

Abstract
We have developed hybrid P450 BM3 enzymes consisting of a Ru(II)-diimine photosensitizer covalently attached to non-native single cysteine residues of P450 BM3 heme domain mutants. These enzymes are capable, upon light activation, of selectively hydroxylating lauric acid with 40 times higher total turnover numbers compared to the peroxide shunt.
Cytochromes P450 are heme-thiolate enzymes that utilize molecular dioxygen and two reducing equivalents to catalyze the selective oxidation of unactivated C–H bond in a variety of organic substrates.1 The catalytic cycle involves several well characterized intermediates.2,3 Upon substrate binding and reduction of the resting ferric-aquo enzyme, the ferrous heme compound binds dioxygen to form the first intermediate, an oxy-P450 species. Further one electron reduction and protonation of the oxy-P450 complex lead to a hydroperoxo-Fe(III) intermediate. Protonation of the distal oxygen in the hydroperoxo adduct then results in the scission of the O–O bond and the formation of a recently fully characterized Compound I, a high-valent Fe(IV)-oxo porphyrin radical species responsible for the oxygen atom insertion.3
Interest in these proteins arises in part from the desire to harness their synthetic potential for biotechnological applications.4 However, progress has been hampered by the limited stability and activity of most P450s as well as the need for an expensive cofactor (NAD(P)H) and a dependence on reductase proteins as electron carriers.5 Several approaches have been undertaken to overcome these challenges. Early reports demonstrated the limited peroxygenase activity of the P450 heme domain with hydrogen peroxide to carry out the oxidation under the peroxide shunt.6a Since then, a directed evolution approach has led to enzymes with enhanced stability and peroxygenase activity.6b Other systems have focused on NADPH recycling strategies7 as well as alternative methods to provide the necessary electrons to the heme domain.8–12 Electrochemical methods have been employed to shuttle electrons to the heme active site using various electron mediators.9 A very inexpensive Zn/Co(III) sepulchrate system also showed great promise with enhanced activity of the heme domain.10 Recent studies have used light-activated processes to initiate P450 reactions. The light-harvesting ability of photosystem I (PS1) coupled with a membrane bound CYP79A1 was able to perform the light-driven hydroxylation of L-tyrosine.11 In addition, the use of deazaflavins with the full length P450 BM3 enzyme in a recycling photochemical system has been developed for the hydroxylation of lauric acid.12
As an alternative approach to perform light-activated P450 reactions, we have developed hybrid P450 BM3 enzymes consisting of a Ru(II)-diimine photosensitizer attached to heme domain mutants of the archetypal highly active P450 BM3.13 The Ru(II)-diimine compound was covalently attached to non-native single cysteine residues, K97C or Q397C, strategically positioned in close proximity to the heme. A large body of work from the Gray group has pioneered the use of the Ru(II) photosensitizer and flash quench technique in the study of fast electron transfer in various proteins.14 Recently, using the modified Ru(II)-K97C-BM3 heme domain mutant, we have demonstrated photooxidation of the resting ferric-aquo P450 BM3 heme domain to the ferryl Compound II species under flash quench oxidative conditions.15
Herein, we report the ability of the hybrid enzymes, Ru-K97C-BM3 and Ru-Q397C-BM3, to catalyze the hydroxylation of lauric acid under flash quench reductive conditions (Fig. 1). Our results show that the hybrid enzymes have 40 times higher total turnover numbers (TON)16 compared to the wild type heme domain (BM3-WT) using the peroxide shunt.
Fig. 1.
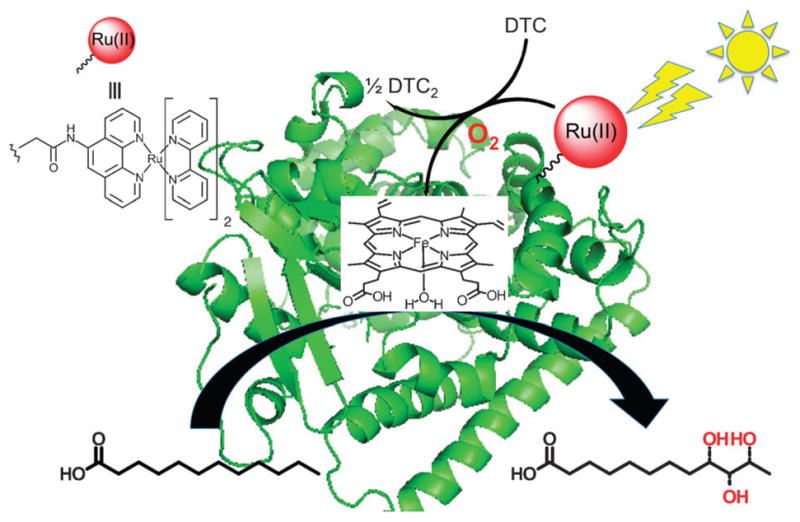
Hybrid P450 BM3 enzymes composed of a [Ru(bpy)2-(5-CH2CONHPhen]2+ photosensitizer covalently attached to nonnative single cysteine residues of P450 BM3 heme domain mutants in order to carry out the selective hydroxylation of lauric acid under visible light irradiation and in the presence of a sacrificial electron donor, sodium diethyldithiocarbamate (DTC).
Large scale synthesis and purification of the hybrid enzymes were performed following reported procedures.15 Briefly, the hybrid enzymes (Ru-K97C-BM3 and Ru-Q397C-BM3) were assembled by covalently attaching the photosensitizer Ru(II)-diimine to non-native single cysteine residues of P450 BM3 heme domain mutants (K97C and Q397C, respectively). These BM3 mutants were overexpressed in E. coli and purified taking advantage of the His6-tag. The iodoacetamide derivative of the Ru(II) photosensitizer, Ru(II)(bpy)2(5-ICH2CONHPhen), was synthesized17 from iodoacetic acid, 5-aminophenanthroline and Ru(bpy)2Cl2 and reacted readily in aqueous solution with the sulfhydryl group of the single cysteine P450 BM3 mutants under reducing conditions (300 μM dithiothreitol). The hybrid enzymes were further purified using an anion exchange column (Fig. S1†). The purified hybrid enzymes were characterized by UV-vis and fluorescence spectroscopic techniques as well as mass spectrometry (Fig. S2–S4†).
Upon light excitation into the metal-to-ligand charge transfer (3MLCT) absorption band, the long-lived Ru(II) excited state can be quenched in situ by a sacrificial electron donor to create a highly reductive Ru(I) species with an E0 = −1.26 V vs. NHE.14,18 This reductant generated via bimolecular reductive flash quench technique has been used to reduce various active site and model complexes.19 Among several sacrificial reductive quenchers investigated, sodium diethyldithiocarbamate (DTC) fulfills the desired requirements. It is an irreversible sacrificial electron donor, inexpensive, highly water soluble and capable of quenching the Ru(II)* emission (Fig. S4†).18b,19a Following the photocatalytic cycle highlighted in Scheme 1, we investigated the ability of the hybrid enzymes to catalyze the hydroxylation of lauric acid under constant visible light irradiation.
Scheme 1.
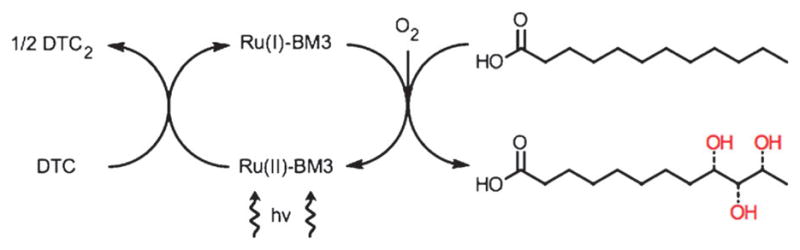
Proposed photocatalytic cycle using hybrid P450 BM3 enzymes under flash quench reductive conditions.
Typically, the activity of the hybrid enzymes was determined as follows: an aerated solution of the hybrid enzyme (3 μM), in the presence of excess lauric acid (1.5 mM) and DTC (100 mM), was continually irradiated with visible light. The formation of hydroxylated products was monitored by gas chromatography after extraction of the reaction mixture and derivatization of the acid and alcohol groups.6 The peroxygenase activity of the wild type heme domain (BM3-WT) under the peroxide shunt was used as a control reaction. Using 10 mM H2O2, the BM3-WT heme domain is rapidly deactivated and leads to only a few total turnovers (Table 1, entry 1).6a
Table 1.
Control and reaction conditions for the Ru-Q397C-BM3 and Ru-K97C-BM3 hybrid enzymes and BM3-WT with their respective total turnover numbers (TON)
| Entry | 10 mM H2O2 | 100 mM DTC | Light | TON | |
|---|---|---|---|---|---|
| 1 | WT | + | − | − | <2 |
| 2 | − | + | − | n.d. | |
| 3 | − | + | + | n.d. | |
| 4 | WT/Ru(bpy)32+ | − | − | + | n.d. |
| 5 | − | + | + | <3 | |
| 6 | Ru-K97C-BM3/ | + | − | − | <4 |
| 7 | Ru-Q397C-BM3 | − | + | − | n.d. |
| 8 | − | − | + | n.d. | |
| 9 | − | + | + | 30/40 60/80a |
In the presence of 10 μM catalase;
n.d.: not determined because product concentration below detection limit.
Under the photoreductive conditions, the activity of the hybrid enzymes, Ru-Q397C-BM3 and Ru-K97C-BM3, exceeded the activity of the wild type using the peroxide shunt (Fig. 2). The typical product ratio is maintained between the BM3-WT and the hybrid enzymes, reflecting no detrimental interactions of the label with the enzyme active site. The hybrid enzymes require four components to be present in order to carry out the reaction: (1) the Ru(II) photosensitizer covalently attached to the heme domain; (2) reductive quencher (DTC); (3) visible light excitation and (4) dioxygen as the source of oxygen atom. In the absence of one or more components, very low or no activity of the enzyme is detected. For example, the use of Ru(bpy)32+ in solution with BM3-WT in a 1 : 1 and 10 : 1 ratio and 100 mM DTC resulted in TON similar to the peroxide shunt (Table 1, entry 5). In the absence of DTC or visible light, the hybrid enzymes are unable to catalyze the hydroxylation of lauric acid (Table 1, entries 7 and 8). Moreover, no products were detected when DTC alone was used with BM3-WT under visible light excitation (Table 1, entry 3). Unlike sodium dithionite (E0 = −850 mV vs. Ag/AgCl),8c DTC (E0 = −600 mV vs. Ag/AgCl)20 does not reduce the heme domain and is not able to sustain catalytic activity.
Fig. 2.

Representative chromatograms of trimethylsilylated derivatized hydroxylated products (ω1, ω2, ω3) from reactions of 3 μM BM3 enzymes with 1.5 mM lauric acid for 2 h. 12-Hydroxydodecanoic acid was used as internal standard (I.S., 10 nmol).
The hybrid enzymes followed Michaelis–Menten kinetics, and the kinetics parameters for the hybrid enzymes were determined (Fig. S5–S6†). The hybrid enzymes display a Km of 0.32 and 0.28 ± 0.03 mM and a kcat of 0.95 and 1.12 ± 0.05 min−1 for Ru-K97C-BM3 and Ru-Q397C-BM3, respectively. The initial reaction rates determined after one-minute reaction are higher compared to the BM3-WT with H2O2 or Ru(bpy)32+ (Table S1†).
Investigations into the mechanism of the hybrid enzymes suggest that under the current photoreductive conditions, the proposed Ru(I) species is not able to reduce the ferric heme to initiate the catalytic cycle. Under constant light irradiation, only a small amount of Fe(II)–CO adduct is detected under CO atmosphere and in the presence of lauric acid (Fig. S7†). Instead, the data are more consistent with the generation of reactive oxygen species (ROS) from molecular dioxygen. The reaction rates determined for the hybrid enzymes are comparable to the peroxide shunt rates.6a Photochemical processes between Ru(bpy)32+ and dioxygen are known to generate various reactive oxygen species including singlet oxygen, superoxide anions and hydrogen peroxide.21 While being mostly detrimental to P450 enzymes due to oxidative damage, these ROS can also form various intermediates along the catalytic cycle and therefore be utilized by the hybrid enzymes to perform oxidation reactions.22 Attempts to measure the amount of ROS generated by the hybrid enzymes have been unsuccessful due in part to a high concentration of DTC. Under light irradiation and in the absence of DTC, the reactive oxygen species generated from quenching of the Ru(II)* excited state do not lead to any detectable amount of product in neither the hybrid enzymes nor the WT/Ru(bpy)32+ system (Table 1, entries 4 and 8).
In order to limit the amount of ROS present in solution under the photocatalytic conditions, we probed the protective role of catalase as well as the hybrid enzymes’ exposure time to various light sources. Addition of 10 μM catalase resulted in an additional two-fold increase in TON (Fig. 2). A mercury lamp with UV- and IR-cutoff filters, a blue 465 nm LED and a fluorescent light bulb were employed as visible light sources.23 The mercury lamp consistently gave higher TON (40) than the blue LED (30) and the fluorescent light bulb (25). Light pulsing or on/off light cycles resulted, however, in no significant changes in product formation or protein degradation. Monitoring of the Soret absorbance band at 418 nm during the course of the reaction shows a rate of hybrid enzymes degradation of 0.02 min−1 in the presence of 10 μM catalase (Fig. S8†). Under the current conditions, the hybrid enzymes are stable for more than two hours in contrast with the BM3-WT under the peroxide shunt (Fig. S9†).
Overall, the hybrid enzymes are capable of hydroxylating lauric acid upon light excitation with higher total turnover numbers compared to the peroxide shunt, up to 40-fold increase for the Ru-Q397C-BM3 enzyme. The hybrid enzymes most likely utilize reactive oxygen species generated in situ under the photoreductive conditions to perform the hydroxylation reaction for more than two hours.
Supplementary Material
Acknowledgments
L.C., N.H. and N.-H. T. would like to thank the National Institute of Health (GM095415) for financial support. L.C. thanks the National Science Foundation (0923573) and San José State University for the use of mass spectrometry facilities in the Protein Lab. We are grateful to Prof. Gilles Muller and Prof. Marc D’Alarcao (SJSU) for helpful discussions.
Footnotes
Electronic supplementary information (ESI) available: Spectroscopic and kinetics data for hybrid enzymes. See DOI: 10.1039/c1cc15124j
Notes and references
- 1.Ortiz de Montellano PR. Cytochrome P450: structure, mechanism, and biochemistry. 3. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]; (b) http://drnelson.uthsc.edu/cytochromeP450.html
- 2.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 3.Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 4.(a) Urlacher VB, Lutz-Wahl S, Schmid RD. Appl Microbiol Biotechnol. 2004;64:317–325. doi: 10.1007/s00253-003-1514-1. [DOI] [PubMed] [Google Scholar]; (b) Bernhardt R. J Biotechnol. 2006;124:128–145. doi: 10.1016/j.jbiotec.2006.01.026. [DOI] [PubMed] [Google Scholar]; (c) Hlavica P. Biotechnol Adv. 2009;27:103–121. doi: 10.1016/j.biotechadv.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 5.O’Reilly E, Köhler V, Flitsch SL, Turner NJ. Chem Commun. 2011;47:2490–2501. doi: 10.1039/c0cc03165h. [DOI] [PubMed] [Google Scholar]
- 6.(a) Cirino PC, Arnold FH. Adv Synth Catal. 2002;344:932–937. [Google Scholar]; (b) Cirino PC, Arnold FH. Angew Chem, Int Ed. 2003;42:3299–3301. doi: 10.1002/anie.200351434. [DOI] [PubMed] [Google Scholar]
- 7.(a) Hollmann F, Witholt B, Schmid A. J Mol Catal B: Enzym. 2002;19–20:167–176. [Google Scholar]; (b) Taylor M, Lamb DC, Cannell RJ, Dawson MJ, Kelly SL. Biochem Biophys Res Commun. 2000;279:708–711. doi: 10.1006/bbrc.2000.4002. [DOI] [PubMed] [Google Scholar]
- 8.(a) Munro AW, Girvan HM, McLean KJ. Biochim Biophys Acta, Gen Subj. 2007;1770:345–359. doi: 10.1016/j.bbagen.2006.08.018. [DOI] [PubMed] [Google Scholar]; (b) Gillam EMJ. Chem Res Toxicol. 2008;21:220–231. doi: 10.1021/tx7002849. [DOI] [PubMed] [Google Scholar]; (c) Fang X, Halpert JR. Drug Metab Dispos. 1996;24:1282–1285. [PubMed] [Google Scholar]
- 9.(a) Sadeghi SJ, Fantuzzi A, Gilardi G. Biochim Biophys Acta, Proteins Proteomics. 2011;1814:237–248. doi: 10.1016/j.bbapap.2010.07.010. [DOI] [PubMed] [Google Scholar]; (b) Estabrook RW, Faulkner KM, Shet MS, Fisher CW. Methods Enzymol. 1996;272:44–51. doi: 10.1016/s0076-6879(96)72007-4. [DOI] [PubMed] [Google Scholar]; (c) Udit AK, Arnold FH, Gray HB. J Inorg Biochem. 2004;98:1547–1550. doi: 10.1016/j.jinorgbio.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Nazor J, Dannenmann S, Adjei RO, Fordjour YB, Ghampson IT, Blanusa M, Roccatano D, Schwaneberg U. Protein Eng, Des Sel. 2008;21:29–35. doi: 10.1093/protein/gzm074. [DOI] [PubMed] [Google Scholar]
- 11.Jensen K, Jensen PE, Møller BL. ACS Chem Biol. 2011;6:533–539. doi: 10.1021/cb100393j. [DOI] [PubMed] [Google Scholar]
- 12.Zilly FE, Taglieber A, Schulz F, Hollman F, Reetz MT. Chem Commun. 2009:7152–7154. doi: 10.1039/b913863c. [DOI] [PubMed] [Google Scholar]
- 13.Munro AW, Leys DG, McLean KJ, Marshall KR, Ost TWB, Daff S, Miles CS, Chapman SK, Lysek DA, Moser CC, Page CC, Dutton PL. Trends Biochem Sci. 2002;27:250–257. doi: 10.1016/s0968-0004(02)02086-8. [DOI] [PubMed] [Google Scholar]
- 14.(a) Gray HB, Winkler JR. Proc Natl Acad Sci U S A. 2005;102:3534–3539. doi: 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gray HB, Winkler JR. Q Rev Biophys. 2003;36:341–372. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]
- 15.Ener ME, Lee YT, Winkler JR, Gray HB, Cheruzel L. Proc Natl Acad Sci U S A. 2010;107:18783–18786. doi: 10.1073/pnas.1012381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Total turnover number is defined as the number of moles of the hydroxylated lauric acid produced per mole of enzyme.
- 17.Castellano FN, Dattelbaum JD, Lakowicz JR. Anal Biochem. 1998;255:165–170. doi: 10.1006/abio.1997.2468. [DOI] [PubMed] [Google Scholar]
- 18.(a) Hoffman MZ, Bolletta F, Moggi L, Hug GL. J Phys Chem Ref Data. 1989;18:219–543. [Google Scholar]; (b) Deronzier A, Meyer TJ. Inorg Chem. 1980;19:2912–2917. [Google Scholar]
- 19.(a) Na Y, Pan J, Wang M, Sun L. Inorg Chem. 2007;46:3813–3815. doi: 10.1021/ic070234k. [DOI] [PubMed] [Google Scholar]; (b) Roth LE, Nguyen JC, Tezcan FA. J Am Chem Soc. 2010;132:13672–13674. doi: 10.1021/ja1071866. [DOI] [PubMed] [Google Scholar]
- 20.Thackeray JW, Natan MJ, Ng P, Wrighton MS. J Am Chem Soc. 1986;108:3570–3577. [Google Scholar]
- 21.(a) Das A, Joshi V, Kotkar D, Pathak VS, Swayambunathan V, Kamat PV, Ghosh PK. J Phys Chem A. 2001;105:6945–6954. [Google Scholar]; (b) Kurimura Y, Yokota H, Muraki Y. Bull Chem Soc Jpn. 1981;54:2450. [Google Scholar]; (c) Ruggi A, van Leeuwen FWB, Velders A. Coord Chem Rev. 2011;255:2542–2554. [Google Scholar]
- 22.(a) Hayashi S, Yasui H, Sakurai H. DrugMetab Pharmacokinet. 2005;20:14–23. doi: 10.2133/dmpk.20.14. [DOI] [PubMed] [Google Scholar]; (b) Hlavica P. Eur J Biochem. 2004;271:4335–4360. doi: 10.1111/j.1432-1033.2004.04380.x. [DOI] [PubMed] [Google Scholar]
- 23.Output powers measured at the sample were 0.75 W, 0.55 W and 0.45W for the respective light sources.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.