

Abstract
In humans, mutations in the Dystrophin Glycoprotein Complex (DGC) cause muscular dystrophies (MDs) that are associated with muscle loss, seizures and brain abnormalities leading to early death. Using Drosophila as a model to study MD we have found that loss of Dystrophin (Dys) during development leads to heat-sensitive abnormal muscle contractions that are repressed by mutations in Dys's binding partner, Dystroglycan (Dg). Hyperthermic seizures are independent from dystrophic muscle degeneration and rely on neurotransmission, which suggests involvement of the DGC in muscle-neuron communication. Additionally, reduction of the Ca2+ regulator, Calmodulin or Ca2+ channel blockage rescues the seizing phenotype, pointing to Ca2+ mis-regulation in dystrophic muscles. Also, Dys and Dg mutants have antagonistically abnormal cellular levels of ROS, suggesting that the DGC has a function in regulation of muscle cell homeostasis. These data show that muscles deficient for Dys are predisposed to hypercontraction that may result from abnormal neuromuscular junction signaling.
Muscular dystrophy (MD) patients have progressive muscle weakening and loss and so far no cure exists to treat these fatal disorders. Many forms of MDs are associated with abnormalities in the evolutionary conserved Dystrophin Glycoprotein Complex (DGC) that links the extracellular matrix (ECM) to the cytoskeleton1. Dystrophin (Dys) is the main component of the DGC required for muscle stability and is found at extrasynaptic and synaptic regions of muscle fibers where it is required for neuromuscular junction (NMJ) development2. Dys structurally links cytoskeletal actin to the ECM via the glycoprotein Dystroglycan (Dg), where Dys also binds several other proteins (two syntrophins, two dystrobrevins, and four sarcoglycans)3,4. Improper association between muscle and the surrounding basal lamina is found in a variety of MDs and cardiomyopathies5,6,7.
The DGC has been also shown to play a role in cellular signaling processes that require Dg extracellular binding to Laminin. For example, G-protein binding to Syntrophin activates PI3K/Akt signaling in a manner that is dependent upon Laminin-Dg interaction8, as well Syntrophin phosphorylation has been shown to be dependent upon this interaction resulting in Syntrophin-Grb2-Sos1-Rac1-Pak1-JNK signaling9. Additionally, the DGC is involved in TRPC channel activation at the muscle sarcolemma where Syntrophin regulates cation influx via anchoring the store-operated channels that are critical for normal calcium homeostasis in muscle cells10,11.
Recently Drosophila has been shown to be a suitable genetic model to study the DGC12. Initial characterization of the DGC in Drosophila has portrayed that components studied so far are evolutionary conserved13 and DGC deficiencies lead to age dependent muscle degeneration, reduced mobility, dilated cardiomyopathy and a shorter life span12,14,15. Using this Drosophila MD model we previously performed in vivo genetic screens in ageing dystrophic muscles to find novel DGC interactors16. These interactors were divided into three main functional groups: mechanosignaling, cellular stress response, and neuron-muscle communication. While the role of the DGC in providing muscle integrity has been extensively studied, its function in muscle-neuron communication has not been fully defined.
Since our previous data showed that the DGC mutants and the interactors found in the screen show mobility defects that are intensified by higher temperatures, we decided to analyze the reason for this behavioral defect. Temperature induced walking disability can originate from muscle or neuron malfunction. Here we measured the muscle activity on live dystrophic animals and showed that Drosophila Dys mutants have muscle defects resulting in seizures. These seizures robustly occur at elevated temperatures that seem to enhance an underlying physiological defect. On further inspection we have determined that this phenotype is only displayed when Dys is downregulated during development of muscles, implying a developmental requirement for Dys. Surprisingly, the seizure phenotype is not shared by, and repressed by mutations in, the binding partner of Dys, Dg, possibly due to malfunctioning of the NMJ. In addition, we show that a deficiency in the Ca2+ regulator Calmodulin (Cam) and calcium channel blockers rescue the Dys phenotype, suggesting that Ca2+ plays a role in dystrophic seizures. Under stress Dys and Dg mutants have altered levels of ROS suggesting that these proteins play a role in maintaining cellular homeostasis. These results reveal novel dynamics between two components of the DGC and their interaction.
Results
In a previous report we showed that the DGC is involved in the stress response pathway and that energetic stress and changes in the ambient temperature accelerate dystrophic muscle degeneration16. Therefore we decided to test if dystrophic animals show abnormal cellular homeostasis. Since elevated temperatures enhance cellular responses17,18, we measured the amount of lipid peroxidation products which are the result of reactive oxygen species (ROS) of animals kept for three days at 29°C (higher than ambient, but not heat shock temperature). Dys mutants had a strikingly larger amount of lipid peroxidation products than did wt animals (Fig. 1a). Inversely, Dg mutants have significantly decreased ROS levels relative to control. Though heterozygous single mutants (Dys/+ and Dg/+) do not have changed ROS levels, the levels are significantly decreased in double heterozygous mutants (Dg/+; Dys/+) indicating that these two proteins interact in maintaining proper ROS cellular levels (Fig. 1a).
Figure 1. Dystrophin mutants have elevated ROS and hyperthermic seizures.

(a) Assessment of cellular lipid peroxidation products normalized to protein content in animals housed at 29°C for three days. The axis represents the raw molar absorption after blank subtraction and normalization to protein content. Statistical analysis was done using a one-tailed Student's t-test and the error bars represent the standard deviation. All comparisons are made against OregonR. Exact experimental values and P-values are given in Supplementary Table 1. (b) Electrical output from IFM vs. temperature during seizures of DysDf/DfKX23 and dsDys/24B-Gal4 mutants compared to Control. Stars indicate the average start temperature where the error bar indicates the standard deviation. Arrows indicate where plots in panel c are taken from. (c) Electrical output relative to time where the region examined correlates with the numbered arrows in panel b. Output is displayed over a 0.5 s time period emphasizing the increase in frequency and decrease in amplitude as the seizure progresses. (d) All tested Dys classical and RNAi alleles (levels of Dys mRNA downregulation are shown in Supplementary Table 2) had hyperthermic seizures with similar seizure indices tested in a two-tailed Kruskal-Wallis test where P = 0.316 excluding data from dsDys/D42-Gal4 animals (Supplementary Figure 1, Table 1). Seizure indices of dystrophic mutants are significantly higher when compared to the appropriate control (separated by vertical lines). One to one comparative statistics were done using a one-tailed Mann-Whitney U-test. Error bars indicate the 25th and 75th percentiles of the data spread considering that if an animal does not seize then the index is 0. The Si range of some controls have zero values in the 25th and 75th percentile range, thus it appears that there is no error bar.*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Cellular homeostasis depends on Ca2+ levels and a protein regulating proper calcium levels, Calmodulin (Cam) was found to interact with Dys leading to muscle degeneration16. We analyzed ROS levels of animals heterozygous for both Cam and Dys (Cam/+; Dys/+) and observed a significant decrease in ROS levels indicating that these two proteins genetically interact (Fig. 1a), suggesting that Dys-dependent cellular stress depends on Ca2+ levels.
Next, we applied higher than ambient temperatures to Dys classical and RNAi mutants to examine in vivo dystrophic muscle function in increased temperature conditions. This revealed a complex physiological defect that can be measured by electrophysiological recordings from indirect flight muscles (IFMs). All Dys mutant animals at temperatures ranging anywhere from 30–46°C started to have abnormal muscle contractions (seizures) (Fig. 1b–d, Supplementary Fig. 1). These seizures slowly decreased in amplitude while increasing in frequency. When a seizure occurred a seizure index (Si) was calculated taking into consideration the amplitude and duration of the seizure as well as the temperature seizing started at. If an animal did not have a seizure then the Si was assigned a value of zero (Table 1). Using different Dys allelic combinations we observed that 60–100% of dystrophic animals had seizures with the median Si ranging from 1.6 to 2.4, while the median Si for control animals was 0 (Table 1).
Table 1. Seizure Indices (Si).
| Genotype | n | % seizedŦ | Avg. Ts±sd (°C)* | Avg. Amax±sd (mV)* | Avg. Area±sd (pixel × 10-3)* | Median Si** (25th/75th percentiles) | +P-value |
|---|---|---|---|---|---|---|---|
| OregonR | 7 | 29 | 40.7±6.1 | 12.2±13.4 | 2.9 ± 2.4 | 0.0 (0.0/0.9) | 0.944 (χ2 = 0.115) |
| w1118;CyO/+; TM6/+ | 4 | 25 | 46.4 | 2.6 | 1.3 | 0.0 (0.0/0.4) | |
| w1118;CyO/Pin | 4 | 25 | 38.3 | 14.3 | 6.9 | 0.0 (0.0/0.6) | |
| wt*** | 15 | 24 | 42.1±4.1 | 14.0±13.8 | 4.5±2.9 | 0.0 (0.0/0.8) | - |
| DysDf/DfKX23 | 5 | 80 | 37.0±3.1 | 6.0±5.2 | 2.4 ± 2.0 | 1.8 (1.7/2.2) | 0.028* |
| DfKX23/+ | 4 | 100 | 43.0±3.3 | 11±6.8 | 4.9 ± 5.4 | 1.8 (1.5/2.2) | 0.011* |
| DysDf/Dys8-2 | 10 | 60 | 40.8±6.4 | 13±6.4 | 4.2 ± 2.9 | 1.6 (0.0/2.2) | 0.045* |
| Dys8-2/+ | 5 | 80 | 40.2±8.2 | 3.3±1.7 | 1.4 ± 0.8 | 1.7 (1.3/1.8) | 0.045* |
| DysDf | 9 | 89 | 35.2±4.7 | 7.7±6.4 | 4.9 ± 3.7 | 2.4 (2.2/2.4) | 2.6 × 10−4*** |
| DysDf/+ | 8 | 75 | 39.6±3.5 | 8.7±5.3 | 2.4 ± 2.5 | 1.7 (1.1/1.8) | 0.050* |
| ◊Dg−/− | 9 | 11 | 45.3 | 5.6 | 1.3 | 0.0 (0.0/0.0) | 0.19 |
| DgO86/+;DysDf/+ | 10 | 20 | 38.0±7.6 | 5.2±2.9 | 2.8 ± 0.3 | 0.0 (0.0/0.0) | 0.50 |
| ♦parats1/+; DysDf/+ | 5 | 0 | 0 | 0 | - | - | 0.28 |
| corak08713 | 8 | 0 | - | - | - | - | 0.15 |
| corak08713/+; DysDf/+ | 8 | 12.5 | 36.0°C | 5.2 | 5.6 | 0.0 (0.0/0.0) | 0.41 |
| ♦Camn339/+; DysDf/+ | 7 | 0 | - | - | - | - | 0.19 |
| tub-Gal4/+ | 10 | 20 | 42.5±5.5 | 2.6±0.1 | 1.6±0.4 | 0.0 (0.0/0.0) | - |
| tub-Gal4::dsDys/+ | 10 | 70 | 40.1±5.8 | 4.5±2.1 | 2.3 ±1.8 | 1.7 (0.2/1.9) | 0.020* |
| tub-Gal4::dsDg/+ | 8 | 50 | 43.6±2.6 | 4.1±2.1 | 1.2 ±0.7 | 0.7 (0.0/1.6) | 0.19 |
| tub-Gal4::dsDys/dsDg £ | 6 | 50 | 44.2±5.3 | 12±13.4 | 0.8 ±0.2 | 0.7 (0.0/1.4) | 0.29 |
| tub-Gal80ts/+, tub-Gal4::dsDys/+ – Put to 29°C after eclosion | 14 | 29 | 45.7±2.4 | 10±5.8 | 2.1 ± 1.2 | 0.0 (0.0/0.9) | 0.47 |
| tub-Gal80ts/+, tub-Gal4::dsDys/+ – Put to 29°C as L3 larvae | 13 | 77 | 40.1±4.9 | 7.9±5.2 | 4.2 ± 4.2 | 2.0 (1.7/2.2) | 3.7 × 10−4*** |
| tub-Gal80ts/+, tub-Gal4::dsDg/+ – Put to 29°C after eclosion | 5 | 0 | - | - | - | - | 0.40 |
| tub-Gal80ts/+, tub-Gal4::dsDys/dsDg – Put to 29°C after eclosion | 6 | 17 | 48.2 | 5.1 | 0.4 | 0.0 (0.0/0.0) | 0.34 |
| parats1/+; tub-Gal4::dsDys/+ | 5 | 0 | 0 | 0 | - | - | 0.40 |
| CyO(Pin)/+;24B-Gal4/+ | 4 | 0 | - | - | - | - | - |
| dsDys/24B-Gal4 | 5 | 100 | 41.5±6.3 | 5.8±4.1 | 3.1 ± 2.7 | 1.8 (1.7/1.8) | 0.0079** |
| dsDg/24B-Gal4 | 5 | 0 | - | - | - | - | 1.0 |
| D42-Gal4/+ | 4 | 0 | - | - | - | - | - |
| dsDys/D42-Gal4 | 5 | 40 | 37.8±8.2 | 5.7±5.2 | 2.3 ± 0.8 | 0.0 (0.0/1.8) | 0.28 |
| dsDg/D42-Gal4 | 5 | 0 | - | - | - | - | 1.0 |
n = number of animals measured for the specified genotype
Ŧpercent of animals measured that had a seizure
*Calculated using data from animals that seized only
**Index calculated by integrating the area of the graph of voltage vs. temperature during a seizure, taking the natural logarithm of this number, dividing by the temperature that the seizure started, and then multiplying times ten. A Si of zero was assigned to animals that did not seize
***There is no difference between animals of genotypes OregonR, w1118;CyO/+;TM6/+ or w1118;CyO/Pin as assessed using a Kruskal-Wallis test. These animals were pooled and termed WT.
+P-values are relative to the control animals at the beginning of each shaded section, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001
◊Five animals were of genotype DgO86/O55, two were of genotype DgO86 and two were of genotype DgO86/+ where one animal of genotype DgO86/O55 had a seizure
£Four animals were of genotype tub-Gal4::dsDg/dsDys and two animals were of genotype tub-Gal4::dsDys/dsDg
♦Animals of genotype parats1/Y and Camn339/+ did not have seizures (n = 2)
To understand the origin of this phenotype, we downregulated Dys in different tissues using specific Gal4 drivers and Dys RNAi. Animals with ubiquitous and mesodermal Dys downregulation showed a strikingly similar phenotype to that seen with the allelic mutations (Fig. 1b,d). Contrarily, the indices were not significantly greater compared to control when Dys RNAi was expressed using a motoneuron driver (Fig. 1d). From these data we conclude that seizures are a result of the lack of Dys in muscle tissue, but not in motoneurons.
The occurrence of temperature-sensitive seizures could be due to a problem arising upon Dys deficiency during muscle establishment or maintenance. To allow for developmental stage-specific downregulation of Dys, RNAi transgenic constructs were combined with the Gal80 temperature-sensitive system that suppresses expression of constructs until the animals are transferred to 29°C. When Dys RNAi animals were shifted to 29°C after muscle development and synapse maturation is completed, a significant decrease in seizure indices took place compared to a Dys deficit in development (Fig. 2a,b). This evidence shows that Dys is required during development to prevent heat induced seizures.
Figure 2. Dystrophic seizures are dependent upon neuronal input that require Dys during development.

(a) Electrical output of animals shifted to the restrictive temperature as late L3 larvae (left) and as adults (right). Dys protein reduction levels using the Gal80 temperature sensitive system are given in Supplementary Table 3 and Supplementary Figure 2. (b) Seizure indices for RNAi mutants after being shifted to the restrictive temperature at different life stages. A Mann-Whitney U-test was used to compare seizure indices upon Dys downregulation in adulthood to Dys downregulation in development (P = 0.007). Dys downregulation in adulthood resulted in a Si that is not significantly different from control (tub-Gal4/+, P = 0.47, Table 1). In addition, animals of the genotype tub-Gal80ts/+;tub-Gal4/+ shifted as adults did not have seizures (n = 2). (c) Bar graphs showing relative fold increase in the frequency of muscle degeneration of Dys RNAi mutants targeting Dys throughout the lifetime or shifted to the restrictive temperature as adult. The percent of muscle degeneration has been normalized to that in control animals under the same conditions (Supplementary Table 4). Statistics for muscle degeneration were done using a one-tailed chi-square test (χ2 = 2.62, P = 0.11). (d) Histological sections of thoracic muscles. Arrows indicate areas of degeneration. (e) The parats1 allele abolishes seizures in DysDf/+ and tub-Gal4::dsDys/+ animals (red plot). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Animals expressing Dys RNAi during development have properly developed muscles, which degenerate only as the animal ages12. To determine if previously shown age-dependent muscle degeneration is a result of the seizure activity described here, Dys RNAi animals were allowed to develop at a permissive temperature (no RNAi expression) and aged at the restrictive temperature after adult muscle and NMJ formation. Although the animals no longer had seizures, they exhibited muscle degeneration similar to animals with temporally unrestricted Dys downregulation (Fig. 2c,d, Supplementary Table 4). Importantly, seizing is not the result of aging since wt older animals still do not seize and older Dys mutants have a reduced occurrence of seizures (∼50%). Perhaps, degenerating muscle is unable to respond in the same manner as young intact muscle. This result implies that the absence of Dys influences muscle degeneration and seizures independently.
The seizures in Dys mutants could be due to either muscle autonomous mis-regulation or inability to properly respond to synaptic input. To test if a neuronal contribution is required we used a previously described temperature-sensitive mutation in the paralytic gene that abolishes neuronal action potentials at temperatures above 29°C in adults (Fig. 2e)19,20. Opposite to Dys heterozygous and RNAi mutants, the introduction of the parats1 allele into a dystrophic background prevented seizures suggesting that neuronal transmission is required to trigger contractions in dystrophic muscles.
We initially believed that mutations in Dys's binding partner, Dg would phenocopy the dystrophic seizure phenotype. Surprisingly, Dg loss-of-function mutants exhibit no seizing activity (Fig. 3a,b, Table 1). Ubiquitous Dg RNAi expression produced some small low occurrence seizures, but specific expression in the mesoderm and motoneurons caused no seizures (Supplementary Fig. 3). Even more, Dg/+;Dys/+ transheterozygous animals did not have heat-induced contractions (Fig. 3a,b, Table 1). This reveals that seizure susceptibility at high temperatures can be reduced by loss of one copy of Dg.
Figure 3. Loss of Dystroglycan, Coracle or Calmoduline represses the seizure phenotype of Dystrophin mutants.

(a) Seizure indices show that Dg and cora mutants do not have seizures (Table 1) and mutations in Dg, Cam or cora suppress the seizure phenotype in Dys transheterozygous animals (P = 0.032, 0.006 and 0.017 respectively when compared to Dys/+ animals). Animals carrying RNAi constructs against both Dys and Dg also showed a 50% decrease in seizure activity when downregulated during development and 17% when downregulated after development indicating a repression of the phenotype by downregulation of Dg (P = 0.072 and 0.017 respectively compared to Dys RNAi mutant, Table 1). Statistics were done using a one-tailed Mann-Whitney U-test. (b) Electrical output from IFM vs. temperature. (c) In control animals Dg (magenta) is localized to the NMJ. Dys mutants have a decreased amount of Dg at the NMJ, but some Dg localization is still observed. Dg mutants have no Dg localized to the NMJ. Transheterozygous animals (Dg/+;Dys/+, cora/+;Dys/+) show a significant decrease in Dg staining at the NMJ as well. The overall structure and presence of NMJs does not appear to be altered in the mutants as can be seen via Dlg staining (green). (d) In adult abdominal NMJs DGluRIIA is localized similarly in Dys mutants as in the wild type (OregonR) animals; however, Dg loss-of-function mutants have a decreased amount of the receptor. Statistics on relative intensities were done using a Student's t-test compared to OregonR where the error bars represent the average deviation from the mean. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
It has been shown that at Drosophila larval NMJs Dys is not localized in the absence of Dg, while Dg localization is only partially dependent upon Dys20. Accordingly, we found that at adult dystrophic NMJs Dg is reduced (Fig. 3c,d), while in transheterozygous mutants (Dg/+;Dys/+) Dg is missing from the NMJ similar to Dg loss-of-function mutants (Fig. 3c,d, Supplementary Table 5). Based on this evidence, we propose that the presence of Dg at the NMJ is necessary for Dys mutants to have temperature-induced seizures. This conclusion is further supported by the fact that animals expressing both Dg and Dys RNAi (causing Dys and Dg levels to be reduced but not abolished, Supplementary Table 1,6) have lower seizure occurrence than Dys RNAi mutants (Table 1).
Since Dys and Dg are the key components of the DGC, integrity of which is crucial for proper muscle function and maintenance, we wanted to understand why Dg mutants do not have seizures while Dys mutants do. First, we considered that Dg mutants do not have temperature-sensitive seizures due to compensatory mechanisms. Integrins are likely candidates, as alpha (7B) integrin has been shown to compensate for Dg absence in mediating cell-extracellular matrix attachment21. If integrins are making up for loss of Dg, then transheterozygous animals might have enhanced seizures. However, the reduction of both integrin and Dg (mys1/+;Dg/+) leads to a similar phenotype as mys1/+ heterozygous animals, indicating that there is no genetic interaction between these two proteins in seizure occurrence (Supplementary Fig. 4).
Drosophila NMJs are glutamatergic, and previously it has been shown that Dg is required for proper localization of postsynaptic glutamate receptors at larval NMJs20,22. Since the amount of glutamate receptors influences muscle-neuron communications, we next analyzed the localization of glutamate receptor type II A (DGlulRIIA) at the adult NMJ. We found that Dg mutants have an approximately 40% decrease in the levels of the glutamate receptor at the NMJ (Fig. 3e,f). Conversely, this receptor in Dys mutants is localized like in wt animals (Fig. 3e,f) which is in accordance with previous reports on larvae20,23. This indicates that the seizing phenotype observed in Dys mutants is not due to mislocalization of DGlulRIIA.
Next, in order to analyze if the rescue of the seizing phenotype observed in Dys/+;Dg/+ transheterozygous mutants is due to interaction of Dys and Dg at the NMJ we used mutants of Coracle (Cora), a protein that is localized to the NMJ. Cora is the Drosophila homologue of mammalian brain 4.1 proteins20 and it has been shown that Cora and Dg protein concentration at the NMJ are co-dependent20. Since in cora/+;Dys/+ mutants Dg is not properly localized to the adult NMJ (Fig. 3c,d), it would be expected that cora mutants should behave the same as Dg mutants and rescue dystrophic hyperthermic seizures, if they depend on DGC function at NMJs. Hypomorphic cora mutant animals exhibit a mild heat-induced muscle contraction phenotype of their own (Fig. 4a,b), but this activity is not of the nature described here, therefore no seizure indices were calculated for these animals. Data derived from Dys and cora transheterozygous animals (cora/+;Dys/+) mimicked that of the cora mutants and, similar to Dg reduction, rescued dystrophic seizures (Fig. 3a,b). Based on these data and previous reports of increased neurotransmitter release at dystrophic larval NMJs23,24, we suggest that hyperthermic seizures are derived from the improper function of the DGC at the NMJ.
Figure 4. Cora mutants have a mild seizure phenotype that does not look like and overrides the dystrophic phenotype.

Mutation in the cora gene results in a distinctive electrophysiological phenotype from that seen with Dys heterozygous mutations. (a) corak08713 mutants have electrical output that is not of a consistent frequency. Animals of genotype corak08713/+; DysDf/+ have a phenotype similar to the cora mutant. The output is recorded over 1 second. (b) Examples of the two types of electrical activity measured in corak08713, DysDf/+ and corak08713/+; DysDf/+ animals. The output is recorded over 10 seconds.
Release of neurotransmitter at the NMJ results in depolarization of the muscle membrane causing release of Ca2+ from the SR required for muscle contraction. Thus, we examined how the Ca2+ binding protein, Calmodulin, which binds to DGC components dystrophin and syntrophins in mammals25, affects the dystrophic seizure phenotype. In Drosophila there is only one gene encoding Cam that is homologous to the human CAM2 gene with 97% identity26. Introduction of a Cam null mutation into a Dys mutant background (Cam/+;Dys/+) rescued hyperthermic seizures (Fig. 3a,b). Since Cam regulates levels of Ca2+, our data implies that Ca2+ levels are important for the dystrophic seizure activity.
Previous work has demonstrated that there is an increase in Ca2+ release from the sarcoplasmic reticulum (SR) in DMD human myotubes27. To assess by which mechanism Ca2+ plays a role in the observed seizures, Dys mutants were fed overnight various Ca2+ channel blockers [Nifedipine (dihydropyridine channel), 2-APB (inositol 1,4,5-triphosphate receptors, IP3R) and Ryanodine (Ryanodine receptors, RyR)] and assayed for temperature induced seizures. To prevent total inhibition of muscle contraction a low drug concentration was used to block individual channels at one time. Animals fed Nifedipine did not show less seizure activity than controls, but both 2-APB and Ryanodine treated flies showed a decrease in seizure activity, with Ryanodine having the most dramatic effect (Fig. 5a,b, Supplementary Table 7). This shows that Ca2+ released via IP3R and RyR activated channels plays a role in hyperthermic seizures.
Figure 5. Elevated Ca2+ from the SR leads to hyperthermic seizures.
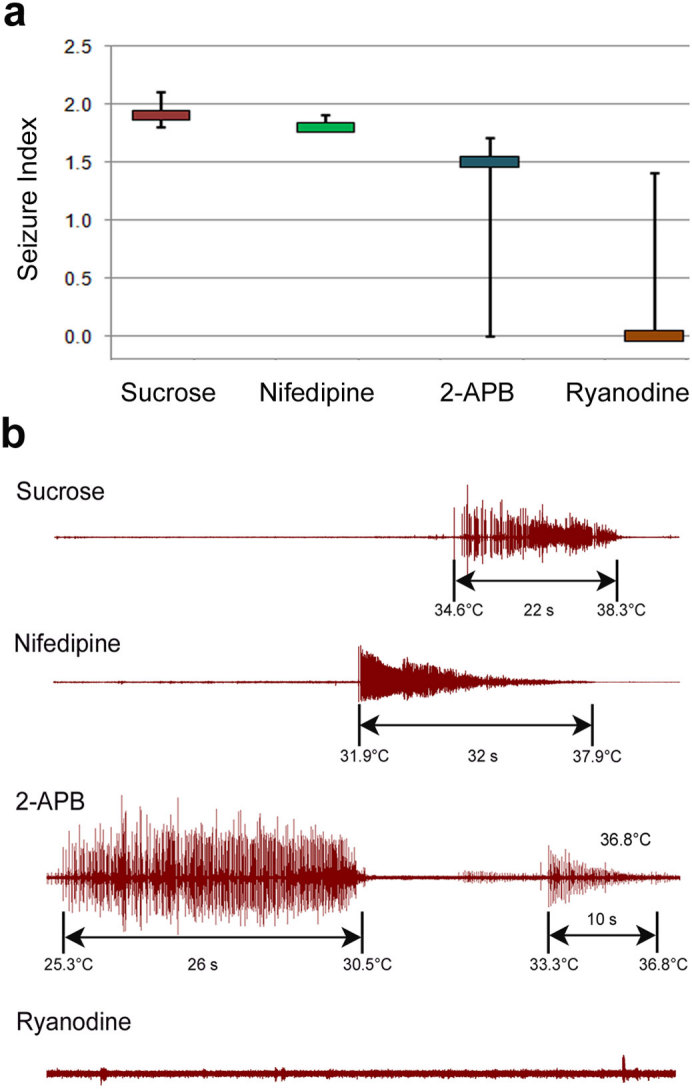
(a) Seizure indices of DysDf mutants after being fed various Ca2+ channel blockers. The control animals were only fed 5% sucrose and the other drugs were given in a 5% sucrose solution. Statistics were calculated using a one-tailed Mann-Whitney U-test where the following P-values were obtained by comparing to sucrose fed animals: Nifedipine: P = 0.421, 2-APB: P = 0.107 and Ryanodine: P = 0.078. (b) Electrical output from dystrophic heated muscles after being fed overnight with sucrose (control), L-type calcium channel blocker (Nifedipine), Ryanodine SR fast calcium channel blocker or IP3R SR slow calcium channel blocker (2-APB). 5–6 animals were measured per condition. Seizure onset and subsiding temperatures are noted as well as seizure duration. Animals fed Nifedipine did not show a decrease in seizure activity. Animals fed 2-APB had additional seizure activity that was occurring spontaneously before heating began that was different in its electrical pattern that would be consistent with the observed phenotype of typical Dys mutants. This is shown here where output occurs for almost 30 s. This is later followed by a seizure at a reasonable temperature that looks like what would be expected for a dystrophic mutant. This extra output was only seen with animals fed this channel blocker, and in general the occurrence of seizures was less than controls (Supplementary Table 5). Animals fed Ryanodine still had seizures 33% of the time, but in general there was a decrease in the seizure indices associated with these animals, therefore we considered this reagent to have a therapeutic effect. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Discussion
Muscle seizures accompany multiple human neurological and muscular disorders. Seizures can occur when a group of neurons becomes hyperexcited and discharge action potentials irregularly without suppression, which causes muscle fibers to contract inappropriately. The uncontrolled action potential is dependent upon K+ and Na+ channel permeability or Ca2+ mis-regulation28. Ryanodine receptors that are regulators of fast Ca2+ release from the sarcoplasmic reticulum have been found to play a role in the disease pathology of seizures29. Additionally, inositol 1,4,5-triphosphate receptors (IP3Rs) which regulate slow Ca2+ release from the SR are necessary in the central nervous system, causing epilepsy when mutated30. Seizures can be alleviated by manipulation of ion channels or by blocking neurotransmission.
It has been shown that Laminin binds directly to voltage gated Ca2+ channels (CaV) at the presynapse in mice, specifically P/Q- and N-type channels, and this binding induces vesicle clustering31. Laminin also binds the transmembrane protein Dg providing a direct link between Dys and the presynaptic motoneuron in mammals. Importantly, not only does the motoneuron send signals to the muscle, but also retrograde signaling exists, where a signal travels from the muscle back to the presynaptic neuron. The current pathway by which synaptic retrograde signaling communicates is not known, however Dys has been implicated previously to play a role in the process23,24. If we assume that hyperthermic seizures reported here are related to the role of Dys in retrograde signaling, then the CaV-Laminin-Dg signaling cascade could explain why the P/Q- and N-type, but not the L-type Ca2+ channel blocker affected Dys seizures. Similarly, the reduction of Dg or Cora reduced the seizure occurrence possibly by preventing propagation of signaling via loss of communication with Laminin. Another pathway that plays a role in retrograde signaling at the NMJ32 is TGF-β and interestingly, mutants of the TGF-β pathway have a hyperthermic seizure phenotype similar to Dys mutants (Supplementary Fig. 5). However, we did not observe a genetic interaction between the DGC and TGF-β pathway components suggesting that they might act in parallel.
Dys and Dg have been reported to have opposing functions in control of neurotransmitter release at the NMJ. Dg mutants show a decrease and Dys loss of function and heterozygous mutants an increase in release of neurotransmitter, however both mutants do not show a change in response to altered neurotransmitter levels20,22,23,24. These opposing phenotypes are similar to what we report here, where Dys mutants have seizures and Dg mutants do not. Additionally, Dys mutants have an increase in synaptic vesicle docking sites (T-bars) at larval NMJs23, which could explain our data showing the developmental requirement for Dys. Once the NMJ is established with a normal number of active sites, animals would not be prone to seizures.
Our data also show that Dys and Dg mutants have altered cellular homeostasis. In vertebrates, multiple metabolic disorders have been implicated in seizure activity; for example, mitochondrial encephalopathy, the most common neurometabolic disorder, presents various symptoms including seizures33 and mice that are partially deficient for mitochondrial superoxide dismutase have an increased incidence of spontaneous seizures34. Additionally, mdx mice, a model for MD has sustained oxidative stress in skeletal muscle35,36,37. In Drosophila, it has been shown that Dg mutant larvae have an altered state of cellular homeostasis and are sensitive to ambient temperature. A constant increase in mitochondrial oxidative metabolism, caused by a Dg hypomorphic mutation, results in a change in thermoregulatory behavior38. In addition, it was reported that suboptimal temperatures and energetic stress accelerate age-dependent muscular dystrophy in both, Dys and Dg mutants16. Now we show that Dys and Dg mutants have antagonistically abnormal cellular levels of ROS.
ROS are derived from elemental oxygen (O2), and ROS cascades begin with the superoxide anion radical. Sources for superoxide anion radicals include xanthine oxidase, prostanoid metabolism, catecholamine autooxidation, NAD(P)H oxidase activity and NO synthase. These radicals are generated in normal muscle, and the rate of generation is increased by muscle contraction39. In Duchenne MD the absence of dystrophin at the sarcolemma delocalizes and downregulates neuronal nitric oxide synthase (nNOS), which in turn leads to increase in inducible nitric oxide synthase (nNOS) that generates excessive NO•40. This mechanism can explain extremely high ROS levels in Dys mutants. This high level of ROS in dystrophic Drosophila can be alleviated by transheterozygous interaction with Dg and Cam, which indicates a genetic interaction between Dys and these two genes in control of cellular homeostasis.
Our study provides the first in vivo measurements on dystrophic animals showing that they have hyperthermic seizures that are dependent upon neurotransmission. Dystrophin is required during development, since Dys downregulation in adulthood, after muscles and NMJs are already established precludes hyperthermic seizures. Our data suggest that the DGC has a role in signaling at the NMJ: reduction of Dg, a protein that binds Dys12 and regulates localization of the NMJ specific proteins, prevents dystrophic seizure occurrence. Seizures are associated with abnormal Ca2+ release from the SR; introduction of a mutation of Ca2+ mediator Calmodulin and supply of calcium channel blockers reduce seizures. Taken together, our data show that the DGC acts at the muscle side of the NMJ to regulate muscle cell homeostasis in response to neuronal signaling and implies that Dys is involved in muscle-neuron communication.
Methods
Fly strains and genetics
Fly stocks were maintained at 25°C on a standard cornmeal-agar diet unless otherwise stated. Fly strains used in this study are: UAS-DysC-RNAi (dsDys), tub-Gal4::dsDys/TM6, UAS-DgRNAi (dsDg), tub-Gal4::dsDg/TM3 (described previously12 and recombined onto chromosomes with the tub-Gal4 driver where indicated), loss of function (lof) mutants DysDf (deletion of the Dys gene generated by outcrossing the deficiency Df(3R)Exel618441), DgO86/CyO, DgO55/CyO42, DfKX23/Ser,TM3 (Df(3R)Dl-KX23 - partial deletion of Dys gene, Bloomington ID: 2411 ), Dys8-2/TM6 (deletion of N-terminal region causing a loss of at least one long isoform12), tub-Gal4/TM3, 24B-Gal4, D42-Gal4, act-Gal4, tub-Gal80ts, OregonR. Alleles used in this study are: parats1 (temperature sensitive, lof), Camn339 (lof), mys1 (lof), corak08713 (hypomorph), tkv1 (hypomorph) and Mad12 (lof). The RNAi line directed against tkv (tkvRNAi) was obtained from the Vienna Drosophila RNAi Center. Unless otherwise stated, lines were obtained from Bloomington Stock Center.
Immunohistochemistry
Adult flies were immobilized on a tissue culture dish using Vaseline. The thorax and head were removed and the abdomen was opened in saline buffer (115 mM NaCl, 5 mM KCl, 6 mM CaCl2•2H2O, 1 mM MgCl2•6H2O, 4 mM NaHCO3, 1 mM NaH2PO4•1H2O, 5 mM trehalose, 75 mM sucrose, and 5 mM N-Tris [hydroxymethyl] methyl-2-aminoethane sulfonic acid). The tissue was then fixed without agitation for 10 minutes at room temperature in 4% formaldehyde. Tissue was fixed for 5 minutes in methanol for the anit-DGluRIIA antibody. Antibody staining was performed as described before17 except with the use of saline buffer. The following antibodies were used: rabbit anti-Dg42 (1∶1000), mouse anti-Dlg (1∶50; Developmental Studies Hybridoma Bank), mouse anti-DGluRIIA (1∶50; Developmental Studies Hybridoma Bank), Alexa 488 goat anti-mouse and Alexa 568 goat anti-rabbit (Invitrogen). Samples were then mounted on slides in 70% glycerol, 2% NPG, 1× PBS and analyzed using a confocal microscope (Leica TCS SP5). Quantification of staining intensities was done using Leica software (LAS AF version 2.1.1). The mean pixel value in the area of interest (NMJ) and in the same size area of the background was calculated. The background level was subtracted from the value found in the area of interest. Reported intensities were normalized to control and the Student's t-test was performed for statistical analysis.
Electrophysiology
IFM recordings were done on adult flies raised at 25°C. Flies were 3–5 days old unless otherwise indicated. Platinum electrodes were fashioned via KOH etching. The recording electrode was inserted into the lateral thorax with the ground electrode inserted into the abdomen. Temperature was shifted from room temperature (∼25°C) up to ∼50°C using an infrared lamp. Signals generated from the IFMs were amplified and digitized with the Micro 1401 data acquisition unit (Cambridge Electronic Design) and analyzed with Spike 2 software (Cambridge Electronic Devices). Temperature was monitored using an analogue thermometer (DKT200, 0–5 V, −20°C−80°C, ±0.3°C, Driesen Kern GmbH) where approximately every ten seconds resulted in a 1°C T increase. Plots were made of voltage output vs. temperature. All plots were scaled the same and converted into binary JPEG images. A macro was written using ImageJ software (NIH) to count the number of pixels of the voltage on the graph during the seizure. When a seizure occurred, a seizure index (Si) was calculated to reflect the temperature that the seizure started at (Ts), the amplitude of the seizure, and the duration of the seizure. Since the Ts was deemed to be more informative than the size of the seizure, a Si was calculated by taking the natural logarithm of the pixel count and dividing by the seizure start temperature. This number was then multiplied by 10.
If no seizure occurred then the index was set to zero because the Ts could not be determined if there was no seizure. Only seizures that occurred at lower than 49°C were considered because at very high temperatures even control animals start having contractions (Fig. 1b).
Statistics
Since our seizure electrophysiological data are not normally distributed, we used non-parametric methods for statistical analysis. Values are reported as M (x/y) where M = median, y = 25th percentile rank and x = 75th percentile rank. For determination of agreement between groups of data, the Kruskal-Wallis test was applied resulting in a chi-squared statistic. For comparison between two data sets a one-tailed Mann-Whitney U-test was used. Graphical representations of data show the median value of the data spread and error bars correspond to the 25th and 75th percentiles. Statistical analysis of muscle degeneration was done using a one-tailed chi-squared test. Statistical analysis of biochemical assays and immunohistochemical signal intensity was done using a one-tailed Student's t-test.
Histology
For analysis of indirect flight muscle (IFM) morphology, 10 μm paraffin-embedded sections were cut from fly thoraxes. In order to prepare Drosophila muscle sections, the fly bodies were immobilized in collars in the required orientation and fixed in Carnoy fixative solution (6∶3∶1 = Ethanol∶Chloroform∶Acetic acid) at 4°C overnight. Tissue dehydration and embedding in paraffin was performed as described previously43. Histological sections were prepared using a Hyrax M25 (Zeiss) microtome and stained with hematoxylin and eosin. All chemicals for these procedures were obtained from Sigma Aldrich. Muscle analysis was done using a light microscope (Zeiss). The frequency of muscle degeneration was quantified as a ratio of degenerated muscles to the total number of analyzed muscles.
Lipid peroxidation detection
Flies were kept at 29°C for three days after eclosion and emersed in liquid N2. Cellular extracts were prepared via homogenization of 20–25 whole flies in cell lysis buffer and then boiling the homogenates for 5 min at 100°C followed by centrifuging on high for 5 min. Protein levels in the extracts were determined via the BCA protein assay (Pierce Chemical). 100 μl of fly extract was combined with 300 μl acetic acid (20%, pH 3.5), 300 μl TBA (0.8% w/v in 0.5 M NaOH) and 40 μl SDS (8% w/v). The mixture was heated for 45 min at 100°C to allow for adduct formation. Adducts were then extracted into 1-butanol and the absorption was measured at 540 nm along with appropriate blanks. The raw absorption values minus any absorbance from the blank samples were then normalized to protein content (μg/ml). Three replicates were measured from four or more independent extracts. Data was compared using a one-tailed Student's t-test. Heterozygous animals were the result of crossing mutant stocks to OregonR.
Drug assays
Flies (DysDf genotype) raised at 25°C that were 3–7 days old were put overnight in vials containing filter paper soaked in 5% sucrose and either 500 μM 2-APB, 20 μM Ryanodine or 3.6 mM Nifedipine. Controls were only kept on sucrose. Electrophysiological measurements of IFMs were done the following day. When testing for drug lethality, OregonR wild type flies were used with lethality rates as follows: sucrose (1/10 dead), 2-APB (3/10 dead), Ryanodine (0/10 dead), Nifedipine (0/10 dead). When testing was conducted on Dys mutants there was a much higher lethality rate with both 2-APB and Ryanodine as follows: sucrose (4/15 dead), 2-APB (10/15 dead), Ryanodine (9/15 dead), Nifedipine (1/15 dead).
Reverse transcription quantitative PCR
Quantitative reverse transcription (RT-qPCR) was performed on total RNA derived from whole adult animals. RNAs were extracted from flies with the RNeasy Mini kit (Qiagen), followed by reverse transcription using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems) following the manufacturer's protocols. mRNA amounts were tested with RpL32 as an endogenous control for q-PCR using the Fast SYBR® Green master mix on a Step One Plus 96 well system (Applied Systems). The reactions were incubated at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 54°C for 30 s. All reactions were run in triplicate with appropriate blank controls. The threshold cycle (CT) is defined as the fractional cycle number at which the fluorescence passes the fixed threshold. Primers were used as follows: RpL32 forward - AAGATGACCATCCGCCCAGC; RpL32 reverse – GTCGATACCCTTGGGCTTGC; Dg forward – ACTCAAGGACGAGAAGCCGC; Dg reverse – ATGGTGGTGGCACATAATCG; Dys forward – GTTGCAGACACTGACCGACG; Dys reverse - CGAGGGCTCTATGTTGGAGC. The ΔCT value is determined by subtracting the average RpL32 CT value from the average Dys/Dg CT value. The ΔΔCT value is calculated by subtracting the ΔCT of the control sample from the ΔCT of the suspect sample. The relative amount of mRNA is then determined using the expression 2−ΔΔCT and the fold reduction is determined using the expression 2ΔΔCT. Errors for RNAi transgenes were determined starting with the standard deviation of the raw CT values and performing appropriate regression analysis.
Author Contributions
AKM designed and performed experiments, analyzed data, wrote the manuscript, MMK performed experiments, analyzed data, RW and MCG helped with electrophysiology and discussed data, HRS designed experiments, analyzed data, wrote the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
We would like to thank H. Ruohola-Baker and R. Ray for fly stocks and reagents, N. Glyvuk, Y. Tsytsyura, G. Vorbrüggen, A. Yatsenko and A. König for comments on the manuscript. This work was funded by Max Planck Society.
References
- Barresi R. & Campbell K. P. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci 119, 199–207 (2006). [DOI] [PubMed] [Google Scholar]
- Grady R. M. et al.. Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin–glycoprotein complex. Neuron 25, 279–293, S0896-6273(00)80894-6 [pii] (2000). [DOI] [PubMed] [Google Scholar]
- Kanagawa M. & Toda T. The genetic and molecular basis of muscular dystrophy: roles of cell-matrix linkage in the pathogenesis. J Hum Genet 51, 915–926, 10.1007/s10038-006-0056-7 (2006). [DOI] [PubMed] [Google Scholar]
- Sciandra F. et al.. Dystroglycan and muscular dystrophies related to the dystrophin-glycoprotein complex. Ann Ist Super Sanita 39, 173–181 (2003). [PubMed] [Google Scholar]
- Lim L. E. & Campbell K. P. The sarcoglycan complex in limb-girdle muscular dystrophy. Curr.Opin.Neurol. 11, 443–452 (1998). [DOI] [PubMed] [Google Scholar]
- Matsumura K. & Campbell K. P. Dystrophin-glycoprotein complex: its role in the molecular pathogenesis of muscular dystrophies. Muscle Nerve 17, 2–15 (1994). [DOI] [PubMed] [Google Scholar]
- Hayashi Y. K. et al.. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet 19, 94–97, 10.1038/ng0598-94 (1998). [DOI] [PubMed] [Google Scholar]
- Xiong Y., Zhou Y. & Jarrett H. W. Dystrophin glycoprotein complex-associated Gbetagamma subunits activate phosphatidylinositol-3-kinase/Akt signaling in skeletal muscle in a laminin-dependent manner. J Cell Physiol 219, 402–414, 10.1002/jcp.21684 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y. W., Thomason D. B., Gullberg D. & Jarrett H. W. Binding of laminin alpha1-chain LG4-5 domain to alpha-dystroglycan causes tyrosine phosphorylation of syntrophin to initiate Rac1 signaling. Biochemistry 45, 2042–2052 (2006). [DOI] [PubMed] [Google Scholar]
- Vandebrouck A. et al.. Regulation of capacitative calcium entries by alpha1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. Faseb J 21, 608–617 (2007). [DOI] [PubMed] [Google Scholar]
- Sabourin J. et al.. Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophin-dependent complex in skeletal mouse myotubes. J Biol Chem 284, 36248–36261 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbata H. R. et al.. Dissecting muscle and neuronal disorders in a Drosophila model of muscular dystrophy. EMBO J. 26, 481–493 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greener M. J. & Roberts R. G. Conservation of components of the dystrophin complex in Drosophila. FEBS Lett 482, 13–18 (2000). [DOI] [PubMed] [Google Scholar]
- Taghli-Lamallem O. et al.. Dystrophin deficiency in Drosophila reduces lifespan and causes a dilated cardiomyopathy phenotype. Aging Cell 7, 237–249 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikian M. J. et al.. Reduced life span with heart and muscle dysfunction in Drosophila sarcoglycan mutants. Hum.Mol.Genet. 16, 2933–2943 (2007). [DOI] [PubMed] [Google Scholar]
- Kucherenko M. M., Marrone A. K., Rishko V. M., Magliarelli Hde F. & Shcherbata H. R. Stress and muscular dystrophy: a genetic screen for dystroglycan and dystrophin interactors in Drosophila identifies cellular stress response components. Dev Biol 352, 228–242 (2011). [DOI] [PubMed] [Google Scholar]
- Klose M. K., Atwood H. L. & Robertson R. M. Hyperthermic preconditioning of presynaptic calcium regulation in Drosophila. J.Neurophysiol. 99, 2420–2430 (2008). [DOI] [PubMed] [Google Scholar]
- Montana E. S. & Littleton J. T. Characterization of a hypercontraction-induced myopathy in Drosophila caused by mutations in Mhc. J.Cell Biol. 164, 1045–1054 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. F. & Ganetzky B. Genetic alteration of nerve membrane excitability in temperature-sensitive paralytic mutants of Drosophila melanogaster. Nature 286, 814–816 (1980). [DOI] [PubMed] [Google Scholar]
- Bogdanik L. et al.. Muscle dystroglycan organizes the postsynapse and regulates presynaptic neurotransmitter release at the Drosophila neuromuscular junction. PLoS.One. 3, e2084 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote P. D., Moukhles H. & Carbonetto S. Dystroglycan is not required for localization of dystrophin, syntrophin, and neuronal nitric-oxide synthase at the sarcolemma but regulates integrin alpha 7B expression and caveolin-3 distribution. J Biol Chem 277, 4672–4679 (2002). [DOI] [PubMed] [Google Scholar]
- Wairkar Y. P., Fradkin L. G., Noordermeer J. N. & DiAntonio A. Synaptic defects in a Drosophila model of congenital muscular dystrophy. J Neurosci 28, 3781–3789 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Plas M. C. et al.. Dystrophin is required for appropriate retrograde control of neurotransmitter release at the Drosophila neuromuscular junction. J.Neurosci. 26, 333–344 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilgram G. S., Potikanond S., van der Plas M. C., Fradkin L. G. & Noordermeer J. N. The RhoGAP crossveinless-c interacts with Dystrophin and is required for synaptic homeostasis at the Drosophila neuromuscular junction. J Neurosci 31, 492–500 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan R., Massom L. R. & Jarrett H. W. Calmodulin specifically binds three proteins of the dystrophin-glycoprotein complex. Biochem.Biophys.Res.Commun. 185, 753–759 (1992). [DOI] [PubMed] [Google Scholar]
- Heiman R. G. et al.. Spontaneous avoidance behavior in Drosophila null for calmodulin expression. Proc.Natl.Acad.Sci.U.S.A 93, 2420–2425 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deval E. et al.. Na(+)/Ca(2+) exchange in human myotubes: intracellular calcium rises in response to external sodium depletion are enhanced in DMD. Neuromuscul.Disord. 12, 665–673 (2002). [DOI] [PubMed] [Google Scholar]
- Surtees R. Inherited ion channel disorders. Eur J Pediatr 159 Suppl 3, S199–203 (2000). [DOI] [PubMed] [Google Scholar]
- Kushnir A., Betzenhauser M. J. & Marks A. R. Ryanodine receptor studies using genetically engineered mice. FEBS Lett 584, 1956–1965 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M. et al.. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature 379, 168–171 (1996). [DOI] [PubMed] [Google Scholar]
- Nishimune H., Sanes J. R. & Carlson S. S. A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature 432, 580–587 (2004). [DOI] [PubMed] [Google Scholar]
- Keshishian H. & Kim Y. S. Orchestrating development and function: retrograde BMP signaling in the Drosophila nervous system. Trends Neurosci 27, 143–147 (2004). [DOI] [PubMed] [Google Scholar]
- Tucker E. J., Compton A. G. & Thorburn D. R. Recent advances in the genetics of mitochondrial encephalopathies. Curr Neurol Neurosci Rep 10, 277–285 (2010). [DOI] [PubMed] [Google Scholar]
- Liang L. P. & Patel M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2(−/+) mice. Free Radic Biol Med 36, 542–554 (2004). [DOI] [PubMed] [Google Scholar]
- Dudley R. W. et al.. Dynamic responses of the glutathione system to acute oxidative stress in dystrophic mouse (mdx) muscles. Am J Physiol Regul Integr Comp Physiol 291, R704–710 (2006). [DOI] [PubMed] [Google Scholar]
- Tidball J. G. & Wehling-Henricks M. The role of free radicals in the pathophysiology of muscular dystrophy. J Appl Physiol 102, 1677–1686 (2007). [DOI] [PubMed] [Google Scholar]
- Whitehead N. P., Pham C., Gervasio O. L. & Allen D. G. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J Physiol 586, 2003–2014 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K. et al.. Changes in temperature preferences and energy homeostasis in dystroglycan mutants. Science 323, 1740–1743 (2009). [DOI] [PubMed] [Google Scholar]
- Reid M. B. Invited Review: redox modulation of skeletal muscle contraction: what we know and what we don't. J Appl Physiol 90, 724–731 (2001). [DOI] [PubMed] [Google Scholar]
- Bellinger A. M. et al.. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 15, 325–330 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforou C. P., Greer C. E., Challoner B. R., Charizanos D. & Ray R. P. The detached locus encodes Drosophila Dystrophin, which acts with other components of the Dystrophin Associated Protein Complex to influence intercellular signalling in developing wing veins. Dev Biol 313, 519–532 (2008). [DOI] [PubMed] [Google Scholar]
- Deng W. M. et al.. Dystroglycan is required for polarizing the epithelial cells and the oocyte in Drosophila. Development 130, 173–184 (2003). [DOI] [PubMed] [Google Scholar]
- Kucherenko M. M. et al.. Paraffin-embedded and frozen sections of Drosophila adult muscles. J Vis Exp, 2438 [pii]10.3791/2438 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information