

Abstract
Identifying the genetic variants that increase the risk of type 2 diabetes (T2D) in humans has been a formidable challenge. Adopting a genome-wide association strategy, we genotyped 1161 Finnish T2D cases and 1174 Finnish normal glucose-tolerant (NGT) controls with >315,000 single-nucleotide polymorphisms (SNPs) and imputed genotypes for an additional >2 million autosomal SNPs. We carried out association analysis with these SNPs to identify genetic variants that predispose to T2D, compared our T2D association results with the results of two similar studies, and genotyped 80 SNPs in an additional 1215 Finnish T2D cases and 1258 Finnish NGT controls. We identify T2D-associated variants in an intergenic region of chromosome 11p12, contribute to the identification of T2D-associated variants near the genes IGF2BP2 and CDKAL1 and the region of CDKN2A and CDKN2B, and confirm that variants near TCF7L2, SLC30A8, HHEX, FTO, PPARG, and KCNJ11 are associated with T2D risk. This brings the number of T2D loci now confidently identified to at least 10.
Type 2 diabetes (T2D) is a disease characterized by insulin resistance and impaired pancreatic beta-cell function that affects >170 million people worldwide (1). With first-degree relatives having ~3.5 times as much risk as compared to individuals in the general middle-aged population (2), hereditary factors, together with lifestyle and behavioral factors, play an important role in determining T2D risk (3). To date, intense efforts to identify genetic risk factors in T2D have met with only limited success. This study, reports from our collaborators (4-6), and the recently published work of Sladek et al. (7) describe results of genome-wide association (GWA) studies that further define the genetic architecture of T2D and identify biological pathways involved in T2D pathogenesis.
We genotyped 1161 Finnish T2D cases and 1174 Finnish NGT controls on 317,503 SNPs on the Illumina HumanHap300 BeadChip in stage 1 of a two-stage GWA study of T2D (8). These samples are from the Finland–United States Investigation of Non–Insulin-Dependent Diabetes Mellitus Genetics (FUSION) (9, 10) and Finrisk 2002 (11) studies (tables S1 and S2A). Among the 317,503 GWA SNPs, 315,635 had ≥10 copies of the less common allele [minor allele frequency (MAF) > 0.002] and passed quality-control criteria (8). We tested these 315,635 SNPs for association with T2D using a model that is additive on the log-odds scale (Table 1 and tables S3 and S4) (8). We observed a modest excess (41 observed versus 31.6 expected; P = 0.19) of SNPs with P values < 10−4 (fig. S1). These results argue against the existence of multiple common SNPs with a large impact on T2D disease risk but are consistent with the presence of multiple common SNPs that each confer modest risk. The results also suggest that the matching of cases and controls by birth province, sex, and age (8) has been successful; in support of this conclusion, the genomic control (12) correction value is 1.026.
Table 1.
Confirmed T2D susceptibility loci based on all available data from the FUSION, DGI, and WTCCC/UKT2D samples.
| FUSION | Chr | Position (bp) |
Genes | Risk allele / nonrisk allele |
FUSION Stage 1 + 2 control risk allele freq. |
FUSION stage 1
|
FUSION stage 2
|
FUSION stage 1 + 2
|
DGI All Data
|
WTCCC/UKT2D All Data
|
FUSION-DGI- WTCCC/UKT2D All Data |
Total sample size for 80% power** |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) |
P | OR (95% CI) |
P | OR (95% CI) |
P | OR (95% CI) |
P | OR (95% CI) |
P | OR (95% CI) |
P | |||||||
| New T2D Loci | ||||||||||||||||||
| rs4402960 | 3 | 186,994,389 | IGF2BP2 | T/G | 0.30 | 1.28 (1.13–1.45) | 1.2×10−4 | 1.08 (0.96–1.22) | 0.22 | 1.18 (1.08–1.28) | 2.1×10−4 | 1.17 (1.11–1.23) | 1.7×10−9 | 1.11 (1.05–1.16) | 1.6×10−4 | 1.14 (1.11–1.18) | 8.9×10−16 | ~4300 |
| rs7754840* | 6 | 20,769,229 | CDKAL1 | C/G | 0.36 | 1.16 (1.02–1.30) | 0.021 | 1.08 (0.96–1.22) | 0.20 | 1.12 (1.03–1.22) | 0.0095 | 1.08 (1.03–1.14) | 2.4×10−3 | 1.16 (1.10–1.22) | 1.3×10−8 | 1.12 (1.08–1.16) | 4.1×10−11 | ~5300 |
| rs10811661 | 9 | 22,124,094 | CDKN2A/B | T/C | 0.85 | 1.17 (0.98–1.39) | 0.082 | 1.22 (1.04–1.44) | 0.015 | 1.20 (1.07–1.36) | 0.0022 | 1.20 (1.12–1.28) | 5.4×10−8 | 1.19 (1.11–1.28) | 4.9×10−7 | 1.20 (1.14–1.25) | 7.8×10−15 | ~3900 |
| rs9300039† | 11 | 41,871,942 | C/A | 0.89 | 1.52 (1.24–1.87) | 6.0×10−5 | 1.45 (1.19–1.77) | 2.7×10−4 | 1.48 (1.28–1.71) | 5.7×10−8 | 1.16¶ (0.95–1.42) | 0.12 | 1.13# (0.99–1.29) | 0.068 | 1.25 (1.15–1.37) | 4.3×10−7 | ~3400 | |
| rs8050136 | 16 | 52,373,776 | FTO | A/C | 0.38 | 1.03 (0.92–1.16) | 0.58 | 1.18 (1.05–1.33) | 0.0063 | 1.11 (1.02–1.20) | 0.016 | 1.03¶ (0.91–1.17) | 0.25 | 1.23 (1.18–1.32) | 7.3×10−14 | 1.17 (1.12–1.22) | 1.3×10−12 | ~2700 |
| Previously published T2D association | ||||||||||||||||||
| rs1801282 | 3 | 12,368,125 | PPARG | C/G | 0.82 | 1.30 (1.11–1.53) | 0.0011 | 1.08 (0.93–1.26) | 0.33 | 1.20 (1.07–1.33) | 0.0014 | 1.09 (1.01–1.16) | 0.019 | 1.23# (1.09–1.41) | 0.0013 | 1.14 (1.08–1.20) | 1.7×10−6 | ~6400 |
| rs13266634 | 8 | 118,253,964 | SLC30A8 | C/T | 0.61 | 1.22 (1.08–1.38) | 0.0010 | 1.14 (1.02–1.28) | 0.026 | 1.18 (1.09–1.29) | 7.0×10−5 | 1.07 (1.0–1.16) | 0.047 | 1.12 (1.05–1.18) | 7.0×10−5 | 1.12 (1.07–1.16) | 5.3×10−8 | ~5100 |
| rs1111875‡ | 10 | 94,452,862 | HHEX | C/T | 0.52 | 1.13 (1.01–1.27) | 0.039 | 1.06 (0.94–1.19) | 0.34 | 1.10 (1.01–1.19) | 0.026 | 1.14 (1.06–1.22) | 1.7×10−4 | 1.13 (1.07–1.19) | 4.6×10−6 | 1.13 (1.09–1.17) | 5.7×10−10 | ~4200 |
| rs7903146§ | 10 | 114,748,339 | TCF7L2 | T/C | 0.18 | 1.39 (1.20–1.61) | 1.2×10−5 | 1.30 (1.12–1.50) | 3.5×10−4 | 1.34 (1.21–1.49) | 1.3×10−8 | 1.38 (1.31–1.46) | 2.3×10−31 | 1.37# (1.25–1.49) | 6.7×10−13 | 1.37 (1.31–1.43) | 1.0×10−48 | ~1000 |
| rs5219∥ | 11 | 17,366,148 | KCNJ11 | T/C | 0.46 | 1.20 (1.07–1.36) | 0.0022 | 1.04 (0.92–1.16) | 0.55 | 1.11 (1.02–1.21) | 0.013 | 1.15 (1.09–1.21) | 1.0×10−7 | 1.15# (1.05–1.25) | 0.0013 | 1.14 (1.10–1.19) | 6.7×10−11 | ~3700 |
| Total sample size | 2,335 | 2,473 | 4,808 | 13,781 | 13,965 | 32,544 | ||||||||||||
| Number of cases/controls | 1,161/1,174 | 1,215/1,258 | 2,376/2,432 | 6,529/7,252 | 5,681/8,284 | 14,586/17,968 | ||||||||||||
rs10946398 WTCCC/UKT2D (r2 = 1).
Multimarker tag for rs9300039 DGI and rs1514823 WTCCC/UKT2D (r2 = 0.965).
rs5015480 WTCCC GWA only (r2 = 1).
rs7901695 WTCCC/UKT2D (r2 = 0.849).
rs5215 WTCCC/UKT2D (r2 = 0.995).
DGI GWA samples.
WTCCC GWA samples.
Approximate total sample size for 80% power to detect T2D SNP association at significance level 0.05 is based on the FUSION control risk allele frequency and the risk ratio calculated from FUSION-DGI-WTCCC/UKT2D all-data analyses, assuming 0.10 T2D prevalence. The sample sizes vary slightly from those of (4) because study-specific allele frequencies were used in the calculations.
Analysis of our Illumina HumanHap300 data allowed us to query much of the known SNP variation in the genome. To increase this proportion, we developed an imputation method (8, 13) that uses genotype data and linkage disequilibrium (LD) information from the HapMap Centre d’Etude du Polymorphisme Humain (Utah residents with ancestry from northern and western Europe) (CEU) samples to predict genotypes of autosomal SNPs not genotyped in our subjects. A total of 2.09 million HapMap CEU SNPs (14) had imputed MAF >1% in FUSION and passed our imputation quality-control criteria. In the HapMap CEU sample, imputed SNPs passing these criteria increased coverage of SNPs with MAF >1% from 71.9 to 89.1% at an r2 threshold of 0.8.
To increase the statistical power to detect T2D predisposing variants, we compared our stage 1 results to GWA results from the Diabetes Genetics Initiative (DGI) and the Wellcome Trust Case Control Consortium (WTCCC). We selected 82 SNPs for FUSION stage 2 follow-up genotyping based on evidence from: (i) FUSION-genotyped and FUSION-imputed SNPs; (ii) a combined analysis of GWA results from FUSION, DGI, and WTCCC; and (iii) previous T2D association results. For (i) and (ii), we used a prioritization algorithm that advantaged SNPs based on genome annotation (8) (table S7) and gave preference to genotyped SNPs over nearby imputed SNPs. We successfully genotyped 80 of the 82 SNPs in our stage 2 sample of 1215 Finnish T2D cases and 1258 Finnish NGT controls (8) (table S2B) and carried out joint analysis of the combined FUSION stage 1 + 2 sample (table S5). DGI (4) and United Kingdom T2D Genetics Consortium (UKT2D) (5) investigators also followed up DGI and WTCCC GWAs by genotyping replication samples.
We confirmed well-established T2D associations with TCF7L2, PPARG, and KCNJ11 (Table 1) (15-18). SNPs in TCF7L2 reached genome-wide significance in the FUSION stage 1 + 2 sample [odds ratio (OR) = 1.34, P = 1.3 × 10−8] and in the FUSION-DGI-WTCCC/UKT2D “all-data” (i.e., all GWA and follow-up samples) meta-analysis (OR = 1.37, P = 1.0 × 10−48) (Table 1 and table S5). PPARG Pro12→Ala12 (rs1801282) and KCNJ11 Glu23→Lys23 (rs5219) were not genotyped in the FUSION GWA, but nearby SNPs showed some evidence for T2D association, as did the imputed genotypes for the coding variants. All-data meta-analysis resulted in genome-wide significant T2D association with KCNJ11 Glu23→Lys23 (OR = 1.14, P = 6.7 × 10−11) and strong evidence for PPARG Pro12→Ala12 (OR = 1.14, P = 1.7 × 10−6). The PPARG and KCNJ11 results emphasize the value of combining data across studies and suggest that other T2D-associated loci remain to be found.
The combined samples from the three studies provide evidence for seven additional T2D loci. For the first three of these loci, we had strong evidence in the FUSION stage 1 GWA data and, for the latter four, our FUSION stage 1 evidence was more modest.
A cluster of variants in the IGF2BP2 (insulin-like growth factor 2 mRNA binding protein 2) region was associated with T2D in our stage 1 sample (e.g., rs1470579 with OR = 1.27, P = 1.6 × 10−4) (Fig. 1A). The all-data meta-analysis for rs4402960 resulted in genome-wide significance (OR = 1.14, P = 8.9 × 10−16). Including the rs4402960 genotype as a covariate essentially eliminates evidence for T2D association for other variants in the cluster (Fig. 1A), which is consistent with all SNPs representing the same T2D-predisposing variant(s). IGF2BP2 is a paralog of IGF2BP1, which binds to the 5′ untranslated region of the insulin-like growth factor 2 (IGF2) mRNA and regulates IGF2 translation (19). IGF2 is a member of the insulin family of polypeptide growth factors involved in the development, growth, and stimulation of insulin action. The most strongly associated IGF2BP2 SNPs are located in a 50-kb region within intron 2 (Fig. 1A); diabetes-predisposing variants may therefore affect regulation of IGF2BP2 expression.
Fig. 1.
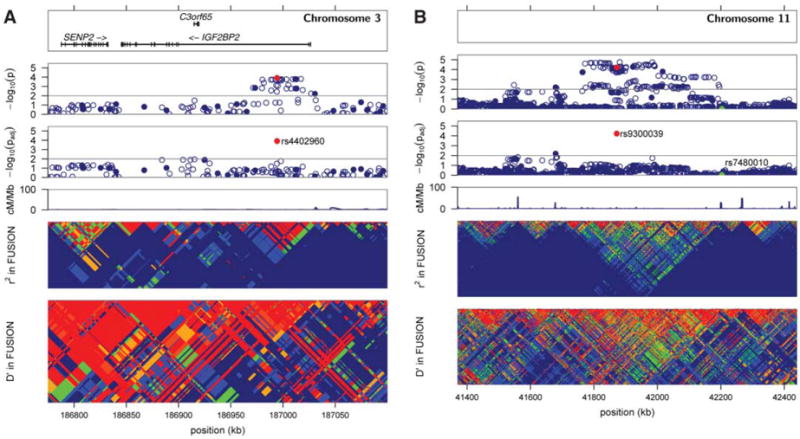
Plots of T2D association and LD in FUSION stage 1 samples for regions surrounding IGF2BP2 (A) and rs9300039 (B). (A) and (B) each contain six panels. The top panels display RefSeq genes; there are none in the rs9300039 region. The second panels (i.e., directly below the top panels) show the T2D association −log10 P values in FUSION stage 1 samples for SNPs genotyped in the GWA panel (closed blue circles) or imputed (open blue circles). The third panels show T2D association −log10 P values for each SNP in a logistic regression model correcting for the reference SNP [indicated by the red circle for rs4402960 in (A) and for rs9300039 in (B)]. SNP rs7480010, reported by Sladek et al. (7), is also labeled in the rs9300039 plot (B) (green circle). A decrease in the −log10 P value from the second to the third panels indicates that the association signal of the tested SNPs can be explained, at least in part, by the reference SNP. In both regions, the reference SNP was chosen for convenience; the choice of another strongly associated SNP nearby would have resulted in a similar picture. The fourth panels show recombination rate in centimorgans per megabase for the HapMap CEU sample (14). The fifth and sixth panels show LD r2 and D’ based on FUSION stage 1–genotyped and FUSION stage 1–imputed data.
SNP rs13266634, a nonsynonymous Arg325→Trp325 variant in the pancreatic beta-cell–specific zinc transporter SLC30A8 (20), showed (through our annotation-based algorithm) evidence for T2D association in stage 1 (Table 1 and fig. S2). Modest evidence in stage 2 resulted in stronger evidence in our stage 1 + 2 sample (OR = 1.18, P = 7.0 × 10−5) (Table 1 and table S5). Subsequent DGI and UKT2D genotyping resulted in strong evidence in the combined samples (OR = 1.12, P = 5.3 × 10−8). Sladek et al. (7) recently reported independent T2D association evidence with the same allele in two French samples (P = 1.8 × 10−5 and P = 5.0 × 10−7). SLC30A8 transports zinc from the cytoplasm into insulin secretory vesicles (20, 21), where insulin is stored as a hexamer bound with two Zn2+ ions before secretion (22). Variation in SLC30A8 may affect zinc accumulation in insulin granules, affecting insulin stability, storage, or secretion. In high-glucose conditions, overexpression of SLC30A8 in insulinoma (INS-1E) cells enhanced glucose-induced insulin secretion (21).
SNP rs9300039 in an intergenic region on chromosome 11 showed evidence for T2D association in stage 1 (Table 1 and Fig. 1B); genotyping our stage 2 sample resulted in near genome-wide significance in our stage 1 + 2 sample (OR = 1.48, P = 5.7 × 10−8) (Table 1 and tables S3 and S5). In the WTCCC and DGI scans, the nearby SNP rs1514823 (r2 = 0.97 with rs9300039) provided weak evidence for T2D association with the appropriate allele; combining results across all three studies gave OR = 1.25 and P = 4.3 × 10−7. Fifty-six imputed SNPs and two more genotyped SNPs spanning 219 kb are in LD with rs9300039 and show substantial evidence for T2D association (P < 10−4) in our stage 1 sample (table S3 and Fig. 1B). Including the genotype for rs9300039 as a covariate essentially eliminates evidence for T2D association with the remaining SNPs (Fig. 1B). This region includes three sets of spliced Expressed Sequence Tags but no annotated genes. The identification of a T2D-associated variant >1 Mb from the nearest annotated gene highlights the value of a genome-wide approach. Sladek et al. (7) reported strongly associated SNPs in two nearby regions on chromosome 11. SNP rs7480010 near hypothetical gene LOC387761 is 331 kb centromeric to rs9300039. LD between rs9300039 and rs7480010 is essentially zero (r2 = 0.00063 and D’ = 0.036), and rs7480010 showed little evidence for association in our stage 1 + 2 sample (OR = 1.03, P = 0.54). Sladek et al. (7) also reported T2D association with three intronic variants of EXT2, located ~2.4 Mb centromeric of rs9300039; we found no evidence for association with EXT2 SNPs.
SNP rs4712523, located within intron 5 of CDKAL1, showed modest evidence for T2D association in our FUSION stage 1 sample, which strengthened slightly in our combined stage 1 + 2 sample (OR = 1.12, P = 0.0073) (table S5). Nearby SNPs in strong LD with rs4712523 including rs7754840 showed modest evidence for T2D association in the DGI scan and considerably stronger evidence in the WTCCC scan. Including strong DGI and UKT2D replication data resulted in genome-wide significance (OR = 1.12, P = 4.1 × 10−11 for rs7754840) in the all-data meta-analysis (Table 1). CDKAL1 [cyclin-dependent kinase 5 (CDK5) regulatory subunit associated protein–1–like 1] shares protein domain similarity with CDK5 regulatory subunit–associated protein 1 (CDK5RAP1), which specifically inhibits activation of CDK5 by CDK5 regulatory subunit 1 (CDK5R1) (23). Using quantitative reverse transcription polymerase chain reaction analysis of a panel of RNA samples from human tissues and cells, we detected the highest expression of CDKAL1 in skeletal muscle and brain cells, as well as in 293T and HepG2 cells (fig. S3A). The associated SNPs within intron 5, or SNPs in LD with them, may regulate expression of CDKAL1 and so affect the expression of CDK5. CDK5 and CDK5R1 activity is influenced by glucose and may influence beta-cell processes (24, 25); overactivity of CDK5 in the pancreas may lead to beta-cell degeneration, especially under glucotoxic conditions (26).
SNP rs10811661 near cyclin-dependent kinase inhibitors CDKN2A and CDKN2B showed modest evidence for T2D association in our stage 1 + 2 sample (OR = 1.20, P = 0.0022) (Table 1 and table S5) and showed genome-wide significance in the all-data meta-analysis (OR = 1.20, P = 7.8 × 10−15). SNP rs10811661 is located upstream of CDKN2A and CDKN2B, may have a long-range effect on one of these genes, or may influence a gene not yet annotated. CDKN2A and CDKN2B inhibit the activity of CDK4 and CDK6. In mice, Cdk4 activity has been shown to influence beta-cell proliferation and mass, with loss of Cdk4 leading to diabetes (27, 28). We find CDKN2A to be expressed at high levels in islets, adipocytes, brain, and pancreas and at even higher levels in 293T, HeLa, and HepG2 cells (fig. S3B); CDKN2B is expressed in islets and adipocytes and, to a lesser degree, in small intestine, colon, 293T, and HepG2 cells (fig. S3C). CDKN2A and CDKN2B are also tumor suppressor genes and may play a role in aging (29).
SNPs rs1111875 and rs7923837 showed modest evidence of T2D association in the FUSION and DGI scans, much stronger evidence in the WTCCC scan, and genome-wide significant evidence (OR = 1.13, P = 5.7 × 10−10 for rs1111875) in the all-data meta-analysis. These SNPs are in LD (r2 = 0.70) in a region that includes HHEX (hematopoietically expressed homeobox), which is critical for development of the ventral pancreas (30), the insulin-degrading enzyme gene IDE, and the kinesin-interacting factor 11 gene KIF11. Sladek et al. (7) recently reported independent genome-wide significant evidence for T2D association with these SNPs.
The WTCCC/UKT2D groups identified evidence for T2D and body mass index (BMI) associations with a set of SNPs including rs8050136 in the FTO region; the T2D association appears to be mediated through a primary effect on adiposity (5, 6, 31). We observed modest evidence for association with T2D in the combined FUSION stage 1 + 2 sample (OR = 1.11, P = 0.016) (Table 1 and table S5).
T2D can be a component of a larger syndrome of metabolic abnormalities, and we were interested to assess the effects of T2D-related traits on our association results. We repeated our T2D association analysis for the 10 SNPs in Table 1 with one of several variables included as an additional covariate. Adjustment for BMI strengthened T2D association with TCF7L2 and SLC30A8, weakened association with rs9300039 and FTO, and had little effect on the other loci. The effect of waist circumference was similar to that of BMI; blood pressure variables had essentially no effect.
We previously carried out T2D linkage analysis in the families of many of our stage 1 cases (10). None of the 10 loci in Table 1 had large T2D logarithm of the odds (LOD) scores, although those for FTO and TCF7L2 were 0.63 and 0.60 and so were nominally significant. LOD scores for six of the 10 loci were greater than 0.2, as compared to 2.2 that would be expected for random genome locations. This suggests enrichment for T2D-associated loci in regions with modest evidence of T2D linkage (P = 0.01) but that the power of the linkage approach was insufficient to distinguish these signals from background noise.
The ability to construct a list of ten robust and replicated T2D-associated loci (Table 1) represents a landmark in efforts to identify genetic variants that predispose to complex human diseases, although the specific predisposing variants and even the relevant genes remain to be defined. We examined the combined risk of T2D based on these 10 loci in our stage 1 + 2 sample by constructing a logistic regression model and predicting T2D risk for each person (8). We found a fourfold variation in T2D risk from the lowest to highest predicted risk groups, which is of potential interest for a personalized preventive-medicine program (Fig. 2). However, these predictions from our data may be biased as compared to predictions based on the general population, likely owing to the overestimation of ORs due to the “winner’s curse,” enrichment for familial T2D cases, and exclusion of individuals with impaired glucose tolerance or impaired fasting glucose.
Fig. 2.
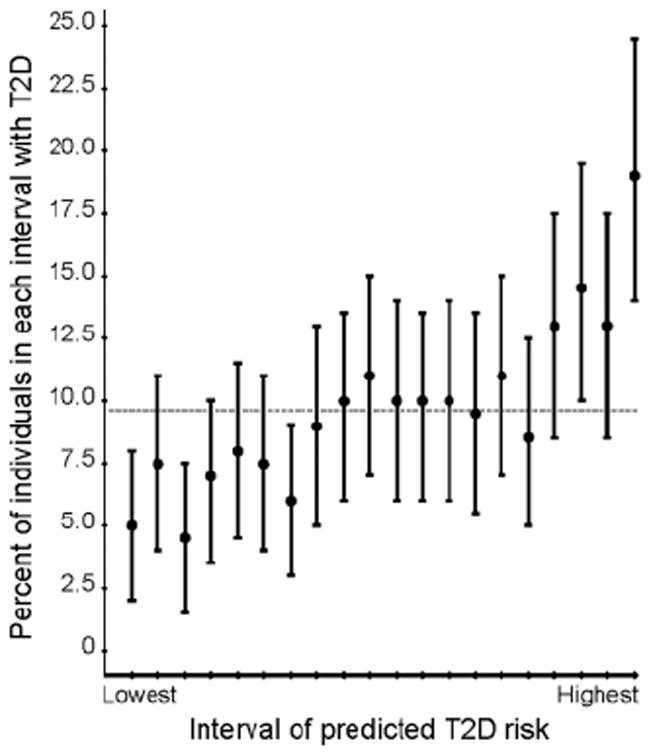
Prediction of T2D risk in the FUSION sample with the use of 10 T2D susceptibility variants. T2D cases and NGT controls with complete genotype data were included in the analysis. To obtain a sample with a T2D prevalence of ~10%, we included nine copies of each of 2176 NGT controls and one copy of each of 2102 T2D cases. The predicted risk for each individual was estimated from a logistic regression model containing the 10 risk variants listed in Table 1. The proportion of T2D cases is shown for 20 equal intervals of predicted T2D risk. We constructed 95% confidence intervals (CIs) for the proportion of T2D cases in each interval using the original sample of 2102 cases and 2176 controls. The constructed sample T2D prevalence (0.096) is shown as a horizontal line. The proportion of T2D cases increases from ~5% in the lowest to 20% in the highest predicted risk categories.
Thirty years ago, James V. Neel labeled T2D as “the geneticist’s nightmare” (32), predicting that the discovery of genetic factors in T2D would be thoroughly challenging. Until recently, his prediction has proven true. Although large samples and collaboration among three groups were required, we can confidently state that new diabetes risk factors have been identified. Each gene discovery points to a pathway that contributes to pathogenesis, and all of these proteins and their relevant pathways represent potential drug targets for the prevention or treatment of diabetes. Based on the number of other interesting results observed in these studies, it is likely that there are additional T2D-predisposing loci to be found. Even though much remains to be done, we are at last awakening from Jim Neel’s nightmare.
Supplementary Material
Acknowledgments
We thank the Finnish citizens who generously participated in this study; our colleagues from the DGI, WTCCC, and UKT2D for sharing prepublication data from their studies; S. Enloe of FUSION and E. Kwasnik, J. Gearhart, J. Romm, M. Zilka, C. Ongaco, A. Robinson, R. King, B. Craig, and E. Hsu of CIDR for expert technical work; and D. Leja of NHGRI for expert assistance with a figure. Support for this research was provided by NIH grants DK062370 (M.B.), DK072193 (K.L.M.), HL084729 (G.R.A.), HG002651 (G.R.A.), and U54 DA021519; National Human Genome Research Institute intramural project number 1 Z01 HG000024 (F.S.C.); a postdoctoral fellowship award from the American Diabetes Association (C.J.W.); a Wenner-Gren Fellowship (L.P.O.); and a Calvin Research Fellowship (R.P.). Genome-wide genotyping was performed by the Johns Hopkins University Genetic Resources Core Facility (GRCF) SNP Center at CIDR with support from CIDR NIH (contract N01-HG-65403) and the GRCF SNP Center.
Footnotes
Supporting Online Material www.sciencemag.org/cgi/content/full/1142382/DC1
Author Contributions
Materials and Methods
Figs. S1 to S3
Tables S1 to S7
References
References and Notes
- 1.Wild S, Roglic G, Green A, Sicree R, King H. Diabetes Care. 2004;27:1047. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Rich SS. Diabetes. 1990;39:1315. doi: 10.2337/diab.39.11.1315. [DOI] [PubMed] [Google Scholar]
- 3.Kaprio J, et al. Diabetologia. 1992;35:1060. doi: 10.1007/BF02221682. [DOI] [PubMed] [Google Scholar]
- 4.Diabetes Genetics Initiative. Science. 2007;316:1331. doi: 10.1126/science.1142358. published online 26 April 2007. [DOI] [PubMed] [Google Scholar]
- 5.Zeggini E, et al. Science. 2007;316:1336. doi: 10.1126/science.1142364. published online 26 April 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The Wellcome Trust Case Control Consortium. Nature. in press. [Google Scholar]
- 7.Sladek R, et al. Nature. 2007;445:881. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 8.Materials and methods are available as supporting material on Science Online.
- 9.Valle T, et al. Diabetes Care. 1998;21:949. doi: 10.2337/diacare.21.6.949. [DOI] [PubMed] [Google Scholar]
- 10.Silander K, et al. Diabetes. 2004;53:821. doi: 10.2337/diabetes.53.3.821. [DOI] [PubMed] [Google Scholar]
- 11.Saaristo T, et al. Diabetes Vasc Dis Res. 2005;2:67. doi: 10.3132/dvdr.2005.011. [DOI] [PubMed] [Google Scholar]
- 12.Devlin B, Roeder K. Biometrics. 1999;55:997. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Scheet P, Ding J, Abecasis GR. submitted for publication; manuscript available from G.R.A (goncalo@umich.edu) [Google Scholar]
- 14.International HapMap Consortium. Nature. 2005;437:1299. [Google Scholar]
- 15.Grant SF, et al. Nat Genet. 2006;38:320. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 16.Deeb SS, et al. Nat Genet. 1998;20:284. doi: 10.1038/3099. [DOI] [PubMed] [Google Scholar]
- 17.Altshuler D, et al. Nat Genet. 2000;26:76. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 18.Gloyn AL, et al. Diabetes. 2003;52:568. doi: 10.2337/diabetes.52.2.568. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen J, et al. Mol Cell Biol. 1999;19:1262. doi: 10.1128/mcb.19.2.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chimienti F, Devergnas S, Favier A, Seve M. Diabetes. 2004;53:2330. doi: 10.2337/diabetes.53.9.2330. [DOI] [PubMed] [Google Scholar]
- 21.Chimienti F, et al. J Cell Sci. 2006;119:4199. doi: 10.1242/jcs.03164. [DOI] [PubMed] [Google Scholar]
- 22.Dunn MF. Biometals. 2005;18:295. doi: 10.1007/s10534-005-3685-y. [DOI] [PubMed] [Google Scholar]
- 23.Ching YP, Pang AS, Lam WH, Qi RZ, Wang JH. J Biol Chem. 2002;277:15237. doi: 10.1074/jbc.C200032200. [DOI] [PubMed] [Google Scholar]
- 24.Ubeda M, Kemp DM, Habener JF. Endocrinology. 2004;145:3023. doi: 10.1210/en.2003-1522. [DOI] [PubMed] [Google Scholar]
- 25.Wei FY, et al. Nat Med. 2005;11:1104. doi: 10.1038/nm1299. [DOI] [PubMed] [Google Scholar]
- 26.Ubeda M, Rukstalis JM, Habener JF. J Biol Chem. 2006;281:28858. doi: 10.1074/jbc.M604690200. [DOI] [PubMed] [Google Scholar]
- 27.Rane SG, et al. Nat Genet. 1999;22:44. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 28.Tsutsui T, et al. Mol Cell Biol. 1999;19:7011. doi: 10.1128/mcb.19.10.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim WY, Sharpless NE. Cell. 2006;127:265. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Bort R, Martinez-Barbera JP, Beddington RS, Zaret KS. Development. 2004;131:797. doi: 10.1242/dev.00965. [DOI] [PubMed] [Google Scholar]
- 31.Frayling TM, et al. Science. 2007;316:889. doi: 10.1126/science.1141634. published online 12 April 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neel JV. In: The Genetics of Diabetes Mellitus. Creutzfeldt W, Köbberling J, Neel JV, editors. Springer; Berlin: 1976. pp. 1–11. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.