity through apoptosis originated from the interaction of the cytotoxic 5,8-dihydroxy-1,4-naphthoquinone (dhnq) with [(p-cymene)RuCl(μ-Cl)]2 which was enhanced in the nanocage due to increased nuclearity and by the presence of the extended π-conjugated system.
5. MISCELLENEOUS
5.1. Self-selection and Self-sorting in Coordination-Driven Self-Assembly
Of late, there has been a paradigm shift from supramolecular pre-organization to more complex systems where the system undergoes self-organization through self-selection and self-sorting to generate one or more well-defined and ordered supramolecular architectures.14,643-647 Self-selections of structurally ‘instructed’ components allow controlled assemblies from complex mixtures of components following specific programs. Nature provides an amazingly wide array of successful examples of complex structures designed through self-organization using non-covalent interactions. For example, in the biosynthesis of lipid membranes, proteins, nucleic acids, protein aggregates and virus particles, nature utilizes the non-covalent informations stored within the structural and electronic properties of the components to self-assemble them into the preferred biomolecules. In fact self-organization is the driving force that led up to the evolution of the biological world from inanimate matter.
Coordination-driven self-assembly paradigms have been extensively used by the research groups of Lehn,643 Raymond,644 Albrecht,645 Stang,646 Severin,647 and Nitschke14 over the past decade to develop and understand the phenomena of self-organization that occurs in nature and biological systems. Wide ranges of complex synthetic mixtures were investigated that undergo varying degrees of self-organization. The facile kinetic reversibility of metal-ligand coordination that drives systems to the formation of thermodynamically favorable collections of supramolecular ensembles is the cornerstone of such synthetic strategies. Several other factors such as temperature, concentration, association constants of complementary pairs, and the presence of competitors, however, also play a vital role in controlling the extent of self-organization phenomena in these systems. Depending on the extent of self-organization occurring in a complex mixture of subunits, the process can range from a statistical mixture of different supramolecules, to amplified organization, where some supramolecules are formed in greater ratios than others, to absolute self-organization and the exclusive formation of discrete supramolecules (Figure 40).646
Figure 40.
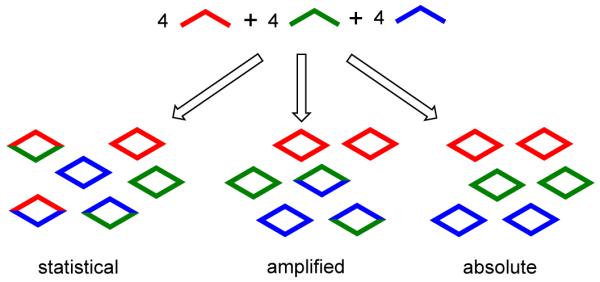
Representation of differing extents of self-organization phenomena that can occur within a complex mixture of subunits.
Coordination-driven self-assembly, with its highly modular character, along with the extensive library of individual molecular subunits of varying size, geometry, and physical properties that have been prepared in recent years, has allowed the study of self-organization in both 2D and 3D supramolecular architectures. In this paradigm, by combining multiple rigid acceptor and donor subunits, the directional nature of the coordination-driven self-assembly protocols can be used to study the phenomena of self-selection and self-sorting under mild conditions. In one of the earliest demonstration of self-selection, in 1993, Lehn et al.648 described the spontaneous formation of metal helices from a mixture of ligands triggered by a metal ion. Upon treatment with Cu(I) ions, mixtures of oligo(2,2′)-bipyridine strands containing 2-5 bipyridine units led to the formation of double helicates without significant crossover. Similarly, when a mixture of the two different tris-bypyridine ligands were allowed to react with Cu(I) and Ni(II) ions, only the a double helicate and a triple helicate were formed. The process represents a self-recognition involving the preferential binding of like metal ions by like ligand strands in a mixture to selectively assemble into the corresponding helicates. Raymond et al.649 have shown that “naked” metal ions can act as a trigger for the self-organized assembly of rationally designed bis(catecholamide) ligands of different lengths (499-501). These structurally “instructed” rigid bis(catecholamide) ligands, in the presence of the Ga(III) ions, undergo facile self-organization, resulting in a highly ordered system of helicates (502-504) in solution (Scheme 140). Albrecht and coworkers have also observed that rigid oligo-p-phenylene type dicatechol ligands having different lengths undergo template-directed self-organization to dinuclear Ti(IV)-based triple stranded M2L3 helicates in the presence of appropriate counterions (M = Li, Na, K or NH4+).650,651 Ligands having similar geometric preferences do not lead to self-organization. Self-assembly of heterodinuclear triple stranded helicates was achieved based on metal selection by “geometric discrimination”.652
Scheme 140.

One of the earliest examples of the self-sorting of molecular components resulting in the absolute self-organization into discrete supramolecular rectangles via the directional bonding approach, was demonstrated by Hupp and coworkers.108 The combination of a metal precursor with two rigid linear linkers of differing lengths, should, in the absence of any driving bias, result in a statistical mixture of two molecular squares of different sizes and a rectangle. However, the combination of the octahedral Re(CO)5Cl as the corner unit and linear pyridyl linkers of different lengths namely pyrazine and 4,4′-bipyridine led to the exclusive formation of only two types of molecular squares rather than a statistical mixture of squares and rectangle.
Stang et al. extensively studied self-organization of platinum-based 2D and 3D metallacycles and metallacages.646 Square-planar platinum(II) metal acceptors of varying angles between the coordination sites ranging from 0° to 90° with electron-rich pyridyl donors were used as molecular subunits. It was shown that by a careful modulation of the intrinsic geometric parameters of the molecular subunits (angle, size, and relative orientation of coordination centers), absolute self-organization could be achieved without introducing any external effectors and the process could be applied to a diverse set of coordinative supramolecules. As shown in Scheme 141, the use of square-planar platinum(II) acceptors 75, 12 and 505 having 0°, 60° and 90° angles between their coordination sites in conjunction with a 180° linear pyridyl donor ligand, 4,4′-bipyridine in 1:1:1:3 molar ratio resulted in the absolute self-organization into the discrete supramolecular rectangle 76, triangle 26 and square 506, respectively.653 Although a random combination of different subunits was observed initially as determined by NMR and ESI-mass spectrometry, prolonged reaction led to fully equilibrated mixtures of the discrete individual assemblies. The major driving force for the selective formation of the discrete polygons was the angular differences in the organoplatinum acceptor units. The size of the resulting assembly can also direct the absolute self-organization of 2D and 3D supramolecular ensembles.654 For example, mixing two pyridyl donors of different lengths, 4,4′-bipyridine and 1,2-bis(4-pyridyl)ethyne (507), with diplatinum acceptors 75 and 12 having 0° and 60° angles leads to molecular rectangles (76, 508) and triangles (26, 509) respectively (Scheme 142).
Scheme 141.

Scheme 142.
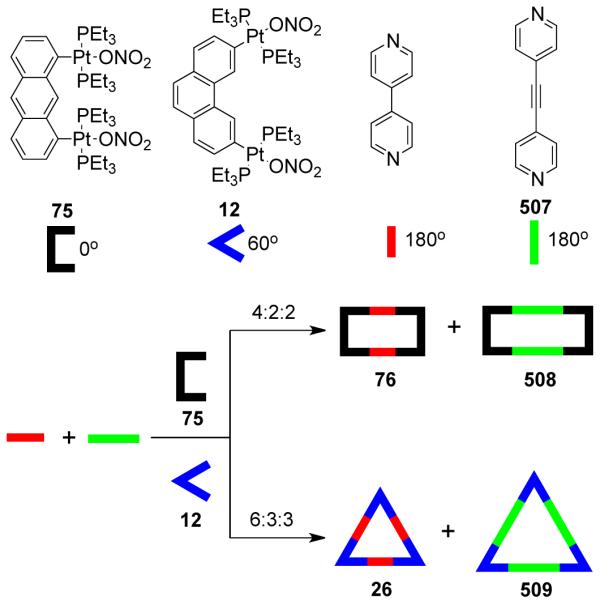
The combination of different subunits of varying size, shape and dimensions also leads to absolute self-organization into discrete supramolecules.655 A complex 7:2:2:2 mixture of 0° diplatinum(II) acceptor (75) with three donors, 4,4′-bipyridine, 1,4-bis(4-pyridylethynyl)benzene and tris(4-pyridyl)methanol (313) differing in size, shape and orientation of the coordination sites has been successfully assembled exclusively into discrete structures – rectangles (76 and 78) and a distorted trigonal prism (511) demonstrating the strength and utility of the directional bonding approach in studying self-organization (Scheme 143). Besides these structural and geometric factors, the reaction temperature and choice of solvent also determines the extent of self-organization.655 It is interesting to note that in all the examples cited above, absolute self-organization occurs in acetone/water (1:1) solvent mixture, but not in dichloromethane.
Scheme 143.

Similarly, the formation of discrete 3D polyhedra from multiple subunits in a complex mixture was also investigated. 656 The self-recognition process was found to be strongly dependent upon the structural information stored within individual building blocks, i.e., geometry, directionality, and rigidity. In addition, the thermodynamic stability of the products also played a great role in the self-recognition process. Self-recognition and self-assembly were achieved as the kinetically labile Pt-N bond allowed for the dynamic exchange of complementary molecular components by internal rearrangement, exchange, extrusion, and incorporation of different components, through which intermediate assemblies are self-selected and proof-read in order to generate the most thermodynamically preferred 3D cages. In fact, constitutional dynamic exchange of complementary molecular components in coordination-driven self-assembly was recently shown using ESI-mass spectrometry for molecular polygons.657 Treatment of 1H and 2D isotope-labeled 4,4′-bipyridines with 60° diplatinum(II) acceptors (12) gave the corresponding molecular triangles. Mixing these two homoisotopic triangles resulted in ligand exchange to give heteroisotopic triangles. Similarly, mixing of homoisotopic rectangles generated from 60° diplatinum(II) acceptors (75) and 1H and 2D isotope-labeled 4,4′-bipyridines also resulted in heteroisotopic rectangles. Formation of these heteroisotopic polygons was established on the basis of quantitative mass spectroscopic results. The exchange process was found to be dependent upon the temperature, solvent, and the nature of the counteranions.
Self-organization during self-assembly of a series of linear bispyridyl donors functionalized with hydrophobic alkyl (C6H13, C12H25, and C18H37) chains or hydrophilic (di-, tri- or tetra)ethylene glycol chains with complementary 0° di-Pt(II) acceptors into supramolecular rectangles was also explored.658 The connectivity and location of functional groups on the bispyridyl ligands ensured that they did not interfere sterically or electronically with their respective binding sites. Employing carefully controlled reaction conditions so that the only means of self-organization during self-assembly was through “second-order” effects arising from the distal functional groups themselves, it was observed that increasing the chain length drove the self-assembly process toward amplified self-organization.
Ambidentate ligands having two different binding sites have been used to demonstrate absolute self-sorting in the self-selection of a single linkage isomeric product. The combination of molecular clip 75 with ambidentate pyridyl carboxylate ligands 512-514 led to the assembly of the most symmetrical isomers, 515-517, with iso-oriented (i.e. each Pt atom is bound to a pyridyl nitrogen and a carboxylate oxygen atom), ligands, despite the possibility of formation of two isomeric rectangles (Scheme 144).659 Similarly, self-selection for the symmetric supramolecular rectangles was observed when molecular clip 75 was treated with linear ambidentate ligand, N-(4-pyridyl)isonicotinamide 30, in a 1:1 ratio.112 It was also found that steric interactions among the substituents present on the subunits in a complex mixture of ditopic ligands could drive the process of self-organized assembly from statistical to absolute organization.660 By preparing a number of unsymmetric bis(4-pyridyl)acetylene ligands with one of the two pyridyl moieties substituted with 2,6-dimethyl-, 2-chloro-, and 3,5-dichloro-substituents and treating them with the 90° cis-(PMe3)2Pd(NO3)2 acceptor, it was shown that the asymmetry of the linear donors leads to four different supramolecular squares, depending upon the relative orientations of each squares’ constituent donors. In keeping with the previous observation of molecular rectangles, in case of molecular triangles also, due to the different bonding connectivity of the ambidentate ligands, two linkage isomeric triangles were expected. However, when the 90° heterobimetallic cis-(dppf)Pd(OTf)2 (32; dppf = diphenylphosphinoferrocene) acceptor was treated with unsymmetrical ambidentate ligand, sodium isonicotinate, a symmetric molecular triangle was obtained as exclusive product.59 Similarly, sodium pyrimidinecarboxylate in conjunction with 32 led to the self-selection for the symmetrical isomer.58 As determined from 31P and 1H NMR, ESI-mass spectrometry, and X-ray crystallography, of the two possible linkage isomers, the most symmetrical one, in which all the ambidentate ligands are iso-oriented, was observed. The driving force for such absolute self-sorting is the enthalpic gain achieved by heteroligation at each Pd center. Furthermore, by orienting the ligands in an alternating fashion, the geometric strains on the triangles are relieved. The combined effect of these driving factors leads to the thermodynamically preferred metallacycles.
Scheme 144.
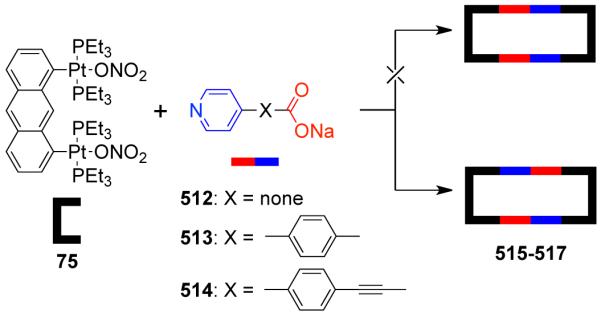
Schmittel et al. have demonstrated multicomponent integrative self-sorting leading to the formation of trapezoids,661 triangles,662 and ladders.663 For example, the isosceles trapezoid 521 was constructed through sequential addition of the specifically designed ligands - two bisphenanthroline ligands 518 and 519 with varied length serve as the shorter edge of the trapezoid while the phenethroline-terpyridine hybrid ligand, 520, serves as the longer edge of trapezoid 521 (Scheme 145).661 First, a mixture of 519 and 520 were treated with Zn(II) followed by the addition of 518 and Cu(I). NMR and ESI-mass spectrometric studies confirmed the exclusive formation of the macrocycle. Steric effects, π–π interactions, electronic effects, and metal-ion interactions play a vital role in controlling the self-sorting process. Similarly, a molecular triangle assembles from five-components via self-sorting from related building units.662
Scheme 145.

Recently, the same group also demonstrated the assembly of a trisheterometallic scalene triangle with three different self-assembled corners from an eight-component algorithm via self-sorting.664 The combination of a phenanthroline-porphyrin hybrid ligand (522), terpyridine-phenanthroline ligand (523), phenanthroline-pyridine hybrid (524), Zn2+ and Cu+ in 1:1:1:1:1 ratio under refluxed condition for 2h in acetonitrile/DCM (2:1) led to the formation of scalene triangle [CuZn(522)(523)(524)](OTf)2(PF6) (525; Scheme 146). The exclusive formation of triangle 525 was established by ESI-mass spectrometry, 1H NMR, DOSY, DPV and elemental analysis. Steric and electronic effects, π-π interactions, and metal-ion interactions drive the three-fold completive self-sorting process, which leads to the assembly of a single species in solution.
Scheme 146.

Schalley et al. have also successfully demonstrated the concept of integrative self-sorting with a hetero[3]pseudorotaxane as a model system.665 The synthesis of a hetero[3]rotaxane with an efficient cascade-stoppering system was achieved from a four component mixture. In a similar study666 of a four component self-sorting system of crown ethers and ammonium ions, six building blocks were designed in which two identical or different binding sites are incorporated. These building blocks, when mixed in many different ways, led to distinctly different pseudorotaxane assemblies. The self-sorting process integrates all building blocks in specific places so that this approach permits one to exert positional control and can widely influence the resulting assemblies with respect to the details of their structures.
In an interesting study, the same group recently demonstrated a thermodynamically controlled self-assembly of hetero-bimetallic metallosupramolecular macrocycles through self-sorting caused by different ancillary ligands present on the cis-blocked Pd(II) and Pt(II) metal centers.667 The self-sorting of metallosupramolecular macrocycles was investigated using a tetratopic ligand (527) that has two different metal-binding sites, a central 2,2′-bipyridine core, which act as a chelate ligand, and two terminal pyridine coordination sites, which mediate assembly formation. Treatment of 527 with M(dppp)(OTf)2 [M = Pd (4), Pt (52); dppp = diphenylphosphinopropane] and/or M(dppe)(OTf)2 [M = Pd (526), Pt (325); dppe = diphenylphosphinoethane] in all four possible combinations led to a high fidelity self-sorting upon equilibration for three days resulting in the generation of two self-sorted hetero-bimetallic (528 and 529) and two homo-bimetallic (530 and 531) macrocycles (Scheme 147). 31P NMR and tandem mass spectrometric experiments provided the evidence for the formulation of the macrocyles. In all four macrocycles, (dppp)MII hinge coordinates to the pyridines. The (dppe)MII is displaced from the pyridine and binds exclusively to the bipyridine sites when thermodynamic equilibrium is achieved. Thus, a difference of merely one methylene group incorporated in the ancillary ligands induced high fidelity self-sorting.
Scheme 147.

Osuka and Kim have investigated high fidelity homochiral self-sorting in porphyrin-based systems where meso-pyridyl-appended Zn(II) porphyrins assemble to form macrocycles and cages of different dimensions. Due to the restricted rotation around the meso-meso linkages, 4-pyridyl-appended meso-meso linked Zn(II) diporphyrins, exist as both R and S enantiomers in solution, leading to formation of corresponding homochiral (R)- and (S)-porphyrin boxes through self-sorting.520 The homochiral self-sorting behaviors of meso-cinchomeronimide (3,4-pyridinedicarboximide)-appended Zn(II)-porphyrin and 10,10′-cinchomeronimide-appended meso-meso linked Zn(II)-diporphyrins in noncoordinating solvents were also investigated.668 While the former undergoes complete assembly to yield its trimer, the latter led to trimeric, tetrameric, and pentameric rings, through rigorous homochiral self-sorting processes, owing to the presence of three stable conformers with respect to the orientation of the pyridyl nitrogen atoms. Similarly, meso-meso’-bis(5-azaindol-2-yl)-appended Zn(II)-porphyrin formed a trimeric fluorescent assembly that consisted of three single atropisomers due to the facile rotation of the 5-azaindol-2-yl substituent and effective trapping via self-sorting assembling by complementary coordination.669
Severin and coworkers have demonstrated the self-sorting process to generate dynamic combinatorial libraries based on half-sandwich complexes having triangular topologies.647 A combinatorial assembly of three Ru half-sandwich complexes having sterically different π-ligands: a small benzene ligand, a large 1,3,5-triisopropybenzene ligand and a very large hexamethylbenzene ligand with 2,3-dihydroxypyridine, gave only three species in significant amount out of 10 possible products.670 From a thermodynamic point of view, the trinuclear metallamacrocycles with the smallest steric congestion i.e. the compound with three (benzene)Ru fragments, is expected to be most stable. However, in the equilibrated mixture, all three macrocycles contained only one (benzene)Ru fragment (Scheme 148). This demonstrated that a higher thermodynamic stability of a dynamic combinatorial library member does not necessarily correlate to an elevated concentration. A more complex mixture containing half-sandwich metal building blocks [{Ru(p-cymene)Cl2}2], and [{Ir(Cp*)Cl2}2] and two different dihydroxypyridine ligands in buffered aqueous solution form a dynamic combinatorial library with unique network topology.671 Upon mixing, these complexes undergo scrambling reactions to give only eight out of a possible 24 macrocycles (Scheme 149). The eight different complexes can be divided into two partially orthogonal sub-libraries. Within these sub-libraries, an exchange of metal fragments and ligands is possible, but communication between the sub-libraries is restricted to an exchange of metal fragments.
Scheme 148.
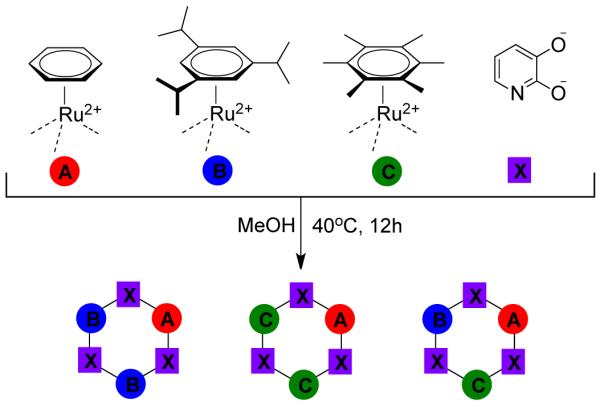
Scheme 149.
Nitschke and coworkers have investigated self-sorting phenomena by using the dynamic nature of both metal-ligand coordination and reversible imine bond formation.14 For example, when 2-formylpyridine (275) and benzaldehyde-2-sulfonate (532) were mixed with a diamine (533) in aqueous medium, a complex library of ligands was generated in dynamic equilibrium with the starting materials.672 However, addition of CuBF4 triggered a collapse of the library leading to the formation of only two self-sorted supramolecules 534 and 535 (Scheme 150). Formation of any other structures is both entropically and enthalpically unfavorable.
Scheme 150.

Similar self-organization phenomena was observed when 2-aminoquinoline and 4-chloroaniline were mixed with the phenanthroline dialdehyde to generate a dynamic library of ligands which subsequently collapse to form dicopper and tricopper helicates upon addition of copper(I).673 Use of two different metals, Cu(I) and Fe(II), also induced a dynamic combinatorial library of 11 structures to self-organize into only two discrete supramolecules.674 The efficiency and selectivity of self-organization could be correlated with the electronic nature of the substituted amine subunits, which were studied using Hammett σpara values, and on the coordination geometries of the metal ions used.675
5.2. Surface Confined Ensembles
Over the past decade, a great deal of attention has been paid to assembling metallosupramolecular macrocycles and metallacages on solid supports to understand the factors that govern and control the assembly process on surfaces and accordingly develop materials with novel properties.20-22 As delineated in previous sections, metallosupramolecular assemblies having novel electronic, magnetic, photophysical, redox and catalytic properties have been extensively investigated in solution using a wide variety solution-phase analytical techniques. However, in the solution-phase, supramolecular assemblies generally lack coherence in their orientation, alignment and packing order, prohibiting the large-scale uniformity and coordination required for advanced functional materials. On the other hand, assembling supramolecules on surfaces imparts a high degree of coherence and uniformity for fabrication of nanostructures and nanodevices for material applications. Various substrate such as SiO2, Cu(100), gold, indium-tin oxide (ITO), highly ordered pyrolytic graphite (HOPG) have been used to prepare metallosupramolecular adlayers. Scanning probe techniques, in particular scanning tunneling microscopy (STM) and atomic force microscopy (AFM) have been commonly used to study the formation and properties of such surface-confined self-assembled molecular systems.
Hupp and coworkers have extensively studied film- and membrane-based porous materials derived from surface-confined metallosupramolecular assemblies for molecular sieving and chemosensing.20 In their initial studies in this area, microporous thin films composed of discrete Re(I)-based metallosupramolecular squares were prepared on inert conductive platforms such as gold, glassy carbon, ITO, and platinum to act as electrodes to evaluate the size-selective transport of small molecules and complexes. 676, 677 Re(I)-based porphyrin and pyrazine-containing molecular squares were assembled on permeable polyester membranes and their thickness (~ 25 nm), morphology and crystallinity was characterized by low-resolution optical microscopy and by AFM measurements.269,678 These film-coated electrodes, when exposed to solutions containing redox-active probe molecules and complexes of various sizes, led to size-selective transport across the membrane as determined from their voltammetric responses. Only those species capable of permeating the film showed faradic current flow. More robust films have been obtained by assembling phosphonate-functionalized Re(I) porphyrinic squares on ZrP-modified ITO and SiO2 surfaces.679,680 The high affinity of Zr(IV) for phosphonate allowed for layer-by-layer assembly either by direct attachment of the octa-phosphonated squares to metal-oxide surfaces or by first derivatizing the supporting surfaces with zirconium binding ligands. This layer-by layer assembly approach allowed precise control over the film thickness as well as the morphology by overridding the defects associated with vapor deposition. Interdigitation of phosphonated porhpyrinic molecular squares with phosphonated perylenediimides through layer-by-layer assembly on ZrP coated ITO surfaces has led to bulk heterojunctions. When employed in photovoltaics, these molecularly-interlaced heterojunctions generate photocurrent with visible light.270,271 In aqueous I3−/I− solutions these photoelectrochemical cells produce cathodic photocurrents, in contrast to generally observed anodic photocurrents with conventional dye-senstized solar cells. Microporous thin films (~ 50 to 400 nm) composed of discrete, cavity-containing Mn(I) and Re(I)-based molecular rectangles assembled on electrically conductive ITO-coated glass slides yielded a collection of needle-shaped structures that were readily visualized via low-resolution optical microscopy and AFM measurements.681 These rectangle-based thin film materials displayed both size- and shape-selective membrane transport behavior when placed in contact with aqueous solutions of redox-active probes. Only those probes that were short and narrow in at least one dimension were allowed to permeate through the membrane. The shape selectivity, however, was found to be through intramolecular rather than intermolecular cavities. Quantitative and semiquantitative measurement using rotating disk electrode (RDE) voltammetry and scanning electrochemical microscopy (SECM), respectively showed that the transport was roughly two orders of magnitude slower than observed in thin films of molecular squares featuring similar-sized cavities.
Wan, Stang and coworkers have investigated the self-organization of supramolecular metallacycles and metallacages on Au(111) and HOPG surfaces by scanning tunneling microscopy (STM).21 Well-ordered self-organized architectures with three metallosupramolecular assemblies, a square, a rectangle, and a three-dimensional cage, on Au(111) surfaces were fabricated.682 The self-assembled adlayers were prepared by immersing the Au beads in an ethanol solution of the respective assemblies. These supramolecular assemblies adsorb on Au(111) surfaces and self-organize to form highly ordered adlayers with distinct conformations that are consistent with their chemical structures. For example, the metallosupramolecular square, [Pt(dppp)(4,4’-bipyridine)]4(PF6)8 spontaneously adsorbed on the Au(111) surface to give regular rows of squares lying flat on the surface and extending in the A and B directions, forming 90° angles (Figure 41). The dimensions of each molecular square were determined from the STM image and found to be 2.1 ± 0.1 nm on each side, consistent with the size previously determined from X-ray crystallography.292 Molecular rectangles, [{(1,8-bis[trans-Pt(PEt3)2)anthracene}2(4,4’-bipyridine)2]4+ (76) also assembled into well-ordered arrays on Au(111) surfaces. Similarly, adsorption of bis-terpyridine-based [2 × 2] grid-type Zn(II) and Co(II) assemblies on HOPG surfaces led to well-organized adlayers.683,684
Figure 41.

(a) Space-filling model of molecular square [Pt(dppp)(4,4’-bipyridine)]4(PF6)8, (b) High-resolution STM images of the adlayer of square on Au(111), (c) Structural model of the adlayer.
Schalley and coworkers also studied the assembly of a metallosupramolecular square [Pt(en)(4,4′-bipyridine)]4(NO3)8 (en = ethylenediamine), on Cu(100) surfaces modified with a chloride adsorbate layer.685 Chloride-covered Cu(100) surfaces were prepared by adsorbing chloride anions on positively polarized Cu(100) electrode from chloride-containing electrolytes. The adsorbed chloride layer adopted the symmetry of the copper substrate and essentially retained its full charge upon adsorption. The negatively charged chloride layer caused the cationic squares to adsorb and lie flat on the electrode surface. While adsorbate-substrate interactions were driven by attractive electrostatic interactions, lateral ordering was governed by van der Waals interactions. However, due to the acidic conditions used to prepare the surface, the square slowly opened up and chainlike oligomers co-adsorb. On HOPG surfaces the molecular squares showed a striped pattern in the STM images. Self-organization of Fréchet-type dendron-functionalized Pd(II) and Pt(II)-based metallosupramolecular squares on mica and HOPG surfaces were also investigated by the same group.251 AFM studies on the materials revealed two distinctly different morphological arrays of adsorbed self-assembled dendronized squares depending on the solvent in which the sample was applied to the surface. When ethanol solutions of squares were used, tower-like aggregates appeared both on mica and HOPG surfaces. When ethanol/acetone solutions of the dendronized squares were used for deposition, the mica surface showed similar tower-like aggregation. However, on a HOPG surface, a mixed solvent solution showed formation of monolayers. Thus, a balance between surface-dendrimer and dendrimer-dendrimer interactions plays a vital role in driving such self-organization.
In addition to 2D metallacycles, 3D metallacages were also shown to form well-ordered adlayers on Au(111).682 The distorted trigonal prism, [(1,8-bis(trans-Pt(PEt3)2)anthracene)3(tris(4-pyridyl)methanol)2](PF6)5(NO3) self-organized into two-dimensional array with regular molecular rows. The cages oriented themselves in such a way that their propeller “blades” were interdigitated along the A direction while along the A–B diagonal, the faces of their bis-Pt(II) anthracenyl moieties aligned roughly parallel. Such an arrangement maximized intermolecular van der Waals interactions thereby providing stability to the adlayers. Self-organization of a supramolecular chiral trifacial box (401) on Au(111) showed domain separated monolayers.686 The self-assembled adlayers were prepared by suspending gold beads in a racemic mixture of the trifacial box, 401, in ethanol. Upon adsorption of the racemic mixture on Au(111), each enantiomer formed separate, well-ordered, locally chiral domains of MMM and PPP helical enantiomers. The supramolecules lie edge-down on the Au(111) surface while projecting one of their three flat faces upwards. A similar manifestation of chirality into separate enentiomericall pure domains on Cu(100) surfaces modified with a chloride adsorbate layer was also observed with Pt(II)-chiral molecular rhomboids, 198.216 STM experiments revealed spontaneous separation of enantiomers at the surface resulting in enantiomerically pure domains when a racemic mixture was used. Only one well-defined orientation with respect to the underlying substrate was observed when enantiomerically pure rhomboids were adsorbed.
The nature of the substrate used for deposition of the supramolecular assemblies was found to profoundly affect the morphology of self-organization on surfaces. For example, the supramolecular rectangles (79) shown in Figure 42a self-assemble in an “face-on” geometry on Au(111) surfaces, while on HOPG surface the same molecules self-assemble in an “edge-on” geometry.687 When gold beads were immersed in a 10−6 M solution of 79 in toluene, a uniform, well-ordered molecular adlayer with few defects was observed. STM images showed that the rectangles lie flat on the Au(111) surface, forming linear chains (Figure 42b). In contrast, on HOPG surface, the long edges of the rectangle stand on the surface, forming a 2D molecular network (Figure 42d). The observation that supramolecular rectangle 79 adopted two different orientations on Au(111) and HOPG surfaces demonstrated that molecular adlayers of coordination-driven self-assemblies could be dominated by the nature of their solid supports. The substrate-adsorbate interaction is much stronger in the case of Au(111) due to the strong Au–π interactions that are maximized when the rectangles lie flat on the surface. The self-organization on HOPG surfaces, on the other hand, was primarily governed by intermolecular π–π and van der Waals interations between the adjacent rectangles, forcing them to adopt a close-packed 2D molecular network.
Figure 42.

(a) Space-filling models of molecular rectangle 79, (b) High-resolution STM images of the adlayer of 79 on Au(111), (c) Structural model of the adlayer on Au(111), (d) High-resolution STM images of the adlayer of 79 on HOPG, (e) Structural model of the adlayer on HOPG surface.
Molecular templates have been found to promote the monodispersion of a well-ordered array of supramolecular assemblies that otherwise form disordered adlayers on surfaces. For example, use of the triple-armed amphiphile, 1,3,5-tris(10-carboxydecyloxy)-benzene (TCDB) as template for coadsorption with supramolecular rectangles [{(1,8-bis[trans-Pt(PEt3)2)anthracene}2(4,4’-bipyridine)2]4+ (76) led to the formation of well distributed and monodispersed adlayers on a HOPG surface, which otherwise form disordered adlayers.688 The judicious choice of template is vital as various interactions such as tetplate-template, adsorbate-template, template-surface, and adsorbate-surface along with appropriate size complementarities act as driving forces for templation and monodispersion. An ethanolic solution containing a 1 : 3 mixture of TCDB and molecular rectangle (76) deposited on a HOPG surface gave a well-ordered assembly. STM images showed that the rectangles were entrapped in the molecular template, thus forming the ordered adlayer. Similarly, use of shape-persistent arylene-ethynylene-butadiynylene macrocycles as templates for codeposition with molecular square, [Pt(dppp)(4,4’-bipyridine)]4(PF6)8 or molecular rectangle [(1,8-bis(trans-Pt(PEt3)2)anthracene)2(4,4’-bipyridine)2]4+ (76) allowed for 2D ordered arrays on HOPG surfaces. 689 For each underlying shape-persistent macrocycle, one metallacycle has been detected, indicating that the structural information of the macrocycle layer was perfectly transformed to the guest molecules.
Dalcanale, Reinhoudt and coworkers have reported the generation of cavitand-based coordination cages directly on surfaces by using self-assembled monolayers on gold.690,691 The formation of the molecular cages was monitored directly by atomic force microscopy (AFM). Self-assembled monolayers of a cavitand, functionalized with four cyano groups at the apical positions on the upper rim, and four alkylthioether groups on the lower rim, were prepared by adsorption on gold surfaces at 60° C from an EtOH/CHCl3 solution for 12 h.690 Monolayers of self-assembled coordination cages were obtained by soaking the self-assembled monolayers of cavitands in a solution of [M(dppp)(OTf)2] (M = Pd, Pt) and tetracyano substituted cavitand 465. The self-assembly process was monitored by directly measuring the height profile of these cages by means of AFM using substrates containing internal height references prepared by microcontact printing. Similarly, larger cavitand-based coordination cages were assembled on gold surfaces through metal induced coordination by using a cavitand having four tolylpyridyl groups at the bridgehead positions on its upper rim, and four 11-mercaptoundecanol groups on its lower rim.691 Reversible disassembly of the resulting coordination cages could also be achieved by exposing the monolayers of the cages to a solution of triethylamine in ethanol for 1 h. Triethylamine was able to shift the equilibrium towards the formation of [Pd(dppp)(NEt3)2(OTf)2] and free cavitand by competing with the Pd coordination centers. AFM studies indicated that more than half of the cages were disassembled, leaving free cavitand molecules.
5.3. Novel Characterization methods
The growth of coordination-driven self-assembly over the last decade has been spurred to some extent by the immense technological advancement made in the characterization tools for supramolecular emsembles. The development of novel characterization methods helped to answer questions associated with these complex architectures held together by coordination bonds. It is equally true that problems faced by supramolecular chemists with characterization served as the impetus for the development of new methodologies and improvement of existing characterization tools to a great extent. Thus, this symbiotic relationship has worked to the benefit of both supramolecular chemistry and analytical chemistry. Today, many novel methods as well as conventional tools, which have improved remarkably in terms of sensitivity, resolution and precision, are at hand for the characterization of supramolecular ensembles. We present here in the following sections a brief overview on some of the novel techniques that are being increasingly used in recent years in the context of elucidating the structures and properties of coordination-driven metallacycles and metallacages.
5.3.1. Mass Spectrometry
Mass spectrometry is perhaps one of the most extensively used tools for the characterization of coordination-driven supramolecular assemblies. In addition to analytical characterization of supramolecules with respect to their exact masses, isotopic patters, charge states, stoichiometries, and impurities, mass spectrometry can provide a wealth of informations about the structures, fragmentation pathways, reactivities, and thermodynamic properties of supramolecular ensembles.692,693 Many of the commonly available ionization techniques such as fast atom bombardment (FAB) and matrix-assisted laser desorption/ionization (MALDI) are incompatible with supramolecular assemblies that contain non-covalent bonds because these ionization methods are intrinsically harsh and impart large internal energy to the ions of the analyte molecule, consequently leading to fragmentation to a significant extent.
Electrospray ionization (ESI),694 which is a soft ionization technique, has emerged in the last decade as an indispensable tool for analyzing supramolecular assemblies that contain non-covalent bonds.695 ESI works by generating charged droplets through dispersion by electrospray from a solution of the desired analyte. The small droplets, through evaporation of solvents, shrink and as a result of coulombic repulsion undergo fission into even smaller droplets until desolvated ions are formed. The advantage of ESI is that it keeps the internal energy of the ions low and thus not only suppresses extensive fragmentation, but also make the intact ionization of non-covalent emsembles feasible. Moreover, the analyte can be ionized from almost any suitable solution, provided that a charge is present in the complex or can be delivered to it during ionization. The combination of ESI sources with time of flight (TOF) mass analyzer (ESI-TOF-MS) was developed to increase the ion sepration time so that much higher mass accuracy and resolution could be achieved. Similarly, combination of ESI sources with Fourier transform– ion cyclotron resonance (FT-ICR) systems allows one to investigate the isotope patterns in a broad distribution of different charge states with high accuracy and resolution. These sophisticated techniques, especially, ESI-MS, have been extensively used to analyze a wide variety of supramolecular assemblies.693
Coldspray ionization–mass spectrometry (CSI-MS), a gentler alternative to ESI-MS, was developed to characterize labile self-assembled species.696 The fundamental difference between ESI-MS and CSI-MS is that in CSI, the ion source and desolvation chamber operate at low temperature. While ESI-MS is not suitable for these compounds because of their instability, CSI-MS affords multiply charged molecular ions with attached solvent molecules and thus, analytical data on molecular mass, charge state, isotope pattern is easily obtained. Fujita and coworkers have used this technique to characterize a large number of metallacycles and metallacages.696
Another softer variant of ESI-MS, sonicspray ionization–mass spectrometry (SSI-MS) allows fragile compounds to remain unfragmented during ionization and results in singly charged species.697,698 The two ionization techniques differ in the way the solvent droplets are formed, the solution concentrations and flow rates required, the magnitude of the applied voltage, and the method of ion production. In SSI, the ion source involves two capillaries, one nested inside the other. While the inner capillary introduces a solution of analyte, the outer capillary provides a coaxial nitrogen gas flow to facilitate droplet nebulization. SSI-MS is typically operated at atmospheric pressure and without applying an electric potential to the source. The soft-ionization method reduces the fragmentation of non-covalent assemblies, fragile analytes, and thermodynamically labile emsembles. For example, transition metal (Fe3+, Co2+ and Cu2+) induced, resorcinarene-based coordination cages were characterized via SSI-mass spectrometry.699 In contrast to ESI-MS, which shows multiply charged peaks, SSI-MS spectra showed monocharged ion peaks, making the identification of the cages straightforward.
In recent years, researchers have increasingly focused on using mass spectrometry as a tool for studying various aspects coordination-driven supramolecular systems such as – host-guest chemistry, fragmentation and reactivity in solution and gas phase, and structural elucidation, through tandem mass spectroscopic experiments.693 Müller et al. have studied the encapsulation of guest cations within the cavity of a metallosupramolecular cage having M6L4 truncated tetrahedral topology.490 A remarkable difference between ESI-FT-ICR-mass spectra of M6L4 tetrahedron, [{(CdCl)3(H6Br3L)}4]8+ (H6Br3L = tris[(5-bromo-2-hydroxybenzylidene)amino]guanidinium) encapsulating NEt4+ or NEt3H+ ions as templating guest was observed. When NEt4+ was encapsulated it exactly filled up the internal space and signals for each charge state accompanied by successive losses of of NEt4Cl or by exchange of NEt4+ against H+ appeared in the mass spectrum. The simulated isotopic pattern also agreed with the experimental one. When NEt3H+ acted as guest, an additional signal due to water molecule was observed indicating that NEt3H+ leaves enough space for an additional water molecule, which is co-encapsulated in the cavity of the tetrahedron.
Tandem MS experiments have also led to the elucidation of structure and the energetics of fragmentation process in metallosupramolecular assemblies. Fragmentation pathways of the metallosupramolecular square,25 [Pt(dppp)(4,4’-bipyridine)]4(OTf)8 have been studied by Schalley et al. by carefully mass-selecting triply-charged squares in the gas phase via tandem MS experiments.700 Infrared multi-photon dissociation (IRMPD) spectra of Pt(II)-squares in their +3 charged state in the gas-phase have been recorded on a ESI-FT-ICR-mass spectrometer. The mass spectrum showed a doubly charged 3 : 3 complex (m/z 1443) and a singly charged 1 : 1 complex (m/z 912) of Pt(II)-corners and bipyridines as primary products. No doubly charged 2 : 2 complex was observed indicating that no fragmentation into two half-squares occured, although the same number of metal-nitrogen bonds needed to be broken (Scheme 151). Instead, a singly charged half-square was formed from the 3 : 3 complex (m/z 1974) as a secondary fragmentation product. The reason for the preference of one pathway over the other has been explained by invoking a rear-attack mechanism. When irradiated by the IR lasers, the internal energy of the ions increases, leading to an opening of the first bond. This gives rise to a chain-like conformer where an uncomplexed pyridine end can attack at the third metal center, thus forming a new M-N bond while facilitating the cleavage of a M-N bond to form species having m/z of 1443 and 912. Although not observed in the solution-phase by NMR experiments, triangles were formed under the gas phase conditions. To understand the effect of charge repulsion on the fragmentation pathway, tandem MS experiments performed with +5 charged states of the same squares showed strong dependencies of fragmentation patterns on the parent ions’ charge states.701 For species with a lower number of charges, expulsions of edge ligands prevailed, whereas charge separation pathways dominated over the dissociation pathways for more highly charged species.
Scheme 151.

Tandem MS experiments carried out Pt(II)-and Pd(II)-based chiral molecular rhomboids also provided access to fragmentation processes that are difficult to study in the condensed phase.216 To understand the fragmentation behavior of the rhomboids in the gas phase, the ions of interest were mass-selected in the FT-ICR cell and subjected to a collision-induced decay (CID) experiment. CID experiments revealed that doubly charged 2 : 2 complexes fragment into two identical, singly charged species followed by C-H and C-C bond activation mediated by coordinatively unsaturated metal centers. The observed β-hydrogen shift was analogous to ones encountered in transition-metal-mediated catalytic processes such as the Heck reaction.
Ion mobility spectrometry–mass spectroscopy (IMS-MS),702 a widely used method for studying conformations of protein and other large biomolecules,703 has been recently used to obtain the structures of coordination-driven metallosupramolecular assemblies that cannot be unambiguously characterized through traditional methods such as X-ray crystallography or NMR spectroscopy. Wesdemiotis, Newcome and coworkers used travelling wave IMS-MS to obtain qualitative conformational information and high-resolution mass spectra for hexacadmium metallacycles. 704, 705 Traveling wave IMS-MS, which enables two-dimensional gas-phase separation and complete deconvolution of the isotope patterns of ions with the same m/z value, facilitates the structural analysis for self-assembled supramolecules.706 With travelling wave IMS-MS, ions with different charges and shapes are separated based on their drift time in the ion mobility device, thus avoiding isomer superposition prevalent in regular ESI-MS or Fourier transform mass spectroscopy. The separated structures or charge states are subsequently characterized by their mass spectra and their fragmentation patterns in tandem mass spectra. Bowers and coworkers have used drift cell IMS-MS to obtain absolute cross sections of a number of coordination-driven supramolecular assemblies. 707 Ion-mobility studies on a molecular rectangle,109 [(1,8-bis(trans-Pt(PEt3)2)anthracene)2(4,4’-bipyridine)2](PF6)4, and a molecular triangle,55 [(3,6-bis(trans-Pt(PEt3)2)phenanthrene)3(4,4’-bipyridine)3](PF6)6, gave cross sections that matched well with the values obtained from their corresponding single crystal X-ray structures. For a trifacial prismatic box,463 [((cis-Pt(PEt3)2)6(tetrakis(4-pyridyl)cyclobutadienecyclopentadienylcobalt(I))3](OTf)12, the experimental cross section values obtained from IMS-MS agreed well with the calculated cross sections from molecular modeling studies. These studies indicate that IMS-MS can be a powerful new tool in supramolecular structure determination and is complementary to existing methods such as X-ray and NMR.
5.3.2. Solution-Phase X-ray Measurement
One of the major challenges that was and is still faced by supramolecular chemists is the inability to characterize the supramolecules structurally. The structural complexity, conformational flexibility and nanoscale dimensions of supramolecular architectures often makes them difficult to crystallize and therefore limits the use of classical single crystal X-ray diffraction methods. Although multinuclear NMR and mass spectroscopy provides information about the general features and composition of the supramolecular architectures, these techniques do not provive direct structural information. Application of multi-dimensional NMR techniques, widely used to deduce conformations of biomolecules, is limited in supramolecular assemblies due to the presence of repetitive, chemically analogous groups in the building blocks.
Solution-phase X-ray measurements provide an extraordinarily useful means to characterize supramolecular architectures whose structures and sizes cannot be determined by traditional crystallographic, spectroscopic and mass spectrometric methods. In addition, this method provides insight into the formation, shape and structure of the supramolecular species in solution. Since many of the applications of supramolecular assemblies are in solution-phase, the knowledge of their structural conformations in solution, which can sometime significantly vary from their solid-state structures, can better explain the fundamental principles involved at the atomic scale. X-ray solution scattering in both the small-angle (SAXS) and wide-angle (WAXS) regimes have been extensively used to understand structural conformations of biological macromolecules such as proteins in solution.708 While SAXS allows for low-resolution structural characterization of macromolecules in solution, WAXS afford high-resolution pattern that approach that of conventional X-ray crystallographic measurements thus providing atomic-scale insight into the structure and conformational dynamics of macromolecules in solution.
In recent years, solution-phase X-ray scattering experiments in conjunction with coordinate-based molecular modeling have been used to elucidate solution-phase structures of coordination-driven self-assembled architectures. Wide-angle X-ray scattering (WAXS) have been used to obtain direct structural information on supramolecular Pt(II)-based molecular triangle,55 [(3,6-bis(trans-Pt(PEt3)2)phenanthrene)3(4,4′-bipyridine)3](PF6)6, molecular rectangle,109 [(1,8-bis(trans-Pt(PEt3)2)anthracene)2(4,4′-bipyridine)2](PF6)4, and trigonal prism,417 [(1,8-bis(trans-Pt(PEt3)2)anthracene)3(tris(4-pyridyl)methanol)2](PF6)4 in solution. 709 X-ray scattering patterns recorded for the low-angle region (0.12-2.48 Å−1) that compared well with their calculated patterns indicated that the supramolecules retained their shape when dissolved in nitromethane solution. Scattering patterns obtained from large-angle region (0.19-16 Å−1) gave platinum-platinum distances and coordination number and the results correlated well with the theoretical calculations. Although the solution-phase structures of these assemblies are similar to that in the crystalline solid-state, they are significantly less rigid in solution as indicated by smaller diagonal platinum-platinum interaction as compared to single crystal X-ray data.
Tiede and coworkers determined the solution-phase structure of a series of Re(I)-based supramolecular squares using wide-angle X-ray scattering (WAXS) experiments.710 WAXS patterns recorded for the molecular squares showed strong interference pattern corresponding to rhenium spacing within the discrete squares. The observed interference patterns are markers of overall macrocycle structure, molecular motions, configurational flexibility, and solvent interactions. The radius of gyration (Rg) values obtained from scattering patterns agreed well with the sizes of the squares calculated from molecular modeling studies. The Re–Re edge distances determined from pair distribution function (PDF) patterns also matched well with the model and solid-state X-ray structures. The solution-phase WAXS pattern recorded for small and conformationally rigid molecular square,711 [(Re(CO)3Cl)(pyrazine)]4 showed a good match with the scattering pattern calculated from the single-crystal X-ray structure. Interestingly, the analogous molecular square, [(Re(CO)3Cl)(4,4′-bipyridine)]4, which has two different X-ray structures – one with a flat geometry711 and other with a puckered butterfly form,676 showed a solution-phase structure that differs from either of the two solid-state structures. The solution-phase structure appeared to be a bimodal distribution of conformers that are interrelated by a butterfly type motion spanning about 25° in the improper Re–Re–Re–Re torsion angle.
Hupp and coworkers have used solution-phase X-ray scattering measurements to obtain structures of porphyrin-based supramolecular assemblies in solution. Small angle X-ray scattering (SAXS) patterns obtained for a series of supramolecular porphyrinic prisms assembled from Zn(II)-containing porphyrin monomers, dimers or trimers gave radii of gyration (Rg) values that agreed well with the values calculated from modeling studies.434 WAXS measurements to obtain information about interatomic spacing within the assemblies showed Zn-Zn interactions unique to prism assemblies. Likewise, the structures of other porphyrinic assemblies such as a tetrameric Zn(II)-porphyrin assembly276 and cavity-tailored porphyrin boxes517 in solution were ascertained using solution-phase X-ray scattering measurements. Similarly, Tiede et al. were able to structurally characterize a hexameric, diphenylene-linked Zn(II)-porphyrin macrocycle and the corresponding host-guest complex formed by inclusion of a tripyridyl guest in solution using X-ray scattering measurements.712,713 X-ray scattering patterns coupled with pair distance function (PDF) analyses revealed that the herameric assembly is structurally non-rigid and exist as a distribution of conformers. The host assembly underwent an expansion upon guest inclusion.
5.4. Photophysical Studies using Laser Spectroscopy
Self-assembly, which provides a facile way to assemble a large number of molecules into highly organized supramolecular architectures, has been applied to assemble chromophores and electroactive molecules for use as molecular devices and machines in nanoscale dimensions.714 These novel materials have found applications in molecular electronics, photonics and photovoltaics. In these multi-chromophoric assemblies, collective excitations at multiple sites contribute to the observed optical behavior. A wide variety of two- and three-dimensional supramolecular systems containing perylene imide and diimide dyes, and porphyrins have been assembled to study the electron transport and charge separation processes.715 Recently, various spectroscopic techniques such as transient absorption, two photon absorption, steady-state fluorescence, and time-resolved fluorescence have been used to explore the charge transfer and photophysical properties of self-assembled metallacycles and metallacages.
The crystal structure of the LH2 light-harvesting antenna system of purple photosynthetic bacteria Rhodopseudomonas acidophila revealed a circularly arranged chromophoric assembly of bacteriochlorophylls, leading to considerable efforts to develop biomimetic artificial photosynthetic systems that may offer insights into the photophysics of energy collection and transfer in these proteins.715 Multi-porphyrin ensembles can function as light-harvesting antennae as in natural photosynthetic systems if sufficiently fast electron transfer between the porphyrin subunits can be achieved. The most common approach to achieve efficient electron transfer is to design ensembles that have photoactive porphyrin moieties and electron deficient chromophores. Photophysical studies on self-assembled Zn(II)-trisporphyrin prisms having 1,4-diazabicyclo[2.2.2]octane (DABCO), 4,4′-bipyridine and 5,15-(4-pyridyl)-10,20-phenyl-porphyrin (4′-trans-DPyP) as pillars have shown efficient photoinduced energy transfer from the triporphyrin unit to the pillars upon excitation of both components within the assemblies.515 Luminescence experiments showed that when 4′-trans-DPyP was used as a pillar, a highly efficient photoinduced electron transfer (96% efficiency) from the trisporphyrin units to the 4′-trans-DPyP pillars occurred. The analogous trigonal prisms having ditopic N-donor ligands based on perylene tetracarboxylic acid bisimide also showed efficient photoinduced electron transfer from the Zn porphyrin units to the axially coordinated bisimide pillars, occuring from the singlet excited states localized on both chromophores.437
Photoinduced energy transfer processes have also been observed in self-assembled porphyrin/perylene bisimide ensembles.438 The metallosupramolecular ensembles (ZnMC)2(PBI)2 were prepared by axial coordination of N,N’-dipyridyl-functionalized perylene bisimide (PBI) dyes to the zinc ion centers of two [2 + 2] porphyrin metallacycles (ZnMC) [trans,cis,cis-RuCl2(CO)2(Zn·4′-cis-DPyP)]2). These complexes were structurally characterized by single crystal X-ray crystallography.716 Ultrafast absorption techniques showed that the observed photophysical behaviors depended on the substituents present at the bay area of the PBI pillars. When a tert-butylphenoxy substituent was present, efficient quenching of both perylene bisimide and the zinc porphyrin chromophores was observed, leading to a charge separated state, PBI−-Zn+, in which a perylene bisimide unit was reduced and zinc porphyrin was oxidized. In contrast, PBI bearing pyrrolidinyl substituents led to singlet energy transfer from the Zn porphyrin chromophores to the perylene bisimide units with an efficiency of 0.7. This value was due to a competing quenching mechanism, leading to the charge separated state, PBI−-Zn+.438
Wasielewski and coworkers investigated energy transfer processes in a cyclic self-assembled Zn(II)-porphyrin tetramer by transient absorption measurements using femtosecond laser pulses. A cyclic tetramer self-assembled from meso-ethynylpyridyl-functionalized Zn(II)-porphyrin showed efficient electron transfer between the porphyrin units.276 The electron transfer rate was found to be ~8 times faster than the analogous Zn(II)-porphyrin tetramers.717,718 A self-assembled cyclic zinc chlorophyll tetramer exhibited an ultrafast intramolecular energy transfer rate as determined from femtosecond singlet-singlet annihilation and transient absorption anisotropy studies.719 The cyclic tetramer upon excitation with 120 fs laser pulses yielded its lowest singlet excited state in which the rate of energy transfer was ~20 times faster than the tetramer in the ground state. However, this ultrafast energy transfer rate is still ~10 times slower than what is observed in the photosynthetic proteins. This difference is attributed to the larger transition dipole moment found in chlorophyll as compared to porphyrin. Thus, subtle changes in the structures of assemblies can profoundly affect electron transfer rates.
Extending this concept, butadyine-linked Zn(II)-chlorophyll and porphyrin dimers were assembled into trigonal prisms to study the photophysics of electron transfer dynamics.720 The butadiyne-linked dimers, upon treatment with three-fold symmetric ligands such as triethynylpyridine, assembled into prismatic structures in which the dimers comprised the faces of the prism. The incorporation of butadiyne linkages increased the ability of these dimers to absorb the solar spectrum over a broad range of wavelengths. The prismatic architecture restricted the rotation of the macrocycles about the joining butadiyne bond. Photoexcitation of these prismatic assemblies, using femtosecond transient absorption spectroscopy, revealed efficient, through-space energy transfer between the macrocyclic dimers within the prisms. Use of a three-fold symmetric ligand possessing longer arms gave larger trigonal prisms, resulting in more efficient energy transfers between the prism faces.
Recently, ultrafast excited state relaxation dynamics of a number of Pt(II)-containing self-assembled metallacycles and metallacages were studied using femtosecond measurements of fluorescence upconversion and transient absorption spectra.721 In case of the metallacycles, the charge transfer dynamics and intersystem crossing were found to be dependent on the geometries and dimensions of the metallacycles. For rectangular anthracene-containing metallacycles, the electronic coupling between adjacent ligands was relatively weak, whereas for the triangular phenanthrene-containing structures, there was a clear interaction between the conjugated ligand and the metal center. The photophysical properties of the coordination-driven self-assembled metallosupramolecular rhomboids having donor ligands 1,2-bis(3-pyridyl)ethyne and 1,4-bis(3-pyridyl)-1,3-butadiyne were also investigated by the use of both time-resolved fluorescence spectroscopy and quantum chemistry calculations.722 Steady-state absorption and fluorescence as well as the time-correlated single photon counting techniques revealed that the nature of the excited states for these metallosupramolecular rhomboids varies with the acetylene chain length of the donor ligands and with the different conformers. Thus, the results demonstrate that differences in the dimensionality and structure of the metallacycles results in different optical properties, which may be utilized in the design of nonlinear optical materials and potential longer-lived excited state materials for further electronic applications.
6. CONCLUSIONS AND PROGNOSTICATION
As described above, coordination-driven self-assembly is a vibrant, active field. It is evident that considerable progress has been made since the beginnings of the field some two decades ago. Early work involved the self-assembly of helicates, grids and other infinite ensembles. Out of this area evolved the self-assembly of finite closed systems, both two-dimensional and three-dimensional with well-defined shapes and sizes that is the topic of this review. Early work in this field focused on the development of rational methodologies for the self-assembly of predesigned systems along with their characterization. At the start, only architechtures based on just two components were prepared primarily involving Pt and Pd pyridine interactions. However, other transition metals and different donor units were rapidly adopted. As it was and still is a challenge to get suitable single crystals of these large systems with cavities filled with solvent for X-ray structure analysis, proper characterization was and remains a challenge. Multinuclear, high-resolution NMR and electrospray mass spectrometry are the primary and essential tools for proper characterization along with X-ray and more recently synchrotron X-ray methods.
As seen in this review, considerable progress has been made in our understanding of these abiological, coordination-based self-assembly processes. Moreover, multi-component self-assembly along with self-sorting and self-organization has been developed. However, the greatest progress, mostly in the last half dozen years has been in the uses and applications of these self-assembled metallacycles and metallacages. Both 2D and 3D systems have been used in chemosensing and host-guest chemistry. Particularly interesting are Fujita’s use of 3D cages for the encapsulation of reactive species as described above. However, the most recent and arguably interesting applications have been in catalysis, use as microreactors and biological applications. In particular Raymond and Bergman have elegantly exploited the cavities of self-assembled 3D cages for enzyme-like catalysis as detailed in section 4.5.2. Likewise, Fujita and coworkers have used 3D cages as nano reaction vassels to carry out unusual reactions as described in the preceding sections. In the last couple of years the biological applications of self-assembled metallacycles and metallacages have been described. Particularly relevant are the anti-tumor studies of Therrien and coworkers and the investigation of Sleiman and coworkers on nucleic acid interactions.
It is clear that coordination-driven self-assembly will continue to be an active area of research and an important component of supramolecular chemistry and nanoscience. Future developments are likely to involve the formation and characterization of even more complex ensembles and their applications. In particular we are likely to see further advances in multi-component self-assembly of functionalized systems. Emphasis will likely be on applications and uses both in material science and biochemical and biomedical areas. More sophisticated sensors as well as better and more useful catalytic applications will undoubtedly emerge. Likewise, the biomedical applications of these systems, such as potential anti-tumor agents, protein and nucleic acid interactions etc. are new vista that will see increasing activity. Self-assembly in general, and coordination-driven self-assembly in particular, will play an important role in the “bottom-up” manufacturing of novel materials in the evolving nanotechnology arena. Likewise, similarity and evolving synergy between coordination-driven self-assembly of metallacages and MOFs will lead to new developments and applications.
Scheme 74.

Acknowledgements
PJS thanks the NIH and NSF for continued financial support over the years of our studies in coordination-driven self-assembly. PSM thanks the Department of Science and Technology (DST), India, for the financial support. We thank Dr. Timothy R Cook for help with editing the manuscript.
Biography
Biographies

Rajesh Chakrabarty was born and grew up in Manipur, India. He received his PhD from Gauhati University working with Prof. Birinchi K Das focusing on homogeneous and heterogeneous catalysis using metal-oxo clusters. He then moved to Indian Institute of Science, Bangalore as a CSIR Research Associate to work with Dr. Partha Sarathi Mukherjee in the field of supramolecular chemistry. Since the fall of 2009, Rajesh is working in the group of Prof. Peter J Stang as postdoctoral fellow at the University of Utah. His current research interests include functionalization of supramolecules for biological applications.

Partha Sarathi Mukherjee was born in 1973 in West Bengal, India. He earned his B.Sc with honours in chemistry from Burdwan University in 1995 and M.Sc in chemistry in 1998 from Jadavpur University. He did his PhD from the Indian Association for the Cultivation of Science in 2002 working with Prof. N. Ray Chaudhuri and his postdoctoral from University of Utah with Prof. Peter J Stang. In 2004 he moved to University of Goettingen to work with Prof. Herbert W Roesky as a Humboldt fellow. He has been at the Indian Institute of Science, Bangalore since July 2005 where he is currently an Associate Professor. His current research interests are in the areas of self-assembly of functional molecular architectures, organometallic materials and magnetic clusters/polyclusters. He is the recipient of young scientist medal of the Indian National Science Academy, Associateship of the Indian Academy of Sciences, and Microsoft Research outstanding young faculty award. Dr. Mukherjee has published 78 original papers in peer-reviewed journals.

Peter J Stang is a Distinguished Professor of Chemistry at the University of Utah, as well as the Editor of the Journal of Americal Chemical Society. He is a member of the US National Academy of Sciences and a Foreign Member of the Chinese (CAS) and Hungarian Academies of Sciences. He was recently listed by the Times Higher Education among the 100 top chemists, who had the highest citation impact scores (citations/papers for the decade of 2000-2010).
References
- 1.Pedersen CJ. Angew. Chem. Int. Ed. Engl. 1988;27:1021. [Google Scholar]
- 2.Lehn J-M. Angew. Chem. Int. Ed. Engl. 1988;27:89. [Google Scholar]
- 3.Cram DJ. Angew. Chem. Int. Ed. Engl. 1988;27:1009. [Google Scholar]
- 4.(a) Steed JW, Turner DR, Wallace KJ. Core Concepts in Supramolecular Chemistry and Nanochemistry. John Wiley & Sons; West Sussex: 2007. [Google Scholar]; (b) Steed JW, Atwood JL. Supramolecular Chemistry. John Wiley & Sons; West Sussex: 2000. [Google Scholar]
- 5.Lehn J-M. Supramolecular Chemistry: Concepts and Perspectives. VCH; Weinheim: 1995. [Google Scholar]
- 6.Sauvage J-P, Dietrich-Buchecker C. Molecular Catenanes, Rotaxanes, and Knots: A Journey Through the World of Molecular Topology. Wiley-VCH; Weinheim: 1999. [Google Scholar]
- 7.For recent reviews see: Beves JE, Leigh DA. Nat. Chem. 2010;2:708. doi: 10.1038/nchem.745. Fang L, Olson MA, Benítez D, Tkatchouk E, Goddard WA, III, Stoddart JF. Chem. Soc. Rev. 2010;39:17. doi: 10.1039/b917901a. Durot S, Reviriego F, Sauvage J-P. Dalton Trans. 2010;39:10557. doi: 10.1039/c0dt00457j. Faiz JA, Heitz V, Sauvage J-P. Chem. Soc. Rev. 2009;38:422. doi: 10.1039/b710908n. Crowley JD, Goldup SM, Lee A-L, Leigh DA, McBurney RT. Chem. Soc. Rev. 2009;38:1530. doi: 10.1039/b804243h. Balzani V, Credi A, Venturi M. Chem. Soc. Rev. 2009;38:1542. doi: 10.1039/b806328c. Stoddart JF. Chem. Soc. Rev. 2009;38:1802. doi: 10.1039/b819333a. Champin B, Mobian P, Sauvage J-P. Chem. Soc. Rev. 2007;36:358. doi: 10.1039/b604484k. Sauvage J-P. Chem. Commun. 2005:1507. doi: 10.1039/b500680p. Bonnet S, Collin J-P, Koizumi M, Mobian P, Sauvage J-P. Adv. Mater. 2006;18:1239. Dietrich-Buchecker C, Colasson BX, Sauvage J-P. Top. Curr. Chem. 2005;249:261. Ruben M, Rojo J, Romero-Salguero FJ, Uppadine LH, Lehn J-M. Angew. Chem. Int. Ed. 2004;43:3644. doi: 10.1002/anie.200300636.
- 8.Reviews: Stang PJ. J. Org. Chem. 2009;74:2. doi: 10.1021/jo801682d. Northrop BH, Chercka D, Stang PJ. Tetrahedron. 2008;64:11495. doi: 10.1016/j.tet.2008.08.062. Seidel SR, Stang PJ. Acc. Chem. Res. 2002;35:972. doi: 10.1021/ar010142d. Leininger S, Olenyuk B, Stang PJ. Chem. Rev. 2000;100:853. doi: 10.1021/cr9601324. Stang PJ. Chem. Eur. J. 1998;4:19. Olenyuk B, Fechtenkötter A, Stang PJ. J. Chem. Soc., Dalton Trans. 1998:1707. Stang PJ, Olenyuk B. Acc. Chem. Res. 1997;30:502.
- 9.Reviews: Caulder DL, Raymond KN. J. Chem. Soc., Dalton Trans. 1999:1185. Caulder DL, Raymond KN. Acc. Chem. Res. 1999;32:975. Caulder DL, Brückner C, Powers RE, König S, Parac TN, Leary JA, Raymond KN. J. Am. Chem. Soc. 2001;123:8923. doi: 10.1021/ja0104507.
- 10.Reviews: Fujita M, Tominaga M, Hori A, Therrien B. Acc. Chem. Res. 2005;38:371. doi: 10.1021/ar040153h. Fujita M, Umemoto K, Yoshizawa M, Fujita N, Kusukawa T, Biradha K. Chem. Commun. 2001:509. Fujita M. Struct. Bonding (Berlin) 2000;96:177. Fujita M. Chem. Soc. Rev. 1998;27:417.
- 11.Reviews: Oliveri CG, Ulmann PA, Wiester MJ, Mirkin CA. Acc. Chem. Res. 2008;41:1618. doi: 10.1021/ar800025w. Gianneschi NC, Masar MS, III, Mirkin CA. Acc. Chem. Res. 2005;38:825. doi: 10.1021/ar980101q. Holliday BJ, Mirkin CA. Angew. Chem. Int. Ed. 2001;40:2022.
- 12.Reviews: Cotton FA, Lin C, Murillo CA. Proc. Natl. Acad. Sci. USA. 2002;99:4810. doi: 10.1073/pnas.012567599. Cotton FA, Lin C, Murillo CA. Acc. Chem. Res. 2001;34:759. doi: 10.1021/ar010062+.
- 13.De S, Mahata K, Schmittel M. Chem. Soc. Rev. 2010;39:1555. doi: 10.1039/b922293f. [DOI] [PubMed] [Google Scholar]
- 14.Nitschke JR. Acc. Chem. Res. 2007;40:103. doi: 10.1021/ar068185n. [DOI] [PubMed] [Google Scholar]
- 15.Reviews: Wiester MJ, Ulmann PA, Mirkin CA. Angew. Chem. Int. Ed. 2011;50:114. doi: 10.1002/anie.201000380. Breiner B, Clegg JK, Nitschke JR. Chem. Sci. 2011;2:51. Laughrey Z, Gibb BC. Chem. Soc. Rev. 2011;40:363. doi: 10.1039/c0cs00030b. Saalfrank RW, Maid H, Scheurer A. Angew Chem. Int. Ed. 2008;47:8794. doi: 10.1002/anie.200702075. Ruben M, Lehn J-M, Müller P. Chem. Soc. Rev. 2006;35:1056. doi: 10.1039/b517267p. Schmittel M, Kalsani V. Top. Curr. Chem. 2005;245:1. Collin J-P, Heitz V, Sauvage J-P. Top. Curr. Chem. 2005;262:29. You C-C, Dobrawa R, Saha-Möller CR, Würthner F. Top. Curr. Chem. 2005;258:39.
- 16.Reviews: Yoshizawa M, Fujita M. Bull. Chem. Soc. Jpn. 2010;83:609. Yoshizawa M, Klosterman JK, Fujita M. Angew. Chem. Int. Ed. 2009;48:3418. doi: 10.1002/anie.200805340. Maurizot V, Yoshizawa M, Kawano M, Fujita M. Dalton Trans. 2006:2750. doi: 10.1039/b516548m.
- 17.Reviews: Pluth MD, Bergman RG, Raymond KN. Acc. Chem. Res. 2009;42:1650. doi: 10.1021/ar900118t. Fiedler D, Leung DH, Bergman RG, Raymond KN. Acc. Chem. Res. 2005;38:351. doi: 10.1021/ar040152p. Davis AV, Yeh RM, Raymond KN. Proc. Natl. Acad. Sci. USA. 2002;99:4793. doi: 10.1073/pnas.052018299.
- 18.van Leeuwen PWNM. Supramolecular Catalysis. Wiley-VCH; Weinheim: 2008. [Google Scholar]
- 19.Reviews: Meeuwissen J, Reek JNH. Nat. Chem. 2010;2:615. doi: 10.1038/nchem.744. Koblenz TS, Wassenaar J, Reek JNH. Chem. Soc. Rev. 2008;37:247. doi: 10.1039/b614961h. Kleij AW, Reek JNH. Chem. Eur. J. 2006;12:4218. doi: 10.1002/chem.200500875. Wilkinson MJ, van Leeuwen PWNM, Reek JNH. Org. Biomol. Chem. 2005;3:2371. doi: 10.1039/b503407h.
- 20.Reviews: Hupp JT. Struct. Bonding (Berlin) 2006;121:145. Snurr RQ, Hupp JT, Nguyen ST. AIChE J. 2004;50:1090. Nguyen ST, Gin DL, Hupp JT, Zhang X. Proc. Natl. Acad. Sci. USA. 2001;98:11849. doi: 10.1073/pnas.201373898. Dinolfo PH, Hupp JT. Chem. Mater. 2001;13:3113.
- 21.Li S-S, Northrop BH, Yuan Q-H, Wan L-J, Stang PJ. Acc. Chem. Res. 2009;42:249. doi: 10.1021/ar800117j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kudernac T, Lei S, Elemans JAAW, De Feyter S. Chem. Soc. Rev. 2009;38:402. doi: 10.1039/b708902n. [DOI] [PubMed] [Google Scholar]
- 23.Fujita M, Yazaki J, Ogura K. J. Am. Chem. Soc. 1990;112:5645. [Google Scholar]
- 24.Fujita M, Yazaki J, Ogura K. Chem. Lett. 1991:1031. [Google Scholar]
- 25.Stang PJ, Cao DH. J. Am. Chem. Soc. 1994;116:4981. [Google Scholar]
- 26.Fujita M, Oguro D, Miyazawa M, Oka H, Yamaguchi K, Ogura K. Nature. 1995;378:469. [Google Scholar]
- 27.Yu S-Y, Kusukawa T, Biradha K, Fujita M. J. Am. Chem. Soc. 2000;122:2665. [Google Scholar]
- 28.Fujita N, Biradha K, Fujita M, Sakamoto S, Yamaguchi K. Angew. Chem. Int. Ed. 2001;40:1718. [PubMed] [Google Scholar]
- 29.Bar AK, Chakrabarty R, Mostafa G, Mukherjee PS. Angew. Chem. Int. Ed. 2008;47:8455. doi: 10.1002/anie.200803543. [DOI] [PubMed] [Google Scholar]
- 30.Review: Pitt MA, Johnson DW. Chem. Soc. Rev. 2007;36:1441. doi: 10.1039/b610405n.
- 31.Fujita M, Aoyagi M, Ogura K. Inorg. Chim. Acta. 1996;246:53. [Google Scholar]
- 32.Fujita M, Ibukuro F, Hagihara H, Ogura K. Nature. 1994;367:720. [Google Scholar]
- 33.Fujita M, Ibukuro F, Seki H, Kamo O, Imanari M, Ogura K. J. Am. Chem. Soc. 1996;118:899. [Google Scholar]
- 34.Fujita M, Ibukuro F, Yamaguchi K, Ogura K. J. Am. Chem. Soc. 1995;117:4175. [Google Scholar]
- 35.Lu J, Turner DR, Harding LP, Byrne LT, Baker MV, Batten SR. J. Am. Chem. Soc. 2009;131:10372. doi: 10.1021/ja9041912. [DOI] [PubMed] [Google Scholar]
- 36.Schmitz M, Leininger S, Fan J, Arif AM, Stang PJ. Organometallics. 1999;18:4817. [Google Scholar]
- 37.Habicher T, Nierengarten J-F, Gramlich V, Diederich F. Angew. Chem. Int. Ed. 1998;37:1916. [Google Scholar]
- 38.Chatterjee B, Noveron JC, Resendiz MJE, Liu J, Yamamoto T, Parker D, Cinke M, Nguyen CV, Arif AM, Stang PJ. J. Am. Chem. Soc. 2004;126:10645. doi: 10.1021/ja0388919. [DOI] [PubMed] [Google Scholar]
- 39.Yang H-B, Das N, Huang F, Hawkridge AM, Díaz DD, Arif AM, Finn MG, Muddiman DC, Stang PJ. J. Org. Chem. 2006;71:6644. doi: 10.1021/jo0608117. [DOI] [PubMed] [Google Scholar]
- 40.Review: Zangrando E, Casanova M, Alessio E. Chem. Rev. 2008;108:4979. doi: 10.1021/cr8002449.
- 41.Murray HH, Raptis RG, Fackler JP., Jr. Inorg. Chem. 1988;27:26. [Google Scholar]
- 42.Raptis RG, Fackler JP., Jr. Inorg. Chem. 1990;29:5003. [Google Scholar]
- 43.For examplex see: Dias HVR, Diyabalanage HVK, Rawashdeh-Omary MA, Franzman MA, Omary MA. J. Am. Chem. Soc. 2003;125:12072. doi: 10.1021/ja036736o. Dias HVR, Diyabalanage HVK, Eldabaja MG, Elbjeirami O, Rawashdeh-Omary MA, Omary MA. J. Am. Chem. Soc. 2005;127:7489. doi: 10.1021/ja0427146. Omary MA, Elbjeirami O, Gamage CSP, Sherman KM, Dias HVR. Inorg. Chem. 2009;48:1784. doi: 10.1021/ic8021326. Dias HVR, Gamage CSP, Keltner J, Diyabalanage HVK, Omari I, Eyobo Y, Dias NR, Roehr N, McKinney L, Poth T. Inorg. Chem. 2007;46:2979. doi: 10.1021/ic062374k.
- 44.Rawashdeh-Omary MA, Rashdan MD, Dharanipathi S, Elbjeirami O, Ramesh P, Dias HVR. Chem. Commun. 2011;47:1160. doi: 10.1039/c0cc03964k. [DOI] [PubMed] [Google Scholar]
- 45.Dias HVR, Singh S, Campana CF. Inorg. Chem. 2008;47:3943. doi: 10.1021/ic800396t. [DOI] [PubMed] [Google Scholar]
- 46.Dias HVR, Gamage CSP. Angew. Chem. Int. Ed. 2007;46:2192. doi: 10.1002/anie.200604585. [DOI] [PubMed] [Google Scholar]
- 47.Hall JR, Loeb SJ, Shimizu GKH, Yap GPA. Angew. Chem. Int. Ed. 1998;37:121. [Google Scholar]
- 48.Espinet P, Soulantica K, Charmant JPH, Orpen AG. Chem. Commun. 2000:915. doi: 10.1021/ic990634a. [DOI] [PubMed] [Google Scholar]
- 49.Vicente J, Chicote M-T, Alvarez-Falcón MM, Jones PG. Chem. Commun. 2004:2658. doi: 10.1039/b409921d. [DOI] [PubMed] [Google Scholar]
- 50.Lippert B, Miguel PJS. Chem. Soc. Rev. 2011 DOI: 10.1039/c1cs15090a. [Google Scholar]
- 51.Schnebeck R-D, Freisinger E, Lippert B. Chem. Commun. 1999:675. [Google Scholar]
- 52.Schnebeck R-D, Freisinger E, Glahé F, Lippert B. J. Am. Chem. Soc. 2000;122:1381. [Google Scholar]
- 53.Hwang S-H, Moorefield CN, Fronczek FR, Lukoyanova O, Echegoyen L, Newkome GR. Chem. Commun. 2005:713. doi: 10.1039/b409348h. [DOI] [PubMed] [Google Scholar]
- 54.Romero FM, Ziessel R, Dupont-Gervais A, Van Dorsselaer A. Chem. Commun. 1996:551. [Google Scholar]
- 55.Kryschenko YK, Seidel SR, Arif AM, Stang PJ. J. Am. Chem. Soc. 2003;125:5193. doi: 10.1021/ja030018k. [DOI] [PubMed] [Google Scholar]
- 56.Qin Z, Jennings MC, Puddephatt RJ. Chem. Commun. 2001:2676. [Google Scholar]
- 57.Qin Z, Jennings MC, Puddephatt RJ. Inorg. Chem. 2003;42:1956. doi: 10.1021/ic020322z. [DOI] [PubMed] [Google Scholar]
- 58.Bar AK, Chakrabarty R, Chi K-W, Batten SR, Mukherjee PS. Dalton Trans. 2009:3222. doi: 10.1039/b900118b. [DOI] [PubMed] [Google Scholar]
- 59.Ghosh S, Turner DR, Batten SR, Mukherjee PS. Dalton Trans. 2007:1869. doi: 10.1039/b702353g. [DOI] [PubMed] [Google Scholar]
- 60.Teo P, Koh LL, Hor TSA. Inorg. Chem. 2008;47:6464. doi: 10.1021/ic8007238. [DOI] [PubMed] [Google Scholar]
- 61.Teo P, Koh LL, Hor TSA. Chem. Commun. 2007:2225. doi: 10.1039/b618309c. [DOI] [PubMed] [Google Scholar]
- 62.Park K-M, Kim S-Y, Heo J, Whang D, Sakamoto S, Yamaguchi K, Kim K. J. Am. Chem. Soc. 2002;124:2140. doi: 10.1021/ja011654q. [DOI] [PubMed] [Google Scholar]
- 63.Schweiger M, Seidel SR, Arif AM, Stang PJ. Angew. Chem. Int. Ed. 2001;40:3467. doi: 10.1002/1521-3773(20010917)40:18<3467::aid-anie3467>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 64.Tzeng B-C, Kuo J-H, Lee Y-C, Lee G-H. Inorg. Chim. Acta. 2008;361:2515. [Google Scholar]
- 65.Berben LA, Faia MC, Crawford NRM, Long JR. Inorg. Chem. 2006;45:6378. doi: 10.1021/ic060570l. [DOI] [PubMed] [Google Scholar]
- 66.Shan N, Vickers SJ, Adams H, Ward MD, Thomas JA. Angew. Chem. Int. Ed. 2004;43:3938. doi: 10.1002/anie.200460022. [DOI] [PubMed] [Google Scholar]
- 67.Shan N, Ingram JD, Easun TL, Vickers SJ, Adams H, Ward MD, Thomas JA. Dalton Trans. 2006:2900. doi: 10.1039/b516080b. [DOI] [PubMed] [Google Scholar]
- 68.Schmittel M, Mahata K. Chem. Commun. 2008:2550. doi: 10.1039/b801462k. [DOI] [PubMed] [Google Scholar]
- 69.Schmittel M, Mahata K. Inorg. Chem. 2009;48:822. doi: 10.1021/ic8021084. [DOI] [PubMed] [Google Scholar]
- 70.Würthner F, You C-C, Saha-Möller CR. Chem. Soc. Rev. 2004;33:133. doi: 10.1039/b300512g. [DOI] [PubMed] [Google Scholar]
- 71.Stang PJ, Olenyuk B, Fan J, Arif AM. Organometallics. 1996;15:904. [Google Scholar]
- 72.Whiteford JA, Lu CV, Stang PJ. J. Am. Chem. Soc. 1997;119:2524. [Google Scholar]
- 73.Stang PJ, Cao DH, Chen K, Gray GM, Muddiman DC, Smith RD. J. Am. Chem. Soc. 1997;119:5163. [Google Scholar]
- 74.Stang PJ, Fan J, Olenyuk B. Chem. Commun. 1997:1453. [Google Scholar]
- 75.Stang PJ, Chen K, Arif AM. J. Am. Chem. Soc. 1995;117:8793. [Google Scholar]
- 76.Manna J, Kuehl CJ, Whiteford JA, Stang PJ, Muddiman DC, Hofstadler SA, Smith RD. J. Am. Chem. Soc. 1997;119:11611. [Google Scholar]
- 77.Goeb S, Prusakova V, Wang X, Vézinat A, Sallé M, Castellano FN. Chem. Commun. 2011;47:4397. doi: 10.1039/c1cc10239g. [DOI] [PubMed] [Google Scholar]
- 78.Sun SS, Lees AJ. Inorg. Chem. 2001;40:3154. doi: 10.1021/ic0101681. [DOI] [PubMed] [Google Scholar]
- 79.Würthner F, Sautter A. Chem. Commun. 2000:445. [Google Scholar]
- 80.You CC, Würthner F. J. Am. Chem. Soc. 2003;125:9716. doi: 10.1021/ja029648x. [DOI] [PubMed] [Google Scholar]
- 81.Sautter A, Kaletas BK, Schmid DG, Dobrawa R, Zimine M, Jung G, van Stokkum IHM, Cola LD, Williams RM, Würthner F. J. Am. Chem. Soc. 2005;127:6719. doi: 10.1021/ja0448216. [DOI] [PubMed] [Google Scholar]
- 82.Kraft S, Hanuschek E, Beckhaus R, Haase D, Saak W. Chem. Eur. J. 2005;11:969. doi: 10.1002/chem.200400880. [DOI] [PubMed] [Google Scholar]
- 83.Theilmann O, Saak W, Haase D, Beckhaus R. Organometallics. 2009;28:2799. [Google Scholar]
- 84.Janzen DE, Patel KN, VanDerveer DG, Grant GJ. Chem. Commun. 2006:3540. doi: 10.1039/b606753k. [DOI] [PubMed] [Google Scholar]
- 85.Teo P, Koh LL, Hor TSA. Inorg. Chem. 2003;42:7290. doi: 10.1021/ic034690u. [DOI] [PubMed] [Google Scholar]
- 86.Ghosh S, Mukherjee PS. Inorg. Chem. 2009;48:2605. doi: 10.1021/ic802254f. [DOI] [PubMed] [Google Scholar]
- 87.Shanmugaraju S, Bar AK, Chi K-W, Mukherjee PS. Organometallics. 2010;29:2971. [Google Scholar]
- 88.Gontier E, Bellec N, Brignou P, Gohier A, Guerro M, Roisnel T, Lorcy D. Org. Lett. 2010;12:2386. doi: 10.1021/ol1007422. [DOI] [PubMed] [Google Scholar]
- 89.Gianneschi NC, Mirkin CA, Zakharov LN, Rheingold AL. Inorg. Chem. 2002;41:5326. doi: 10.1021/ic025875o. [DOI] [PubMed] [Google Scholar]
- 90.Liu X, Stern CL, Mirkin CA. Organometallics. 2002;21:1017. [Google Scholar]
- 91.Fujita M, Sasaki O, Mitsuhashi T, Fujita T, Yazaki J, Yamaguchi K, Ogura K. Chem. Commun. 1996:1535. [Google Scholar]
- 92.Weilandt T, Troff RW, Saxell H, Rissanen K, Schalley CA. Inorg. Chem. 2008;47:7588. doi: 10.1021/ic800334k. [DOI] [PubMed] [Google Scholar]
- 93.Holló-Sitkei E, Tárkányi G, Párkányi L, Megyes T, Besenyei G. Eur. J. Inorg. Chem. 2008:1573. [Google Scholar]
- 94.Ferrer M, Mounir M, Rossell O, Ruiz E, Maestro MA. Inorg. Chem. 2003;42:5890. doi: 10.1021/ic034489j. [DOI] [PubMed] [Google Scholar]
- 95.Ferrer M, Gutiérrez A, Mounir M, Rossell O, Ruiz E, Rang A, Engeser M. Inorg. Chem. 2007;46:3395. doi: 10.1021/ic062373s. [DOI] [PubMed] [Google Scholar]
- 96.Ferrer M, Pedrosa A, Rodríguez L, Rossell O, Vilaseca M. Inorg. Chem. 2010;49:9438. doi: 10.1021/ic101150p. [DOI] [PubMed] [Google Scholar]
- 97.Sautter A, Schmid DG, Jung G, Würthner F. J. Am. Chem. Soc. 2001;123:5254. doi: 10.1021/ja004360y. [DOI] [PubMed] [Google Scholar]
- 98.You C-C, Würthner F. J. Am. Chem. Soc. 2003;125:9716. doi: 10.1021/ja029648x. [DOI] [PubMed] [Google Scholar]
- 99.Würthner F, Sautter A. Org. Biol. Chem. 2003;1:240. doi: 10.1039/b208582h. [DOI] [PubMed] [Google Scholar]
- 100.Schweiger M, Seidel SR, Arif AM, Stang P. J. Inorg. Chem. 2002;41:2556. doi: 10.1021/ic0112692. [DOI] [PubMed] [Google Scholar]
- 101.Yamamoto T, Arif AM, Stang PJ. J. Am. Chem. Soc. 2003;125:12309. doi: 10.1021/ja0302984. [DOI] [PubMed] [Google Scholar]
- 102.Yu S-Y, Huang H-P, Li S-H, Jiao Q, Li Y-Z, Wu B, Sei Y, Yamaguchi K, Pan Y-J, Ma H-W. Inorg. Chem. 2005;41:9471. doi: 10.1021/ic0509332. [DOI] [PubMed] [Google Scholar]
- 103.Sun Q-F, Wong KM-C, Liu L-X, Huang H-P, Yu S-Y, Yam VW-W, Li Y-Z, Pan Y-J, Yu K-C. Inorg. Chem. 2008;47:2142. doi: 10.1021/ic701344p. [DOI] [PubMed] [Google Scholar]
- 104.Kraus T, Buděšínský M, Cvačka J, Sauvage J-P. Angew. Chem. Int. Ed. 2006;45:258. doi: 10.1002/anie.200503163. [DOI] [PubMed] [Google Scholar]
- 105.Voignier J, Frey J, Kraus T, Buděšínský M, Cvačka J, Heitz V, Sauvage J-P. Chem. Eur. J. 2011;17:5404. doi: 10.1002/chem.201003592. [DOI] [PubMed] [Google Scholar]
- 106.Champin B, Sartor V, Sauvage J-P. New J. Chem. 2008;32:1048. [Google Scholar]
- 107.Thanasekaran P, Liao R-T, Liu Y-H, Rajendran T, Rajagopal S, Liu K-L. Coord. Chem. Rev. 2005;249:1085. [Google Scholar]
- 108.Slone RV, Benkstein KD, Bélanger S, Hupp JT, Guzei IA, Rheingold AL. Coord. Chem. Rev. 1998;171:221. [Google Scholar]
- 109.Kuehl CJ, Huang SD, Stang PJ. J. Am. Chem. Soc. 2001;123:9634. doi: 10.1021/ja0114355. [DOI] [PubMed] [Google Scholar]
- 110.Kaim W, Schwederski B, Dogan A, Fiedler J, Kuehl CJ, Stang PJ. Inorg. Chem. 2002;41:4025. doi: 10.1021/ic020122n. [DOI] [PubMed] [Google Scholar]
- 111.Resendiz MJE, Noveron JC, Disteldorf H, Fischer S, Stang PJ. Org. Lett. 2004;6:651. doi: 10.1021/ol035587b. [DOI] [PubMed] [Google Scholar]
- 112.Ghosh S, Mukherjee PS. Dalton Trans. 2007:2542. doi: 10.1039/b617513a. [DOI] [PubMed] [Google Scholar]
- 113.Addicott C, Oesterling I, Yamamoto T, Müllen K, Stang PJ. J. Org. Chem. 2005;70:797. doi: 10.1021/jo048239b. [DOI] [PubMed] [Google Scholar]
- 114.Ghosh S, Chakrabarty R, Mukherjee PS. Inorg. Chem. 2009;48:549. doi: 10.1021/ic801381p. [DOI] [PubMed] [Google Scholar]
- 115.Kim D, Paek JH, Jun M-J, Lee JY, Kang SO, Ko J. Inorg. Chem. 2005;44:7886. doi: 10.1021/ic0508369. [DOI] [PubMed] [Google Scholar]
- 116.Sommer RD, Rheingold AL, Goshe AJ, Bosnich B. J. Am. Chem. Soc. 2001;123:3940. doi: 10.1021/ja004279v. [DOI] [PubMed] [Google Scholar]
- 117.Benkstein KD, Hupp JT, Stern CL. J. Am. Chem. Soc. 1998;120:12982. [Google Scholar]
- 118.Benkstein KD, Stern CL, Splan KE, Johnson RC, Walters KA, Vanhelmont FWM, Hupp JT. Eur. J. Inorg. Chem. 2002:2818. [Google Scholar]
- 119.Yan H, Süss-Fink G, Neels A, Stoeckli-Evans H. J. Chem. Soc., Dalton Trans. 1997:4345. [Google Scholar]
- 120.Suzuki H, Tajima N, Tatsumi K, Yamamoto Y. Chem. Commun. 2000:1801. [Google Scholar]
- 121.Review: Therrien B. Eur. J. Inorg. Chem. 2009:2445.
- 122.Mattsson J, Govindaswamy P, Renfrew AK, Dyson PJ, Stěpnička P, Süss-Fink G, Therrien B. Organometallics. 2009;28:4350. [Google Scholar]
- 123.Barry NPE, Furrer J, Freudenreich J, Süss-Fink G, Therrien B. Eur. J. Inorg. Chem. 2010:725. [Google Scholar]
- 124.Barry NPE, Furrer J, Therrien B. Helv. Chim. Acta. 2010;93:1313. [Google Scholar]
- 125.Han Y-F, Jia W-G, Lin Y-J, Jin G-X. Organometallics. 2008;27:5002. [Google Scholar]
- 126.Han Y-F, Fei Y, Jin G-X. Dalton Trans. 2010;39:3976. doi: 10.1039/b925098k. [DOI] [PubMed] [Google Scholar]
- 127.Han Y-F, Lin Y-J, Jia W-G, Wang G-L, Jin G-X. Chem. Commun. 2008:1807. doi: 10.1039/b717554j. [DOI] [PubMed] [Google Scholar]
- 128.Barry NPE, Therrien B. Inorg. Chem. Commun. 2009;12:465. [Google Scholar]
- 129.Yu W-B, Han Y-F, Lin Y-J, Jin G-X. Chem. Eur. J. 2011;17:1863. doi: 10.1002/chem.201002986. [DOI] [PubMed] [Google Scholar]
- 130.Yu W-B, Han Y-F, Lin Y-J, Jin G-X. Organometallics. 2011 DOI: 10.1021/om200189j. [Google Scholar]
- 131.Beauchamp DA, Leob SJ. Chem. Commun. 2002:2484. [Google Scholar]
- 132.Yue NLS, Jennings MC, Puddephatt RJ. Inorg. Chem. 2005;44:1125. doi: 10.1021/ic048549c. [DOI] [PubMed] [Google Scholar]
- 133.Lindner E, Zong R, Eichele K, Weisser U, Ströbele M. Eur. J. Inorg. Chem. 2003:705. [Google Scholar]
- 134.Stang PJ, Persky NE, Manna J. J. Am. Chem. Soc. 1997;119:4777. [Google Scholar]
- 135.Leininger S, Schmitz M, Stang PJ. Org. Lett. 1999;1:1921. doi: 10.1021/ol9911425. [DOI] [PubMed] [Google Scholar]
- 136.Zhao L, Ghosh K, Zheng Y, Lyndon MM, Williams TI, Stang PJ. Inorg. Chem. 2009;48:5590. doi: 10.1021/ic900649m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hasenknopf B, Lehn J-M, Kneisel BO, Baum G, Fenske D. Angew. Chem. Int. Ed. Engl. 1996;35:1838. [Google Scholar]
- 138.Hasenknopf B, Lehn J-M, Boumediene N, Dupont-Gervais A, Van Dorsselaer A, Kneisel B, Fenske D. J. Am. Chem. Soc. 1997;119:10956. [Google Scholar]
- 139.Campos-Fernández CS, Clérac R, Koomen JM, Russell DH, Dunbar KR. J. Am. Chem. Soc. 2001;123:773. doi: 10.1021/ja002960r. [DOI] [PubMed] [Google Scholar]
- 140.Campos-Fernández CS, Schottel BL, Chifotides HT, Bera JK, Basca J, Koomen JM, Russell DH, Dunbar KR. J. Am. Chem. Soc. 2005;127:12909. doi: 10.1021/ja052108q. [DOI] [PubMed] [Google Scholar]
- 141.Mamula O, von Zelewsky A, Bernardinelli G. Angew. Chem. Int. Ed. 1998;37:289. doi: 10.1002/(SICI)1521-3773(19980216)37:3<289::AID-ANIE289>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 142.Baxter PNW, Khoury RG, Lehn J-M, Baum G, Fenske D. Chem. Eur. J. 2000;6:4140. doi: 10.1002/1521-3765(20001117)6:22<4140::aid-chem4140>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 143.Coronado E, Galan-Mascaros JR, Gaviña P, Martí-Gastaldo C, Romero FM, Tatay S. Inorg. Chem. 2008;47:5197. doi: 10.1021/ic8000569. [DOI] [PubMed] [Google Scholar]
- 144.Review: Eryazici I, Moorefield CN, Newkome GR. Chem. Rev. 2008;108:1834. doi: 10.1021/cr0781059.
- 145.Newkome GR, Cho TJ, Moorefield CN, Baker GR, Cush R, Russo PS. Angew. Chem. Int. Ed. 1999;38:3717. [PubMed] [Google Scholar]
- 146.Newkome GR, Cho TJ, Moorefield CN, Cush R, Russo PS, Godínez LA, Saunders MJ, Mohapatra P. Chem. Eur. J. 2002;8:2946. doi: 10.1002/1521-3765(20020703)8:13<2946::AID-CHEM2946>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 147.Newkome GR, Cho TJ, Moorefield CN, Mohapatra PP, Godínez LA. Chem. Eur. J. 2004;10:1493. doi: 10.1002/chem.200305267. [DOI] [PubMed] [Google Scholar]
- 148.Perera S, Li X, Guo M, Wesdemiotis C, Moorefield CN, Newkome GR. Chem. Commun. 2011;47:4658. doi: 10.1039/c1cc10649j. [DOI] [PubMed] [Google Scholar]
- 149.Hwang S-H, Wang P, Moorefield CN, Godínez LA, Manríquez J, Bustos E, Newkome GR. Chem. Commun. 2005:4672. doi: 10.1039/b509662f. [DOI] [PubMed] [Google Scholar]
- 150.Wang G-L, Lin Y-J, Jin G-X. Chem. Eur. J. 2011;17:5578. doi: 10.1002/chem.201003167. [DOI] [PubMed] [Google Scholar]
- 151.Farrell JR, Mirkin CA, Guzei IA, Liable-Sands LM, Rheingold AL. Angew. Chem. Int. Ed. 1998;37:465. doi: 10.1002/(SICI)1521-3773(19980302)37:4<465::AID-ANIE465>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 152.Liu X, Eisenberg AH, Stern CL, Mirkin CA. Inorg. Chem. 2001;40:2940. doi: 10.1021/ic010393i. [DOI] [PubMed] [Google Scholar]
- 153.Dixon FM, Eisenberg AH, Farrell JR, Mirkin CA, Liable-Sands LM, Rheingold AL. Inorg. Chem. 2000;39:3432. doi: 10.1021/ic000062q. [DOI] [PubMed] [Google Scholar]
- 154.Masar MS, III, Ovchinnikov MV, Mirkin CA, Zakharov LN, Rheingold AL. Inorg. Chem. 2003;42:6851. doi: 10.1021/ic034393p. [DOI] [PubMed] [Google Scholar]
- 155.Eisenberg AH, Dixon FM, Mirkin CA, Stern CL, Incarvito CD, Rheingold AL. Organometallics. 2001;20:2052. [Google Scholar]
- 156.Masar MS, III, Mirkin CA, Stern CL, Zakharov LN, Rheingold AL. Inorg. Chem. 2004;43:4693. doi: 10.1021/ic049658u. [DOI] [PubMed] [Google Scholar]
- 157.Eisenberg AH, Ovchinnikov MV, Mirkin CA. J. Am. Chem. Soc. 2003;125:2836. doi: 10.1021/ja027936n. [DOI] [PubMed] [Google Scholar]
- 158.Das N, Mukherjee PS, Arif AM, Stang PJ. J. Am. Chem. Soc. 2003;125:13950. doi: 10.1021/ja037788g. [DOI] [PubMed] [Google Scholar]
- 159.Mukherjee PS, Das N, Kryschenko YK, Arif AM, Stang PJ. J. Am. Chem. Soc. 2004;126:2464. doi: 10.1021/ja039235b. [DOI] [PubMed] [Google Scholar]
- 160.Das N, Ghosh A, Arif AM, Stang PJ. Inorg. Chem. 2005;44:7130. doi: 10.1021/ic050919p. [DOI] [PubMed] [Google Scholar]
- 161.Bar AK, Gole B, Ghosh S, Mukherjee PS. Dalton Trans. 2009:6701. doi: 10.1039/b911622m. [DOI] [PubMed] [Google Scholar]
- 162.Das N, Arif AM, Stang PJ, Sieger M, Sarkar B, Kaim W, Fiedler J. Inorg. Chem. 2005;44:5798. doi: 10.1021/ic0481834. [DOI] [PubMed] [Google Scholar]
- 163.Severin K. Coord. Chem Rev. 2003;245:3. [Google Scholar]
- 164.Piotrowski H, Polborn K, Hilt G, Severin K. J. Am. Chem. Soc. 2001;123:2699. doi: 10.1021/ja005804t. [DOI] [PubMed] [Google Scholar]
- 165.Lehaire M-L, Scopelitti R, Hardeis L, Polborn K, Mayer P, Severin K. Inorg. Chem. 2004;43:1609. doi: 10.1021/ic035328i. [DOI] [PubMed] [Google Scholar]
- 166.Mirtschin S, Krasniqi E, Scopelliti R, Severin K. Inorg. Chem. 2008;47:6375. doi: 10.1021/ic800410x. [DOI] [PubMed] [Google Scholar]
- 167.Brasey T, Scopelliti R, Severin K. Inorg. Chem. 2005;44:160. doi: 10.1021/ic048619f. [DOI] [PubMed] [Google Scholar]
- 168.Review: Bray DJ, Clegg JK, Lindoy LF, Schilter D. Adv. Inorg. Chem. 2006;59:1.
- 169.Clegg JK, Lindoy LF, McMurtrie JC, Schilter D. Dalton Trans. 2005:857. doi: 10.1039/b418870e. [DOI] [PubMed] [Google Scholar]
- 170.Clegg JK, Bray DJ, Gloe K, Gloe K, Hayter MJ, Jolliffe KA, Lawrance GA, Meehan GV, McMurtrie JC, Lindoy LF, Wenzel M. Dalton Trans. 2007:1719. doi: 10.1039/b701575e. [DOI] [PubMed] [Google Scholar]
- 171.Clegg JK, Lindoy LF, Moubaraki B, Murray KS, McMurtrie JC. Dalton Trans. 2004:2417. doi: 10.1039/B403673E. [DOI] [PubMed] [Google Scholar]
- 172.Clegg JK, Bray DJ, Gloe K, Gloe K, Jolliffe KA, Lawrance GA, Lindoy LF, Meehan GV, Wenzel M. Dalton Trans. 2008:1331. doi: 10.1039/b716653b. [DOI] [PubMed] [Google Scholar]
- 173.Clegg JK, Lindoy LF, McMurtrie JC, Schilter D. Dalton Trans. 2006:3114. doi: 10.1039/b517274h. [DOI] [PubMed] [Google Scholar]
- 174.Clegg JK, Gloe K, Hayter MJ, Kataeva O, Lindoy LF, Moubaraki B, McMurtrie JC, Murray KS, Schilter D. Dalton Trans. 2006:3977. doi: 10.1039/b606523f. [DOI] [PubMed] [Google Scholar]
- 175.Clegg JK, Iremonger SS, Hayter MJ, Southon PD, Macquart RB, Duriska MB, Jensen P, Turner P, Jolliffe KA, Kepert CJ, Meehan GV, Lindoy LF. Angew. Chem. Int. Ed. 2010;49:1075. doi: 10.1002/anie.200905497. [DOI] [PubMed] [Google Scholar]
- 176.Dinolfo PH, Williams ME, Stern CL, Hupp JT. J. Am. Chem. Soc. 2004;126:12989. doi: 10.1021/ja0473182. [DOI] [PubMed] [Google Scholar]
- 177.Benkstein KD, Hupp JL, Stern CL. Angew. Chem. Int. Ed. 2000;39:2891. doi: 10.1002/1521-3773(20000818)39:16<2891::aid-anie2891>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 178.Manimaran B, Rajendran T, Lu Y-L, Lee G-H, Peng S-M, Lu K-L. J. Chem. Soc., Dalton Trans. 2001:515. [Google Scholar]
- 179.Gupta D, Rajakannu P, Shankar B, Shanmugam R, Hussain F, Sarkar B, Sathiyendiran M. Dalton Trans. 2011;40:5433. doi: 10.1039/c0dt01227k. [DOI] [PubMed] [Google Scholar]
- 180.Benkstein KD, Hupp JT, Stern CL. Inorg. Chem. 1998;37:5404. doi: 10.1021/ic980513v. [DOI] [PubMed] [Google Scholar]
- 181.Woessner SM, Helms JB, Shen Y, Sullivan BP. Inorg. Chem. 1998;37:5406. doi: 10.1021/ic9805168. [DOI] [PubMed] [Google Scholar]
- 182.Rajendran T, Manimaran B, Liao R-T, Lin R-J, Thanasekaran P, Lee G-H, Peng S-M, Liu Y-H, Chang I-J, Rajagopal S, Lu K-L. Inorg. Chem. 2003;42:6388. doi: 10.1021/ic034099x. [DOI] [PubMed] [Google Scholar]
- 183.Rajendran T, Manimaran B, Lee F-Y, Lee G-H, Peng S-M, Wang CM, Lu K-L. Inorg. Chem. 2000;39:2016. doi: 10.1021/ic9912474. [DOI] [PubMed] [Google Scholar]
- 184.Sun S-S, Lees AJ. J. Am. Chem. Soc. 2000;122:8956. [Google Scholar]
- 185.Cotton FA, Daniels LM, Lin C, Murillo CA. J. Am. Chem. Soc. 1999;121:4538. [Google Scholar]
- 186.Cotton FA, Lin C, Murillo CA. Inorg. Chem. 2001;40:478. doi: 10.1021/ic001016t. [DOI] [PubMed] [Google Scholar]
- 187.Cotton FA, Daniels LM, Lin C, Murillo CA, Yu S-Y. J. Chem. Soc., Dalton Trans. 2001:502. [Google Scholar]
- 188.Angaridis P, Berry JF, Cotton FA, Murillo CA, Wang X. J. Am. Chem. Soc. 2003;125:10327. doi: 10.1021/ja036095x. [DOI] [PubMed] [Google Scholar]
- 189.Bonar-Law RP, McGrath TD, Singh N, Bickley JF, Steiner A. Chem. Commun. 1999:2457. [Google Scholar]
- 190.Bickley JF, Bonar-Law RP, Femoni C, MacLean EJ, Steiner A, Teat SJ. J. Chem. Soc., Dalton Trans. 2000:4025. [Google Scholar]
- 191.Schiavo SL, Pocsfalvi G, Serroni S, Cardiano P, Piraino P. Eur. J. Inorg. Chem. 2000:1371. [Google Scholar]
- 192.Cotton FA, Lin C, Murillo CA. Inorg. Chem. 2001;40:575. doi: 10.1021/ic000963z. [DOI] [PubMed] [Google Scholar]
- 193.Cotton FA, Lin C, Murillo CA. Inorg. Chem. 2001;40:472. doi: 10.1021/ic0010151. [DOI] [PubMed] [Google Scholar]
- 194.Cotton FA, Jin J-Y, Li Z, Liu CY, Murillo CA. Dalton Trans. 2007:2328. doi: 10.1039/b617932k. [DOI] [PubMed] [Google Scholar]
- 195.Bera JK, Angaridis P, Cotton FA, Petrukhina MA, Fanwick PE, Walton RA. J. Am. Chem. Soc. 2001;123:1515. [Google Scholar]
- 196.Cotton FA, Liu CY, Murillo CA, Wang X. Inorg. Chem. 2006;45:2619. doi: 10.1021/ic0521473. [DOI] [PubMed] [Google Scholar]
- 197.Cotton FA, Murillo CA, Yu R. Dalton Trans. 2006:3900. doi: 10.1039/b604928a. [DOI] [PubMed] [Google Scholar]
- 198.Chisholm MH, Patmore NJ, Reed CR, Singh N. Inorg. Chem. 2010;49:7116. doi: 10.1021/ic1009237. [DOI] [PubMed] [Google Scholar]
- 199.Khoshbin MS, Ovchinnikov MV, Mirkin CA, Zakharov LN, Rheingold AL. Inorg. Chem. 2005;44:496. doi: 10.1021/ic048975y. [DOI] [PubMed] [Google Scholar]
- 200.Reviews: Lee SJ, Lin W. Acc. Chem. Res. 2008;41:521. doi: 10.1021/ar700216n. Leigh DA, Pérez EM. Top. Curr. Chem. 2006;265:185. Amabilino DB, Veciana J. Top. Curr. Chem. 2006;265:253. Mamula O, von Zelewsky A. Coord. Chem. Rev. 2003;242:87.
- 201.Enders D, Jaeger K-E, editors. Assymetric Synthesis with Chemical and Biological Methods. Wiley-VCH; New York: 2007. [Google Scholar]
- 202.Stang PJ, Olenyuk B, Arif AM. Organometallics. 1995;14:5281. [Google Scholar]
- 203.Olenyuk B, Whiteford JA, Stang PJ. J. Am. Chem. Soc. 1996;118:8221. [Google Scholar]
- 204.Stang PJ, Olenyuk B. Angew. Chem. Int. Ed. Engl. 1996;35:732. [Google Scholar]
- 205.Fan J, Whitefold JA, Olenyuk B, Levin MD, Stang PJ, Fleischer EB. J. Am. Chem. Soc. 1999;121:2741. [Google Scholar]
- 206.Heo J, Jeon Y-M, Mirkin CA. J. Am. Chem. Soc. 2007;129:7712. doi: 10.1021/ja0716812. [DOI] [PubMed] [Google Scholar]
- 207.Lee SJ, Lin W. J. Am. Chem. Soc. 2002;124:4554. doi: 10.1021/ja0256257. [DOI] [PubMed] [Google Scholar]
- 208.Lee SJ, Kim JS, Lin W. Inorg. Chem. 2004;43:6579. doi: 10.1021/ic0497937. [DOI] [PubMed] [Google Scholar]
- 209.Lee SJ, Hu A, Lin W. J. Am. Chem. Soc. 2002;124:12948. doi: 10.1021/ja028099s. [DOI] [PubMed] [Google Scholar]
- 210.Zhang L, Niu Y-H, Jen AK-Y, Lin W. Chem. Commun. 2005:1002. doi: 10.1039/b413708f. [DOI] [PubMed] [Google Scholar]
- 211.Lee SJ, Luman CR, Castellano FN, Lin W. Chem. Commun. 2003:2124. doi: 10.1039/b307727f. [DOI] [PubMed] [Google Scholar]
- 212.Jiang H, Lin W. J. Am. Chem. Soc. 2003;125:8084. doi: 10.1021/ja034402t. [DOI] [PubMed] [Google Scholar]
- 213.Jiang H, Hu A, Lin W. Chem. Comm. 2003:96. [PubMed] [Google Scholar]
- 214.Hua J, Lin W. Org. Lett. 2004;6:861. doi: 10.1021/ol036111v. [DOI] [PubMed] [Google Scholar]
- 215.Jiang H, Lin W. J. Am. Chem. Soc. 2004;126:7426. doi: 10.1021/ja049145m. [DOI] [PubMed] [Google Scholar]
- 216.Jeong KS, Kim SY, Shin U-S, Kogej M, Hai NTM, Broekmann P, Jeong N, Kirchner B, Reiher M, Schalley CA. J. Am. Chem. Soc. 2005;127:17672. doi: 10.1021/ja053781i. [DOI] [PubMed] [Google Scholar]
- 217.Jeong KS, Kim SY, Oh Y, Min DW, Kim J, Jeong N. CrystEngComm. 2007;9:273. [Google Scholar]
- 218.Wisser B, Chamayou A-C, Miller R, Scherer W, Janiak C. CrystEngComm. 2008;10:461. [Google Scholar]
- 219.Rang A, Engeser M, Maier NM, Nieger M, Lindner W, Schalley CA. Chem. Eur. J. 2008;14:3855. doi: 10.1002/chem.200800113. [DOI] [PubMed] [Google Scholar]
- 220.Rang A, Nieger M, Engeser M, Lützen A, Schalley CA. Chem. Commun. 2008:4789. doi: 10.1039/b806916f. [DOI] [PubMed] [Google Scholar]
- 221.Das N, Ghosh A, Singh OM, Stang PJ. Org. Lett. 2006;8:1701. doi: 10.1021/ol060365+. [DOI] [PubMed] [Google Scholar]
- 222.Yeh RM, Raymond KN. Inorg. Chem. 2006;45:1130. doi: 10.1021/ic0515711. [DOI] [PubMed] [Google Scholar]
- 223.Yamanari K, Fukuda I, Kawamoto T, Kushi Y, Fuyuhiro A, Kubota N, Fukuo T, Arakawa R. Inorg. Chem. 1998;37:5611. doi: 10.1021/ic9802146. [DOI] [PubMed] [Google Scholar]
- 224.Zhang Y, Wang S, Enright GD, Breeze SR. J. Am. Chem. Soc. 1998;120:9398. [Google Scholar]
- 225.Liu C-M, Nordlander E, Schmeh D, Shoemaker R, Pierpont CG. Inorg. Chem. 2004;43:2114. doi: 10.1021/ic0354264. [DOI] [PubMed] [Google Scholar]
- 226.Cotton FA, Murillo CA. Eur. J. Inorg. Chem. 2006:4209. [Google Scholar]
- 227.Cotton FA, Murillo CA, Wang X, Yu R. Inorg. Chem. 2004;43:8394. doi: 10.1021/ic0492183. [DOI] [PubMed] [Google Scholar]
- 228.Cotton FA, Murillo CA, Stiriba S-E, Wang X, Yu R. Inorg. Chem. 2005;44:8223. doi: 10.1021/ic051282c. [DOI] [PubMed] [Google Scholar]
- 229.Cotton FA, Murillo CA, Yu R. Inorg. Chem. 2005;44:8211. doi: 10.1021/ic051149k. [DOI] [PubMed] [Google Scholar]
- 230.Reviews: Kumar A, Sun S-S, Lees AJ. Coord. Chem. Rev. 2008;252:922. Cooke MW, Chartrand D, Hanan GS. Coord. Chem. Rev. 2008;252:903. Amijs CHM, van Klink GPM, Van Koten G. Dalton Trans. 2006:308. doi: 10.1039/b505354d. Lützen A. Angew. Chem. Int. Ed. 2005;44:1000. doi: 10.1002/anie.200462272.
- 231.Northrop BH, Yang H-B, Stang PJ. Chem. Commun. 2008:5896. doi: 10.1039/b811712h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Chi K-W, Addicot C, Stang PJ. J. Org. Chem. 2004;69:2910. doi: 10.1021/jo049883t. [DOI] [PubMed] [Google Scholar]
- 233.Huang F, Yang H-B, Das N, Maran U, Arif AM, Gibson HW, Stang PJ. J. Org. Chem. 2006;71:6623. doi: 10.1021/jo061007n. [DOI] [PubMed] [Google Scholar]
- 234.Pirondini L, Dalcanle E. In: Modern Supramolecular Chemistry: Stretegies for Macrocycle Synthesis. Diederich F, Stang PJ, Tykwinski RR, editors. Wiley-VCH; Weinheim: 2008. p. 233. [Google Scholar]
- 235.Menozzi E, Busi M, Massera C, Ugozzoli F, Zuccaccia D, Macchioni A, Dalcanale E. J. Org. Chem. 2006;71:2617. doi: 10.1021/jo052460m. [DOI] [PubMed] [Google Scholar]
- 236.Jude H, Sinclair DJ, Das N, Sherburn MS, Stang PJ. J. Org. Chem. 2006;71:4155. doi: 10.1021/jo060133o. [DOI] [PubMed] [Google Scholar]
- 237.Jude H, Disteldorf H, Fischer S, Wedge T, Hawkridge AM, Arif AM, Hawthorne MF, Muddiman DC, Stang PJ. J. Am. Chem. Soc. 2005;127:12131. doi: 10.1021/ja053050i. [DOI] [PubMed] [Google Scholar]
- 238.Das N, Stang PJ, Arif AM, Campana CF. J. Org. Chem. 2005;70:10440. doi: 10.1021/jo0517085. [DOI] [PubMed] [Google Scholar]
- 239.Northrop BH, Glöckner A, Stang PJ. J. Org. Chem. 2008;73:1787. doi: 10.1021/jo702380b. [DOI] [PubMed] [Google Scholar]
- 240.Thanasekaran P, Wu J-Y, Manimaran B, Rajendran T, Chang I-J, Rajagopal S, Lee G-H, Peng S-M, Lu K-L. J. Phys. Chem. A. 2007;111:10953. doi: 10.1021/jp0742315. [DOI] [PubMed] [Google Scholar]
- 241.You C-C, Hippius C, Grüne M, Würthner F. Chem. Eur. J. 2006;12:7510. doi: 10.1002/chem.200600413. [DOI] [PubMed] [Google Scholar]
- 242.Yang H-B, Ghosh K, Northrop BH, Zheng Y-R, Lyndon MM, Muddiman DC, Stang PJ. J. Am. Chem. Soc. 2007;129:14187. doi: 10.1021/ja073744m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Ghosh K, Yang H-B, Northrop BH, Lyndon MM, Zheng Y-R, Muddiman DC, Stang PJ. J. Am. Chem. Soc. 2008;130:5320. doi: 10.1021/ja711502t. [DOI] [PubMed] [Google Scholar]
- 244.Reviews: Lee CC, MacKay JA, Fréchet JMJ, Szoka FC. Nat. Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171. Boas U, Heegaard PMH. Chem. Soc. Rev. 2004;33:43. doi: 10.1039/b309043b.
- 245.Reviews: Méry D, Astruc D. Coord. Chem. Rev. 2006;250:1965. Berger A, Gebbink RJMK, van Koten G. Top. Organomet. Chem. 2006;20:1. Ribaudo F, van Leeuwen PWNM, Reek JNH. Top. Organomet. Chem. 2006;20:39. Chandler BD, Gilbertson JD. Top. Organomet. Chem. 2006;20:97. Hajji C, Haag R. Top. Organomet. Chem. 2006;20:149. van Heerbeeck R, Kamer PCJ, van Leeuwen PWNM, Reek JNH. Chem. Rev. 2002;102:3717. doi: 10.1021/cr0103874. Grayson SM, Fréchet JMJ. Chem. Rev. 2001;101:3819. doi: 10.1021/cr990116h.
- 246.Reviews: Astruc D, Daniel M-C, Ruiz J. Top. Organomet. Chem. 2006;20:121. Venturi M, Serroni S, Juris A, Campagna S, Balzani V. Top. Curr. Chem. 1998;197:193. Balzani V, Campagna S, Denti G, Juris A, Serroni S, Venturi M. Acc. Chem. Res. 1998;31:26.
- 247.Yang H-B, Das N, Huang F, Hawkridge AM, Muddiman DC, Stang PJ. J. Am. Chem. Soc. 2006;128:10014. doi: 10.1021/ja063377z. [DOI] [PubMed] [Google Scholar]
- 248.Yang H-B, Hawkridge AM, Huang SD, Das N, Bunge SD, Muddiman DC, Stang PJ. J. Am. Chem. Soc. 2007;129:2120. doi: 10.1021/ja066804h. [DOI] [PubMed] [Google Scholar]
- 249.Yang H-B, Northrop BH, Zheng Y-R, Ghosh K, Lyndon MM, Muddiman DC, Stang PJ. J. Org. Chem. 2009;74:3524. doi: 10.1021/jo900067v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Yang H-B, Northrop BH, Zheng Y-R, Ghosh K, Stang PJ. J. Org. Chem. 2009;74:7067. doi: 10.1021/jo9013589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Baytekin HT, Sahre M, Rang A, Engeser M, Schulz A, Schalley CA. Small. 2008;4:1823. doi: 10.1002/smll.200800135. [DOI] [PubMed] [Google Scholar]
- 252.Yang H-B, Ghosh K, Zhao Y, Northrop BH, Lyndon MM, Muddiman DC, White HS, Stang PJ. J. Am. Chem. Soc. 2008;130:839. doi: 10.1021/ja710349j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ghosh K, Zhao Y, Yang H-B, Northrop BH, White HS, Stang PJ. J. Org. Chem. 2008;73:8553. doi: 10.1021/jo801692v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Ghosh K, Hu J, Yang HB, Northrop BH, White HS, Stang PJ. J. Org. Chem. 2009;74:4828. doi: 10.1021/jo900580w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Zhao G-Z, Chen L-J, Wang C-H, Yang H-B, Ghosh K, Zheng Y-R, Lyndon MM, Muddiman DC, Stang PJ. Organometallics. 2010;29:6137. [Google Scholar]
- 256.Xu X-D, Yang H-B, Zheng Y-R, Ghosh K, Lyndon MM, Muddiman DC, Stang PJ. J. Org. Chem. 2010;75:7373. doi: 10.1021/jo101648p. [DOI] [PubMed] [Google Scholar]
- 257.Chan Y-T, Moorefield CN, Soler M, Newkome GR. Chem. Eur. J. 2010;16:1768. doi: 10.1002/chem.200902988. [DOI] [PubMed] [Google Scholar]
- 258.Zhao L, Ghosh K, Zheng Y-R, Stang PJ. J. Org. Chem. 2009;74:8516. doi: 10.1021/jo9019607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Reviews: Beletskaya I, Tyurin VS, Tsivadze AY, Guilard R, Stern C. Chem. Rev. 2009;109:1659. doi: 10.1021/cr800247a. Scandola F, Chiorboli C, Prodi A, Iengo E, Alessio E. Coord. Chem. Rev. 2006;250:1471. doi: 10.1002/cphc.200600065. Rose E, Andrioletti B, Zrig S, Quelquejeu-Ethève M. Chem. Soc. Rev. 2005;34:573. doi: 10.1039/b405679p. Iengo E, Zangrando E, Alessio E. Eur. J. Inorg. Chem. 2003:2371. doi: 10.1002/chem.200304795.
- 260.Drain CM, Lehn J-M. J. Chem. Soc., Chem. Commun. 1994:2313. [Google Scholar]
- 261.Iengo E, Milani B, Zangrando E, Geremia S, Alessio E. Angew. Chem. Int. Ed. 2000;39:1096. doi: 10.1002/(sici)1521-3773(20000317)39:6<1096::aid-anie1096>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 262.Iengo E, Zangrando E, Minatel R, Alessio E. J. Am. Chem. Soc. 2002;124:1003. doi: 10.1021/ja016162s. [DOI] [PubMed] [Google Scholar]
- 263.Iengo E, Zangrando E, Bellini M, Alessio E, Prodi A, Chiorboli C, Scandola F. Inorg. Chem. 2005;44:9752. doi: 10.1021/ic051210l. [DOI] [PubMed] [Google Scholar]
- 264.Iengo E, Scandola F, Alessio E. Struct. Bond. (Berlin) 2006;121:105. [Google Scholar]
- 265.Lee SJ, Hupp JT. Coord. Chem. Rev. 2006;250:1710. [Google Scholar]
- 266.Mines GA, Tzeng B-C, Stevenson KJ, Li J, Hupp JT. Angew. Chem. Int. Ed. 2002;41:154. doi: 10.1002/1521-3773(20020104)41:1<154::aid-anie154>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 267.Chang SH, Chung K-B, Slone RV, Hupp JT. Synth. Metals. 2001;117:215. [Google Scholar]
- 268.Slone RV, Hupp JT. Inorg. Chem. 1997;36:5422. [Google Scholar]
- 269.Czaplewski KF, Hupp JT, Snurr RQ. Adv. Mater. 2001;13:1895. [Google Scholar]
- 270.Splan KE, Massari AM, Hupp JT. J. Phys. Chem. B. 2004;108:4111. [Google Scholar]
- 271.Martinson ABF, Massari AM, Lee SJ, Gurney RW, Splan KE, Hupp JT, Nguyen ST. J. Electrochem. Soc. 2006;153:A527. [Google Scholar]
- 272.Maeda C, Kim P, Cho S, Park JK, Lim JM, Kim D, Vura-Weis J, Wasielewski MR, Shinokubo H, Osuka A. Chem. Eur. J. 2010;16:5052. doi: 10.1002/chem.200903195. [DOI] [PubMed] [Google Scholar]
- 273.Park M, Yoon M-C, Yoon ZS, Hori T, Peng X, Aratani N, Hotta J, Uji-i H, Sliwa M, Hofkens J, Osuka A, Kim D. J. Am. Chem. Soc. 2007;129:3539. doi: 10.1021/ja065813n. [DOI] [PubMed] [Google Scholar]
- 274.Yang J, Park M, Yoon ZS, Hori T, Peng X, Aratani N, Dedecker P, Hotta J, Uji-i H, Sliwa M, Hofkens J, Osuka A, Kim D. J. Am. Chem. Soc. 2008;130:1879. doi: 10.1021/ja075701b. [DOI] [PubMed] [Google Scholar]
- 275.Splan KE, Keefe MH, Massari AM, Walters KA, Hupp JT. Inorg. Chem. 2002;41:619. doi: 10.1021/ic010992p. [DOI] [PubMed] [Google Scholar]
- 276.Jensen RA, Kelley RF, Lee SJ, Wasielewski MR, Hupp JT, Tiede DM. Chem. Commun. 2008:1886. doi: 10.1039/b718628b. [DOI] [PubMed] [Google Scholar]
- 277.Reviews: Haak RM, Wezenberg SJ, Kleij AW. Chem. Commun. 2010;46:2713. doi: 10.1039/c001392g. Gupta KC, Sutar AK. Coord. Chem. Rev. 2008;252:1420. McGarrigle EM, Gilheany DG. Chem. Rev. 2005;105:1563. doi: 10.1021/cr0306945. Cozzi PG. Chem. Soc. Rev. 2004;33:410. doi: 10.1039/b307853c. Larrow JF, Jacobsen EN. Top. Organomet. Chem. 2004;6:123. Katsuki T. Adv. Synth. Catal. 2002;344:131.
- 278.Reviews: Kleij AW. Chem. Eur. J. 2008;14:10520. doi: 10.1002/chem.200801149. Kleij AW. Dalton Trans. 2009:4635. doi: 10.1039/b902866h.
- 279.Wezenberg SJ, Kleij AW. Angew. Chem. Int. Ed. 2008;47:2354. doi: 10.1002/anie.200702468. [DOI] [PubMed] [Google Scholar]
- 280.See for some examples: Clever GH, Söltl Y, Burks H, Spahl W, Carell T. Chem. Eur. J. 2006;12:8708. doi: 10.1002/chem.200600558. Clever GH, Polborn K, Carell T. Angew. Chem. Int. Ed. 2005;44:7204. doi: 10.1002/anie.200501589. Czlapinski JL, Sheppard TL. Chem. Commun. 2004:2468. doi: 10.1039/b410493e. Escudero-Adán EC, Benet-Buchholz J, Kleij AW. Inorg. Chem. 2008;47:4256. doi: 10.1021/ic702257e.
- 281.Sun S-S, Stern CL, Nguyen ST, Hupp JT. J. Am. Chem. Soc. 2004;126:6314. doi: 10.1021/ja037378s. [DOI] [PubMed] [Google Scholar]
- 282.Splan KE, Massari AM, Morris GA, Sun S-S, Reina E, Nguyen ST, Hupp JT. Eur. J. Inorg. Chem. 2003:2348. [Google Scholar]
- 283.Sun S-S, Anspach JA, Lees AJ. Inorg. Chem. 2002;41:1862. doi: 10.1021/ic010998e. [DOI] [PubMed] [Google Scholar]
- 284.Kleij AW, Kuil M, Tooke DM, Lutz M, Spek AL, Reek JNH. Chem. Eur. J. 2005;11:4743. doi: 10.1002/chem.200500227. [DOI] [PubMed] [Google Scholar]
- 285.Wezenberg SJ, Escudero-Adán EC, Benet-Buchholz J, Kleij AW. Inorg. Chem. 2008;47:2925. doi: 10.1021/ic800033v. [DOI] [PubMed] [Google Scholar]
- 286.Kleij AW, Kuil M, Tooke DM, Spek AL, Reek JNH. Inorg. Chem. 2007;46:5829. doi: 10.1021/ic700408v. [DOI] [PubMed] [Google Scholar]
- 287.Salassa G, Castilla AM, Kleij AW. Dalton Trans. 2011;40:5236. doi: 10.1039/c1dt10069f. [DOI] [PubMed] [Google Scholar]
- 288.Maverick AW, Buckingham SC, Yao Q, Bradbury JR, Stanley GG. J. Am. Chem. Soc. 1986;108:7430. [Google Scholar]
- 289.Maverick AW, Klavetter FE. Inorg. Chem. 1984;23:4129. [Google Scholar]
- 290.Fujita M, Yazaki J, Ogura K. Tetrahedron Lett. 1991;32:5589. [Google Scholar]
- 291.Fujita M, Nagao S, Iida M, Ogata K, Ogura K. J. Am. Chem. Soc. 1993;115:1574. [Google Scholar]
- 292.Stang PJ, Cao DH, Saito S, Arif AM. J. Am. Chem. Soc. 1995;117:6273. [Google Scholar]
- 293.Whiteford JA, Stang PJ, Huang SD. Inorg. Chem. 1998;37:5595. doi: 10.1021/ic980727c. [DOI] [PubMed] [Google Scholar]
- 294.Chang S-Y, Um M-C, Uh H, Jang H-Y, Jeong K-S. Chem. Commun. 2003:2026. doi: 10.1039/b306497b. [DOI] [PubMed] [Google Scholar]
- 295.Jeong K-S, Cho YL, Chang S-Y, Park T-Y, Song JU. J. Org. Chem. 1999;64:9459. [Google Scholar]
- 296.Chen Y-Q, Wang X-Z, Shao X-B, Hou J-L, Chen X-Z, Jiang X-K, Li Z-T. Tetrahedron. 2004;60:10253. [Google Scholar]
- 297.Sato H, Tashiro K, Shinmori H, Osuka A, Murata Y, Komatsu K, Aida T. J. Am. Chem. Soc. 2005;127:13086. doi: 10.1021/ja052993c. [DOI] [PubMed] [Google Scholar]
- 298.Vajpayee V, Kim H, Mishra A, Mukherjee PS, Stang PJ, Lee MH, Kim HK, Chi K-W. Dalton Trans. 2011 doi: 10.1039/c0dt01481h. DOI: 10.1039/c0dt01481h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Heo J, Mirkin CA. Angew. Chem. Int. Ed. 2006;45:941. doi: 10.1002/anie.200503343. [DOI] [PubMed] [Google Scholar]
- 300.Grote Z, Lehaire M-L, Scopelliti R, Severin K. J. Am. Chem. Soc. 2003;125:13638. doi: 10.1021/ja036621k. [DOI] [PubMed] [Google Scholar]
- 301.Grote Z, Scopelliti R, Severin K. J. Am. Chem. Soc. 2004;126:16959. doi: 10.1021/ja044874n. [DOI] [PubMed] [Google Scholar]
- 302.Piotrowski H, Hilt G, Schulz A, Mayer P, Polborn K, Severin K. Chem. Eur. J. 2001;7:3196. doi: 10.1002/1521-3765(20010803)7:15<3196::aid-chem3196>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 303.Lehaire M-L, Scopelitti R, Piotrowski H, Severin K. Angew. Chem. Int. Ed. 2002;41:1419. doi: 10.1002/1521-3773(20020415)41:8<1419::aid-anie1419>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 304.Rochat S, Grote Z, Severin K. Org. Biomol. Chem. 2009;7:1147. doi: 10.1039/b820592b. [DOI] [PubMed] [Google Scholar]
- 305.Qin Z, Jennings MC, Puddephatt RJ. Inorg. Chem. 2002;41:3967. doi: 10.1021/ic020227q. [DOI] [PubMed] [Google Scholar]
- 306.Yue N, Qin Z, Jennings MC, Eisler DJ, Puddephatt RJ. Inorg. Chem. Commun. 2003;6:1269. [Google Scholar]
- 307.Yue NLS, Eisler DJ, Jennings MC, Puddephatt RJ. Inorg. Chem. Commun. 2005;8:31. [Google Scholar]
- 308.Sathiyendiran M, Liao R-T, Thanasekaran P, Luo T-T, Venkataramanan NS, Lee G-H, Peng S-M, Lu K-L. Inorg. Chem. 2006;45:10052. doi: 10.1021/ic061886w. [DOI] [PubMed] [Google Scholar]
- 309.Waiter CJ, Anderson HL, Sanders JKM. J. Chem. Soc., Chem. Commun. 1993:458. [Google Scholar]
- 310.Mackay LG, Wylie RS, Sanders JKM. J. Am. Chem. Soc. 1994;116:3141. [Google Scholar]
- 311.Merlau ML, del Pilar Mejia M, Nguyen ST, Hupp JT. Angew. Chem. Int. Ed. 2001;40:4239. doi: 10.1002/1521-3773(20011119)40:22<4239::AID-ANIE4239>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 312.Fritsky IO, Ott R, Krämer R. Angew. Chem. Int. Ed. 2000;39:3255. doi: 10.1002/1521-3773(20000915)39:18<3255::aid-anie3255>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 313.Fritsky IO, Ott R, Pritzkow H, Krämer R. Chem. Eur. J. 2001;7:1221. doi: 10.1002/1521-3765(20010316)7:6<1221::aid-chem1221>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 314.Gianneschi NC, Bertin PA, Nguyen ST, Mirkin CA, Zakharov LN, Rheingold AL. J. Am. Chem. Soc. 2003;125:10508. doi: 10.1021/ja035621h. [DOI] [PubMed] [Google Scholar]
- 315.Gianneschi NC, Nguyen ST, Mirkin CA. J. Am. Chem. Soc. 2005;127:1644. doi: 10.1021/ja0437306. [DOI] [PubMed] [Google Scholar]
- 316.Yoon HJ, Kuwabara J, Kim J-H, Mirkin CA. Science. 2010;330:66. doi: 10.1126/science.1193928. [DOI] [PubMed] [Google Scholar]
- 317.Oliveri CG, Gianneschi NC, Nguyen ST, Mirkin CA, Stern CL, Wawrzak Z, Pink M. J. Am. Chem. Soc. 2006;128:16286. doi: 10.1021/ja0661010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Oliveri CG, Heo J, Nguyen ST, Mirkin CA, Wawrzak Z. Inorg. Chem. 2007;46:7716. doi: 10.1021/ic701424j. [DOI] [PubMed] [Google Scholar]
- 319.Yoon HJ, Heo J, Mirkin CA. J. Am. Chem. Soc. 2007;129:14182. doi: 10.1021/ja077467v. [DOI] [PubMed] [Google Scholar]
- 320.Gianneschi NC, Cho S-H, Nguyen ST, Mirkin CA. Angew. Chem. Int. Ed. 2004;43:5503. doi: 10.1002/anie.200460932. [DOI] [PubMed] [Google Scholar]
- 321.Müller P, Baud C, Ené D, Motallebi S, Doyle MP, Brandes BD, Dyatkin AB, See MM. Helv. Chim. Acta. 1995;78:459. [Google Scholar]
- 322.Reviews: Franceschin M. Eur. J. Org. Chem. 2009:2225. Ou T-M, Lu Y-J, Tan J-H, Huang Z-S, Wong K-Y, Gu L-Q. ChemMedChem. 2008;3:690. doi: 10.1002/cmdc.200700300. Neidle S, Read MA. Biopolymers. 2001;56:195. doi: 10.1002/1097-0282(2000)56:3<195::AID-BIP10009>3.0.CO;2-5. Shafer RH, Smirnov I. Biopolymers. 2001;56:209. doi: 10.1002/1097-0282(2000/2001)56:3<209::AID-BIP10018>3.0.CO;2-Y. Han H, Hurley LH. Trends Pharm. Sci. 2000;21:136. doi: 10.1016/s0165-6147(00)01457-7.
- 323.Kieltyka R, Englebienne P, Fakhoury J, Autexier C, Moitessier N, Sleiman HF. J. Am. Chem. Soc. 2008;130:10040. doi: 10.1021/ja8014023. [DOI] [PubMed] [Google Scholar]
- 324.Mounir M, Lorenzo J, Ferrer M, Prieto MJ, Rossell O, Avilès FX, Moreno V. J. Inorg. Biochem. 2007;101:660. doi: 10.1016/j.jinorgbio.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 325.Fyles TM, Tong CC. New J. Chem. 2007;31:655. [Google Scholar]
- 326.Barry NPE, Edafe F, Dyson PJ, Therrien B. Dalton Trans. 2010;39:2816. doi: 10.1039/b925015h. [DOI] [PubMed] [Google Scholar]
- 327.Vajpayee V, Song YH, Yang YJ, Kang SC, Kim H, Kim IS, Wang M, Stang PJ, Chi K-W. Organometallics. 2011 doi: 10.1021/om200294x. DOI: 10.1021/om200294x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Ang WH, Grote Z, Scopelliti R, Juillerat-Jeanneret L, Severin K, Dyson PJ. J. Organomet. Chem. 2009;694:968. [Google Scholar]
- 329.Saalfrank RW, Stark A, Peters K, von Schnering HG. Angew. Chem. Int. Ed. Engl. 1988;27:851. [Google Scholar]
- 330.Saalfrank RW, Demleitner B, Glaser H, Maid H, Reihs S, Bauer W, Maluenga M, Hampel F, Teichert M, Krautscheid H. Eur. J. Inorg. Chem. 2003:822. [Google Scholar]
- 331.Saalfrank RW, Stark A, Bremer M, Hummel H-U. Angew. Chem. Int. Ed. Engl. 1990;29:311. [Google Scholar]
- 332.Saalfrank RW, Burak R, Breit A, Stalke D, Herbst-Irmer R, Daub J, Porsch M, Bill E, Müther M, Trautwein AX. Angew. Chem. Int. Ed. Engl. 1994;33:1621. [Google Scholar]
- 333.Saalfrank RW, Hörner B, Stalke D, Salbeck J. Angew. Chem. Int. Ed. Engl. 1993;32:1179. [Google Scholar]
- 334.Beissel T, Powers RE, Raymond KN. Angew. Chem. Int. Ed. Engl. 1996;35:1084. [Google Scholar]
- 335.Beissel T, Powers RE, Parac TN, Raymond KN. J. Am. Chem. Soc. 1999;121:4200. [Google Scholar]
- 336.Caulder DL, Powers RE, Parac TN, Raymond KN. Angew. Chem. Int. Ed. 1998;37:1840. [Google Scholar]
- 337.Parac TN, Caulder DL, Raymond KN. J. Am. Chem. Soc. 1998;120:8003. doi: 10.1021/ja0104507. [DOI] [PubMed] [Google Scholar]
- 338.Johnson DW, Raymond KN. Inorg. Chem. 2001;40:5157. doi: 10.1021/ic0102283. [DOI] [PubMed] [Google Scholar]
- 339.Biros SM, Yeh RM, Raymond KN. Angew. Chem. Int. Ed. 2008;47:6062. doi: 10.1002/anie.200801226. [DOI] [PubMed] [Google Scholar]
- 340.Review: Ward MD. Chem. Commun. 2009:4487. doi: 10.1039/b906726b.
- 341.Fleming JS, Mann KLV, Carraz C-A, Psillakis E, Jeffery JC, McCleverty JA, Ward MD. Angew. Chem. Int. Ed. 1998;37:1279. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1279::AID-ANIE1279>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 342.Paul RL, Bell ZR, Jeffery JC, McCleverty JA, Ward MD. Proc. Natl. Acad. Sci. USA. 2002;99:4883. doi: 10.1073/pnas.052575199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Paul RL, Bell ZR, Jeffery JC, Harding LP, McCleverty JA, Ward MD. Polyhedron. 2003;22:781. [Google Scholar]
- 344.Tidmarsh IS, Taylor BF, Hardie MJ, Russo L, Clegg W, Ward MD. New J. Chem. 2009;33:366. [Google Scholar]
- 345.Paul RL, Couchman SM, Jeffery JC, McCleverty JA, Reeves ZR, Ward MD. J. Chem. Soc., Dalton Trans. 2000:845. [Google Scholar]
- 346.Paul RL, Argent SP, Jeffery JC, Harding LP, Lynam JM, Ward MD. Dalton Trans. 2004:3453. doi: 10.1039/B409809A. [DOI] [PubMed] [Google Scholar]
- 347.Bell ZR, Jeffery JC, McCleverty JA, Ward MD. Angew. Chem. Int. Ed. 2002;41:2515. doi: 10.1002/1521-3773(20020715)41:14<2515::AID-ANIE2515>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 348.Glasson CRK, Meehan GV, Clegg JK, Lindoy LF, Turner P, Duriska MB, Willis R. Chem. Commun. 2008:1190. doi: 10.1039/b717740b. [DOI] [PubMed] [Google Scholar]
- 349.Mal P, Schultz D, Beyeh K, Rissanen K, Nitschke JR. Angew. Chem. Int. Ed. 2008;47:8297. doi: 10.1002/anie.200803066. [DOI] [PubMed] [Google Scholar]
- 350.Mal P, Nitschke JR. Chem. Commun. 2010;46:2417. doi: 10.1039/b920745g. [DOI] [PubMed] [Google Scholar]
- 351.Hristova YR, Smulders MMJ, Clegg JK, Breiner B, Nitschke JR. Chem. Sci. 2011;2:638. [Google Scholar]
- 352.Clegg JK, Li F, Jolliffe KA, Meehan GV, Lindoy LF. Chem. Commun. 2011;47:6042. doi: 10.1039/c1cc11167a. [DOI] [PubMed] [Google Scholar]
- 353.Reviews: Albrecht M, Janser I, Fröhlich R. Chem. Commun. 2005:157. doi: 10.1039/b410828k. Albrecht M. Angew. Chem. Int. Ed. 1999;38:3463. doi: 10.1002/(sici)1521-3773(19991203)38:23<3463::aid-anie3463>3.3.co;2-5.
- 354.Brückner C, Powers RE, Raymond KN. Angew. Chem. Int. Ed. 1998;37:1837. [Google Scholar]
- 355.Saalfrank RW, Glaser H, Demleitner B, Hampel F, Chowdhry MM, Schünemann V, Trautwein AX, Vaughan GBM, Yeh R, Davis AV, Raymond KN. Chem. Eur. J. 2002;8:493. doi: 10.1002/1521-3765(20020118)8:2<493::AID-CHEM493>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 356.Amoroso AJ, Jeffery JC, Jones PL, McCleverty JA, Thornton P, Ward MD. Angew. Chem. Int. Ed. Engl. 1995;34:1443. [Google Scholar]
- 357.Paul RL, Amoroso AJ, Jones PL, Couchman SM, Reeves ZR, Rees LH, Jeffery JC, McCleverty JA, Ward MD. J. Chem. Soc., Dalton Trans. 1999:1563. [Google Scholar]
- 358.Yeh RM, Xu J, Seeber G, Raymond KN. Inorg. Chem. 2005;44:6228. doi: 10.1021/ic0505145. [DOI] [PubMed] [Google Scholar]
- 359.Albrecht M, Janser I, Meyer S, Weis P, Fröhlich R. Chem. Commun. 2003:2854. doi: 10.1039/b309495b. [DOI] [PubMed] [Google Scholar]
- 360.Albrecht M, Janser I, Runsink J, Raabe G, Weis P, Fröhlich R. Angew. Chem. Int. Ed. 2004;43:6662. doi: 10.1002/anie.200453975. [DOI] [PubMed] [Google Scholar]
- 361.Saalfrank RW, Maid H, Scheurer A, Heinemann FW, Puchta R, Bauer W, Stern D, Stalke D. Angew. Chem. Int. Ed. 2008;47:8941. doi: 10.1002/anie.200804225. [DOI] [PubMed] [Google Scholar]
- 362.Kusukawa T, Fujita M. J. Am. Chem. Soc. 2002;124:13576. doi: 10.1021/ja020712k. [DOI] [PubMed] [Google Scholar]
- 363.Yamashita K, Kawano M, Fujita M. Chem. Commun. 2007:4102. doi: 10.1039/b712529a. [DOI] [PubMed] [Google Scholar]
- 364.Leininger S, Fan J, Schmitz M, Stang PJ. Proc. Natl. Acad. Sci. USA. 2000;97:1380. doi: 10.1073/pnas.030264697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Stang PJ, Olenyuk B, Muddiman DC, Smith RD. Organometallics. 1997;16:3094. [Google Scholar]
- 366.Schweiger M, Yamamoto T, Stang PJ, Bläser D, Boese R. J. Org. Chem. 2005;70:4861. doi: 10.1021/jo050469i. [DOI] [PubMed] [Google Scholar]
- 367.Zheng Y-R, Zhao Z, Kim H, Wang M, Ghosh K, Pollock JB, Chi K-W, Stang PJ. Inorg. Chem. 2010;49:10238. doi: 10.1021/ic1018373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Granzhan A, Riis-Johannessen T, Scopelliti R, Severin K. Angew. Chem. Int. Ed. 2010;49:5515. doi: 10.1002/anie.201002748. [DOI] [PubMed] [Google Scholar]
- 369.Granzhan A, Schouwey C, Riis-Johannessen T, Scopelliti R, Severin K. J. Am. Chem. Soc. 2011;133:7106. doi: 10.1021/ja200580x. [DOI] [PubMed] [Google Scholar]
- 370.Roche S, Haslam C, Adams H, Heath SL, Thomas JA. Chem. Commun. 1998:1681. [Google Scholar]
- 371.Suzuki K, Tominaga M, Kawano M, Fujita M. Chem. Commun. 2009:1638. doi: 10.1039/b822311d. [DOI] [PubMed] [Google Scholar]
- 372.Natarajan R, Savitha G, Moorthy JN. Cryst. Growth Des. 2005;5:69. [Google Scholar]
- 373.Johannessen SC, Brisbois RG, Fischer JP, Grieco PA, Counterman AE, Clemmer DE. J. Am. Chem. Soc. 2001;123:3818. doi: 10.1021/ja004007s. [DOI] [PubMed] [Google Scholar]
- 374.Klausmeyer KK, Rauchfuss TB, Wilson SR. Angew. Chem. Int. Ed. 1998;37:1694. doi: 10.1002/(SICI)1521-3773(19980703)37:12<1694::AID-ANIE1694>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 375.Klausmeyer KK, Wilson SR, Rauchfuss TB. J. Am. Chem. Soc. 1999;121:2705. [Google Scholar]
- 376.Lang J-P, Xu Q-F, Chen Z-N, Abrahams BF. J. Am. Chem. Soc. 2003;125:12682. doi: 10.1021/ja036995d. [DOI] [PubMed] [Google Scholar]
- 377.Heinrich JL, Berseth PA, Long JR. Chem. Commun. 1998:1231. [Google Scholar]
- 378.Berseth PA, Sokol JJ, Shores MP, Heinrich JL, Long JR. J. Am. Chem. Soc. 2000;122:9655. [Google Scholar]
- 379.Sokol JJ, Shores MP, Long JR. Inorg. Chem. 2002;41:3052. doi: 10.1021/ic0255499. [DOI] [PubMed] [Google Scholar]
- 380.Bell ZR, Harding LP, Ward MD. Chem. Commun. 2003:2432. doi: 10.1039/b307172n. [DOI] [PubMed] [Google Scholar]
- 381.Argent SP, Adams H, Harding LP, Ward MD. Dalton Trans. 2006:542. doi: 10.1039/b515296h. [DOI] [PubMed] [Google Scholar]
- 382.Tidmarsh IS, Faust TB, Adams H, Harding LP, Russo L, Clegg W, Ward MD. J. Am. Chem. Soc. 2008;130:15167. doi: 10.1021/ja805605y. [DOI] [PubMed] [Google Scholar]
- 383.Meng W, Breiner B, Rissanen K, Thoburn JD, Clegg JK, Nitschke JR. Angew. Chem. Int. Ed. 2011;50:3479. doi: 10.1002/anie.201100193. [DOI] [PubMed] [Google Scholar]
- 384.Hiraoka S, Harano K, Shiro M, Ozawa Y, Yasuda N, Toriumi K, Shionoya M. Angew. Chem. Int. Ed. 2006;45:6488. doi: 10.1002/anie.200601431. [DOI] [PubMed] [Google Scholar]
- 385.Harano K, Hiraoka S, Shionoya M. J. Am. Chem. Soc. 2007;129:5300. doi: 10.1021/ja0659727. [DOI] [PubMed] [Google Scholar]
- 386.Ronson TK, Fisher J, Harding LP, Hardie MJ. Angew. Chem. Int. Ed. 2007;46:9086. doi: 10.1002/anie.200703903. [DOI] [PubMed] [Google Scholar]
- 387.Prakash MJ, Zou Y, Hong S, Park M, Bui M-PN, Seong GH, Lah MS. Inorg. Chem. 2009;48:1281. doi: 10.1021/ic802382p. [DOI] [PubMed] [Google Scholar]
- 388.Müller IM, Spillmann S, Franck H, Pietschnig R. Chem. Eur. J. 2004;10:2207. doi: 10.1002/chem.200305564. [DOI] [PubMed] [Google Scholar]
- 389.Li J-R, Timmons DJ, Zhou H-C. J. Am. Chem. Soc. 2009;131:6368. doi: 10.1021/ja901731z. [DOI] [PubMed] [Google Scholar]
- 390.Olenyuk B, Levin MD, Whiteford JA, Shield JE, Stang PJ. J. Am. Chem. Soc. 1999;121:10434. [Google Scholar]
- 391.Levin MD, Stang PJ. J. Am. Chem. Soc. 2000;122:7428. [Google Scholar]
- 392.Paquette LA, Balogh DW, Usha R, Kountz D, Christoph GG. Science. 1981;211:575. doi: 10.1126/science.211.4482.575. [DOI] [PubMed] [Google Scholar]
- 393.Melder J-P, Pinkos R, Fritz H, Prinzbach H. Angew. Chem. Int. Ed. Engl. 1989;28:305. [Google Scholar]
- 394.Olenyuk B, Whiteford JA, Fechtenkötter A, Stang PJ. Nature. 1999;398:796. doi: 10.1038/19740. [DOI] [PubMed] [Google Scholar]
- 395.Argent SP, Adams H, Riis-Johannessen T, Jeffery JC, Harding LP, Ward MD. J. Am. Chem. Soc. 2006;128:72. doi: 10.1021/ja056993o. [DOI] [PubMed] [Google Scholar]
- 396.Al-Rasbi NK, Tidmarsh IS, Argent SP, Adams H, Harding LP, Ward MD. J. Am. Chem. Soc. 2008;130:11641. doi: 10.1021/ja803847w. [DOI] [PubMed] [Google Scholar]
- 397.Fujita M, Nagao S, Ogura K. J. Am. Chem. Soc. 1995;117:1649. doi: 10.1021/ja00110a026. [DOI] [PubMed] [Google Scholar]
- 398.Radhakrishnan U, Schweiger M, Stang PJ. Org. Lett. 2001;3:3141. doi: 10.1021/ol0164684. [DOI] [PubMed] [Google Scholar]
- 399.Mukherjee PS, Das N, Stang PJ. J. Org. Chem. 2004;69:3526. doi: 10.1021/jo049772u. [DOI] [PubMed] [Google Scholar]
- 400.Ghosh S, Mukherjee PS. Tetrahedron Lett. 2006;47:9297. [Google Scholar]
- 401.Fujita M, Yu S-Y, Kusukawa T, Funaki H, Ogura K, Yamaguchi K. Angew. Chem. Int. Ed. 1998;37:2082. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2082::AID-ANIE2082>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 402.Ghosh S, Batten SR, Turner DR, Mukherjee PS. Organometallics. 2007;26:3252. [Google Scholar]
- 403.Wang M, Vajpayee V, Shanmugaraju S, Zheng Y-R, Zhao Z, Kim H, Mukherjee PS, Chi K-W, Stang PJ. Inorg. Chem. 2011;50:1506. doi: 10.1021/ic1020719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Bar AK, Chakrabarty R, Mukherjee PS. Inorg. Chem. 2009;48:10880. doi: 10.1021/ic901633g. [DOI] [PubMed] [Google Scholar]
- 405.Garrison JC, Panzner MJ, Custer PD, Reddy DV, Rinaldi PL, Tessier CA, Youngs WJ. Chem. Commun. 2006:4644. doi: 10.1039/b608991g. [DOI] [PubMed] [Google Scholar]
- 406.Schweiger M, Seidel SR, Schmitz M, Stang PJ. Org. Lett. 2000;2:1255. doi: 10.1021/ol005781n. [DOI] [PubMed] [Google Scholar]
- 407.Zheng Y-R, Ghosh K, Yang H-B, Stang PJ. Inorg. Chem. 2010;49:4747. doi: 10.1021/ic100330c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Cui Y, Ngo HL, Lin W. Inorg. Chem. 2002;41:5940. doi: 10.1021/ic025804p. [DOI] [PubMed] [Google Scholar]
- 409.Kumazawa K, Biradha K, Kusukawa T, Okano T, Fujita M. Angew. Chem. Int. Ed. 2003;42:3909. doi: 10.1002/anie.200351797. [DOI] [PubMed] [Google Scholar]
- 410.Yoshizawa M, Nakagawa J, Kumazawa K, Nagao M, Kawano M, Ozeki T, Fujita M. Angew. Chem. Int. Ed. 2005;44:1810. doi: 10.1002/anie.200462171. [DOI] [PubMed] [Google Scholar]
- 411.Ono K, Yoshizawa M, Kato T, Watanabe K, Fujita M. Angew. Chem. Int. Ed. 2007;46:1803. doi: 10.1002/anie.200604790. [DOI] [PubMed] [Google Scholar]
- 412.Yoshizawa M, Ono K, Kumazawa K, Kato T, Fujita M. J. Am. Chem. Soc. 2005;127:10800. doi: 10.1021/ja053009f. [DOI] [PubMed] [Google Scholar]
- 413.Kuehl CJ, Yamamoto T, Seidel SR, Stang PJ. Org. Lett. 2002;4:913. doi: 10.1021/ol017296d. [DOI] [PubMed] [Google Scholar]
- 414.For examples see: Hiraoka S, Kubota Y, Fujita M. Chem. Commun. 2000:1509. Ikeda A, Udzu H, Zhong Z, Shinkai S, Sakamoto S, Yamaguchi K. J. Am. Chem. Soc. 2001;123:3872. doi: 10.1021/ja003269r. Ikeda A, Yoshimura M, Udzu H, Fukuhara C, Shinkai S. J. Am. Chem. Soc. 1999;121:4296. Liu H-K, Sun W-Y, Ma D-J, Yu K-B, Tang W-X. Chem. Commun. 2000:591.
- 415.Ghosh S, Mukherjee PS. Organometallics. 2008;27:316. [Google Scholar]
- 416.Ghosh S, Gole B, Bar AK, Mukherjee PS. Organometallics. 2009;28:4288. doi: 10.1039/b911622m. [DOI] [PubMed] [Google Scholar]
- 417.Kuehl CJ, Kryschenko YK, Radhakrishnan U, Seidel SR, Huang SD, Stang PJ. Proc. Natl. Acad. Sci. USA. 2002;99:4932. doi: 10.1073/pnas.012540799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Kryschenko YK, Seidel SR, Muddiman DC, Nepomuceno AI, Stang PJ. J. Am. Chem. Soc. 2003;125:9647. doi: 10.1021/ja030209n. [DOI] [PubMed] [Google Scholar]
- 419.Benkstein KD, Hupp JT. Mol. Cryst. Liq. Cryst. 2000;342:151. [Google Scholar]
- 420.Sun S-S, Lees AJ. Chem. Commun. 2001:103. [Google Scholar]
- 421.Dinolfo PH, Coropceanu V, Brédas J-L, Hupp JT. J. Am. Chem. Soc. 2006;128:12592. doi: 10.1021/ja0623764. [DOI] [PubMed] [Google Scholar]
- 422.Manimaran B, Rajendran T, Lu Y-L, Lee G-H, Peng S-M, Lu K-L. Eur. J. Inorg. Chem. 2001:633. [Google Scholar]
- 423.Wu J-Y, Chang C-H, Thanasekaran P, Tsai C-C, Tseng T-W, Lee G-H, Peng S-M, Lu K-L. Dalton Trans. 2008:6110. doi: 10.1039/b809489f. [DOI] [PubMed] [Google Scholar]
- 424.Govindaswamy P, Süss-Fink G, Therrien B. Organometallics. 2007;26:915. [Google Scholar]
- 425.Govindaswamy P, Süss-Fink G, Therrien B. Inorg. Chem. Commun. 2007;10:1489. [Google Scholar]
- 426.Govindaswamy P, Linder D, Lacour J, Süss-Fink G, Therrien B. Chem. Commun. 2006:4691. doi: 10.1039/b610155k. [DOI] [PubMed] [Google Scholar]
- 427.Fan Y-F, Lin Y-J, Jia W-G, Weng L-H, Jin G-X. Organometallics. 2007;26:5848. [Google Scholar]
- 428.Mattsson J, Govindaswamy P, Furrer J, Sei Y, Yamaguchi K, Süss-Fink G, Therrien B. Organometallics. 2008;27:4346. [Google Scholar]
- 429.Freudenreich J, Barry NPE, Süss-Fink G, Therrien B. Eur. J. Inorg. Chem. 2010:2400. [Google Scholar]
- 430.Freudenreich J, Furrer J, Süss-Fink G, Therrien B. Organometallics. 2011;30:942. [Google Scholar]
- 431.Therrien B, Süss-Fink G, Govindaswamy P, Renfrew AK, Dyson PJ. Angew. Chem. Int. Ed. 2008;47:3773. doi: 10.1002/anie.200800186. [DOI] [PubMed] [Google Scholar]
- 432.Shanmugaraju S, Bar AK, Mukherjee PS. Inorg. Chem. 2010;49:10235. doi: 10.1021/ic101823s. [DOI] [PubMed] [Google Scholar]
- 433.Mirtschin S, Slabon-Turski A, Scopelliti R, Velders AH, Severin K. J. Am. Chem. Soc. 2010;132:14004. doi: 10.1021/ja1063789. [DOI] [PubMed] [Google Scholar]
- 434.Lee SJ, Mulfort KL, O’Donnell JL, Zuo X, Goshe AJ, Wesson PJ, Nguyen ST, Hupp JT, Tiede DM. Chem. Commun. 2006:4581. doi: 10.1039/b610025b. [DOI] [PubMed] [Google Scholar]
- 435.Kelley RF, Lee SJ, Wilson TM, Nakamura Y, Tiede DM, Osuka A, Hupp JT, Wasielewski MR. J. Am. Chem. Soc. 2008;130:4277. doi: 10.1021/ja075494f. [DOI] [PubMed] [Google Scholar]
- 436.Youm K-T, Nguyen ST, Hupp JT. Chem. Commun. 2008:3375. doi: 10.1039/b800063h. [DOI] [PubMed] [Google Scholar]
- 437.Oliva AI, Ventura B, Würthner F, Camara-Campos A, Hunter CA, Ballester P, Flamigni L. Dalton Trans. 2009:4023. doi: 10.1039/b819496c. [DOI] [PubMed] [Google Scholar]
- 438.Indelli MT, Chiorboli C, Scandola F, Iengo E, Osswald P, Würthner F. J. Phys. Chem. B. 2010;114:14495. doi: 10.1021/jp101849m. [DOI] [PubMed] [Google Scholar]
- 439.Zheng Y-R, Zhao Z, Wang M, Ghosh K, Pollock JB, Cook TR, Stang PJ. J. Am. Chem. Soc. 2010;132:16873. doi: 10.1021/ja106251f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Zhao L, Northrop BH, Stang PJ. J. Am. Chem. Soc. 2008;130:11886. doi: 10.1021/ja805770w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Stephenson A, Argent SP, Riis-Johannessen T, Tidmarsh IS, Ward MD. J. Am. Chem. Soc. 2011;133:858. doi: 10.1021/ja107403p. [DOI] [PubMed] [Google Scholar]
- 442.Campbell VE, de Hatten X, Delsuc N, Kauffmann B, Huc I, Nitschke JR. Nat. Chem. 2010;2:684. doi: 10.1038/nchem.693. [DOI] [PubMed] [Google Scholar]
- 443.Yamanaka M, Yamada Y, Sei Y, Yamaguchi K, Kobayashi K. J. Am. Chem. Soc. 2006;128:1531. doi: 10.1021/ja0555365. [DOI] [PubMed] [Google Scholar]
- 444.Bar AK, Mostafa G, Mukherjee PS. Inorg. Chem. 2010;49:7647. doi: 10.1021/ic101139s. [DOI] [PubMed] [Google Scholar]
- 445.Schmittel M, He B, Mal P. Org. Lett. 2008;10:2513. doi: 10.1021/ol800796h. [DOI] [PubMed] [Google Scholar]
- 446.Schmittel M, He B. Chem. Commun. 2008:4723. doi: 10.1039/b808988d. [DOI] [PubMed] [Google Scholar]
- 447.Barbour LJ, Orr GW, Atwood JL. Nature. 1998;393:671. [Google Scholar]
- 448.Su C-Y, Cai Y-P, Chen C-L, Zhang H-X, Kang B-S. J. Chem. Soc., Dalton Trans. 2001:359. [Google Scholar]
- 449.Su C-Y, Cai Y-P, Chen C-L, Smith MD, Kaim W, zur Loye H-C. J. Am. Chem. Soc. 2003;125:8595. doi: 10.1021/ja034267k. [DOI] [PubMed] [Google Scholar]
- 450.Liu Z-M, Liu Y, Zheng S-R, Yu Z-Q, Pan M, Su C-Y. Inorg. Chem. 2007;46:5814. doi: 10.1021/ic062270+. [DOI] [PubMed] [Google Scholar]
- 451.Owens TD, Hollander FJ, Oliver AG, Ellman JA. J. Am. Chem. Soc. 2001;123:1539. doi: 10.1021/ic0104205. [DOI] [PubMed] [Google Scholar]
- 452.Chand DK, Biradha K, Fujita M. Chem. Commun. 2001:1652. doi: 10.1039/b104853h. [DOI] [PubMed] [Google Scholar]
- 453.McMorran DA, Steel PJ. Angew. Chem. Int. Ed. 1998;37:3295. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3295::AID-ANIE3295>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 454.Yue NLS, Eisler DJ, Jennings MC, Puddephatt RJ. Inorg. Chem. 2004;43:7671. doi: 10.1021/ic048893+. [DOI] [PubMed] [Google Scholar]
- 455.Barry NPE, Govindaswamy P, Furrer J, Süss-Fink G, Therrien B. Inorg. Chem. Commun. 2008;11:1300. [Google Scholar]
- 456.Barry NPE, Abd Karim NH, Vilar R, Therrien B. Dalton Trans. 2009:10717. doi: 10.1039/b913642h. [DOI] [PubMed] [Google Scholar]
- 457.Han Y-F, Lin Y-J, Weng L-H, Berke H, Jin G-X. Chem. Commun. 2008:350. doi: 10.1039/b711809k. [DOI] [PubMed] [Google Scholar]
- 458.Barry NPE, Austeri M, Lacour J, Therrien B. Organometallics. 2009;28:4894. [Google Scholar]
- 459.Manimaran B, Thanasekaran P, Rajendran T, Liao R-T, Liu Y-H, Lee G-H, Peng S-M, Rajagopal S, Lu K-L. Inorg. Chem. 2003;42:4795. doi: 10.1021/ic034172j. [DOI] [PubMed] [Google Scholar]
- 460.Wang M, Zheng Y-R, Ghosh K, Stang PJ. J. Am. Chem Soc. 2010;132:6282. doi: 10.1021/ja100889h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Zhao Z, Zheng Y-R, Wang M, Pollock JB, Stang PJ. Inorg. Chem. 2010;49:8653. doi: 10.1021/ic1014219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Wang M, Zheng Y-R, Cook TR, Stang PJ. Inorg. Chem. 2011 doi: 10.1021/ic2002157. DOI: 10.1021/ic2002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Caskey DC, Yamamoto T, Addicott C, Shoemaker RK, Vacek J, Hawkridge AM, Muddiman DC, Kottas GS, Michl J, Stang PJ. J. Am. Chem. Soc. 2008;130:7620. doi: 10.1021/ja710715e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Vacek J, Caskey DC, Horinek D, Shoemaker RK, Stang PJ, Michl J. J. Am. Chem. Soc. 2008;130:7629. doi: 10.1021/ja801341m. [DOI] [PubMed] [Google Scholar]
- 465.Yamanoi Y, Sakamoto Y, Kusukawa T, Fujita M, Sakamoto S, Yamaguchi K. J. Am. Chem. Soc. 2001;123:980. doi: 10.1021/ja003043o. [DOI] [PubMed] [Google Scholar]
- 466.Suzuki K, Kawano M, Fujita M. Angew. Chem. Int. Ed. 2007;46:2819. doi: 10.1002/anie.200605084. [DOI] [PubMed] [Google Scholar]
- 467.Liu H-K, Tong X. Chem. Commun. 2002:1316. doi: 10.1039/b201965e. [DOI] [PubMed] [Google Scholar]
- 468.Chand DK, Biradha K, Fujita M, Sakamoto S, Yamaguchi K. Chem. Commun. 2002:2486. [Google Scholar]
- 469.Hong M, Zhao Y, Su W, Cao R, Fujita M, Zhou Z, Chan ASC. J. Am. Chem. Soc. 2000;122:4819. [Google Scholar]
- 470.Moon D, Kang S, Park J, Lee K, John RP, Won H, Seong GH, Kim YS, Kim GH, Rhee H, Lah MS. J. Am. Chem. Soc. 2006;128:3530. doi: 10.1021/ja060051h. [DOI] [PubMed] [Google Scholar]
- 471.Ghosh S, Mukherjee PS. J. Org. Chem. 2006;71:8412. doi: 10.1021/jo061311g. [DOI] [PubMed] [Google Scholar]
- 472.Duriska MB, Neville SM, Moubaraki B, Cashion JD, Halder GJ, Chapman KW, Balde C, Létard J-F, Murray KS, Kepert CJ, Batten SR. Angew. Chem. Int. Ed. 2009;48:2549. doi: 10.1002/anie.200805178. [DOI] [PubMed] [Google Scholar]
- 473.Duriska MB, Neville SM, Lu J, Iremonger SS, Boas JF, Kepert CJ, Batten SR. Angew. Chem. Int. Ed. 2009;48:8919. doi: 10.1002/anie.200903863. [DOI] [PubMed] [Google Scholar]
- 474.Tominaga M, Suzuki K, Kawano M, Kusukawa T, Ozeki T, Sakamoto S, Yamaguchi K, Fujita M. Angew. Chem. Int. Ed. 2004;43:5621. doi: 10.1002/anie.200461422. [DOI] [PubMed] [Google Scholar]
- 475.Moulton B, Lu J, Mondal A, Zaworotko MJ. Chem. Commun. 2001:863. [Google Scholar]
- 476.Sun Q-F, Iwasa J, Ogawa D, Ishido Y, Sato S, Ozeki T, Sei Y, Yamaguchi K, Fujita M. Science. 2010;328:1144. doi: 10.1126/science.1188605. [DOI] [PubMed] [Google Scholar]
- 477.Reviews: Scarso A, Rebek J., Jr. Top. Curr. Chem. 2006;265:1. Mateos-Timoneda MA, Crego-Calama M, Reinhoudt DN. Chem. Soc. Rev. 2004;33:363. doi: 10.1039/b305550g.
- 478.Reviews: Seeber G, Tiedemann BEF, Raymond KN. Top. Curr. Chem. 2006;265:147. Hamilton TD, MacGillivray LR. Cryst. Growth Des. 2004;4:419.
- 479.Saalfrank RW, Löw N, Demleitner B, Stalke D, Teichert M. Chem. Eur. J. 1998;4:1305. [Google Scholar]
- 480.(a) Saalfrank RW, Demleitner B, Glaser H, Maid H, Bathelt D, Hampel F, Bauer W, Teichert M. Chem. Eur. J. 2002;8:2679. doi: 10.1002/1521-3765(20020617)8:12<2679::AID-CHEM2679>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]; (b) Saalfrank RW, Maid H, Scheurer A, Puchta R, Bauer W. Eur. J. Inorg. Chem. 2010:2903. [Google Scholar]
- 481.Fiedler D, Pagliero D, Brumaghim JL, Bergman RG, Raymond KN. Inorg. Chem. 2004;43:846. doi: 10.1021/ic035105s. [DOI] [PubMed] [Google Scholar]
- 482.Terpin AJ, Ziegler M, Johnson DW, Raymond KN. Angew. Chem. Int. Ed. 2001;40:157. [PubMed] [Google Scholar]
- 483.Davis AV, Fiedler D, Ziegler M, Terpin A, Raymond KN. J. Am. Chem. Soc. 2007;129:15354. doi: 10.1021/ja0764815. [DOI] [PubMed] [Google Scholar]
- 484.Ziegler M, Davis AV, Johnson DW, Raymond KN. Angew. Chem. Int. Ed. 2003;42:665. doi: 10.1002/anie.200390183. [DOI] [PubMed] [Google Scholar]
- 485.Frantz R, Grange CS, Al-Rasbi NK, Ward MD, Lacour J. Chem. Commun. 2007:1459. doi: 10.1039/b618092b. [DOI] [PubMed] [Google Scholar]
- 486.Argent SP, Riis-Johannessen T, Jeffery JC, Harding LP, Ward MD. Chem. Commun. 2005:4647. doi: 10.1039/b509239f. [DOI] [PubMed] [Google Scholar]
- 487.Hamilton TD, Bučar D-K, MacGillivray LR. Chem. Commun. 2007:1603. doi: 10.1039/b700748e. [DOI] [PubMed] [Google Scholar]
- 488.Hamilton TD, Papaefstathiou GS, MacGillivray LR. J. Am. Chem. Soc. 2002;124:11606. doi: 10.1021/ja0278281. [DOI] [PubMed] [Google Scholar]
- 489.Müller IM, Robson R, Separovic F. Angew. Chem. Int. Ed. 2001;40:4385. doi: 10.1002/1521-3773(20011203)40:23<4385::aid-anie4385>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 490.Müller IM, Möller D, Schalley CA. Angew. Chem. Int. Ed. 2005;44:480. doi: 10.1002/anie.200461800. [DOI] [PubMed] [Google Scholar]
- 491.Müller IM, Möller D. Angew. Chem. Int. Ed. 2005;44:2969. doi: 10.1002/anie.200463034. [DOI] [PubMed] [Google Scholar]
- 492.Childs LJ, Alcock NW, Hannon MJ. Angew. Chem. Int. Ed. 2002;41:4244. doi: 10.1002/1521-3773(20021115)41:22<4244::AID-ANIE4244>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 493.Liu T, Liu Y, Xuan W, Cui Y. Angew. Chem. Int. Ed. 2010;49:4121. doi: 10.1002/anie.201000416. [DOI] [PubMed] [Google Scholar]
- 494.Jeong KS, Kim YS, Kim YJ, Lee E, Yoon JH, Park WH, Park YW, Jeon S-J, Kim ZH, Kim J, Jeong N. Angew. Chem. Int. Ed. 2006;45:8134. doi: 10.1002/anie.200603622. [DOI] [PubMed] [Google Scholar]
- 495.Ayabe M, Yamashita K, Sada K, Shinkai S, Ikeda A, Sakamoto S, Yamaguchi K. J. Org. Chem. 2003;68:1059–1066. doi: 10.1021/jo020575+. [DOI] [PubMed] [Google Scholar]
- 496.Kubota Y, Biradha K, Fujita M, Sakamoto S, Yamaguchi K. Bull. Chem. Soc. Jpn. 2002;75:559. [Google Scholar]
- 497.Hiraoka S, Fujita M. J. Am. Chem. Soc. 1999;121:10239. [Google Scholar]
- 498.Kamiya N, Tominaga M, Sato S, Fujita M. J. Am. Chem. Soc. 2007;129:3816. doi: 10.1021/ja0693082. [DOI] [PubMed] [Google Scholar]
- 499.Kikuchi T, Sato S, Fujita M. J. Am. Chem. Soc. 2010;132:15930. doi: 10.1021/ja108334g. [DOI] [PubMed] [Google Scholar]
- 500.Ikemi M, Kikuchi T, Matsumura S, Shiba K, Sato S, Fujita M. Chem. Sci. 2010;1:68. doi: 10.1021/jacs.5b06184. [DOI] [PubMed] [Google Scholar]
- 501.Ghosh K, Hu J, White HS, Stang PJ. J. Am. Chem. Soc. 2009;131:6695. doi: 10.1021/ja902045q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Tominaga M, Suzuki K, Murase T, Fujita M. J. Am. Chem. Soc. 2005;127:11950. doi: 10.1021/ja054069o. [DOI] [PubMed] [Google Scholar]
- 503.Sato S, Iida J, Suzuki K, Kawano M, Ozeki T, Fujita M. Science. 2006;313:1273. doi: 10.1126/science.1129830. [DOI] [PubMed] [Google Scholar]
- 504.Suzuki K, Takao K, Sato S, Fujita M. J. Am. Chem. Soc. 2010;132:2544. doi: 10.1021/ja910836h. [DOI] [PubMed] [Google Scholar]
- 505.Murase T, Sato S, Fujita M. Angew. Chem. Int. Ed. 2007;46:1083. doi: 10.1002/anie.200603561. [DOI] [PubMed] [Google Scholar]
- 506.Kikuchi T, Murase T, Sato S, Fujita M. Supramol. Chem. 2008;20:81. [Google Scholar]
- 507.Suzuki K, Kawano M, Sato S, Fujita M. J. Am. Chem. Soc. 2007;129:10652. doi: 10.1021/ja073629b. [DOI] [PubMed] [Google Scholar]
- 508.Suzuki K, Sato S, Fujita M. Nat. Chem. 2010;2:25. doi: 10.1038/nchem.446. [DOI] [PubMed] [Google Scholar]
- 509.Suzuki K, Takao K, Sato S, Fujita M. Angew. Chem. Int. Ed. 2011;50:4858. doi: 10.1002/anie.201006965. [DOI] [PubMed] [Google Scholar]
- 510.Murase T, Sato S, Fujita M. Angew. Chem. Int. Ed. 2007;46:5133. doi: 10.1002/anie.200700793. [DOI] [PubMed] [Google Scholar]
- 511.Suzuki K, Iida J, Sato S, Kawano M, Fujita M. Angew. Chem. Int. Ed. 2008;47:5780. doi: 10.1002/anie.200801700. [DOI] [PubMed] [Google Scholar]
- 512.Li D, Zhou W, Landskron K, Sato S, Kiely CJ, Fujita M, Liu T. Angew. Chem. Int. Ed. 2011;50:5182. doi: 10.1002/anie.201007829. [DOI] [PubMed] [Google Scholar]
- 513.Li D, Zhang J, Landskron K, Liu T. J. Am. Chem. Soc. 2008;130:4226. doi: 10.1021/ja710820a. [DOI] [PubMed] [Google Scholar]
- 514.Ballester P, Oliva AI, Costa A, Deyà PM, Frontera A, Gomila RM, Hunter CA. J. Am. Chem. Soc. 2006;128:5560. doi: 10.1021/ja060608t. [DOI] [PubMed] [Google Scholar]
- 515.Flamigni L, Ventura B, Oliva AI, Ballester P. Chem. Eur. J. 2008;14:4214. doi: 10.1002/chem.200800026. [DOI] [PubMed] [Google Scholar]
- 516.Slagt VF, van Leeuwen PWNM, Reek JNH. Angew. Chem. Int. Ed. 2003;42:5619. doi: 10.1002/anie.200352525. [DOI] [PubMed] [Google Scholar]
- 517.Lee SJ, Mulfort KL, Zuo X, Goshe AJ, Wesson PJ, Nguyen ST, Hupp JT, Tiede DM. J. Am. Chem. Soc. 2008;130:836. doi: 10.1021/ja077661h. [DOI] [PubMed] [Google Scholar]
- 518.Aratani N, Kim D, Osuka A. Acc. Chem. Res. 2009;42:1922. doi: 10.1021/ar9001697. [DOI] [PubMed] [Google Scholar]
- 519.Tsuda A, Nakamura T, Sakamoto S, Yamaguchi K, Osuka A. Angew. Chem. Int. Ed. 2002;41:2817. doi: 10.1002/1521-3773(20020802)41:15<2817::AID-ANIE2817>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 520.Hwang I-W, Kamada T, Ahn TK, Ko DM, Nakamura T, Tsuda A, Osuka A, Kim D. J. Am. Chem. Soc. 2004;126:16187. doi: 10.1021/ja046241e. [DOI] [PubMed] [Google Scholar]
- 521.Review: Cram DJ. Nature. 1992;356:29.
- 522.Reviews: Purse BW, Rebek J., Jr. Proc. Natl. Acad. Sci. USA. 2005;102:10777. doi: 10.1073/pnas.0501731102. Warmuth R, Yoon J. Acc. Chem. Res. 2001;34:95. doi: 10.1021/ar980082k. Jasat A, Sherman JC. Chem. Rev. 1999;99:931. doi: 10.1021/cr960048o.
- 523.Jacopozzi P, Dalcanale E. Angew. Chem. Int. Ed. Engl. 1997;36:613. [Google Scholar]
- 524.Fochi F, Jacopozzi P, Wegelius E, Rissanen K, Cozzini P, Marastoni E, Fisicaro E, Manini P, Fokkens R, Dalcanale E. J. Am. Chem. Soc. 2001;123:7539. doi: 10.1021/ja0103492. [DOI] [PubMed] [Google Scholar]
- 525.Zuccaccia D, Pirondini L, Pinalli R, Dalcanale E, Macchioni A. J. Am. Chem. Soc. 2005;127:7025. doi: 10.1021/ja042265+. [DOI] [PubMed] [Google Scholar]
- 526.Cuminetti N, Ebbing MHK, Prados P, de Mendoza J, Dalcanale E. Tetrahedron Lett. 2001;42:527. [Google Scholar]
- 527.Gruppi F, Boccini F, Elviri L, Dalcanale E. Tetrahedron. 2009;65:7289. [Google Scholar]
- 528.Kobayashi K, Yamada Y, Yamanaka M, Sei Y, Yamaguchi K. J. Am. Chem. Soc. 2004;126:13896. doi: 10.1021/ja0465799. [DOI] [PubMed] [Google Scholar]
- 529.Lim CW, Hong J-I. Tetrahedron Lett. 2000;41:3113. [Google Scholar]
- 530.Park SJ, Hong J-I. Chem. Commun. 2001:1554. doi: 10.1039/b104897j. [DOI] [PubMed] [Google Scholar]
- 531.Park SJ, Shin DM, Sakamoto S, Yamaguchi K, Chung YK, Lah MS, Hong J-I. Chem. Commun. 2003:998. doi: 10.1039/b212855a. [DOI] [PubMed] [Google Scholar]
- 532.Park SJ, Shin DM, Sakamoto S, Yamaguchi K, Chung YK, Lah MS, Hong J-I. Chem. Eur. J. 2005;11:235. doi: 10.1002/chem.200400801. [DOI] [PubMed] [Google Scholar]
- 533.Haino T, Kobayashi M, Chikaraishi M, Fukazawa Y. Chem. Commun. 2005:2321. doi: 10.1039/b502598b. [DOI] [PubMed] [Google Scholar]
- 534.Haino T, Kobayashi M, Fukazawa Y. Chem. Eur. J. 2006;12:3310. doi: 10.1002/chem.200500976. [DOI] [PubMed] [Google Scholar]
- 535.Fox OD, Dalley NK, Harrison RG. J. Am. Chem. Soc. 1998;120:7111. [Google Scholar]
- 536.Fox OD, Dalley NK, Harrison RG. Inorg. Chem. 1999;38:5860. [Google Scholar]
- 537.Fox OD, Leung JF-Y, Hunter JM, Dalley NK, Harrison RG. Inorg. Chem. 2000;39:783. doi: 10.1021/ic991235i. [DOI] [PubMed] [Google Scholar]
- 538.Harrison RG, Burrows JL, Hansen LD. Chem. Eur. J. 2005;11:5881. doi: 10.1002/chem.200500299. [DOI] [PubMed] [Google Scholar]
- 539.Pirondini L, Bertolini F, Cantadori B, Ugozzoli F, Massera C, Dalcanale E. Proc. Natl. Acad. Sci. USA. 2002;99:4911. doi: 10.1073/pnas.072612199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Pirondini L, Bonifazi D, Cantadori B, Braiuca P, Campagnolo M, Zorzi RD, Geremia S, Diederich F, Dalcanale E. Tetrahedron. 2006;62:2008. [Google Scholar]
- 541.Pinalli R, Cristini V, Sottili V, Geremia S, Campagnolo M, Caneschi A, Dalcanale E. J. Am. Chem. Soc. 2004;126:6516. doi: 10.1021/ja038694+. [DOI] [PubMed] [Google Scholar]
- 542.Ikeda A, Shinkai S. Chem. Rev. 1997;97:1713. doi: 10.1021/cr960385x. [DOI] [PubMed] [Google Scholar]
- 543.Baldini L, Ballester P, Casnati A, Gomila RM, Hunter CA, Sansone F, Ungaro R. J. Am. Chem. Soc. 2003;125:14181. doi: 10.1021/ja036758a. [DOI] [PubMed] [Google Scholar]
- 544.Cotton FA, Lei P, Lin C, Murillo CA, Wang X, Yu S-Y, Zhang Z-X. J. Am. Chem. Soc. 2004;126:1518. doi: 10.1021/ja0396899. [DOI] [PubMed] [Google Scholar]
- 545.Fox OD, Drew MGB, Beer PD. Angew. Chem. Int. Ed. 2000;39:136. doi: 10.1002/(sici)1521-3773(20000103)39:1<135::aid-anie135>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 546.Fox OD, Cookson J, Wilkinson EJS, Drew MGB, MacLean EJ, Teat SJ, Beer PD. J. Am. Chem. Soc. 2006;128:6990. doi: 10.1021/ja060982t. [DOI] [PubMed] [Google Scholar]
- 547.Fox OD, Drew MGB, Wilkinson EJS, Beer PD. Chem. Commun. 2000:391. [Google Scholar]
- 548.Schröder T, Brodbeck R, Letzel MC, Mix A, Schnatwinkel B, Tonigold M, Volkmer D, Mattay J. Tetrahedron Lett. 2008;49:5939. [Google Scholar]
- 549.Reviews: Rebek J., Jr. Acc. Chem. Res. 2009;42:1660. doi: 10.1021/ar9001203. Ajami D, Rebek J., Jr. Supramol. Chem. 2009;21:103. doi: 10.1080/10610278.2010.510188. Rebek J., Jr. Angew. Chem. Int. Ed. 2005;44:2068. doi: 10.1002/anie.200462839. Palmer LC, Rebek J., Jr. Org. Biomol. Chem. 2004;2:3051. doi: 10.1039/B412510J.
- 550.Pluth MD, Raymond KN. Chem. Soc. Rev. 2007;36:161. doi: 10.1039/b603168b. [DOI] [PubMed] [Google Scholar]
- 551.Pluth MD, Johnson DW, Szigethy G, Davis AV, Teat SJ, Oliver AG, Bergman RG, Raymond KN. Inorg. Chem. 2009;48:111. doi: 10.1021/ic8012848. [DOI] [PubMed] [Google Scholar]
- 552.Leung DH, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2008;130:2798. doi: 10.1021/ja075975z. [DOI] [PubMed] [Google Scholar]
- 553.Sgarlata C, Mugridge JS, Pluth MD, Tiedemann BEF, Zito V, Arena G, Raymond KN. J. Am. Chem. Soc. 2010;132:1005. doi: 10.1021/ja9056739. [DOI] [PubMed] [Google Scholar]
- 554.Pluth MD, Tiedemann BEF, van Halbeek H, Nunlist R, Raymond KN. Inorg. Chem. 2008;47:1411. doi: 10.1021/ic7020885. [DOI] [PubMed] [Google Scholar]
- 555.Davis AV, Raymond KN. J. Am. Chem. Soc. 2005;127:7912. doi: 10.1021/ja051037s. [DOI] [PubMed] [Google Scholar]
- 556.Davis AV, Fiedler D, Seeber G, Zahl A, van Eldik R, Raymond KN. J. Am. Chem. Soc. 2006;128:1324. doi: 10.1021/ja056556+. [DOI] [PubMed] [Google Scholar]
- 557.Riddell IA, Smulders MMJ, Clegg JK, Nitschke JR. Chem. Commun. 2011;47:457. doi: 10.1039/c0cc02573a. [DOI] [PubMed] [Google Scholar]
- 558.Biros SM, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2007;129:12094. doi: 10.1021/ja075236i. [DOI] [PubMed] [Google Scholar]
- 559.Hastings CJ, Pluth MD, Biros SM, Bergman RG, Raymond KN. Tetrahedron. 2008;64:8362. [Google Scholar]
- 560.Glasson CRK, Clegg JK, McMurtrie JC, Meehan GV, Lindoy LF, Motti CA, Moubaraki B, Murray KS, Cashion JD. Chem. Sci. 2011;2:540. [Google Scholar]
- 561.Review: Klosterman JK, Yamauchi Y, Fujita M. Chem. Soc. Rev. 2009;38:1714. doi: 10.1039/b901261n.
- 562.Murase T, Otsuka K, Fujita M. J. Am. Chem. Soc. 2010;132:7864. doi: 10.1021/ja103109z. [DOI] [PubMed] [Google Scholar]
- 563.Ono K, Yoshizawa M, Kato T, Fujita M. Chem. Commun. 2008:2328. doi: 10.1039/b801701h. [DOI] [PubMed] [Google Scholar]
- 564.Ono K, Klosterman JK, Yoshizawa M, Sekiguchi K, Tahara T, Fujita M. J. Am. Chem. Soc. 2009;131:12526. doi: 10.1021/ja904875y. [DOI] [PubMed] [Google Scholar]
- 565.Yamauchi Y, Yoshizawa M, Fujita M. J. Am. Chem. Soc. 2008;130:5832. doi: 10.1021/ja077783+. [DOI] [PubMed] [Google Scholar]
- 566.Sato S, Morohara O, Fujita D, Yamaguchi Y, Kato K, Fujita M. J. Am. Chem. Soc. 2010;132:3670. doi: 10.1021/ja100325b. [DOI] [PubMed] [Google Scholar]
- 567.Yamauchi Y, Hanaoka Y, Yoshizawa M, Akita M, Ichikawa T, Yoshio M, Kato T, Fujita M. J. Am. Chem. Soc. 2010;132:9555. doi: 10.1021/ja103180z. [DOI] [PubMed] [Google Scholar]
- 568.Yamauchi Y, Yoshizawa M, Akita M, Fujita M. Proc. Natl. Acad. Sci. USA. 2009;106:10435. doi: 10.1073/pnas.0810319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Yamauchi Y, Yoshizawa M, Akita M, Fujita M. J. Am. Chem. Soc. 2010;132:960. doi: 10.1021/ja904063r. [DOI] [PubMed] [Google Scholar]
- 570.Osuga T, Murase T, Ono K, Yamauchi Y, Fujita M. J. Am. Chem. Soc. 2010;132:15553. doi: 10.1021/ja108367j. [DOI] [PubMed] [Google Scholar]
- 571.Ono K, Yoshizawa M, Akita M, Kato T, Tsunobuchi Y, Ohkoshi S, Fujita M. J. Am. Chem. Soc. 2009;131:2782. doi: 10.1021/ja8089894. [DOI] [PubMed] [Google Scholar]
- 572.Ozaki Y, Kawano M, Fujita M. Chem. Commun. 2009:4245. doi: 10.1039/b906674h. [DOI] [PubMed] [Google Scholar]
- 573.Nakabayashi K, Ozaki Y, Kawano M, Fujita M. Angew. Chem. Int. Ed. 2008;47:2046. doi: 10.1002/anie.200704924. [DOI] [PubMed] [Google Scholar]
- 574.Nakabayashi K, Kawano M, Yoshizawa M, Ohkoshi S, Fujita M. J. Am. Chem. Soc. 2004;126:16694. doi: 10.1021/ja044445p. [DOI] [PubMed] [Google Scholar]
- 575.Nakabayashi K, Kawano M, Kato T, Furukawa K, Ohkoshi S, Hozumi T, Fujita M. Chem. Asian J. 2007;2:164. doi: 10.1002/asia.200600293. [DOI] [PubMed] [Google Scholar]
- 576.Nakabayashi K, Kawano M, Fujita M. Angew. Chem. Int. Ed. 2005;44:5322. doi: 10.1002/anie.200501568. [DOI] [PubMed] [Google Scholar]
- 577.Fiedler D, Leung DH, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2004;126:3674. doi: 10.1021/ja039225a. [DOI] [PubMed] [Google Scholar]
- 578.Yoshizawa M, Tamura M, Fujita M. Angew. Chem. Int. Ed. 2007;46:3874. doi: 10.1002/anie.200700103. [DOI] [PubMed] [Google Scholar]
- 579.Cram DJ, Tanner ME, Thomas R. Angew. Chem. Int. Ed. Engl. 1991;30:1024. [Google Scholar]
- 580.Ziegler M, Brumaghim JL, Raymond KN. Angew. Chem. Int. Ed. 2000;39:4119. doi: 10.1002/1521-3773(20001117)39:22<4119::aid-anie4119>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 581.Brumaghim JL, Michels M, Raymond KN. Eur. J. Org. Chem. 2004:4552. [Google Scholar]
- 582.Brumaghim JL, Michels M, Pagliero D, Raymond KN. Eur. J. Org. Chem. 2004:5115. [Google Scholar]
- 583.Dong VM, Fiedler D, Carl B, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2006;128:14464. doi: 10.1021/ja0657915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Fiedler D, Bergman RG, Raymond KN. Angew. Chem. Int. Ed. 2006;45:745. doi: 10.1002/anie.200501938. [DOI] [PubMed] [Google Scholar]
- 585.Pluth MD, Fiedler D, Mugridge JS, Bergman RG, Raymond KN. Proc. Natl. Acad. Sci. USA. 2009;106:10438. doi: 10.1073/pnas.0809806106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Kawano M, Kobayashi Y, Ozeki T, Fujita M. J. Am. Chem. Soc. 2006;128:6558. doi: 10.1021/ja0609250. [DOI] [PubMed] [Google Scholar]
- 587.Yoshizawa M, Kusukawa T, Fujita M, Yamaguchi K. J. Am. Chem. Soc. 2000;122:6311. doi: 10.1021/ja010875t. [DOI] [PubMed] [Google Scholar]
- 588.Yoshizawa M, Kusukawa T, Fujita M, Sakamoto S, Yamaguchi K. J. Am. Chem. Soc. 2001;123:10454. doi: 10.1021/ja010875t. [DOI] [PubMed] [Google Scholar]
- 589.Sawada T, Yoshizawa M, Sato S, Fujita M. Nat. Chem. 2009;1:53. doi: 10.1038/nchem.100. [DOI] [PubMed] [Google Scholar]
- 590.Sawada T, Fujita M. J. Am. Chem. Soc. 2010;132:7194. doi: 10.1021/ja101718c. [DOI] [PubMed] [Google Scholar]
- 591.Mal P, Breiner B, Rissanen K, Nitschke JR. Science. 2009;324:1697. doi: 10.1126/science.1175313. [DOI] [PubMed] [Google Scholar]
- 592.Yoshizawa M, Tamura M, Fujita M. Science. 2006;312:251. doi: 10.1126/science.1124985. [DOI] [PubMed] [Google Scholar]
- 593.Nishioka Y, Yamaguchi T, Yoshizawa M, Fujita M. J. Am. Chem. Soc. 2007;129:7000. doi: 10.1021/ja071591x. [DOI] [PubMed] [Google Scholar]
- 594.Murase T, Horiuchi S, Fujita M. J. Am. Chem. Soc. 2010;132:2866. doi: 10.1021/ja9107275. [DOI] [PubMed] [Google Scholar]
- 595.Horiuchi S, Nishioka Y, Murase T, Fujita M. Chem. Commun. 2010;46:3460. doi: 10.1039/c003191g. [DOI] [PubMed] [Google Scholar]
- 596.Yoshizawa M, Takeyama M, Kusukawa T, Fujita M. Angew. Chem. Int. Ed. 2002;41:1347. doi: 10.1002/1521-3773(20020415)41:8<1347::aid-anie1347>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 597.Karthikeyan S, Ramamurthy V. Tetrahedron Lett. 2005;46:4495. [Google Scholar]
- 598.Takaoka K, Kawano M, Ozeki T, Fujita M. Chem. Commun. 2006:1625. doi: 10.1039/b600812g. [DOI] [PubMed] [Google Scholar]
- 599.Karthikeyan S, Ramamurthy V. J. Org. Chem. 2007;72:452. doi: 10.1021/jo0617722. [DOI] [PubMed] [Google Scholar]
- 600.Karthikeyan S, Ramamurthy V. J. Org. Chem. 2006;71:6409. doi: 10.1021/jo0606498. [DOI] [PubMed] [Google Scholar]
- 601.Yoshizawa M, Takeyama Y, Okano T, Fujita M. J. Am. Chem. Soc. 2003;125:3243. doi: 10.1021/ja020718+. [DOI] [PubMed] [Google Scholar]
- 602.Nishioka Y, Yamaguchi T, Kawano M, Fujita M. J. Am. Chem. Soc. 2008;130:8160. doi: 10.1021/ja802818t. [DOI] [PubMed] [Google Scholar]
- 603.Furusawa T, Kawano M, Fujita M. Angew. Chem. Int. Ed. 2007;46:5717. doi: 10.1002/anie.200701250. [DOI] [PubMed] [Google Scholar]
- 604.Yamaguchi T, Fujita M. Angew. Chem. Int. Ed. 2008;47:2067. doi: 10.1002/anie.200705139. [DOI] [PubMed] [Google Scholar]
- 605.Yoshizawa M, Miyagi S, Kawano M, Ishiguro K, Fujita M. J. Am. Chem. Soc. 2004;126:9172. doi: 10.1021/ja047612u. [DOI] [PubMed] [Google Scholar]
- 606.Furutani Y, Kandori H, Kawano M, Nakabayashi K, Yoshizawa M, Fujita M. J. Am. Chem. Soc. 2009;131:4764. doi: 10.1021/ja8089075. [DOI] [PubMed] [Google Scholar]
- 607.Leung DH, Fiedler D, Bergman RG, Raymond KN. Angew. Chem. Int. Ed. 2004;43:963. doi: 10.1002/anie.200352772. [DOI] [PubMed] [Google Scholar]
- 608.Leung DH, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2006;128:9781. doi: 10.1021/ja061412w. [DOI] [PubMed] [Google Scholar]
- 609.Fiedler D, Bergman RG, Raymond KN. Angew. Chem. Int. Ed. 2004;43:6748. doi: 10.1002/anie.200461776. [DOI] [PubMed] [Google Scholar]
- 610.Brown CJ, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2009;131:17530. doi: 10.1021/ja906386w. [DOI] [PubMed] [Google Scholar]
- 611.Fiedler D, van Halbeek H, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2006;128:10240. doi: 10.1021/ja062329b. [DOI] [PubMed] [Google Scholar]
- 612.Hastings CJ, Fiedler D, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2008;130:10977. doi: 10.1021/ja8013055. [DOI] [PubMed] [Google Scholar]
- 613.Hastings CJ, Pluth MD, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2010;132:6938. doi: 10.1021/ja102633e. [DOI] [PubMed] [Google Scholar]
- 614.Pluth MD, Bergman RG, Raymond KN. Science. 2007;316:85. doi: 10.1126/science.1138748. [DOI] [PubMed] [Google Scholar]
- 615.Pluth MD, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2008;130:11423. doi: 10.1021/ja802839v. [DOI] [PubMed] [Google Scholar]
- 616.Pluth MD, Bergman RG, Raymond KN. Angew. Chem. Int. Ed. 2007;46:8587. doi: 10.1002/anie.200703371. [DOI] [PubMed] [Google Scholar]
- 617.Pluth MD, Bergman RG, Raymond KN. J. Org. Chem. 2009;74:58. doi: 10.1021/jo802131v. [DOI] [PubMed] [Google Scholar]
- 618.Leung DH, Bergman RG, Raymond KN. J. Am. Chem. Soc. 2007;129:2746. doi: 10.1021/ja068688o. [DOI] [PubMed] [Google Scholar]
- 619.Wang Z,J, Brown CJ, Bergman RG, Raymond KN, Toste FD. J. Am. Chem. Soc. 2011;133:7358. doi: 10.1021/ja202055v. [DOI] [PubMed] [Google Scholar]
- 620.Ito H, Kusukawa T, Fujita M. Chem. Lett. 2000:598. [Google Scholar]
- 621.Yoshizawa M, Sato N, Fujita M. Chem. Lett. 2005;34:1392. [Google Scholar]
- 622.Lee SJ, Cho S-H, Mulfort KL, Tiede DM, Hupp JT, Nguyen ST. J. Am. Chem. Soc. 2008;130:16828. doi: 10.1021/ja804014y. [DOI] [PubMed] [Google Scholar]
- 623.Noh TH, Heo E, Park KH, Jung O-S. J. Am. Chem. Soc. 2011;133:1236. doi: 10.1021/ja1093925. [DOI] [PubMed] [Google Scholar]
- 624.Review: Hannon M. J. Chem. Soc. Rev. 2007;36:280. doi: 10.1039/b606046n.
- 625.Hannon MJ, Painting CL, Jackson A, Hamblin J, Errington W. Chem. Commun. 1997:1807. [Google Scholar]
- 626.Hannon MJ, Moreno V, Prieto MJ, Moldrheim E, Sletten E, Meistermann I, Isaac CJ, Sanders KJ, Rodger A. Angew. Chem. Int. Ed. 2001;40:879. doi: 10.1002/1521-3773(20010302)40:5<879::AID-ANIE879>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 627.Khalid S, Hannon MJ, Rodger A, Rodger PM. Chem. Eur. J. 2006;12:3493. doi: 10.1002/chem.200501168. [DOI] [PubMed] [Google Scholar]
- 628.Meistermann I, Moreno V, Prieto MJ, Moldrheim E, Sletten E, Khalid S, Rodger PM, Peberdy JC, Isaac CJ, Rodger A, Hannon MJ. Proc. Natl. Acad. Sci. USA. 2002;99:5069. doi: 10.1073/pnas.062634499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Uerpmann C, Malina J, Pascu M, Clarkson GJ, Moreno V, Rodger A, Grandas A, Hannon MJ. Chem. Eur. J. 2005;11:1750. doi: 10.1002/chem.200401054. [DOI] [PubMed] [Google Scholar]
- 630.Childs LJ, Malina J, Rolfsnes BE, Pascu M, Prieto MJ, Broome MJ, Rodger PM, Sletten E, Moreno V, Rodger A, Hannon MJ. Chem. Eur. J. 2006;12:4919. doi: 10.1002/chem.200600060. [DOI] [PubMed] [Google Scholar]
- 631.Oleski A, Blanco AG, Boer R, Usón I, Aymamí J, Rodger A, Hannon MJ, Coll M. Angew. Chem. Int. Ed. 2006;45:1227. doi: 10.1002/anie.200503822. [DOI] [PubMed] [Google Scholar]
- 632.Tashiro S, Tominaga M, Kawano M, Therrien B, Ozeki T, Fujita M. J. Am. Chem. Soc. 2005;127:4546. doi: 10.1021/ja044782y. [DOI] [PubMed] [Google Scholar]
- 633.Tashiro S, Tominaga M, Yamaguchi Y, Kato K, Fujita M. Angew. Chem. Int. Ed. 2006;45:241. doi: 10.1002/anie.200502802. [DOI] [PubMed] [Google Scholar]
- 634.Tashiro S, Tominaga M, Yamaguchi Y, Kato K, Fujita M. Chem. Eur. J. 2006;12:3211. doi: 10.1002/chem.200501424. [DOI] [PubMed] [Google Scholar]
- 635.Dolain C, Hatakeyama Y, Sawada T, Tashiro S, Fujita M. J. Am. Chem. Soc. 2010;132:5564. doi: 10.1021/ja100585w. [DOI] [PubMed] [Google Scholar]
- 636.Tashiro S, Kobayashi M, Fujita M. J. Am. Chem. Soc. 2006;128:9280. doi: 10.1021/ja061722e. [DOI] [PubMed] [Google Scholar]
- 637.Hatakeyama Y, Sawada T, Kawano M, Fujita M. Angew. Chem. Int. Ed. 2009;48:8695. doi: 10.1002/anie.200903563. [DOI] [PubMed] [Google Scholar]
- 638.Mattsson J, Zava O, Renfrew AK, Sei Y, Yamaguchi K, Dyson PJ, Therrien B. Dalton Trans. 2010;39:8248. doi: 10.1039/c0dt00436g. [DOI] [PubMed] [Google Scholar]
- 639.Barry NPE, Zava O, Furrer J, Dyson PJ, Therrien B. Dalton Trans. 2010;39:5272. doi: 10.1039/c001521k. [DOI] [PubMed] [Google Scholar]
- 640.Zava O, Mattsson J, Therrien B, Dyson PJ. Chem. Eur. J. 2010;16:1428. doi: 10.1002/chem.200903216. [DOI] [PubMed] [Google Scholar]
- 641.Pitto-Barry A, Barry NPE, Zava O, Deschenaux R, Dyson PJ, Therrien B. Chem. Eur. J. 2011;17:1966. doi: 10.1002/chem.201002634. [DOI] [PubMed] [Google Scholar]
- 642.Vajpayee V, Yang YJ, Kang SC, Kim H, Kim IS, Wang M, Stang PJ, Chi K-W. Chem. Commun. 2011;47:5184. doi: 10.1039/c1cc10167f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Reviews: Lehn J-M. Chem. Soc. Rev. 2007;36:151. doi: 10.1039/b616752g. Lehn J-M. Science. 2002;295:2400. doi: 10.1126/science.1071063. Lehn J-M. Proc. Natl. Acad. Sci. USA. 2002;99:4763. doi: 10.1073/pnas.072065599.
- 644.Ziegler M, Miranda JJ, Anderson UN, Johnson DW, Leary JA, Raymond KN. Angew. Chem. Int. Ed. 2001;40:733. [PubMed] [Google Scholar]
- 645.Reviews: Albrecht M. Top. Curr. Chem. 2004;248:105. Albrecht M, Janser I, Fröhlich R. Chem. Commun. 2005:157. doi: 10.1039/b410828k.
- 646.Northorp BH, Zheng Y-R, Chi K-W, Stang PJ. Acc. Chem. Res. 2009;42:1554. doi: 10.1021/ar900077c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Severin K. Chem. Commun. 2006:3859. doi: 10.1039/b606632c. [DOI] [PubMed] [Google Scholar]
- 648.Krämer R, Lehn J-M, Marquis-Rigault A. Proc. Natl. Acad. Sci. USA. 1993;90:5394. doi: 10.1073/pnas.90.12.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649.Caulder DL, Raymond KN. Angew. Chem. Int. Ed. Engl. 1997;36:1440. [Google Scholar]
- 650.Albrecht M, Schneider M, Röttele H. Angew. Chem. Int. Ed. 1999;38:557. doi: 10.1002/(SICI)1521-3773(19990215)38:4<557::AID-ANIE557>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 651.Albrecht M, Schneider M. Eur. J. Inorg. Chem. 2002:1301. [Google Scholar]
- 652.Albrecht M, Liu Y, Zhu SS, Schalley CA, Fröhlich R. Chem. Commun. 2009:1195. doi: 10.1039/b821819f. [DOI] [PubMed] [Google Scholar]
- 653.Addicott C, Das N, Stang PJ. Inorg. Chem. 2004;43:5335. doi: 10.1021/ic049326p. [DOI] [PubMed] [Google Scholar]
- 654.Zheng Y-R, Yang H-B, Northrop BH, Ghosh K, Stang PJ. Inorg. Chem. 2008;47:4706. doi: 10.1021/ic800038j. [DOI] [PubMed] [Google Scholar]
- 655.Zheng Y-R, Yang H-B, Ghosh K, Zhao L, Stang PJ. Chem. Eur. J. 2009;15:7203. doi: 10.1002/chem.200900230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Yang H-B, Ghosh K, Northrop BH, Stang PJ. Org. Lett. 2007;9:1561. doi: 10.1021/ol070371l. [DOI] [PubMed] [Google Scholar]
- 657.Zheng Y-R, Stang PJ. J. Am. Chem. Soc. 2009;131:3487. doi: 10.1021/ja809788x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658.Northrop BH, Yang H-B, Stang PJ. Inorg. Chem. 2008;47:11257. doi: 10.1021/ic801711q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659.Chi K-W, Addicott C, Arif AM, Stang PJ. J. Am. Chem. Soc. 2004;126:16569. doi: 10.1021/ja045542l. [DOI] [PubMed] [Google Scholar]
- 660.Zhao L, Northrop BH, Zheng Y-R, Yang H-B, Lee HJ, Lee YM, Park JY, Chi K-W, Stang PJ. J. Org. Chem. 2008;73:6580. doi: 10.1021/jo800957r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661.Mahata K, Schmittel M. J. Am. Chem. Soc. 2009;131:16544. doi: 10.1021/ja907185k. [DOI] [PubMed] [Google Scholar]
- 662.Schmittel M, Mahata K. Chem. Commun. 2010;46:4163. doi: 10.1039/c0cc00191k. [DOI] [PubMed] [Google Scholar]
- 663.Schmittel M, Kalsani V, Kishore RSK, Cölfen H, Bats JW. J. Am. Chem. Soc. 2005;127:11544. doi: 10.1021/ja0525096. [DOI] [PubMed] [Google Scholar]
- 664.Mahata K, Saha ML, Schmittel M. J. Am. Chem. Soc. 2010;132:15933. doi: 10.1021/ja108419k. [DOI] [PubMed] [Google Scholar]
- 665.Jiang W, Winkler HDF, Schalley CA. J. Am. Chem. Soc. 2008;130:13852. doi: 10.1021/ja806009d. [DOI] [PubMed] [Google Scholar]
- 666.Jiang W, Schalley CA. Proc. Natl. Acad. Sci. USA. 2009;106:10425. doi: 10.1073/pnas.0809512106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667.Brusilowskij B, Dzyuba EV, Troff RW, Schalley CA. Chem. Commun. 2011;47:1830. doi: 10.1039/c0cc04476h. [DOI] [PubMed] [Google Scholar]
- 668.Kamada T, Aratani N, Ikeda T, Shibata N, Higuchi Y, Wakamiya A, Yamaguchi S, Kim KS, Yoon ZS, Kim D, Osuka A. J. Am. Chem. Soc. 2006;128:7670. doi: 10.1021/ja0611137. [DOI] [PubMed] [Google Scholar]
- 669.Maeda C, Shinokubo H, Osuka A. Org. Lett. 2009;11:5322. doi: 10.1021/ol902294r. [DOI] [PubMed] [Google Scholar]
- 670.Grote Z, Scopelliti R, Severin K. Angew. Chem. Int. Ed. 2003;42:3821. doi: 10.1002/anie.200351623. [DOI] [PubMed] [Google Scholar]
- 671.Saur I, Scopelliti R, Severin K. Chem. Eur. J. 2006;12:1058. doi: 10.1002/chem.200500621. [DOI] [PubMed] [Google Scholar]
- 672.Schultz D, Nitschke JR. Proc. Natl. Acad. Sci. USA. 2005;102:11191. doi: 10.1073/pnas.0502830102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.Hutin M, Frantz R, Nitschke JR. Chem. Eur. J. 2006;12:4077. doi: 10.1002/chem.200600079. [DOI] [PubMed] [Google Scholar]
- 674.Schultz D, Nitschke JR. Angew. Chem. Int. Ed. 2006;45:2453. doi: 10.1002/anie.200504447. [DOI] [PubMed] [Google Scholar]
- 675.Schultz D, Nitschke JR. J. Am. Chem. Soc. 2006;128:9887. doi: 10.1021/ja061841u. [DOI] [PubMed] [Google Scholar]
- 676.Bélanger S, Hupp JT, Stern CL, Slone RV, Watson DF, Carrell TG. J. Am. Chem. Soc. 1999;121:557. [Google Scholar]
- 677.Bélanger S, Hupp JT. Angew. Chem. Int. Ed. 1999;38:2222. [PubMed] [Google Scholar]
- 678.Keefe MH, O’Donnell JL, Bailey RC, Nguyen ST, Hupp JT. Adv. Mater. 2003;15:1936. [Google Scholar]
- 679.Massari AM, Gurney RW, Schwartz CP, Nguyen ST, Hupp JT. Langmuir. 2004;20:4422. doi: 10.1021/la049900+. [DOI] [PubMed] [Google Scholar]
- 680.Libera JA, Gurney RW, Schwartz C, Jin H, Lee T-L, Nguyen ST, Hupp JT, Bedzyk MJ. J. Phys. Chem. B. 2005;109:1441. doi: 10.1021/jp0457038. [DOI] [PubMed] [Google Scholar]
- 681.Williams ME, Benkstein KD, Abel C, Dinolfo PH, Hupp JT. Proc. Natl. Acad. Sci. USA. 2002;99:5171. doi: 10.1073/pnas.082643199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Yuan Q-H, Wan L-J, Jude H, Stang PJ. J. Am. Chem. Soc. 2005;127:16279. doi: 10.1021/ja0549300. [DOI] [PubMed] [Google Scholar]
- 683.Semenov A, Spatz JP, Möller M, Lehn J-M, Sell B, Schubert D, Weidl CH, Schubert US. Angew. Chem. Int. Ed. 1999;38:2547. doi: 10.1002/(sici)1521-3773(19990903)38:17<2547::aid-anie2547>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 684.Ziener U, Lehn J-M, Mourran A, Möller M. Chem. Eur. J. 2002;8:951. doi: 10.1002/1521-3765(20020215)8:4<951::aid-chem951>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 685.Safarowsky C, Merz L, Rang A, Broekmann P, Hermann BA, Schalley CA. Angew. Chem. Int. Ed. 2004;43:1291. doi: 10.1002/anie.200352968. [DOI] [PubMed] [Google Scholar]
- 686.Yuan Q-H, Yan C-J, Yan H-J, Wan L-J, Northrop BH, Jude H, Stang PJ. J. Am. Chem. Soc. 2008;130:8878. doi: 10.1021/ja801934w. [DOI] [PubMed] [Google Scholar]
- 687.Gong J-R, Wan L-J, Yuan Q-H, Bai C-L, Jude H, Stang PJ. Proc. Natl. Acad. Sci. U.S.A. 2005;102:971. doi: 10.1073/pnas.0409145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Li S-S, Yan H-J, Wan L-J, Yang H-B, Northrop BH, Stang PJ. J. Am. Chem. Soc. 2007;129:9268. doi: 10.1021/ja0733282. [DOI] [PubMed] [Google Scholar]
- 689.Chen T, Pan G-B, Wettach H, Fritzsche M, Höger S, Wan L-J, Yang H-B, Northrop BH, Stang PJ. J. Am. Chem. Soc. 2010;132:1328. doi: 10.1021/ja907220f. [DOI] [PubMed] [Google Scholar]
- 690.Levi SA, Guatteri P, van Veggel FCJM, Vancso GJ, Dalcanale E, Reinhoudt DN. Angew. Chem. Int. Ed. 2001;40:1892. [PubMed] [Google Scholar]
- 691.Menozzi E, Pinalli R, Speets EA, Ravoo BJ, Dalcanale E, Reinhoudt DN. Chem. Eur. J. 2004;10:2199. doi: 10.1002/chem.200305570. [DOI] [PubMed] [Google Scholar]
- 692.Kogej M, Schalley CA. In: Analytical Methods in Supramolecular Chemistry. Schalley CA, editor. Wiley-VCH; Weinheim: 2007. p. 104. [Google Scholar]
- 693.Reviews: Baytekin B, Baytekin HT, Schalley CA. Org. Biomol. Chem. 2006;4:2825. doi: 10.1039/b604265a. Schalley CA. Mass Spectrom. Rev. 2001;20:253. doi: 10.1002/mas.10009. Schalley CA. Int. J. Mass Spectrom. 2000;194:11.
- 694.Reviews: Fenn JB. Angew. Chem. Int. Ed. 2003;42:3871. doi: 10.1002/anie.200300605. Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Mass Spectrom. Rev. 1990;9:37.
- 695.Schalley CA, Springer A. Mass Spectrometry and Gas-Phase Chemistry of Non-Covalent Complexes. Wiley; Hoboken: 2009. [Google Scholar]
- 696.Reviews: Sakamoto S, Fujita M, Kim K, Yamaguchi K. Tetrahedron. 2000;56:955. Yamaguchi K. J. Mass Spectrom. 2003;38:473. doi: 10.1002/jms.488.
- 697.Xia Y, Liang X, McLuckey SA. Anal. Chem. 2005;77:3683. doi: 10.1021/ac0481811. [DOI] [PubMed] [Google Scholar]
- 698.Hirabayashi A, Sakairi M, Koizumi H. Anal. Chem. 1994;66:4557. [Google Scholar]
- 699.Gardner JS, Harrison RG, Lamb JD, Dearden DV. New J. Chem. 2006;30:1276. [Google Scholar]
- 700.Schalley CA, Müller T, Linnartz P, Witt M, Schäfer M, Lützen A. Chem. Eur. J. 2002;8:3538. doi: 10.1002/1521-3765(20020802)8:15<3538::AID-CHEM3538>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 701.Engeser M, Rang A, Ferrer M, Gutiérrez A, Baytekin HT, Schalley CA. Int. J. Mass Spectrom. 2006;255-256:185. [Google Scholar]
- 702.Kanu AB, Dwivedi P, Tam M, Matz L, Hill HH., Jr. J. Mass Spectrom. 2008;43:1. doi: 10.1002/jms.1383. [DOI] [PubMed] [Google Scholar]
- 703.Reviews: Bohrer BC, Merenbloom SI, Koeniger SL, Hilderbrand AE, Clemmer DE. Annu. Rev. Anal. Chem. 2008;1:293. doi: 10.1146/annurev.anchem.1.031207.113001. Ruotolo BT, Benesch JLP, Sandercock AM, Hyung S-J, Robinson CV. Nat. protocol. 2008;3:1139. doi: 10.1038/nprot.2008.78.
- 704.Chan Y-T, Li X, Soler M, Wang J-L, Wesdemiotis C, Newkome GR. J. Am. Chem. Soc. 2009;131:16395. doi: 10.1021/ja907262c. [DOI] [PubMed] [Google Scholar]
- 705.Li X, Chan Y-T, Newkome GR, Wesdemiotis C. Anal. Chem. 2011;83:1284. doi: 10.1021/ac1022875. [DOI] [PubMed] [Google Scholar]
- 706.Pringle SD, Giles K, Wildgoose JL, Williams JP, Slade SE, Thalassinos K, Bateman RH, Bowers MT, Scrivens JH. Int. J. Mass Spectrom. 2007;261:1. [Google Scholar]
- 707.Brocker ER, Anderson SE, Northrop BH, Stang PJ, Bowers MT. J. Am. Chem. Soc. 2010;132:13486. doi: 10.1021/ja105702y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708.For recent reviews see: Mertens HDT, Svergun DI. J. Struct. Biol. 2010;172:128. doi: 10.1016/j.jsb.2010.06.012. Makowski L. J. Struct. Funct. Genomics. 2010;11:9. doi: 10.1007/s10969-009-9075-x. Putnam CD, Hammel M, Hura GL, Tainer JAQ. Rev. Biophys. 2007;40:191. doi: 10.1017/S0033583507004635. Koch MHJ, Vachette P, Svergun DIQ. Rev. Biophys. 2003;36:147. doi: 10.1017/s0033583503003871.
- 709.Megyes T, Jude H, Grósz T, Bakó I, Radnai T, Tárkányi G, Pálinkás G, Stang PJ. J. Am. Chem. Soc. 2005;127:10731. doi: 10.1021/ja0523690. [DOI] [PubMed] [Google Scholar]
- 710.O’Donnell JL, Zuo X, Goshe AJ, Sarkisov L, Snurr RQ, Hupp JT, Tiede DM. J. Am. Chem. Soc. 2007;129:1578. doi: 10.1021/ja0659065. [DOI] [PubMed] [Google Scholar]
- 711.Slone RV, Hupp JT, Stern CL, Albrecht-Schmitt TE. Inorg. Chem. 1996;35:4096. doi: 10.1021/ic960464r. [DOI] [PubMed] [Google Scholar]
- 712.Tiede DM, Zhang R, Chen LX, Yu L, Lindsey JS. J. Am. Chem. Soc. 2004;126:14054. doi: 10.1021/ja048209q. [DOI] [PubMed] [Google Scholar]
- 713.Mardis KL, Sutton HM, Zuo X, Lindsey JS, Tiede DM. J. Phys. Chem. A. 2009;113:2516. doi: 10.1021/jp808318x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 714.Balzani V, Credi A, Venturi M. Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld. 2nd Ed. Wiley-VCH; Weinheim: 2008. [Google Scholar]
- 715.Reviews: Wasielewski MR. Acc. Chem. Res. 2009;42:1910. doi: 10.1021/ar9001735. Fukuzumi S. Phys. Chem. Chem. Phys. 2008;10:2283. doi: 10.1039/b801198m. Nakamura Y, Aratani N, Osuka A. Chem. Soc. Rev. 2007;36:831. doi: 10.1039/b618854k. Choi M-S, Yamazaki T, Yamazaki I, Aida T. Angew. Chem. Int. Ed. 2004;43:150. doi: 10.1002/anie.200301665. Imahori H. Org. Biomol. Chem. 2004;2:1425. doi: 10.1039/b403024a.
- 716.Iengo E, Gatti T, Zangrando E, Indelli MT, Scandola F, Alessio E. Chem. Commun. 2011;47:1616. doi: 10.1039/c0cc03513k. [DOI] [PubMed] [Google Scholar]
- 717.Yatskou MM, Koehorst RBM, Donker H, Schaafsma TJ. J. Phys. Chem. A. 2001;105:11425. [Google Scholar]
- 718.Yatskou MM, Koehorst RBM, van Hoek A, Donker H, Schaafsma TJ, Gobets B, van Stokkum I, van Grondelle R. J. Phys. Chem. A. 2001;105:11432. [Google Scholar]
- 719.Kelley RF, Goldsmith RH, Wasielewski MR. J. Am. Chem. Soc. 2007;129:6384. doi: 10.1021/ja071362a. [DOI] [PubMed] [Google Scholar]
- 720.Kelley RF, Lee SJ, Wilson TM, Nakamura Y, Tiede DM, Osuka A, Hupp JT, Wasielewski MR. J. Am. Chem. Soc. 2008;130:4277. doi: 10.1021/ja075494f. [DOI] [PubMed] [Google Scholar]
- 721.Flynn DC, Ramakrishna G, Yang H-B, Northrop BH, Stang PJ, Goodson T., III J. Am. Chem. Soc. 2010;132:1348. doi: 10.1021/ja9082655. [DOI] [PubMed] [Google Scholar]
- 722.Zhao G-J, Northrop BH, Stang PJ, Han K-L. J. Phys. Chem. A. 2010;114:3418. doi: 10.1021/jp911597z. [DOI] [PubMed] [Google Scholar]