

Abstract
Modified tetrapyrroles such as chlorophyll, heme, siroheme, vitamin B12, coenzyme F430, and heme d1 underpin a wide range of essential biological functions in all domains of life, and it is therefore surprising that the syntheses of many of these life pigments remain poorly understood. It is known that the construction of the central molecular framework of modified tetrapyrroles is mediated via a common, core pathway. Herein a further branch of the modified tetrapyrrole biosynthesis pathway is described in denitrifying and sulfate-reducing bacteria as well as the Archaea. This process entails the hijacking of siroheme, the prosthetic group of sulfite and nitrite reductase, and its processing into heme and d1 heme. The initial step in these transformations involves the decarboxylation of siroheme to give didecarboxysiroheme. For d1 heme synthesis this intermediate has to undergo the replacement of two propionate side chains with oxygen functionalities and the introduction of a double bond into a further peripheral side chain. For heme synthesis didecarboxysiroheme is converted into Fe-coproporphyrin by oxidative loss of two acetic acid side chains. Fe-coproporphyrin is then transformed into heme by the oxidative decarboxylation of two propionate side chains. The mechanisms of these reactions are discussed and the evolutionary significance of another role for siroheme is examined.
Keywords: enzymes, metabolic pathway, S-adenosylmethionine
An understanding of biochemical pathways involves a comprehension of the chemical logic underpinning the synthetic process. Biochemical reactions are, in essence, highly controlled and regulated chemical reactions that can also provide insights into transformations for which no laboratory-based chemistry may yet have been described. Such studies are at the heart of chemical biology and are an essential prerequisite for its transformation into synthetic biology. Moreover, from an evolutionary perspective, there are many interesting questions relating to the initial appearance of complex multistep pathways, where theories on retro and patchwork models have been discussed (1, 2). These considerations are pertinent to the synthesis of the modified tetrapyrroles (3), which through their representatives chlorophyll, heme, cobalamin (the biological form of vitamin B12), siroheme, heme d1, and coenzyme F430 are involved in a broad variety of essential life processes from photosynthesis to methane production.
The tetrapyrrolic architecture that underpins the structural similarity between these various metalloprosthetic groups results from a shared, though branched, biosynthetic pathway (4, 5), where the whole family is derived from the macrocyclic primogenitor, uroporphyrinogen III (Fig. 1A). The pathways to heme and chlorophyll via protoporphyrin have been previously characterized. Similarly, the syntheses of siroheme and cobalamin have also been elucidated, progressing via the bis-methylated uroporphyrinogen III derivative precorrin-2 (Fig. 1A). However, the biosynthesis of coenzyme F430 and heme d1 have not been reported. Likewise, little is known about a proposed alternative heme biosynthetic route that involves the transformation of precorrin-2 into heme (6). This paper focuses on this unique heme biosynthetic process and reveals a hitherto unsuspected relationship with both the synthesis of siroheme and heme d1.
Fig. 1.

Pathways and genes of modified tetrapyrrole biosynthesis. (A) Outline syntheses of modified tetrapyrroles highlighting key intermediates along the branched pathway. Known reaction sequences are shown in black whereas those not yet elucidated are highlighted in magenta. (B) The known transformation of uroporphyrinogen III into siroheme and heme is shown. The numbering system for the tetrapyrrole macrocycle is highlighted on uroporphyrinogen III. (C) Precorrin-2 as the template for both d1 heme and heme synthesis is shown, with the latter derived via the alternative heme pathway. In both cases the two methyl groups at C2 and C7, highlighted in red, are derived from AdoMet. The enzymes involved in the transformation of precorrin-2 into d1 heme (Nir enzymes) and heme (Ahb enzymes) are shown. (D) The similarity between some of the Nir proteins implicated in d1 synthesis and proteins found in sulfate-reducing bacteria and Archaea thought to be associated with the Ahb pathway are indicated by black arrows.
Siroheme, the prosthetic group of sulfite and nitrite reductases, is synthesized from uroporphyrinogen III by bis-methylation to give precorrin-2, dehydrogenation to produce sirohydrochlorin and finally ferrochelation (7) (Fig. 1B). Heme d1 is only made by denitrifying bacteria with a cytochrome nitrite reductase cd1 (nirS) for which d1 acts as an essential cofactor, tailored to meet the mechanistic requirements of the reaction (8). Bacteria with nirS always appear to contain, downstream of nirS, a set of contiguous genes necessary for heme d1 biogenesis (9). In Paracoccus pantotrophus and Paracoccus denitrificans, these genes are organized in the operon nirECFD-LGHJN (nirD and nirL occur as a fused gene in Paracoccus species) and are transcribed in the presence of nitric oxide (10). This operon is thought to encode all the enzymes for heme d1 biogenesis. So far only the function of NirE has been unambiguously determined as an S-adenosylmethionine (AdoMet)-dependent uroporphyrinogen methyltransferase (11, 12), a finding that is consistent with previous labeling studies that demonstrate the synthesis of d1 heme must proceed via precorrin-2 (Fig. 1 A and C) (13).
Heme requires little introduction as a prosthetic group and has a diverse range of biological functions. Its synthesis has been elucidated in eukaryotes and most bacteria, where the classic pathway for its biosynthesis is advanced via an ordered and sequential decarboxylation of the majority of the acidic side chains of uroporphyrinogen III, followed by oxidation and ferrochelation (4) (Fig. 1B). As alluded to earlier, in sulfate-reducing bacteria and Archaea, it is proposed that heme is made via a completely different pathway, herein called the alternative heme biosynthesis route. A combination of biochemistry and bioinformatics approaches has provided some clues as to the mode of operation of this route. Thus, feeding labeled methionine to Desulfovibrio vulgaris cultures resulted in the isolation of labeled sirohydrochlorin, 12,18-didecarboxysirohydrochlorin, coproporphyrin III and protoporphyrin IX (6). Similarly, analysis of completely sequenced Archaeal and Desulfovibrio genomes suggest that these organisms possess the genes that encode enzymes for the transformation of 5-aminolevulinic acid to uroporphyrinogen but lack those required for the enzymes necessary for the conversion of uroporphyrinogen into protoheme through the classical pathway (14–17). Altogether, these observations led to the theory of an alternative heme biosynthesis pathway that diverges from the classical pathway at the uroporphyrinogen step via precorrin-2 (Fig. 1 A and C).
Our genome comparison of denitrifying bacteria with some sulfate-reducing bacteria and Archaea that make heme via precorrin-2, revealed the presence of homologues of nirD, and nirJ (see additionally ref. 17). The presence of these orthologues in sulfate-reducing bacteria and Archaea, which do not need d1 heme as they do not have cd1, was intriguing because it implied that they may have a role in the alternative heme biosynthesis route. The similarity between the d1 synthesis genes found in Paracoccus and Desulfovibrio is shown diagrammatically in Fig. 1D, where the alternative heme biosynthesis genes are given the prefix ahb. Taken together, these observations prompted us to investigate the link(s) between the alternate heme and d1 heme biosynthetic pathways.
Results
We speculated that the biosynthesis of heme and heme d1 proceeds via the oxidized derivative of precorrin-2, sirohydrochlorin, and didecarboxysirohydrochlorin, at which point the pathways for the two cofactors would diverge. In both cases it was envisaged that iron would be added at the final stage, as is observed with siroheme and the classic heme pathway. To test the hypothesis that heme d1 synthesis may proceed via sirohydrochlorin, cell-free extracts of Escherichia coli, overproducing either individual P. pantotrophus Nir proteins (D-L, G, H, J, F) or combinations of them generated using our link and lock technology (18), were incubated with this metal-free substrate. Strikingly, when lysates of E. coli, overproducing P. pantotrophus NirDL-G-H, were incubated with sirohydrochlorin an instant change in the appearance of the purple sirohydrochlorin to a blue-green color was observed. The UV-visible spectrum of sirohydrochlorin, which has a Soret band at 378 nm, also changed to a new maximum around 390 nm (Fig. 2A). The shift of 12 nm in the Soret of sirohydrochlorin and the disappearance of its d bands in the region of 590 nm suggested that sirohydrochlorin had undergone a chemical transformation and hence this unique compound was further characterized.
Fig. 2.

Siroheme decarboxylase activity of Nir proteins. (A) UV-visible absorption spectra of sirohydrochlorin (solid line) and didecarboxysiroheme (dashed line), under anaerobic conditions in 50 mM potassium phosphate buffer at pH 8. (B) HPLC traces of tetrapyrrole derivatives observed after incubation of sirohydrochlorin (i–iii), and incubation of siroheme (iv–vi), with the absorbance recorded at 390 nm. (B, i) HPLC trace of sirohydrochlorin. (B, ii) HPLC trace of reaction containing E. coli cell lysate harboring empty expression vector and sirohydrochlorin, note the new compound at 15 min. (B, iii) HPLC trace of reaction containing NirD-LGH and sirohydrochlorin. (B, iv) HPLC trace of siroheme. (B, v) HPLC trace of reaction containing NirE,D-L and siroheme. (B, vi) HPLC trace of reaction containing NirD-LGH and siroheme. Siroheme was used at a final concentration of 50 µM in each assay.
NirDL-G-H Carries an Oxygen-Independent Siroheme Decarboxylase Activity.
HPLC analysis of the NirDL-G-H catalyzed conversion of sirohydrochlorin showed the presence of two major peaks, whose retention time and m/z values differed from that of sirohydrochlorin (Fig. 2B). Significantly, the isotopic mass distribution pattern for the two compounds was consistent with the presence of iron. We surmised that sirohydrochlorin was converted into siroheme via the capture of adventitious iron and then subsequently underwent bis-decarboxylation into didecarboxysiroheme. To test this idea, siroheme was generated enzymatically (trace D, Fig. 2B) and was found to have identical properties to compound 1, observed in trace B, eluting at 15 min (Fig. 2B). Moreover, when siroheme was added to cell lysates containing NirD-L, G, and H, it was fully converted into didecarboxysiroheme (trace F, peak at 22 min). In contrast, when siroheme was incubated with E. coli cell lysate overexpressing NirD-L, monodecarboxysiroheme was observed (trace E, peak at 18 min), suggesting that the reaction occurs in a stepwise manner (Fig. 2B). In control reactions where either sirohydrochlorin or siroheme were incubated with the cell extracts of E. coli carrying empty expression vector-pET3a (Fig. 2B, traces B and D, respectively), no loss of substrate or formation of didecarboxysiroheme was detected. These results provide compelling evidence that the d1 heme biosynthesis pathway proceeds via siroheme and didecarboxysiroheme (Fig. 3). Although sequence analysis highlighted a significant sequence identity (35.3%) and similarity (47.1%) between pairs of NirD-L, NirG, and NirH, these proteins are, on the basis of mutagenesis, functionally nonredundant (9, 19).
Fig. 3.

Deduced pathway from siroheme to d1 and heme. (A) The corrected modified tetrapyrrole pathway is shown with siroheme now acting as an intermediate for heme and d1 heme synthesis. This pathway can be compared to Fig. 1C. (B) The branched pathway from siroheme to d1 and heme is shown together with the enzymes that are thought to be involved with each particular step. Didecarboxysiroheme is generated from siroheme by the sequential decarboxylation of the side chains attached to C12 and C18. For d1 synthesis, didecarboxysiroheme is modified by replacement of the propionate side chains at C3 and C8 with oxo groups in a reaction likely catalyzed by NirJ to generate a pre-d1 intermediate, which is converted into d1 by the introduction of a double bond on the propionate side chain on C17. For heme synthesis, didecarboxysiroheme is modified by the action of AhbC to remove the acetic acid side chains attached to C2 and C7 to give Fe-coproporphyrin. This intermediate is converted into heme by AhbD through the oxidative decarboxylation of propionate side chains on C3 and C8.
Subsequently, we found that extracts of E. coli overproducing NirD (where the C-terminal NirL encoding sequence was removed from the plasmid) or NirH could catalyze a single decarboxylation reaction of siroheme (Table S1). When extracts of E. coli overproducing pairs of NirD-L or NirGH were incubated anaerobically with siroheme, formation of a mixture of mono- and didecarboxysiroheme was observed. However, for total conversion of siroheme to didecarboxysiroheme all four proteins were required (Table S1). The structure of didecarboxysiroheme, arising from the loss of the carboxyl groups from the acetic acid side chains attached to C12 and C18 in siroheme, was confirmed by NMR (Fig. S1 and Table S2).
Siroheme is an Intermediate in both d1 Heme and the Alternate Heme Biosynthesis Pathway.
To investigate if siroheme and didecarboxysiroheme are also intermediates in the alternative heme biosynthesis pathway the homologous genes to nirDL, G, and H, from both Desulfovibrio desulfuricans and D. vulgaris (ahbA-B), were recombinantly produced in E. coli. When incubated with siroheme, E. coli extracts containing AhbA and AhbB resulted in the quantitative formation of decarboxylated siroheme (Table S1). Moreover, an incubation of siroheme with purified AhbA and AhbB resulted in the complete conversion of the substrate into didecarboxysiroheme. Thus it would appear that the decarboxylation of siroheme is also a committed step in the biosynthesis of heme in sulfate-reducing bacteria and Archaea that harbor these genes.
Siroheme is Converted into Heme in the Alternate Heme Biosynthesis Pathway.
Furthermore, incubation of anaerobically prepared cell lysates of D. vulgaris with siroheme formed monodecarboxysiroheme, didecarboxysiroheme, monovinyl Fe-coproporphyrin III, and heme (Fig. S2). This observation is consistent with siroheme undergoing sequential decarboxylations to didecarboxysiroheme, then transformation into Fe-coproporphyrin by the oxidative removal of the acetate side chains attached to C2 and C7 of the macrocycle and finally conversion into heme by the sequential oxidative decarboxylation of the propionic acid side chains attached to C3 and C8 (Fig. 3). All these reactions take place under anaerobic conditions, and thus the final two steps in this pathway are likely to involve some radical chemistry. Consistent with this view is the observation that the remaining two candidate genes identified in the alternative heme biosynthesis pathway, ahbC and ahbD, both encode radical AdoMet enzyme family members.
Role of AhbC and AhbD in Conversion of Didecarboxysiroheme to Heme.
To investigate the role of AhbC and AhbD, the two proteins were recombinantly overproduced in E. coli. However, aggregation problems with the D. desulfuricans AhbC resulted in a search for a more soluble orthologue of the protein. Such a candidate was found in Methanosarcina barkeri. Here the M. barkeri AhbC was found to be recombinantly overproduced in a soluble form in E. coli, and crude cell extracts containing this protein were observed to transform didecarboxysiroheme into Fe-coproporphyrin (Fig. 4). In contrast, the D. desulfuricans AhbD was more amenable to purification and was isolated as a brown solution with a broad peak between 340 to 400 nm in the UV-visible spectrum, a feature typical for 4Fe-4S cluster-containing proteins. This result is in agreement with the presence of a conserved CX3CX2C motif in the primary sequence of AhbD and its high homology to NirJ, the AdoMet-dependent radical enzyme found in P. pantotrophus (20) (Fig. 1 and Fig. S3). When a cell-free extract of E. coli overproducing D. desulfuricans AhbD was incubated under strictly anaerobic conditions in the presence of various cofactors for 4 h at room temperature with Fe-coproporphyrin as substrate, heme, a monovinyl heme derivative of Fe-coproporphyrin together with some unreacted Fe-coproporphyrin were detected by liquid chromotagraphy (LC)-MS (Fig. 5, trace B). In control reactions only background levels of heme and unreacted Fe-coproporphyrin were detected (Fig. 5, trace A and C). Finally, quantitative conversion of Fe-coproporphyrin into heme occurred in extracts of E. coli with D. vulgaris AhbD, AdoMet, and dithionite, thus confirming the role of AhbD as a unique heme synthase.
Fig. 4.
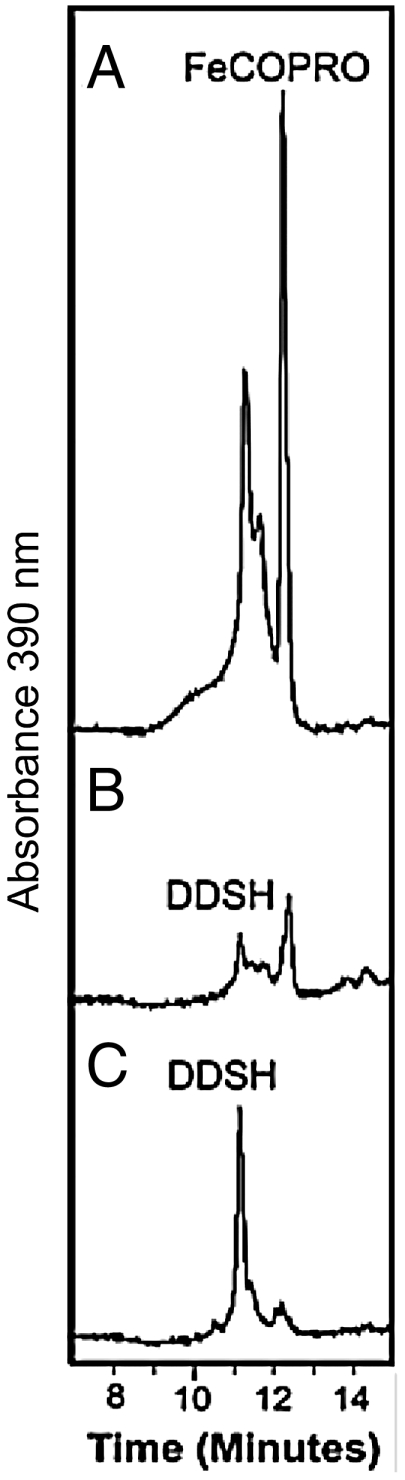
M. barkeri AhbC catalyzes conversion of didicarboxysiroheme into Fe-coproporphyrin III. HPLC traces of the tetrapyrrole derivates observed at 390 nm after anaerobic incubations of (A) E. coli cell-free extracts overexpressing M. barkeri AhbC with didicarboxysiroheme (DDSH), AdoMet and the reducing agent sodium dithionite, (B) E. coli cell-free extracts overexpressing M. barkeri AhbC with DDSH and sodium dithionite, (C) E. coli cell-free extracts with DDSH, AdoMet, and sodium dithionite. AdoMet and sodium dithionite were used at final concentrations of 500 µM and 8.5 mM, respectively. Note that Fe-coproporphyrin III (Fe-Copro) is eluted in two peaks from the HPLC column; both species have the same m/z value of 708 by MS.
Fig. 5.

AhbD catalyzed Fe-coproporphyrin oxidase activity. HPLC traces of observed porphyrin derivatives at 390 nm in reaction containing, (A) Fe-coproporphyrin, cofactor mix, and cell-free extract of E. coli harboring empty expression vector, (B) Fe-coproporphyrin, cofactor mix, and cell-free extract of E. coli overexpressing D. desulfuricans AhbD, and (C) Fe-coproporphyrin, cell-free extract containing D. desulfuricans AhbD without AdoMet in the cofactor mix. Cofactor mix consisted of 500 µM AdoMet and NADPH with 0.3% (vol/vol) Triton X-100 in each assay. Fe-coproporphyrin was used at a final concentration of 25 µM, and is eluted as two close peaks from the HPLC column but with same m/z value of 708 by MS.
Discussion
The results presented herein have established a link between d1 heme and the alternative heme biosynthesis pathway, revealing the surprising finding that both pathways proceed via siroheme and then branch off at the didecarboxysiroheme stage (Fig. 3). Incubations of heme d1 biogenesis enzymes have led to the identification of siroheme as a substrate for d1 synthesis and the detection of an intermediate, didecarboxysiroheme, within the pathway, providing unique insights into how the pathway must operate. The enzyme complex required for the decarboxylation of siroheme involves NirD-L, NirG, and NirH, proteins that are classified as members of the Lrp/AsnC family (21), and recently proposed to serve as regulators (22). In this respect, NirDL, G, and H appear to compose a unique multifunctional enzyme and transcriptional regulator. The reaction catalyzed by this enzyme complex involves the decarboxylation of the acetic acid side chains attached at C12 and C18 of the macrocycle. The mechanism for such a reaction is likely to be similar to that utilized by uroporphyrinogen decarboxylase, where an iminium ion is generated to act as an electron sink (Fig. S4). Although we have detected the presence of a monodecarboxylated intermediate, the order of decarboxylation has not been determined.
The biosynthesis of d1 heme therefore proceeds via siroheme and didecarboxysiroheme, and thereafter via a dioxo intermediate before the introduction of an acrylate function as the final step. We believe that the oxo groups are formed by the action of NirJ, a radical AdoMet enzyme (20), and that the double bond in the propionate side chain attached to C17 may be mediated by NirF, which, being periplasmic, must catalyze the last step. We have shown that the periplasmic NirF is required for d1 heme production whereas two other periplasmic proteins (NirC and NirN) coded for in the biogenesis operon are not (11, 23). Thus we conclude that NirF catalyzes the final step in d1 production unless the product of NirF activity is imported to the cytoplasm for further processing, an energetically expensive possibility. At this stage we do not know what reaction is catalyzed by NirF but the dehydrogenation to give the acrylate side chain of the isobacteriochlorin is a possibility to be addressed in future work (23). Our result that siroheme is an actual intermediate during d1 heme biosynthesis means that a ferrochelatase activity is not coded by the nir operon whose product must work in concert with a siroheme synthesis pathway. It also shows that avoidance of reactive radicals and potential toxicity by inserting iron at the end of the biosynthetic pathway cannot be as important as previously thought.
The similarity between the d1 heme biosynthetic genes nirD-L, G, and H and the putative alternative heme biosynthetic genes ahbA and B immediately suggested that didecarboxysiroheme could also be an intermediate in the latter pathway. This hypothesis was confirmed by the activity of AhbA and AhbB upon incubation with siroheme, which saw the quantitative conversion into didecarboxysiroheme. Moreover, incubation of siroheme with cell-free extracts of D. vulgaris resulted in the appearance not only of didecarboxysiroheme but also Fe-coproporphyrin and heme [and it is also interesting to note that Fe-coproporphyrin is found as a cofactor in the bacterioferritin from D. desulfuricans (24)]. Thus, heme synthesis would appear to be mediated by the conversion of siroheme into didecarboxysiroheme, followed by oxidative loss of the northern acetic acid side chains and then oxidative decarboxylation of the northern propionate side chains to vinyl groups (Fig. 3).
The loss of the acetic acid side chains at C2 and C7 for the synthesis of Fe-coproporphyrin is catalyzed by AhbC, which is a radical AdoMet family member. To promote this reaction the enzyme is likely to generate adenosyl radicals and use these to abstract hydrogen atoms in turn from the C3 or C8 positions thereby producing the respective substrate radical and deoxyadenosine. The radical can then fragment by homolytic cleavage of the C-C bond connecting the side chain acetic acid group to the macrocycle, generating a double bond and an acetate radical (Fig. S5A). This process is analogous to the proposed fragmentation of glutamate in the reaction catalyzed by coenzyme B12-dependent glutamate mutase, where an acrylate molecule and glycyl radical are formed prior to the rearrangement into 3-methylaspartate (25, 26). However, in the case of the reaction catalyzed by AhbC the enzyme presumably converts the acetate radical to acetate by providing a further electron (and proton), which could come from a second predicted Fe-S center on the protein. The mechanism as described consumes two molecules of AdoMet, each of which generates an adenosyl radical. Alternatively, an initially produced adenosyl radical could be recycled by using the methyl group of deoxyadenosine as a source of a hydrogen atom to convert the acetate radical into acetate.
The final reaction in the alternative heme biosynthesis pathway, catalyzed by AhbD, sees the transformation of Fe-coproporphyrin into heme. Overall, this process is similar to the HemN catalyzed oxidative decarboxylation reaction of coproporphyrinogen III to protoporphyrinogen IX that occurs during the classical heme biosynthesis pathway (27, 28). As with HemN (and AhbC), AhbD is also a radical AdoMet family member. By analogy with HemN, the mechanism of AhbD is likely to involve the abstraction of a hydrogen atom from the beta-position of the propionate side chains attached to C3 and C8. The resulting substrate radical can then further oxidize to a carbon cation, which could lead to the vinyl product by loss of CO2 (Fig. S5B). Although a monovinyl intermediate has been identified, it is not known if there is a preferential order for the two oxidative decarboxylations.
The identification of the intermediates of both the heme and heme d1 synthesis pathways highlights some of the ingenious chemistry that takes place. These beguiling reactions include the incorporation of oxo functionalities in place of the northern propionate side chains during d1 synthesis from didecarboxysiroheme and the oxidative loss of the northern acetate side chain from the same branch-point intermediate during heme synthesis. The sequence similarity between NirJ and AhbC not only reflects the proteins being members of the radical AdoMet family but may also reflect recognition of the same substrate. The assignment of function to these enzymes will now permit a more detailed mechanistic investigation into how these transformations are mediated.
Our research also defines a fresh role for siroheme, previously only thought to act as a prosthetic group for assimilatory sulfite and nitrite reductases, as an intermediate in modified tetrapyrrole biosynthesis (Fig. 3A). From an evolutionary perspective, it has previously been suggested that siroheme may have acted as a primordial heme where the isobacteriochlorin ring would have allowed greater conformational flexibility, prior to the selection of porphyrins in the later more oxidizing environments (29). There is a view that denitrification came early in evolution and it is possible that the d1-containing nitrite reductase appeared before both oxygen and the copper nitrite reductase (30). This paper now highlights how heme can be synthesized from siroheme, providing a biosynthetic evolutionary corollary relationship whereby the functional evolution of siroheme into heme is linked by the appearance of a biochemical pathway allowing the physical transformation of one into the other. The seemingly coincidental molecular hijacking of the cofactor for assimilatory nitrite reductase to form the cofactor for dissimilatory nitrite reductase was unanticipated; we cannot rule out the reverse appearance first of d1 heme followed by the hijacking of siroheme by the assimilatory nitrite reductase, which might have emerged once oxidized nitrogen increased as atmospheric oxygen increased. The presence of this branch of tetrapyrrole biosynthesis is consistent with a patchwork assembly model of pathway evolution, and provides a hitherto unsuspected relationship between nitrite assimilation and dissimilation.
Methods
DNA Manipulations.
DNA manipulations were performed by standard methods. Amplifications of genes from P. pantotrophus (GB17), D. desulfuricans (G20), and D. vulgaris Hildenborough were performed by PCR using KOD DNA polymerase (from Thermococcus kodakaraensis) according to supplier’s instructions (Novagen). All constructs generated by PCR were confirmed to be correct by sequencing. Plasmids containing nirED-L, nirGH, and nirD-LGH were constructed by inserting the appropriate amplified P. pantotrophus genes into pET3a using the link and lock method described in ref. 18. The nirH was cloned into pET14b, and thus the recombinant protein contained an N-terminal hexahistidine tag, whereas nirG and nirD-L were cloned into pET3a and pASKIBA13 plus vectors, respectively. Plasmids containing ahbA and ahbB from D. desulfuricans were obtained by cloning into the vectors pKK2233 and pET14b, respectively, whereas a plasmid harboring ahbAB from D. vulgaris was obtained by cloning the genes into pET3a vector using the link and lock method described above.
Recombinant Protein Production.
Cell growth and protein overexpression.
For protein overproduction, the E. coli BL21(DE3) strain was transformed with the appropriate plasmid. The resulting strain was grown with aeration at 37 °C in LB medium containing appropriate antibiotics (50 μg/mL ampicillin, 30 μg/L chloramphenicol, to an A600 0.6–0.8 and the protein expression was induced with 0.4 mM IPTG and cultures were transferred to 16 °C cells for overnight expression. Cells were collected by centrifugation 3,500 × g for 15 min at 4 °C (Beckman Coulter, JI30). The cell pellets from 1 L of LB broth were resuspended in 20 mL of 50 mM Tris·HCl, pH 8.0, containing 0.5 M NaCl. Cells were disrupted by sonication (Sonics Vibracell Ultrasonic processor). Cell debris and insoluble proteins were removed from the soluble cell lysate by centrifugation 35,000 × g for 20 min at 4 °C. A cell extract of sulfate-reducing bacteria was obtained from D. vulgaris Hildenborough frozen cell pellets.
Purification of 5-aminolevulinic acid and succinyl-CoA synthases.
Recombinant Rhodobacter sphaeroides 5-aminolevulinic acid synthase (HemA) was produced and purified as described in ref. 28. Recombinant E. coli succinyl-coenzymeA synthetase (SucCD) was purified as a hexahistidine fusion protein as follows. Cell lysate containing the overexpressed SucCD was applied to the Ni-Sepharose column equilibrated in buffer A (0 mM Tris·HCl, pH 8.0 buffer). The column was washed with 5–10 column volumes of 50 mM imidazole containing buffer A and the proteins were eluted using the 400 mM imidazole containing buffer A. Only SucC (41,400 Da) has a hexahistidine tag but the associated SucD (30,000 Da), which was not tagged, also eluted.
Purification of multienzyme cocktail for sirohydrochlorin production.
For in vitro preparation of sirohydrochlorin a multienzyme method was used as described previously (15) and outlined briefly in SI Text and Fig. S6.
Production of siroheme.
To make siroheme from sirohydrochlorin, a 10-fold excess of FeSO4 was added to a sirohydrochlorin solution and the mixture was left at room temperature for 4 h before purification on the DEAE column. The siroheme was purified on a small DEAE column to assist NMR studies with the siroheme and the didecarboxysiroheme derivative.
High performance LC-MS analysis of siroheme and siroheme derivatives.
The tetrapyrrole intermediates could be purified after separation from the protein by boiling at 80 °C for 10 min and were analyzed by reverse phase chromatography. Samples were resolved on an ACE 5AQ column (2.1 × 150 mm, 5 μ, Advanced Chromatography Technologies) attached to a Agilent 1100 series HPLC equipped with diode array detector and coupled to a micrOTOF-Q (Bruker) mass spectrometer. The column was developed with a binary gradient at a flow rate of 0.2 mL min-1. Solvent A was 0.1% TFA and solvent B was acetonitrile. The column was equilibrated with 5% B. Following sample injection the concentration of B was increased to 20% over 6 min and then to 30% at 25 min and 100% at 35 min where it was held for 5 min before returning to starting conditions. The total length of each run was 50 min. Alternatively, for incubations starting from Fe-coproporphyrin III a faster linear gradient was employed starting at 20% B and reaching 100% B in 30 min. The observed masses for these various intermediates are given in SI Text and shown in Fig. S7.
Supplementary Material
Acknowledgments.
We thank Dr. Michelle Rowe for technical support. This work was supported by Biotechnology and Biological Sciences Research Council Grants BBE0229441 and BB/E024203 (to S.J.F and M.J.W.) and Wellcome Trust Equipment Grant 091163/Z/10/Z (to M.J.H. and M.J.W.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1108228108/-/DCSupplemental.
References
- 1.Horowitz NH. On the evolution of biochemical syntheses. Proc Natl Acad Sci USA. 1945;31:153–157. doi: 10.1073/pnas.31.6.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jensen RA. Enzyme recruitment in evolution of new function. Annu Rev Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- 3.Holliday GL, et al. Evolution of enzymes and pathways for the biosynthesis of cofactors. Nat Prod Rep. 2007;24:972–987. doi: 10.1039/b703107f. [DOI] [PubMed] [Google Scholar]
- 4.Layer G, Reichelt J, Jahn D, Heinz DW. Structure and function of enzymes in heme biosynthesis. Protein Sci. 2010;19:1137–1161. doi: 10.1002/pro.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warren MJ, Raux E, Schubert HL, Escalante-Semerena JC. The biosynthesis of adenosylcobalamin vitamin B12. Nat Prod Rep. 2002;19:390–412. doi: 10.1039/b108967f. [DOI] [PubMed] [Google Scholar]
- 6.Ishida T, et al. A primitive pathway of porphyrin biosynthesis and enzymology in Desulfovibrio vulgaris. Proc Natl Acad Sci USA. 1998;95:4853–4858. doi: 10.1073/pnas.95.9.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raux E, et al. Identification and functional analysis of enzymes required for precorrin-2 dehydrogenation and metal ion insertion in the biosynthesis of sirohaem and cobalamin in Bacillus megaterium. Biochem J. 2003;370:505–516. doi: 10.1042/BJ20021443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rinaldo S, et al. Observation of fast release of NO from ferrous d1 haem allows formulation of a unified reaction mechanism for cytochrome cd1 nitrite reductases. Biochem J. 2011;435:217–225. doi: 10.1042/BJ20101615. [DOI] [PubMed] [Google Scholar]
- 9.Palmedo G, et al. Resolution of the nirD locus for heme d1 synthesis of cytochrome cd1 (respiratory nitrite reductase) from Pseudomonas stutzeri. Eur J Biochem. 1995;232:737–746. [PubMed] [Google Scholar]
- 10.Zajicek RS, Ferguson SJ. The enigma of Paracoccus pantotrophus cytochrome cd1 activation. Biochem Soc Trans. 2005;33:147–148. doi: 10.1042/BST0330147. [DOI] [PubMed] [Google Scholar]
- 11.Storbeck S, et al. The Pseudomonas aeruginosa nirE gene encodes the S-adenosyl-l-methionine-dependent uroporphyrinogen III methyltransferase required for heme d1 biosynthesis. FEBS J. 2009;276:5973–5982. doi: 10.1111/j.1742-4658.2009.07306.x. [DOI] [PubMed] [Google Scholar]
- 12.Zajicek RS, et al. d1 haem biogenesis—assessing the roles of three nir gene products. FEBS J. 2009;276:6399–6411. doi: 10.1111/j.1742-4658.2009.07354.x. [DOI] [PubMed] [Google Scholar]
- 13.Yapbondoc F, Bondoc LL, Timkovich R, Baker DC, Hebbler A. C-methylation occurs during the biosynthesis of heme-d1. J Biol Chem. 1990;265:13498–13500. [PubMed] [Google Scholar]
- 14.Cavallaro G, Decaria L, Rosato A. Genome-based analysis of heme biosynthesis and uptake in prokaryotic systems. J Proteome Res. 2008;7:4946–4954. doi: 10.1021/pr8004309. [DOI] [PubMed] [Google Scholar]
- 15.Lobo SAL, Brindley A, Warren MJ, Saraiva LM. Functional characterization of the early steps of tetrapyrrole biosynthesis and modification in Desulfovibrio vulgaris Hildenborough. Biochem J. 2009;420:317–325. doi: 10.1042/BJ20090151. [DOI] [PubMed] [Google Scholar]
- 16.Panek H, O’Brian MR. A whole genome view of prokaryotic haem biosynthesis. Microbiology-Sgm. 2002;148:2273–2282. doi: 10.1099/00221287-148-8-2273. [DOI] [PubMed] [Google Scholar]
- 17.Storbeck S, et al. A novel pathway for the biosynthesis of heme in Archaea: Genome-based bioinformatic predictions and experimental evidence. Archaea. 2010;2010:175050. doi: 10.1155/2010/175050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGoldrick HM, et al. Identification and characterization of a novel vitamin B12 (cobalamin) biosynthetic enzyme (CobZ) from Rhodobacter capsulatus, containing flavin, heme, and Fe-S cofactors. J Biol Chem. 2005;280:1086–1094. doi: 10.1074/jbc.M411884200. [DOI] [PubMed] [Google Scholar]
- 19.Kawasaki S, Arai H, Kodama T, Igarashi Y. Gene cluster for dissimilatory nitrite reductase (nir) from Pseudomonas aeruginosa: Sequencing and identification of a locus for heme d1 biosynthesis. J Bacteriol. 1997;179:235–242. doi: 10.1128/jb.179.1.235-242.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brindley AA, Zajicek R, Warren MJ, Ferguson SJ, Rigby SE. NirJ, a radical SAM family member of the d1 heme biogenesis cluster. FEBS Lett. 2010;584:2461–2466. doi: 10.1016/j.febslet.2010.04.053. [DOI] [PubMed] [Google Scholar]
- 21.Xiong J, Bauer CE, Pancholy A. Insight into the haem d1 biosynthesis pathway in heliobacteria through bioinformatics analysis. Microbiology-Sgm. 2007;153:3548–3562. doi: 10.1099/mic.0.2007/007930-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oglesby-Sherrouse AG, Vasil ML. Characterization of a heme-regulated non-coding RNA encoded by the prrF Locus of Pseudomonas aeruginosa. PLoS One. 2010;5:e9930. doi: 10.1371/journal.pone.0009930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bali S, Warren MJ, Ferguson SJ. NirF is a periplasmic protein that binds d1 heme as part of its essential role in d1 heme biogenesis. FEBS J. 2010;277:4944–4955. doi: 10.1111/j.1742-4658.2010.07899.x. [DOI] [PubMed] [Google Scholar]
- 24.Romao CV, et al. Iron-coproporphyrin III is a natural cofactor in bacterioferritin from the anaerobic bacterium Desulfovibrio desulfuricans. FEBS Lett. 2000;480:213–216. doi: 10.1016/s0014-5793(00)01939-6. [DOI] [PubMed] [Google Scholar]
- 25.Buckel W, Kratky C, Golding BT. Stabilization of methylene radicals by cob(II)alamin in coenzyme B12 dependent mutases. Chemistry Eur J. 2005;12:352–362. doi: 10.1002/chem.200501074. [DOI] [PubMed] [Google Scholar]
- 26.Marsh ENG, Patterson DP, Li L. Adenosyl radical: Reagent and catalyst in enzyme reactions. ChemBioChem. 2010;11:604–621. doi: 10.1002/cbic.200900777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Layer G, Moser J, Heinz DW, Jahn D, Schubert WD. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of Radical SAM enzymes. EMBO J. 2003;22:6214–6224. doi: 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Layer G, et al. The substrate radical of Escherichia coli oxygen-independent coproporphyrinogen III oxidase HemN. J Biol Chem. 2006;281:15727–15734. doi: 10.1074/jbc.M512628200. [DOI] [PubMed] [Google Scholar]
- 29.Crane BR, Getzoff ED. The relationship between structure and function for the sulfite reductases. Curr Opin Struct Biol. 1996;6:744–756. doi: 10.1016/s0959-440x(96)80003-0. [DOI] [PubMed] [Google Scholar]
- 30.Ducluzeau AL, et al. Was nitric oxide the first deep electron sink? Trends Biochem Sci. 2009;34:9–15. doi: 10.1016/j.tibs.2008.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.