

Abstract
A trans-acting protein interacting with a specific sequence motif proximal to the transcriptional start site of the l-asparaginase promoter has been observed previously (E. Vincze, J. M. Reeves, E. Lamping, K. J. F. Farnden, and P. H. S. Reynolds, Plant Mol. Biol. 26:303-311, 1994). Gel retardation experiments in which protein extracts of Mesorhizobium loti and developing nodules were used suggested a bacterial origin for the repressor binding protein (rep2037). Nodulation tests were performed by using different Fix− Tn5 mutants of M. loti. Analyses of these mutants revealed a correlation between the presence of Mesorhizobium in the nodule-like structures and the ability of nodule protein extracts to bind the repressor binding domain (RBD). Through the use of mutated RBD sequences, the RBD sequence was identified as CTAAAAT. The repressor protein was isolated from M. loti NZP2037 by multiple chromatographic procedures and affinity separation by using concatemers of RBD attached to magnetic beads. Sequencing of the recovered protein resulted in identification of the repressor protein as the sarcosine oxidase α subunit. This was confirmed by expression of the gene encoding the M. loti α subunit of sarcosine oxidase in Escherichia coli. When the expressed peptide was bound to RBD, the gel retardation result was identical to the result obtained with rep2037 from M. loti strain NZP2037.
DNA binding proteins play key roles in replication, recombination, and gene expression. These proteins can be divided into two functional classes: proteins responsible for the replication and orientation of the DNA and proteins directly involved in transcription (e.g., transcriptional activators and repressors). Some proteins recognize specific DNA sequences, whereas others bind to specific DNA structures (25).
In the establishment of effective nitrogen-fixing symbioses between Rhizobium bacteria and leguminous plants, there must be both positive and negative regulation of plant and bacterial expression. Also, in the context of the intriguing and complex process of nodule development, there must be signal exchange between the plant and the bacteria to coordinate the whole process.
Signal exchange between Rhizobium and plants has been extensively studied. At least three different sets of signals are exchanged between plants and rhizobia during nodule formation (2). Plant signals which activate bacterial gene expression have been identified (5, 26), as have bacterial signals which activate the development of leguminous nodules (6-9, 11, 12, 17, 19, 23). During the establishment of an active nitrogen-fixing nodule, a large number of plant genes are activated (5, 26). These genes, often termed nodulins, are responsible for the establishment, maintenance, and functioning of the symbiosis. In addition to the induced expression of either new or organ-specific genes, specific repression of the plant l-asparaginase gene during establishment of symbiosis has been described previously (27).
Upon establishment of an effective symbiosis in the amide-transporting plant Lupinus angustifolius, the enzymatic activity and transcript levels of l-asparaginase were dramatically decreased. Concomitant with this, a specific DNA binding protein was detected in the nodules and was shown to interact with a 59-bp sequence proximal to the transcription start domain. This interaction appeared to be associated with transcriptional repression of the l-asparaginase gene in mature root nodules (27). Such repression offers a means of protecting the end product of ammonia assimilation, l-asparagine, thus allowing subsequent transport of this compound to aerial parts of the plant for use in growth and development. In this report we further define the DNA binding domain (repressor binding domain [RBD]), show that the DNA binding protein, rep2037, is bacterial sarcosine oxidase, and suggest a novel mechanism for repression of the l-asparaginase gene.
MATERIALS AND METHODS
Strain and cultural medium.
Mesorhizobium loti strains NZP2037, MAF303099, and NZP2037, Tn5 mutated strains PN 236, PN 238, and PN 239 (4), and Agrobacterium tumefaciens strain 4404 were cultured in YEB broth (Difco) (5 g of beef extract per liter, 0.5 g of peptone per liter, 0.5 g of yeast extract per liter, 0.5 g of sucrose per liter, 0.25 g of MgSO4 per liter). Incubation was carried out at 28°C with shaking (150 rpm) for 40 h (absorbance at 600 nm, 0.7 to 1.0). After sedimentation with a Sorvall RC5B Plus centrifuge at 6,000 rpm for 10 min, the pellets were washed in 40 ml of phosphate-buffered saline and collected by centrifugation. The pellets were suspended in buffer C (20 mM Tris-HCl [pH 8.0], 25% glycerol, 420 mM NaCl, 1.5 mM MgSO4, 0.2 mM EDTA-Na2, 0.5 mM dithiothreitol, 0.1% Tween 20) and stored at −20°C until they were used. Escherichia coli strain DH5α FT was grown on Luria-Bertani medium and processed as described above.
Gel shift assays.
Gel retardation assays were carried out as described previously (27), except that electrophoresis was performed with Criterion 5% precast TBE gels (Bio-Rad). The 59-bp DNA probe (RBD) was labeled with 32P as described by Marincs and White (18). The binding reaction mixtures (final volume, 15 μl) contained 10 ng of 32P-labeled RBD, 1 μg of poly(dI-dC) and 1 μg of poly(dA-dT) as nonspecific competitor DNA, and 3 μl of protein extract in 5 mM Tris-HCl (pH 8.0)-50 mM KCl-1 mM EDTA-5% glycerol.
Bacteroid preparations.
In all of the previously described gel retardation experiments conducted to characterize the l-asparaginase RBD (27) nuclear extracts prepared by the method of Jensen et al. (13) were used. These extracts were found to be heavily contaminated with bacteroids (Vincze, unpublished results). Therefore, bacteroids free from contaminating nuclei were prepared by using a discontinuous salt density gradient method (20). Protein was extracted from these bacteroid preparations made from nodules on days 7, 11, 13, 15, and 18 after infection of the roots with M. loti NZP2037.
Oligonucleotide sequences.
All primers were phosphorylated at the 5′ end. Oligomers with the following sequences were synthesized (Sigma Genosys Australia): (i) wild-type dimer TCGCTAAAATGCGGTGTCATTATCGCTAAAATTGCGGTGTCAA-3′ and its complement (primer 1); (ii) the same sequence but with the complementary chain biotinylated at the 3′ end (primer 2); and (iii) mutant dimer probes in which the CTAAAAT region (underlined in the wild-type sequence) was replaced with CCAGCTG (single mutant A), the TGTCA region (underlined) was replaced with GACCC (single mutant B), or both regions were replaced with the mutated sequences (double mutant). Concatemers of the wild-type and double mutant dimers were prepared as described previously (11) by using Pwo DNA polymerase.
Purification of DNA binding protein rep2037 from NZP2037.
DNA binding activity was identified by the gel shift assay described above.
Cell pellets from 240-liter cultures of M. loti were extracted in 60-liter batches. Samples were thawed and diluted in 320 ml of buffer C containing a protease inhibitor (P8645; Sigma Chemicals) according to the manufacturer's recommendations. Cells were sonicated on ice (100 W for 3 min with 10-s pulses). Extracts were centrifuged at 40,000 × g for 20 min, and the supernatants were collected. The pellets were reextracted two more times. The supernatants were pooled and concentrated six times by ultrafiltration by using an Amicon filter with a molecular mass cutoff of 20 kDa (type 20 Diafilter). Each concentrate was subjected to ammonium sulfate fractionation. Protein associated with RBD binding activity was detected in the 36 to 47% ammonium sulfate saturation fraction. The precipitate was dissolved in 100 ml of buffer A (20 mM Tris [pH 8], 0.2 mM EDTA, 0.1% Tween 20, 5 mM dithiothreitol,), dialyzed against five changes of 5 liters of buffer A at 4°C, and frozen at −80°C until it was used. This sample was further fractionated by chromatography on heparin Sepharose. Briefly, the sample was applied to a column of heparin Sepharose (100 ml; equilibrated in buffer A; flow rate, 1 ml/min) and eluted with a 0 to 1 M KCl gradient in buffer A (4.5-ml fractions). Fractions containing DNA binding activity were pooled, concentrated (ultrafiltration with PM10 membranes [Amicon Corporation]), and equilibrated with buffer A containing protease inhibitors. Further fractionation was performed with DEAE-Sepharose (Amrad Pharmacia; 100 ml; flow rate, 1 ml/min) equilibrated in buffer A by using a linear 0 to 0.5 M NaCl gradient in buffer A (total volume, 800 ml). Fractions containing DNA binding activity were pooled, concentrated to 20 ml as described previously, dialyzed against buffer A, made to 20% glycerol, and stored at −80°C.
DNA binding activity was further purified by chromatography on Macroprep ceramic hydroxyapatite (Bio-Rad Laboratories, Hercules, Calif.) equilibrated in 10 mM phosphate (pH 7.2) containing 5 mM dithiothreitol, 0.2 mM EDTA, and 0.1% Tween 20. The protein solution was exchanged into equilibration buffer, applied to the column at a rate of 0.5 ml/min, and fractionated by using a 10 to 400 mM phosphate (pH 6.8) gradient. DNA binding substances were collected, exchanged into buffer A containing 20% glycerol and protease inhibitors, and stored at −80°C.
Samples (1 ml, 0.8 mg of protein) were subjected to ion-exchange chromatography at pH 8.0, 8.5, and 9.0 by using 1-ml (bed volume) Hitrap DEAE columns and the AKTA Explorer high-performance liquid chromatography system (flow rate, 0.5 ml/min; fraction size, 0.2 ml; Amersham Pharmacia, Auckland, New Zealand).
Affinity purification of DNA binding protein.
Equimolar concentrations of forward and reverse primers (primers 1 and 2 [see above]) were mixed, heated to 90°C for 2 min, and then cooled to 50°C for 2 min. The duplex was attached to streptavidin-coated magnetic beads (Dynabeads M-280; Dynal Biotech PTY Ltd., South Victoria, Australia) used according to the manufacturer's procedure. Briefly, 2 ml of the beads (10 mg/ml) was transferred into two Eppendorf tubes, and the beads were sedimented with a magnet (Dynal MPC). The beads were washed once with wash buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 2.0 M NaCl) and resuspended in the same buffer to a final concentration of 5 mg/ml. An equal volume of the 3′ biotinylated duplex (30 μg) was added, and the mixture was incubated for 1 h at room temperature with occasional mixing. The beads were washed six times with wash buffer by using a Dynal MPC magnet and stored at 4°C.
The beads were washed six times with 10 mM Tris-HCl (pH 8.0)-1 mM EDTA-2 M NaCl and two times with ligation buffer (52.5 mM Tris-HCl [pH 7.5], 10.5 mM dithiothreitol, 5.2 mM MgCl2, 0.1 mM ATP) prior to ligation. The beads were suspended in ligation buffer (total volume, 0.95 ml) containing 200 mM KCl, 15% polyethylene 6000, and 60 μg of concatemer. T4 DNA ligase (50 μl, 5 U/μl) was added to the suspension, and the mixture was incubated for 3 h at room temperature with occasional swirling to keep the beads in suspension. The beads were collected, washed once with 500 μl of ligation buffer, three times with 500 μl of protein binding buffer (5 mM Tris-HCl [pH 8], 1 mM EDTA [pH 8], 50 mM KCl, 5% glycerol), eight times with 1 ml of elution buffer (protein binding buffer containing 1 M NaCl), and three times with 1 ml of protein binding buffer and finally suspended in 1 ml of protein binding buffer.
Concatemer-labeled beads (collected from 0.3 ml of suspension) were mixed in 0.7 ml of a reaction mixture containing 300 μg of a partially purified preparation of the DNA binding protein, 70 μg of poly(dI-dC), 3.5 μg of poly-l-lysine, protease inhibitors (Roche Complete), 0.1% Tween 20, and 1 mM dithiothreitol. The beads were resuspended in the reaction mixture and incubated on ice for 1 h with occasional swirling to keep the beads in suspension. The beads were separated from the supernatant, and the reaction supernatant was stored on ice. The beads were washed three times with 0.3 ml of protein binding buffer, and the bound proteins were eluted twice with 30 μl of elution buffer. The eluted proteins were stored on ice. The beads were collected, washed once with protein binding buffer, and resuspended in the reaction supernatant. The procedure involving binding and elution with the supernatant from each run was repeated for an additional two cycles. The combined eluates from each cycle were desalted on Micro-spin columns (Sephadex G-25; Amersham Pharmacia). The combined desalted eluate (180 μl) was designated eluate 1. The volume of eluate 1 was adjusted to 0.5 ml with binding buffer, and the preparation was reextracted three times with magnetic beads as described above. The time of incubation was reduced to 30 min. The combined eluates (eluate 2) were treated as described above for eluate 1.
This procedure was repeated up to three more times (yielding eluates 3, 4, and 5), except that the incubation times were reduced to 25 min and 30 μg of the double mutant concatemer was added to the reaction mixture prior to addition of the magnetic beads to remove any nonspecific DNA binding proteins.
Electrophoresis.
Sodium dodecyl sulfate (SDS) gel electrophoresis was performed with 7 and 12% polyacrylamide gels. Proteins were stained with Sypro Ruby (Molecular Probes) used according to the manufacturer's protocols, visualized by exposure to UV light, and photographed by using a Kodak DC120 digital camera.
Peptide sequencing.
Eluate 5 (see above) was electrophoresed on an SDS-polyacrylamide gel and stained with Sypro Ruby. The peptide band (approximately 110 kDa; N terminus blocked) was excised from the gel and subjected to tryptic digestion followed by quantitative time of flight mass spectrometry (MS)-MS analysis (Australian Proteome Analysis Facility, Maquarie University, Sydney, New South Wales, Australia).
Competition experiments.
Competition analyses were carried out by using the gel retardation method described above, except that water was replaced by solutions of unlabeled wild-type, dimer, or mutated probes at concentrations that were 1 to 10 times that of the radioactive probe.
Expression of sarcosine oxidase α subunit in E coli.
DNA was extracted from M. loti strain MAF303099 as previously described (1). DNA manipulations were carried out by using standard procedures (21). Plasmids were isolated with a Qiaprep spin plasmid kit (Qiagen). The gene encoding the α subunit of sarcosine oxidase was obtained by PCR. Genomic DNA isolated from M. loti strain MAF303099 (15) was used as the template with primers 5′ CTTCATCCGCTCCAATCCCAAG (162 bases upstream from the initiation codon) and 5′ CTTCTTGGGTTACGCGACCACGTC (272 bases downstream from the stop codon of the sarcosine oxidase α subunit gene). PCR was performed by using the Expand Long Template PCR system (Roche) and a Hybaid thermocycler according to the manufacturer's instructions. The product of PCR amplification was cloned into the pUC 18 vector (Invitrogen). The insert was partially sequenced, and the regions around the start and stop codons were identified.
E. coli, transformed with the cloned pUC vector, was grown in 100 ml of Luria-Bertani medium containing 100 μg of ampicillin/ml. Cultures were grown at 30°C to an absorbance at 600 nm of approximately 0.4. IPTG (isopropyl-d-thiogalactopyranoside) was added to a final concentration of 0.3 mM in order to induce production of the sarcosine oxidase α subunit. After 4 h of induction, the cells were harvested by centrifugation. The cell pellet was suspended in 4 ml of buffer C and lysed by sonication. The suspension was centrifuged at 17,000 × g, and the supernatant was collected. The sarcosine oxidase α subunit was purified by using affinity-labeled magnetic beads as described above. Cell extracts obtained in the same way from the same strain carrying only the empty pUC 18 vector (without the sarcosine oxidase α subunit gene) were used as a negative control.
RESULTS
Repressor protein originates from the bacteria.
There was no difference between the binding pattern obtained when nuclear extracts were used in the retardation assay and the binding pattern obtained when purified bacteroid protein extracts were used in the retardation assay. The appearance of a DNA binding protein in extracts from bacteroids (Fig. 1) showed the same developmental pattern as that previously reported for repression of l-asparaginase activity and transcript levels during nodule development (27).
FIG. 1.

Gel retardation analysis of protein binding to the 32P-labeled RBD during nodule development. Extracts were prepared from Lotus corniculatus bacteroids infected with NZP2037. Each assay mixture contained 1 ng of radiolabeled 59-bp RBD probe, 2 μg of protein from bacteroid extracts, and 1 μg of poly(dI-dC) and poly(dA-dT) competitor DNA. Lanes 1 to 5 contained bacteroid extracts prepared 7, 11, 13, 15, and 18 days after infection of L. corniculatus with M. loti strain NZP2037, respectively.
Protein isolated from free-living M. loti NZP2037 bound the RBD and yielded a complex whose size was identical to that obtained with a protein extract prepared from nodules formed as a result of plant infection with this strain (Fig. 2, lanes 1 and 2).
FIG. 2.
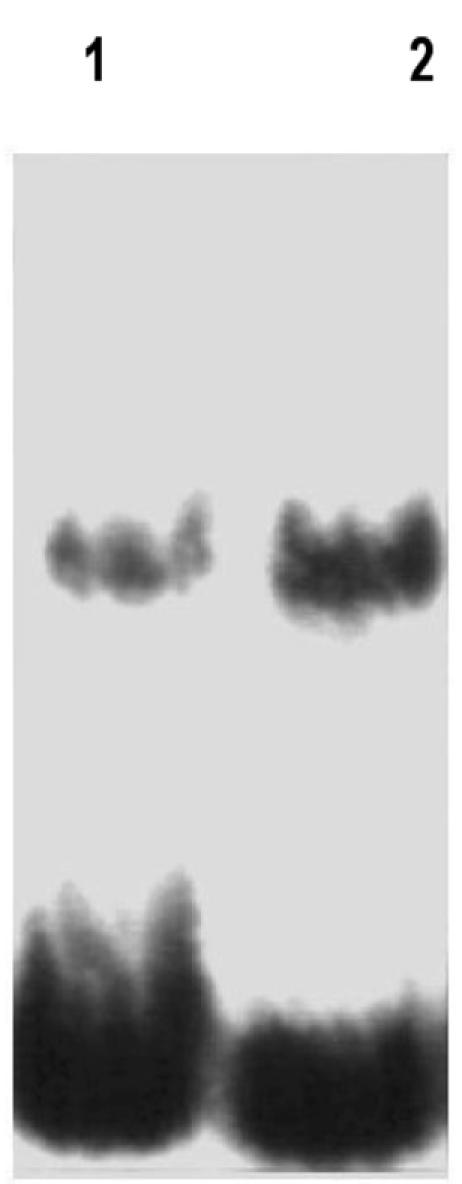
Analysis of rep2037 from free-living M. loti NZP2037 and nodule extracts containing NZP2037 from L. corniculatus by the gel retardation assay. Lane 1, nodule extract; lane 2, M loti NZP2037 extract. Each assay mixture contained 1 ng of radiolabeled 59-bp RBD probe, 2 μg of protein from extracts of nodule or free-living bacteria, and 1 μg of poly(dI-dC) and poly(dA-dT) competitor DNA.
In view of the fact that the DNA binding protein itself originates from the bacteroid, three M. loti Tn5 symbiotic mutants, PN 236, PN 238, and PN 239 (4), were analyzed for repressor production. PN 238 and PN 239 produced nodule-like structures that contained bacteria in the intercellular space between the epidermal and other cortical cells and in infection threads, respectively, whereas no bacteria were visible in the pseudonodules resulting from PN 236 infection (4). Extracts of all three strains of free-living bacteria contained rep2037 activity (results not shown). Protein extracts were obtained from the nodule-like structures resulting from infection of Lotus plants with these bacteria. Extracts of PN 238 and PN 239 nodules showed retardation of the RBD (Fig. 3, lanes 2 and 3). Extracts of the nodules produced by PN 236 did not retard the RBD probe in the assay (Fig. 3, lane 1). Thus, the presence of rep2037 in nodule extracts correlated with the ability of the strains to infect plant tissue. Furthermore, in a DNase I footprinting experiment, protein extracts from free-living M. loti NZP2037 protected the RBD probe in a manner identical to the manner observed previously (27; data not shown).
FIG. 3.
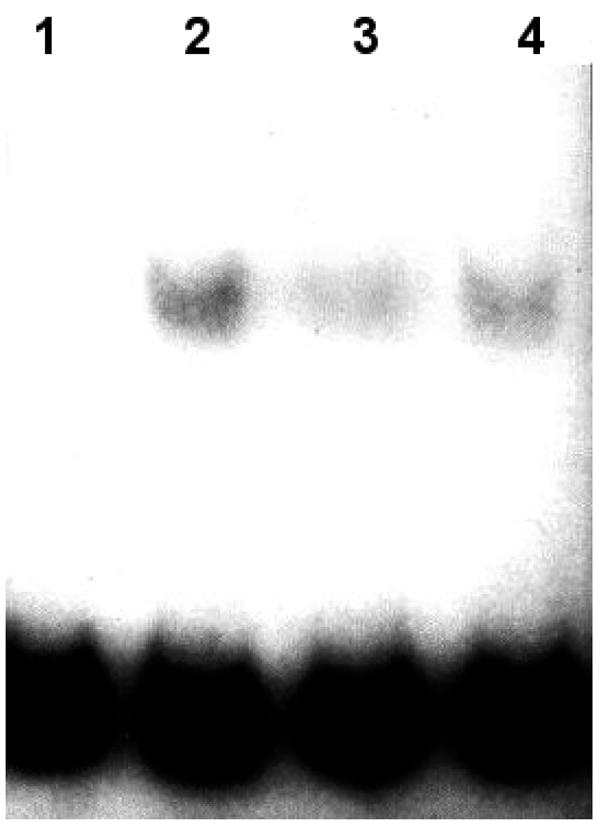
Analysis of complex formation with the 32P-labeled 59-bp RBD probe by using protein extracts from nodules formed with M. loti NZP2037 and M. loti symbiotic mutants. Lane 1, Lotus and PN 236; lane 2, Lotus and PN 238; lane 3, Lotus and PN 239; lane 4, Lotus and NZP2037. Each assay mixture contained 1 ng of radiolabeled 59-bp RBD probe, 2 μg of protein extract, and 1 μg of poly(dI-dC) and poly(dA-dT) competitor DNA.
Extracts of A. tumefaciens bound to the RBD probe and yielded DNA complexes that were the same size as the M. loti NZP2037 DNA complexes, whereas no DNA complexes were observed when extracts of E. coli strain DH5 α FT mixed with RBD were used (results not shown).
These results showed that there is a direct correlation between the presence of bacteria in the nodule structures and retardation of the RBD probe by the nodule protein extract and further confirmed that the repressor protein has a bacterial origin.
Specificity of binding to DNA.
Extracts of free-living NZP2037 treated with proteinase K completely eliminated the binding of rep2037 to the 59-bp DNA fragment of the l-asparaginase promoter (results not shown), indicating that the molecule was a protein.
Vincze et al. (27) proposed that two domains, CTAAAAT and TGACA, in the DNA fragment were likely to be involved in binding to rep2037. Oligonucleotides were synthesized in which one or both of these regions were mutated, labeled with 32P, mixed with extracts containing rep2037, and subjected to gel retardation electrophoresis. Mutation in the CTAAAAT domain eliminated binding to rep2037, whereas mutation in the TGACG region had no effect on binding to rep2037 (Fig. 4).
FIG. 4.
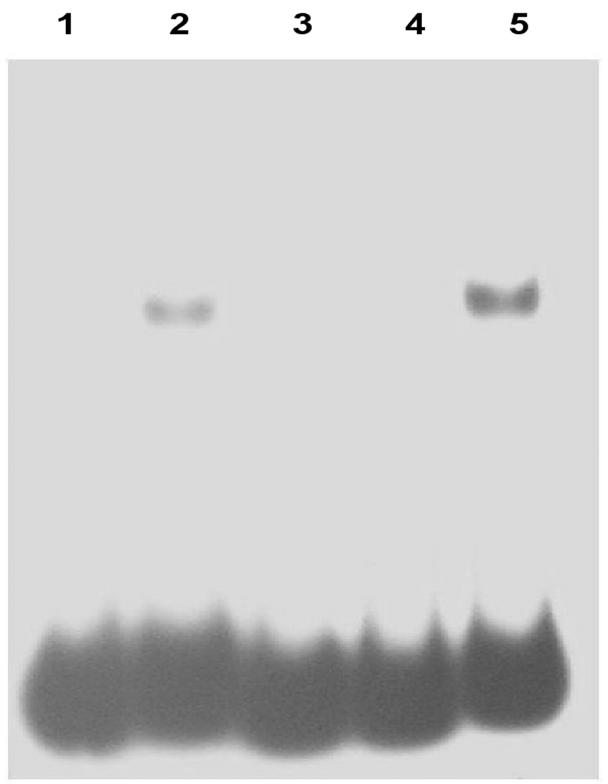
Analysis of formation of complexes of rep2037 with RBD and RBD mutants. Lane 1, wild-type RBD (no rep2037); lane 2, wild-type RBD plus rep2037; lane 3, double mutant (CTAAAAT replaced with CCAGCTG and TGTCA replaced with GACCC) plus rep2037; lane 4, RBD single mutant A (CTAAAAT replaced with CCAGCTG) plus rep2037; lane 5, RBD single mutant B (TGTCA replaced with GACCC) plus rep2037.
Competition analyses of 32P-labeled RBD with the cold wild-type dimer and concatemer for binding to rep2037 were carried out by using gel retardation. Both the wild-type dimer and the concatemer at concentrations that were 1 to 10 times the DNA concentration of RBD exhibited dose-response competition with the 32P-labeled RBD (Fig. 5).
FIG. 5.

Competition experiments performed with cold dimer and concatemer of the RBD probe. (A) Lane 1, 32P-labeled RBD probe; lane 2, 32P-labeled RBD probe plus rep2037; lane 3, 32P-labeled RBD probe plus rep2037 and 1 ng of wild-type dimer; lane 4, 32P-labeled RBD probe plus rep2037 and 5 ng of wild-type dimer; lane 5, 32P-labeled RBD probe plus rep2037 and 25 ng of wild-type dimer. (B) Lane 1, 32P-labeled RBD probe; lane 2, 32P-labeled RBD probe plus rep2037; lane 3, 32P-labeled RBD probe plus rep2037 and 1 ng of wild-type concatemer; lane 4, 32P-labeled RBD probe plus rep2037 and 5 ng of wild-type concatemer; lane 5, 32P-labeled RBD probe plus rep2037 and 25 ng of wild-type concatemer. Each assay mixture contained 1 ng of radiolabeled 59-bp RBD probe, 2 μg of protein containing rep2037, and 1 μg of poly(dI-dC) and poly(dA-dT) competitor DNA. Competition analyses were carried out by using the gel retardation method described in the text, except that water was replaced by solutions of cold wild type, dimer, and cancatmer.
Purification of rep2037.
rep2037 was purified more than 5,000-fold by using ammonium sulfate fractionation, heparin, ion-exchange, and hydroxyapatite chromatography. Further fractionation of 0.8 mg of the hydroxyapatite fraction was carried out with Hitrap DEAE columns (Amersham Pharmacia) at pH 8.0, 8.5, and 9.0. Fractions containing rep2037 were combined for each separation and examined by SDS gel electrophoresis (Fig. 6). Gel retardation analysis (results not shown) indicated that there was strong RBD activity in the pH 8.5 and 9.0 fractions but very low activity in the pH 8 fraction. RBD binding activity correlated with the presence of a protein band at approximately 110 kDa.
FIG. 6.
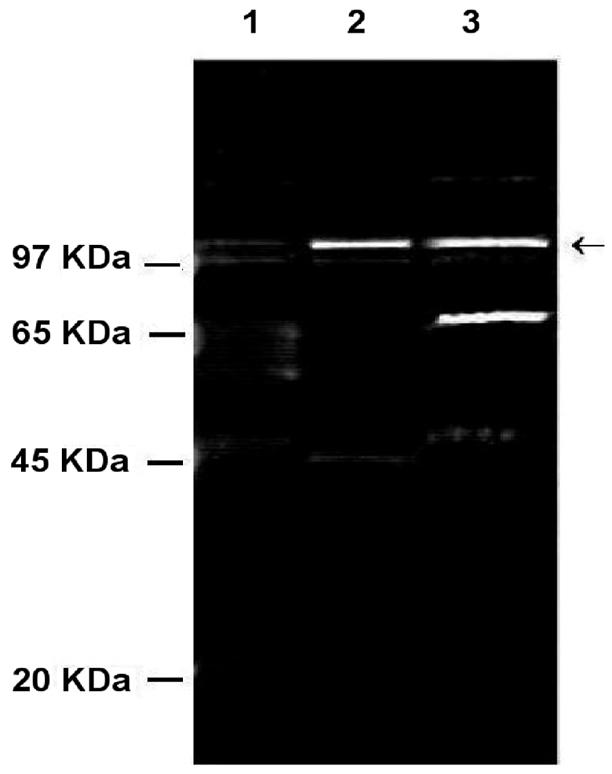
Fractionation of hydroxyapatite-purified rep2037 (1 ml, 0.8 mg of protein) by ion-exchange chromatography (HighTrap DEAE; Amersham Pharmacia) at pH 8.0, 8.5, and 9.0. Samples were separated on an SDS-12% polyacrylamide gel and stained with Sypro Ruby (Molecular Probes). Lane 1, pooled fractions, pH 8.0; lane 2, pooled fractions, pH 8.5; lane 3, pooled fractions, pH 9.0. The arrow indicates the position of the 110-kDa protein that correlated with rep2037 activity.
The hydroxyapatite fractions that contained rep2037 activity were further purified by reaction with concatemers of the DNA binding domain, attached via a biotin-streptavidin bridge to paramagnetic beads. Three rounds of extraction with beads removed all rep2037 binding activity from the bacterial extracts. Elution of the bound protein yielded a single protein band at a molecular mass of approximately 110 kDa on SDS gels (Fig. 7A, lane 3) that bound to the RBD probe (Fig. 7B, lane 5). By using the bead protocols described above, crude ammonium sulfate fractions yielded a peptide having the same molecular mass together with peptides with molecular masses of approximately 27 and 10 kDa (Fig. 7A, lane 2). Extraction of the hydroxyapatite fraction with wild-type RBD coupled to beads totally removed rep2037 (Fig. 7B, lane 4), whereas extraction with beads derivatized with the double mutant concatemers did not remove rep2037 (Fig. 7B, lane 3).
FIG. 7.

Purification of rep2037 by using concatemers of RBD. Concatemers of either wild-type RBD or the RBD double mutant were attached to magnetic particles (Dynal). (A) Purified proteins separated on an SDS-7% polyacrylamide gel. The gel was stained with Sypro Ruby and photographed with a Kodak digital system 120. Lane 1, molecular mass markers (from top to bottom, 97, 65, 45, 20, and <10 kDa); lane 2, purification of rep2037 from a crude ammonium sulfate fraction with RBD concatemer-labeled magnetic beads; lane 3, purification of rep2037 from the hydroxyapatite fraction with RBD concatemer-labeled magnetic beads. (B) Gel retardation analysis of rep2037 activity with RBD. Lane 1, 32P-labeled RBD probe alone; lane 2, 32P-labeled RBD probe plus rep2037 from hydroxyapatite fraction; lane 3, 32P-labeled RBD probe with supernatant of hydroxyapatite fraction previously extracted with the RBD double mutant concatemers coupled to magnetic beads; lane 4, 32P-labeled RBD probe with supernatant of hydroxyapatite fraction previously extracted with the wild-type RBD concatemers coupled to magnetic beads; lane 5, 32P-labeled RBD probe with eluate (panel A, lane 3) of wild-type RBD concatemers coupled to magnetic beads.
Peptide sequence analysis of 110-kDa band.
The 110-kDa band was carefully cut from the gel and subjected to quantitative time of flight MS-MS sequencing. The sequences of three peptides were determined to be GFLSAGAEEPNALVQIVR, VILADEQAEFGGSLR, and VLEEGAQIVEDPK. Blast searches were carried out, and they revealed identity to regions of sarcosine oxidase of M. loti. Tryptic digestion followed by MS revealed peptide masses that corresponded to 24% coverage of the amino acid sequence of sarcosine oxidase.
Purification of the sarcosine oxidase expressed in E. coli strain DH5α FT.
The gene encoding the α subunit of sarcosine oxidase was isolated from M. loti and expressed in E. coli. The peptide was purified by ammonium sulfate fractionation and by using RBD concatemers attached to magnetic beads in the presence of poly(dI-dC). Two rounds of extraction with beads removed all DNA binding activity from the bacterial extracts. Elution of the bound protein yielded a 110-kDa peptide together with an approximately 27-kDa peptide. Uninduced cells yielded only the 27-kDa peptide (Fig. 8A). DNA binding activity (Fig. 8B) correlated with expression of the 110-kDa peptide.
FIG. 8.

Analysis of recombinant sarcosine oxidase. Recombinant and native proteins, purified by using ammonium sulfate fractionation and RBD concatemer-coated magnetic beads, were analyzed by SDS-polyacrylamide gel electrophoresis and by a gel retardation assay. (A) SDS-polyacrylamide gel. The gel was stained with Sypro Ruby and photographed witha Kodak digital 120 system. Lane 1, rep2037 from M. loti; lane 2, protein from lysate of uninduced E. coli strain DH5α FT cells transformed with DNA encoding the sarcosine oxidase α subunit; lane 3, protein from lysate of IPTG-induced E. coli strain DH5α FT cells transformed with DNA encoding the sarcosine oxidase α subunit. (B) Gel retardation analysis. Lane 1, 32P-labeled RBD probe alone; lane 2, 32P-labeled RBD plus purified rep2037 from M. loti; lane 3, 32P-labeled RBD probe plus protein from lysate of uninduced E. coli strain DH5α FT cells transformed with DNA encoding the sarcosine oxidase α subunit (panel A, lane 2); lane 4, 32P-labeled RBD protein from lysate of IPTG-induced E. coli strain DH5α FT cells transformed with DNA encoding the sarcosine oxidase α subunit (panel A, lane 3).
DISCUSSION
Vincze et al. (27) showed that a repressor of l-asparaginase was present in nodule tissue during establishment of an effective nitrogen-fixing symbiosis. The binding domain was shown by DNase I protection experiments to be contained in a 59-bp sequence (RBD) proximal to the transcription start site.
In this study we found that the putative repressor is a protein, which we designated rep2073 and which is encoded by a bacterial gene. In support of this we show here that (i) the characteristic binding can be obtained both with bacteroid extracts and with extracts of the free-living bacteria; (ii) binding activity is observed only in nodule-pseudonodule extracts in which Mesorhizobium can be shown to be present in the nodule structure; and (iii) as determined by gel retardation experiments, the RBD yields complexes that are the same size when they are reacted with extracts of nodule bacteria and when they are reacted with extracts of free-living bacteria.
The proposal that bacteroid proteins may interact directly with plant genes that are important in the symbiosis is not new. Interaction of a rhizobial DNA binding protein with the promoter region of a plant leghemoglobin gene has been described previously (28).
In this study we isolated rep2037, the protein binding to the putative RBD of the l-asparaginase promoter. The protein was partially purified by chromatography on heparin Sepharsose, DEAE Sepharose, and hydroxyapatite, which removed most of the nonspecific DNA binding proteins. Final purification with concatemers of the RBD binding domain resulted in a single major band visible on a Sypro Ruby-stained SDS-polyacrylamide gel. Sequence analysis of three tryptic peptides derived from this major band revealed 100% homology to the α subunit of sarcosine oxidase.
One of the common predictive programs for identification of DNA binding proteins involves identification of helix-turn-helix regions in the primary sequence of the protein (16). Using the program NNPREDICT, we identified three such regions in the sarcosine oxidase α subunit. Blast searches indicated that one of these regions exhibited partial sequence homology with poly(A) binding proteins and several transcription factors. The significance of this obviously needs to be determined. However, our results clearly show that there is specific binding of the sarcosine oxidase α subunit to the promoter region CTAAAAT.
It is not unusual for proteins to move across more than one membrane. Composite signal-target sequences are not unknown (22). For example, importation of proteins from the cytoplasm into thylakoids, the mitochondrial intermembrane space, or the mitochondrial outer membrane is often affected by an N-terminal import sequence followed by an export signal sequence or a stop-transfer sequence. Welters et al. (28) have drawn a parallel between the symbiotic Rhizobium-plant system and the chloroplast-nucleus interaction. Indeed, the question is not so much whether such a scenario is possible, but rather whether a candidate protein has all the necessary targeting and transfer sequences. It is interesting that M. loti strain R7A (24) carries a symbiotic island that contains a type IV secretion system with strong similarity to the pilus of A. tumefaciens, as well as multiple copies of the sarcosine oxidase α subunit.
Bacterial sarcosine oxidases can occur in monomeric, heterodimeric, and heterotetrameric forms (3, 14). The multimeric forms contain an α subunit whose molecular mass is approximately 110 kDa corresponding to the major band sequenced in this study. In M. loti the sarcosine oxidase proteins are encoded by four genes (α, β, δ, and γ subunits) under the control of a single promoter. Sequencing of the genome of M. loti has revealed five copies of the sarcosine oxidase genes. Three of these copies are probably cytoplasmic (two of which have identical DNA sequences), and two other sarcosine oxidase genes are believed to be associated with membranes (15).
Sarcosine oxidase catalyzes the oxidative demethylation of sarcosine, which yields glycine, formaldehyde, and hydrogen peroxide. A possible mechanism by which l-asparaginase is repressed could have resulted from binding of rep2037 to the CTAAAAT promoter site, followed by methylation of the cytosine residues (Mannich type reaction) as a result of localized enzymatic production of formaldehyde.
We therefore postulate that one possible role of sarcosine oxidase is to switch off the l-asparaginase gene by binding to and perhaps methylating the CTAAAAT region of the promoter of l-asparaginase. We noted that the sarcosine oxidase α subunit from A. tumefaciens (70% amino acid identity to the M. loti sarcosine oxidase α subunit) also exhibited strong binding to CTAAAAT. The hypothesis that the sarcosine oxidase α subunit has a more general role in intergenomic interactions is feasible.
Further research will be undertaken to determine the significance of this result. The obvious experiment would be to attempt mutation of the sarcosine oxidase α subunit gene(s) of NZP2037 and test whether the correct phenotypic response is observed in vivo. However, M. loti carries five copies of the sarcosine oxidase genes, and the chances of successfully mutating all of these copies are extremely low.
Based on the published sequence data for the sarcosine oxidase α subunit (strain MAFF303099), there are two identical copies of the soluble gene. Expression of the soluble MAFF303099 sarcosine oxidase α subunit in E. coli gave a strong positive result in the gel retardation assay. Thus, we know that the soluble sarcosine oxidase α subunit shows strong RBD binding activity.
Unlike the soluble sarcosine oxidase α subunit, the sarcosine oxidase α subunit which we purified from strain NZP2037 required the presence of neutral detergents to remain in solution. Although the peptides which we sequenced were identical to the MAFF303099 soluble form, we were unable by using PCR to isolate the gene from our strain NZP2037 with primers derived from the previously published sequences of both soluble and membrane-associated sarcosine oxidase α subunits of the MAFF303099 and R7A strains. However, we were able to isolate the genes from both of these strains by PCR. Attempts to express the membrane-associated sarcosine oxidase α subunit in E. coli failed due to toxicity. Thus, we have no evidence that any of the other sarcosine oxidase α subunits could not substitute for each other, which would lead to inconclusive results. We therefore intend to generate monoclonal antibodies to the sarcosine oxidase α subunit and use an immunological approach to localize the sarcosine oxidase α subunit in developing and mature nodules and to capture DNA-protein complexes.
Acknowledgments
This work was funded by New Zealand Foundation for Research Science and Technology.
REFERENCES
- 1.Al-Samarrai, T. H., N. Zhang, I. L. Lamont, L. Martin, J. Kolbe, M. Wilsher, A. J. Morris, and J. Schmid. 2000. Simple and inexpensive but highly discriminating method for computer-assisted DNA fingerprinting of Pseudomonas aeruginosa. J. Clin. Microbiol. 38:4445-4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Broughton, W. J., S. Jabbouri, and X. Perret. 2000. Keys to symbiotic harmony. J. Bacteriol. 182:5641-5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chlumsky, L. J., A. J. Ramsey, and M. S. Joms. 1993. Preparation and properties of recombinant corynebacterial sarcosine oxidase: evidence for post-translational modification during turnover with sarcosine. Biochemistry 32:11132-11142. [DOI] [PubMed] [Google Scholar]
- 4.Chua, K. Y., C. E. Pankhurst, P. E. MacDonald, D. H. Hopcroft, B. D. W. Jarvis, and D. B. Scott. 1985. Isolation and characterization of transposon Tn5-induced symbiotic mutants of Rhizobium loti. J. Bacteriol. 162:335-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.deBruijn, F. J., and J. Schell. 1992. Regulation of plant genes specifically induced in developing and mature nitrogen-fixing nodules: cis-acting elements and trans-acting factors, p. 241-258. In D. P. S. Vermaed (ed.), Control of plant gene expression. CRC Press Inc., Boca Raton, Fla. [DOI] [PubMed]
- 6.Denarie, J., F. Delelle, and J. C. Prome. 1996. Rhizobium lipo-chitooligosaccharide nodulation factors: signalling molecules mediating recognition and morphogenesis. Annu. Rev. Biochem. 65:503-535. [DOI] [PubMed] [Google Scholar]
- 7.D'Haeze, W., P. Mergaert, J. C. Promé, and M. Holsters. 2000. Nod factor requirements for efficient stem and root nodulation of the tropical legume Sesbania rostrata. J. Biol. Chem. 275:15676-15684. [DOI] [PubMed] [Google Scholar]
- 8.Dunlap, J., E. Minami, A. A. Bhagwat, D. L. Keister, and G. Stacey. 1996. Nodule development induced by mutants of Bradyrhizobium japonicum defective in cyclic β-glucan synthesis. Mol. Plant-Microbe Interact. 9:546-555. [DOI] [PubMed] [Google Scholar]
- 9.Gressent, F., S. Drouillard, N. Mantegazza, E. Samain, R. A. Geremia, H. Canut, A. Niebel, H. Driguez, R. Ranjeva, J. Cullimore, and J. J. Bono. 1999. Ligand specificity of a high-affinity binding site for lipo-chitooligosaccharidic nod factors in Medicago cell suspension cultures. Proc. Natl. Acad. Sci. USA 96:4704-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanin, M., S. Jabbouri, W. J. Broughton, and R. Fellay. 1998. SyrM1 of Rhizobium sp. NGR234 activates transcription of symbiotic loci and controls the level of sulfated Nod factors. Mol. Plant-Microbe Interact. 11:343-350. [Google Scholar]
- 11.Hemat, F., and H. F. McEntee. 1994. A rapid and efficient PCR-based method for synthesizing high-molecular weight multimers of oligonucleotides. Biochem. Biophys. Res. Commun. 205:475-481. [DOI] [PubMed] [Google Scholar]
- 12.Ielpi, L., T. Dylan, G. S. Ditta, D. R. Helinski, and S. W. Stanfield. 1990. The ndvB locus of Rhizobium meliloti encodes a 319-kDa protein involved in the production of P-W2-glucan. J. Biol. Chem. 265:2843-2851. [PubMed] [Google Scholar]
- 13.Jensen, E. Q., K. A. Marker, J. Schell, and F. J. de Bruijn. 1988. Interaction of a nodule-specific trans-acting factor with distinct DNA elements in the soybean 1bc3 5′ upstream region. EMBO J. 7:1265-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalla, K. K., and M. S. Jorns. 1986. Bacterial sarcosine oxidase: comparison of two multisubunit enzymes containig both covalent and noncovalent flavin. Biochemistry 25:6061-6069. [DOI] [PubMed] [Google Scholar]
- 15.Kaneko, T., Y. Nakamura, S. Sato, E. Asamizu, T. Kato, S. Sasamoto, A. Watanabe, K. Idesawa, A. Ishikawa, K. Kawashima, T. Kimura, Y. Kishida, C. Kiyokawa, M. Kohara, M. Matsumoto, A. Matsuno, Y. Mochizuki, S. Nakayama, N. Nakazaki, S. Shimpo, M. Sugimoto, C. Takeuchi, M. Yamada, and S. Tabata. 2000. Complete genome structure of the nitrogen-fixing symbiotic bacterium M. loti. DNA Res. 7:331-338. [DOI] [PubMed] [Google Scholar]
- 16.Kneller, D. G., F. E. Cohen, and R. Langridge. 1999. Improvements in protein secondary structure prediction by an enhanced neural network. J. Mol. Biol. 214:171-182. [DOI] [PubMed] [Google Scholar]
- 17.Loh, J., M. G. Stacey, M. J. Sadowsky, and G. Stacey. 1999. The Bradyrhizobium japonicum nolA gene encodes three functionally distinct proteins. J. Bacteriol. 181:1544-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marincs, F., and D. W. R. White. 1993. Nopaline causes a conformational change in the nocR regulatory protein-nocR promoter complex of A. tumefaciens Ti plasmid PtiT37. Mol. Gen. Genet. 241:65-72. [DOI] [PubMed] [Google Scholar]
- 19.Pueppke, S. G., M. C. Bolanos-Vásquez, D. Werner, M. P. Bec-Ferté, J. C. Promé, and H. B. Krishnan. 1998. Release of flavonoids by the soybean cultivars McCall and Peking and their perception as signals by the nitrogen-fixing symbiont Sinorhizobium fredii. Plant Physiol. 117:599-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robertson, J. G., P. Lyttleton, S. Bullivant, and G. F. Grayston. 1978. Membranes in lupin root nodules. I. The role of Golgi bodies in the biogenesis of infection threads and peribacteroid membranes. J. Cell Sci. 30:129-149. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook, J., E. F. Fritsch., and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed., p. 8.3-11.58, Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 22.Schatz, G., and B. Dobberstein. 1996. Common principles of protein translocation across membranes. Science 271:1519-1526. [DOI] [PubMed] [Google Scholar]
- 23.Stacey, G., J. Sanjuan, S. Luka, T. Dockendorff, and R. W. Carlson. 1995. Signal exchange in the Bradyrhizobium-soybean symbiosis. Soil Biol. Biochem. 27:473-483. [Google Scholar]
- 24.Sullivan, J. T., R. W. C. Trzebiatowski, J. Gouzy, S. D. Brown, R. M. Elliot, D. J. Fleetwood, N. G. McCallum, U. Roosbach, G. S., Stuart, J. E. Weaver, R. J. Webby, F. J. De Bruijn, and C. W. Ronson. 2002. Comparative sequence analysis of the symbiosis island of Mesorhizobium loti strain of R7A. J. Bacteriol. 184:3086-3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Travers, A. 1993. DNA-protein interactions. Chapman & Hall, London, Great Britain.
- 26.van Rhijn, P., and J. Vanderleyden. 1995. The Rhizobium-plant symbiosis. Microbiol. Rev. 59:124-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vincze, E., J. M. Reeves, E. Lamping, K. J. F. Farnden, and P. H. S. Reynolds. 1994. Repression of the l-asparaginase gene during nodule development in Lupinus angustifolius. Plant Mol. Biol. 26:303-311. [DOI] [PubMed] [Google Scholar]
- 28.Welters, P., B. Metz, G., Felix, K., Palme K. Szyglowski, and F. J. deBruijn. 1993. Interaction of a rhizobial DNA-binding protein with the promoter region of a plant leghaemoglobin gene. Plant Physiol. 102:1095-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]