

Abstract
Although various function of chemerin have been suggested, its physiological role remains to be elucidated. Here we show that chemerin-deficient mice are glucose intolerant irrespective of exhibiting reduced macrophage accumulation in adipose tissue. The glucose intolerance was mainly due to increased hepatic glucose production and impaired insulin secretion. Chemerin and its receptor ChemR23 were expressed in β-cell. Studies using isolated islets and perfused pancreas revealed impaired glucose-dependent insulin secretion (GSIS) in chemerin-deficient mice. Conversely, chemerin transgenic mice revealed enhanced GSIS and improved glucose tolerance. Expression of MafA, a pivotal transcriptional factor for β-cell function, was downregulated in chemerin-deficient islets and a chemerin-ablated β-cell line and rescue of MafA expression restored GSIS, indicating that chemerin regulates β-cell function via maintaining MafA expression. These results indicate that chemerin regulates β-cell function and plays an important role in glucose homeostasis in a tissue-dependent manner.
Type-2 diabetes is an emerging health hazard worldwide and contributes to increased morbidity and mortality. It results from the development of insulin resistance and a concomitant impairment of insulin secretion. Particularly for the onset of the disease, impaired insulin secretion is an essential factor. Under these conditions, adipokines secreted from the adipose tissue play a pivotal role1,2. Adipokines such as leptin, adiponectin, and resistin, regulate energy and glucose homeostasis. Adipocytes also secrete inflammatory cytokines, such as tumor necrosis factor-α, interleukin-6 and monocyte chemotactic protein-1 (MCP-1) that are classically considered to be produced by macrophages, and link inflammation and insulin resistance3,4. These adipokines exert their effects in an endocrine, autocrine, or paracrine manner and regulate metabolism.
Chemerin was originally identified in the skin as tazarotene-induced gene 2 (TIG2)5. It is a chemotactic substance that was identified as the ligand for an orphan G protein-coupled receptor, ChemR23/CMKLR1, which shares a similarity to chemokine receptor and is expressed in immature dendritic cells (DCs) and macrophages6. Chemerin promotes chemotaxis of DCs and macrophages6, suggesting that chemerin is associated with a proinflammatory state like other chemokines. On the other hand, ChemR23-deficient mice treated with LPS exhibit greater neutrophil and macrophage recruitment into lung tissue than wild type mice, suggesting that chemerin has an anti-inflammatory function7. Importantly, ChemR23-deficient mice are unresponsive to chemerin, indicating that the chemerin/ChemR23 system plays anti-inflammatory role, and that ChemR23 is the physiological receptor for chemerin7,8. Recently, it has been reported that CCRL2 and GPR1 may also serve as receptors for chemerin. However, CCRL2 does not transduce signals into cells9. Furthermore, chemerin-induced Ca mobilisation via GPR1 is much lower than that of ChemR2310. Thus far, the physiological relevance of these receptors in chemerin function remains unknown.
Recently, several groups have reported that chemerin is an adipokine11,12,13,14 that regulates adipocyte differentiation and lipolysis11,13. Furthermore, chemerin enhances insulin-dependent glucose uptake in adipocytes14. These data suggest that chemerin functions as an adipokine in metabolic regulation. Indeed, serum chemerin levels are associated with BMI, fasting glucose, serum insulin, plasma triglycerides, and serum cholesterol levels12. However, the physiological role of chemerin has not been elucidated. In this study, we investigated the role of chemerin in glucose homeostasis particularly in β-cells using chemerin-deficient and transgenic mice.
Results
Generation of chemerin-deficient mice
To clarify the physiological role of chemerin, we generated chemerin-deficient mice by targeted gene deletion (Supplementary Fig. 1a). The chemerin (−/−) mice showed grossly normal and were fertile. The chemerin (+/+) and (−/−) mice gained weights similarly (Supplementary Fig. 2a). Food intake was not significantly different in chemerin (+/+) and (−/−) mice (Supplementary Fig. 2b). Serum cholesterol and triglyceride levels in chemerin (−/−) mice were comparable to those of the (+/+) mice (Supplementary Fig. 2c, d). However, the serum levels of non-esterified fatty acid (NEFA, free-fatty acids) were significantly lower in (−/−) mice than in (+/+) mice (Supplementary Fig. 2e).
Impaired glucose homeostasis in chemerin-deficient mice
Several reports have suggested that chemerin plays a role in glucose homeostasis in vitro14,15 and in vivo16, therefore we examined glucose metabolism in chemerin (−/−) mice using the intraperitoneal glucose tolerance test (IPGTT). Although the fasting glucose levels were comparable in chemerin (+/+) and (−/−) mice, the blood glucose levels at 15, 30, 60, and 90 min after intraperitoneal glucose administration were significantly higher in chemerin (−/−) mice than in chemerin (+/+) mice (Fig. 1a), indicating that chemerin deficiency leads to impaired glucose tolerance. To further define the tissue-specific alterations in glucose metabolism in chemerin (−/−) mice, we performed euglycemic hyperinsulinemic clamp studies. The glucose infusion rate (GIR) was not significantly altered (Fig. 1b), indicating that the net insulin sensitivity was comparable in chemerin (+/+) and (−/−) mice. The glucose disappearance rate (Rd) was significantly increased in the (−/−) mice, indicating enhanced muscle glucose uptake (Fig. 1c). Although basal hepatic glucose production (BHGP) was not different (Fig. 1d), clamp hepatic glucose production (CHGP) was significantly higher in (−/−) mice than in (+/+) mice (Fig. 1e), indicating that insulin suppression of hepatic glucose production was impaired in chemerin (−/−) mice. Liver weight and histological analyses revealed no obvious differences between chemerin (+/+) and (−/−) mice (Fig. 1f, g). We further analyzed the expression of G6Pase and PEPCK, which are the main regulators of gluconeogenesis in the liver. Consistent with increased CHGP, the abundance of G6Pase and PEPCK mRNAs were significantly higher in the liver of chemerin (−/−) mice than in those of chemerin (+/+) mice (Fig. 1h). Furthermore, the transcriptional coactivator PGC-1α mRNA was upregulated in chemerin (−/−) mice (Fig. 1h). These data indicate that chemerin deficiency enhanced muscle glucose uptake and increased hepatic gluconeogenesis in vivo. Then, as a measure of insulin downstream signaling, we examined Akt phosphorylation in muscle and adipose tissue. Consistent with the results of the clamp studies, Akt phosphorylation was enhanced in the gastrocnemius muscle under insulin-stimulated conditions (Fig. 1i). In contrast, insulin-stimulated Akt phosphorylation was decreased in the epididymal fat (Fig. 1j).
Figure 1. Chemerin-deficient mice exhibit impaired glucose tolerance and enhanced glucose production in the liver.
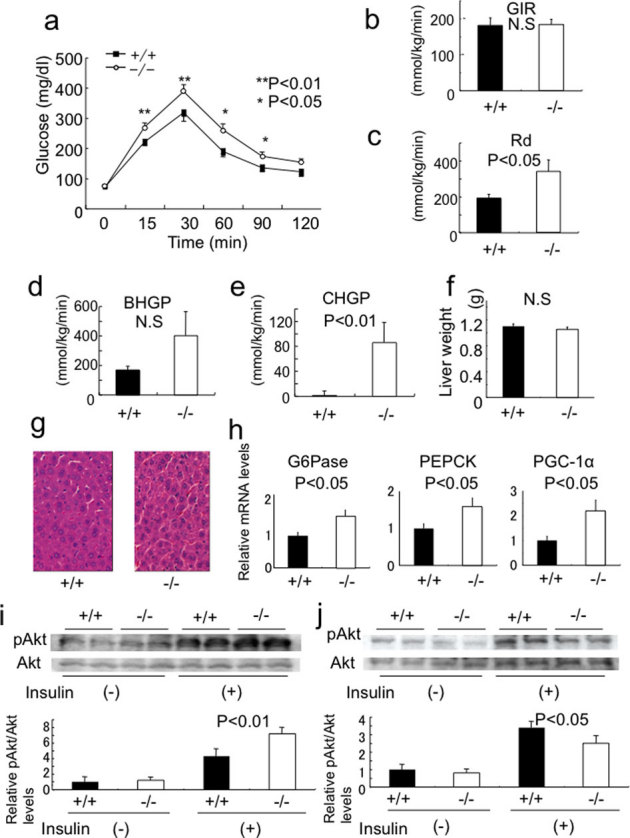
(a) Intraperitoneal glucose tolerance test in 12-week-old chemerin (+/+) and (−/−) mice. (n = 5–6/genotype). (b) Glucose infusion rate (GIR) at steady-state blood glucose, a measure of general insulin sensitivity, was determined by euglycemic clamp studies. Ten to 14 week-old mice were used for these studies. (n = 6/genotype). (c) Steady-state glucose disposable rate (Rd), a measure of insulin-dependent glucose uptake in the muscle, was calculated as described in Supplementary Methods. (d) Basal hepatic glucose production. (e) Clamp hepatic glucose production at the steady-state were calculated. (f) Liver weights of 10 week-old chemerin (+/+) and (−/−) mice. (g) H&E sections of liver in chemerin (+/+) and (−/−) mice. (400×) (h) Relative expression of genes related to glucose production in mice liver during steady-state conditions in clamp studies using quantitative real-time PCR. (n = 6/genotype). (i, j) Activation of insulin signaling in gastrocnemius muscle (i) and epididymal fat (j) of 12-week-old chemerin (+/+) and (−/−) mice. Insulin at a dose of 10 U/kg was intraperitoneally administered, and the mice were sacrificed 15 min later. Lysates of indicated tissues were subjected to immunoblotting analysis for Ser473-phosphorylated forms of Akt. (n = 3/genotype).
Macrophage infiltration into adipose tissue of chemerin-deficient mice
It has been demonstrated that chemerin regulates adipocyte differentiation and function. We next examined the characteristics of adipose tissue in chemerin (−/−) mice. There were no differences in epididymal fat weight between chemerin (+/+) and (−/−) mice (Fig. 2a). Histological analyses of epididymal fat revealed no obvious differences (Fig. 2b). Further, we examined the expression of markers of adipose tissue differentiation, function, and inflammation. There were no obvious differences in the expression of differentiation markers; however, the expression of macrophage markers, CD68 and F4/80, were significantly downregulated in chemerin (−/−) mice (Fig. 2c). Comparably, the number of Mac3-positive cells in the adipose tissue was significantly reduced (Fig. 2d, e), suggesting that macrophage infiltration was suppressed in chemerin (−/−) mice. Serum leptin and adiponectin concentrations were comparable in chemerin (+/+) and (−/−) mice (Fig. 2f).
Figure 2. Phenotypic analysis of adipose tissue in chemerin-deficient mice.
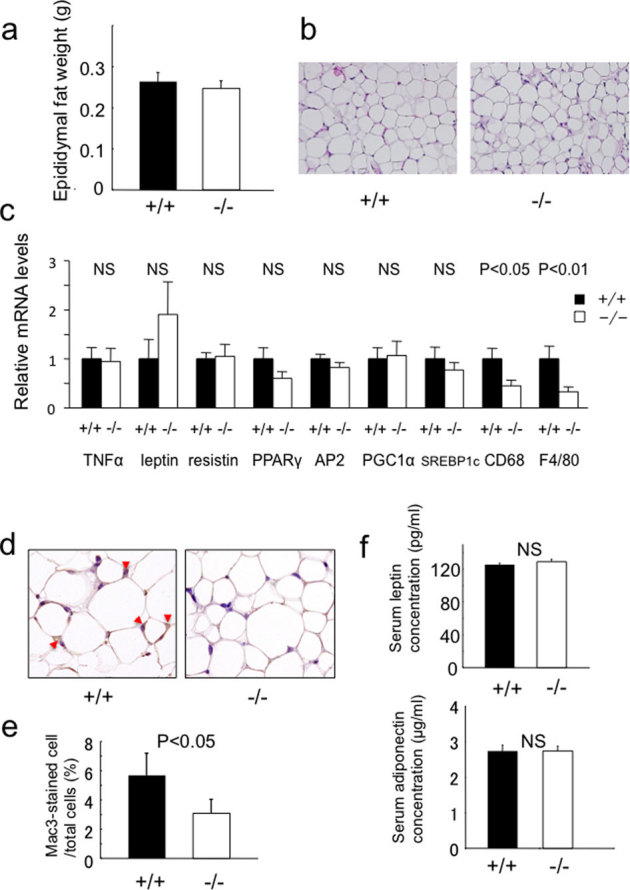
(a) Weights of epididymal fat in 10-week-old chemerin (+/+) and (−/−) mice. (b) H&E sections of epididymal fat tissue from chemerin (+/+) and (−/−) mice. (100×) (c) Expression of adipocyte markers in epididymal adipose tissue. (n = 4–5/genotype). (d) Immunohistochemical detection of Mac3 in epididymal adipose tissue of 10-week-old (+/+) and (−/−) mice. Macrophages are stained brown. (400×) (e) Macrophage infiltration into epididymal fat tissue was quantified as the ratio of Mac3-positive cells to total cells. Data are mean ± SEM. (n = 3/genotype). (f) Serum leptin and adiponectin concentrations in chemerin (+/+) and (−/−) mice. (n = 6/genotype).
Chemerin and its receptor ChemR23 are expressed in β−cells
The results of euglycemic hyperinsulinemic clamp studies suggested that impaired insulin secretion, and not insulin resistance, may be the predominant cause of glucose intolerance in chemerin (−/−) mice. First, we examined the expression of chemerin, its receptor ChemR23, and putative receptors GPR1 and CCRL2 in various tissues, including the pancreas. Intriguingly, chemerin was strongly expressed in the human pancreas (Fig. 3a). ChemR23 was predominantly expressed in WAT, heart, and testis (Fig. 3b). GPR1 was ubiquitously expressed but a relatively high expression was observed in WAT, BAT, and heart (Fig. 3c). CCRL2 was predominantly expressed in the heart (Fig. 3Dd). We further analyzed the expression of these receptors in the pancreas using immunohistochemistry. In the pancreas, ChemR23 and GPR1 were expressed in the islets, in contrast to the scant expression of CCRL2 (Supplementary Figu. 3a). Because ChemR23 has been shown to be the physiological receptor for chemerin7,17, we examined the expression of ChemR23 and chemerin in greater detail. ChemR23 and chemerin were exclusively detected in the β-cells of the islet as compared to insulin (Fig. 3e). Then, we examined the expression of ChemR23 and chemerin in the pancreas of obese and diabetic mouse models. Although there were no obvious changes in the expression of ChemR23 mRNA in these mouse models (Fig. 3f), the expression of chemerin was significantly downregulated in db/db mice and streptozotocin (STZ)-treated diabetic mice (Fig. 3g). Immunohistochemical analysis revealed that, similar to mRNA, the expression of chemerin was downregulated in the islet of db/db and STZ mice (Supplementary Fig. 3b). Interestingly, the expression of ChemR23 was downregulated in HFD, db/db, and STZ mice, suggesting the presence of regulatory mechanisms at the protein level (Supplementary Fig. 3b). Furthermore, the expression of ChemR23 was severely impaired in chemerin (−/−) mice, suggesting that chemerin itself may regulate the expression of its receptor (Fig. 3h).
Figure 3. Chemerin and its receptor expression in β- cells.
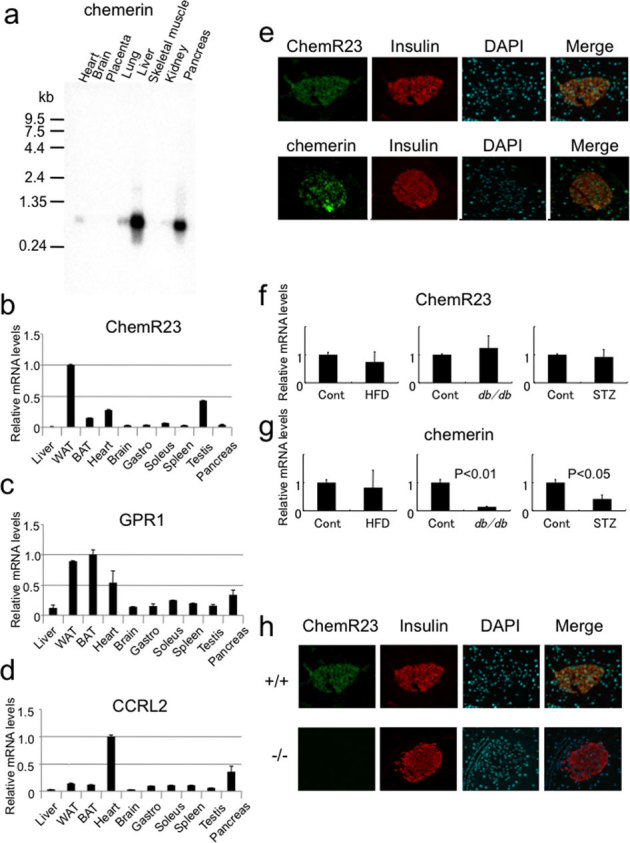
(a) Northern blotting analysis of chemerin in human tissues. Relative mRNA expression of (b) ChemR23, (c) GPR1, and (d) CCRL2 in mouse tissues evaluated by quantitative real time PCR analysis (QRPCR). (e) Immunofluorescence analysis of the chemerin receptor, ChemR23, chemerin and insulin in the mouse islets. (f) QRPCR analysis of ChemR23 and chemerin expressions in pancreas from HFD-fed mice, db/db mice, and streptozotocin (STZ)-induced diabetic mice. (n = 4/group). (h) Immunofluorescence analysis of ChemR23 and insulin in chemerin (+/+) and (−/−) islets.
Chemerin-deficient β−cells exhibit impaired glucose-stimulated insulin secretion
The expression and regulation of ChemR23 and chemerin in β-cells suggest that β-cell may be a direct target for chemerin. First, we analyzed the morphology of the islets in the chemerin (−/−) mice, and detected no obvious morphological changes in the islets (Fig. 4a); no significant differences were observed in the islet areas of chemerin (+/+) and (−/−) mice (Fig. 4b). Then we analyzed the insulin secretion ability of chemerin (−/−) mice in detail. Indeed, serum insulin levels were significantly lower in the chemerin (−/−) mice 30 min after intraperitoneal glucose administration (Fig. 4c). Further, we examined insulin secretion in response to glucose in the islets in vitro. Interestingly, in response to 16.7mM glucose, the isolated islets of (−/−) mice exhibited a marked decrease in insulin secretion compared to those of (+/+) mice (Fig 4d, Supplementary Fig. 4a). There were no differences in the insulin content of (+/+) and (−/−) islets (Fig. 4e). To evaluate the insulin secretion ability in vivo, we analyzed insulin secretion in perfused pancreas. As shown in Figure 4f, perfusion with 16.7 mM glucose elicited substantial insulin secretion in (+/+) mice. In contrast, insulin secretion was significantly reduced in the littermate (−/−) mice. The area under the curve (%AUC) for insulin during glucose stimulation was significantly reduced in the (−/−) mice (Fig. 4g). In response to KCl, a potent secretagogue of insulin secretion, the (−/−) pancreata exhibited a significant decrease in insulin secretion compared to the controls (Fig. 4h). These results indicate that the insulin-secretion capability of β-cells was impaired in chemerin (−/−) mice. To determine whether the action of chemerin was direct, we used MIN6 cells, β-cell line. Immunostaining analysis revealed that ChemR23 was substantially expressed in MIN6 cells (Fig. 4i). The expression of chemerin and ChemR23 protein was also detected in MIN6 cells (Figure 4j). To explore the role of chemerin and ChemR23 in GSIS in MIN6 cells, we performed knockdown experiments (Fig. 4j, Supplementary Fig. 4b, c). Consistent with the results from chemerin (−/−) mice, knockdown of chemerin or ChemR23 in MIN6 cells led to impaired GSIS (Fig. 4k).
Figure 4. Impaired glucose-stimulated insulin secretion (GSIS) in chemerin-deficient pancreas, islets and MIN6 cells.
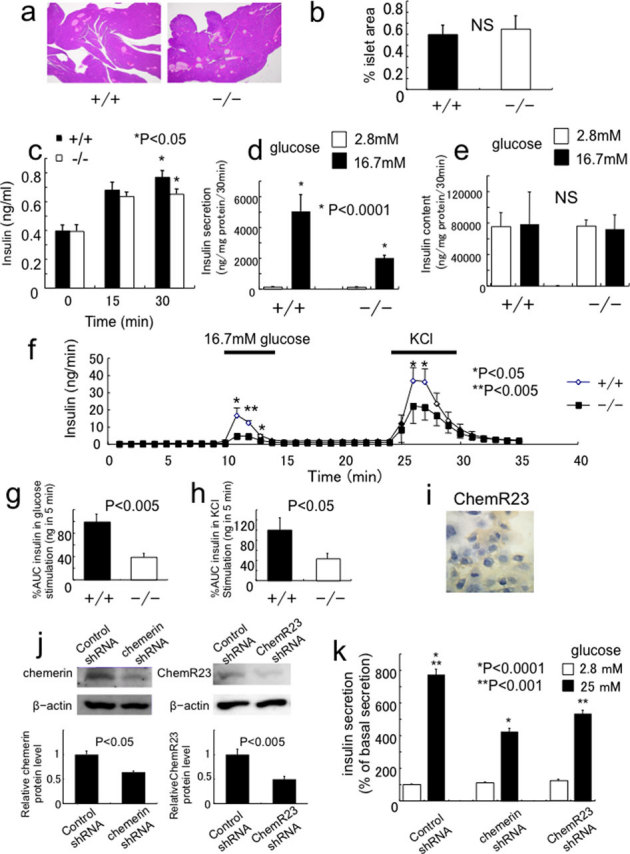
(a) Histological analysis of pancreas by H&E staining in chemerin (+/+) and (−/−) mice. Representative images are shown. (b) Percent islet area in 15-week-old (+/+) and (−/−) mice. (n = 4/genotype). (c) Serum insulin levels after glucose administration in 14-week-old mice. (n = 6/genotype). (d) Insulin secretion in islets isolated from at 12-14 week-old (+/+) and (−/−) mice. (n = 6 /genotype). (e) Insulin content in islets isolated from (+/+) and (−/−) mice. (f) In situ pancreatic perfusion in chemerin (−/−) mice. Insulin release was measured from perfused pancreata of (+/+) and (−/−) mice after the indicated stimuli. (n = 5–6/genotype). (g) The amount of insulin released (area under the curve [AUC]) after stimulation with 16.7 mM glucose. The AUC was significantly decreased in chemerin (−/−) pancreas. (h) The AUC was significantly decreased in chemerin (−/−) pancreas after KCl stimulation. (i) Immunohistochemical analysis of ChemR23 in MIN6 cells. (j) Knockdown of chemerin or ChemR23 in MIN6 cells. (k) GSIS was significantly reduced in chemerin- and ChemR23-knockdown MIN6 cells.
The effect of a high fat diet (HFD) on chemerin-deficient mice and GSIS in chemerin-transgenic mice
We examined the effect of HFD on chemerin (−/−) mice. After 6 weeks of HFD, although we did not detect significant differences in the body weights of chemerin (+/+) and (−/−) mice (Fig. 5a), glucose intolerance deteriorated (Fig. 5b) and insulin secretion tended to decrease in chemerin (−/−) mice at 30 min after intraperitoneal glucose administration (Fig. 5c). To examine the effect of chemerin overexpression in vivo, we generated serum amyloid protein (SAP) promoter-driven chemerin transgenic mice (Supplementary Fig. 1g, h), in which chemerin is overexpressed in the liver. We used this promoter because liver is the most abundant tissue for chemerin expression (Fig 3a). We obtained 2 transgenic mice lines, (Tg1 and Tg2). Immunoblotting analysis revealed that the serum chemerin levels were 1.9- fold and 7.2- fold higher in Tg1 and Tg2 mice, respectively, than in wild-type mice (Fig. 5d, e). We then performed the glucose tolerance test, and analyzed insulin secretion in Tg1 mice. Consistent with the results from chemerin (−/−) mice, chemerin transgenic mice exhibited improved glucose tolerance (Fig. 5f) and enhanced GSIS 15 and 30 min after intraperitoneal glucose administration than in wild-type mice (Fig. 5g).
Figure 5. Effects of HFD on chemerin-deficient mice and GSIS in chemerin-transgenic mice.
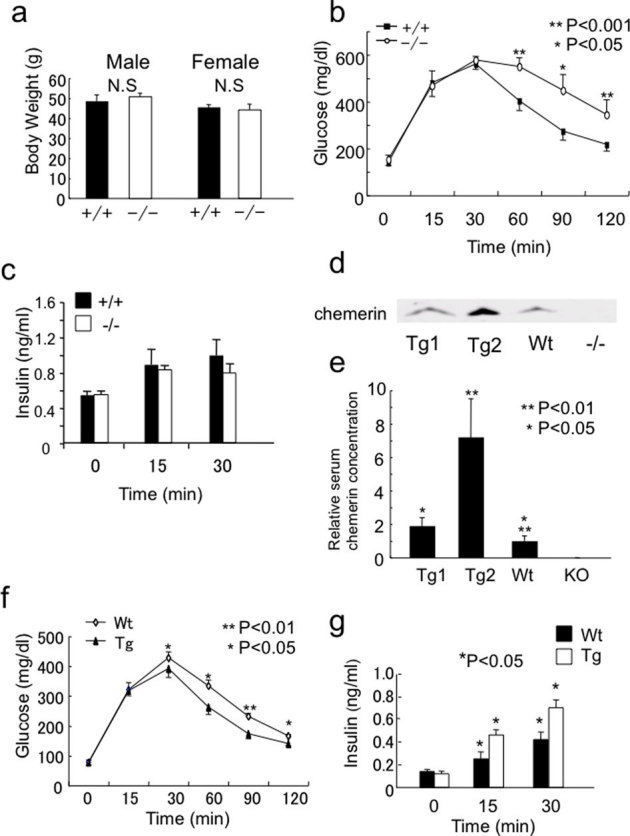
(a) Body weights of chemerin (+/+) and (−/−) mice after HFD. Body weights were determined 6 weeks after high-fat diet feeding from 8 weeks of age. (n = 5-8/genotype). (b) Intraperitoneal glucose tolerance test after 8 weeks of HFD in (+/+) and (−/−) mice. (n = 5/genotype). (c) Serum insulin levels after glucose administration following HFD in (+/+) and (−/−) mice. (n = 5/genotype). (d) Characterization of SAP promoter-driven chemerin transgenic mice. Serum (5 μL each) from Tg1, Tg2, wild-type, and chemerin (−/−) mice was subjected to immunoblotting analysis for chemerin. (e) Quantification of serum chemerin concentrations in chemerin transgenic mice. (f) Intraperitoneal glucose tolerance test in 12-week-old chemerin transgenic and wild-type mice. (n = 5/genotype). (g) Serum insulin levels after glucose administration in chemerin transgenic and wild-type mice. (n = 5/genotype).
Chemerin regulates GSIS by maintaining MafA expression in β−cells
To dissect the molecular mechanisms by which chemerin regulates β-cell function, we compared the expression of genes that regulate β-cell function in islets isolated from chemerin (+/+) and (−/−) mice. Although most of the genes exhibited no changes in expression, MafA was significantly downregulated (by 53%) in the islets of (−/−) mice (Fig. 6a, Supplementary Fig. 4d). Furthermore, the expression of Glut2 was also significantly downregulated in the islets of (−/−) mice. The protein levels of MafA were lower in the chemerin (−/−) islets (Fig. 6b, c). Immunohistochemical analysis revealed that the MafA nuclear staining in (−/−) islets was substantially weaker than that of chemerin (+/+) islet (Fig. 6d). Further, we analyzed whether knockdown of chemerin or ChemR23 in MIN6 cells reduced MafA expression in vitro. Expectedly, MafA expression was downregulated in chemerin- and ChemR23-knockdown MIN6 cells (Fig. 6e, f). These data suggest that chemerin and ChemR23 are necessary for MafA expression in β-cells. To clarify the functional relevance of the changes in MafA expression in chemerin (−/−) β-cells, we rescued MafA expression (Supplementary Fig. 4e) in the isolated islets (Fig. 6g) and MIN6 cells (Fig. 6i) using adenovirus. Although overexpression of MafA did not affect GSIS in the islets from (+/+) mice (Fig. 6h), the rescue of MafA expression in the islets from (−/−) mice partially but significantly improved the impaired GSIS (Fig. 6h). Further, the rescue of MafA expression in chemerin-ablated MIN6 cells also significantly improved GSIS (Figure 6 j).
Figure 6. MafA is regulated by chemerin and rescue of MafA expression in chemerin-deficient islets or chemerin-knockdown MIN6 cells restores GSIS.
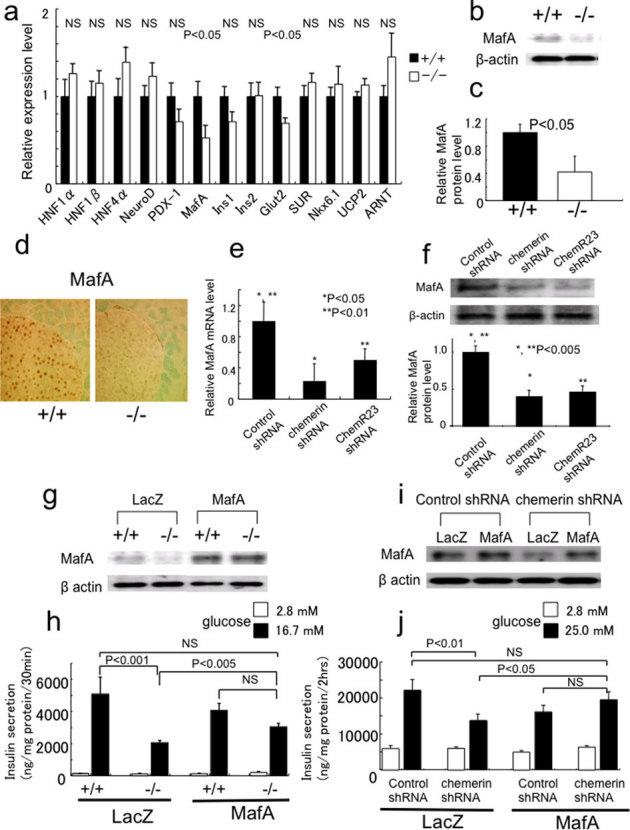
(a) MafA and Glut2 mRNA levels were significantly lower in the islets from 12–14-week-old chemerin (−/−) mice than in those of (+/+) mice. (n = 5–6/genotype). (b) MafA protein expression was downregulated in chemerin (−/−) islets. (c) Quantitative analysis of MafA protein expression in islets from 12–14-week-old chemerin (+/+) and (−/−) mice. (n = 3/genotype). (d) Immunohistochemical analysis of MafA expression in chemerin (+/+) and (−/−) islets. Similar results were obtained from three independent experiments. Representative results are shown. (e, f) MafA mRNA (e) and protein (f) expression were downregulated in chemerin- and ChemR23- knockdown MIN6 cells. (g) Rescue of MafA expression in isolated islets using MafA expressing adenovirus. Islets were infected with control (LacZ) or MafA adenovirus at a MOI of 125. (h) The impaired GSIS in chemerin-deficient islets was partially restored by exogenous MafA expression. Similar results were obtained from 3 independent experiments. Representative results are shown (age, 10–15 weeks; n = 3/genotype). (i) Rescue of MafA expression in chemerin-knockdown MIN6 cells infected with control (LacZ) or MafA adenovirus at a MOI of 8. (j) The impaired GSIS in chemerin-knockdown MIN6 cells was restored by exogenous MafA expression (n = 6).
Discussion
We have shown that chemerin (−/−) mice exhibit glucose intolerance. The glucose clamp studies using chemerin (−/−) mice demonstrated that chemerin regulates hepatic glucose production and muscle glucose uptake. GSIS was impaired in chemerin (−/−) mice, indicating that chemerin is involved in insulin secretion as well as in insulin sensitivity. These results indicate that chemerin plays an important role in glucose homeostasis in a tissue-dependent manner. We demonstrated that ChemR23 and chemerin are substantially expressed in β-cells and chemerin is necessary for insulin secretion in β-cells in vitro and vivo. Conversely, the improved glucose tolerance and enhanced insulin secretion observed in chemerin transgenic mice further support that chemerin regulates β-cell function in vivo. Although it is unknown whether systemically circulating or locally secreted chemerin is important for the regulation of β-cells, the observation that chemerin expression in β-cells is regulated depending on the metabolic status, suggests that locally produced chemerin may function in an autocrine/paracrine manner.
With respect to the mechanism in β-cells, we have shown that MafA expression is regulated by chemerin in vitro and in vivo. MafA has been identified as a basic leucine zipper transcription factor that is essential for β-cell function18. Intriguingly, MafA-deficient mice exhibit glucose intolerance, impaired GSIS with normal insulin content and reduced expressions of Glut2 and Pdx1 in the islets19, thus displayng a striking similarity with chemerin (−/−) mice, which exhibited impaired GSIS with normal insulin content and reduced expression of MafA and Glut2. The functional rescue of GSIS by MafA expression using adenovirus in chemerin (−/−) islets and chemerin-ablated MIN6 cells indicates that chemerin regulates β-cell function at least in part by maintaining MafA expression. However, the rescue of GSIS by MafA was partial, suggesting the existence of other mechanisms.
MafA expression is regulated both at the transcriptional and post-translational levels20. It has been reported that oxidative stress induced by hyperglycemia decreases MafA expression21,22. To exclude the possibility that the reduced MafA expression was due to hyperglycemia-induced oxidative stress, we analyzed the oxidative stress status in chemerin(−/−) islets; however, there were no changes in the oxidative stress status (data not shown). In addition, the results from MIN6 cells suggest a direct regulation of MafA expression by chemerin. The reduction in MafA mRNA in chemerin (−/−) islets suggested that chemerin may regulate MafA transcription. Therefore, we examined whether chemerin stimulated transcriptional activity of the MafA promoter in MIN6 cells; however, an obvious stimulatory effect on transcriptional activity was not observed (data not shown), suggesting that chemerin may regulate the stability of MafA mRNA.
Recently, it has been reported that not only ChemR23, but the orphan G-protein coupled receptors, CCRL2 and GPR1, also serve as chemerin receptors. ChemR23-deficient mice were unresponsive to chemerin with respect to anti-inflammatory effects, indicating that ChemR23 is essential for chemerin function7,8. In contrast, chemerin binds to CCRL2 but does not transduce a signal into cells9, and chemerin-induced Ca mobilisation via GPR1 is much weaker than that of ChemR2310. In this study, we have shown that ChemR23 is expressed in β-cells, and that the ablation of ChemR23 in MIN6 cells decreased MafA expression and impaired GSIS, which is similar to the phenotype of chemerin-ablated MIN6 cells. These data suggest that in β-cells, ChemR23 may be the main receptor for chemerin. On the other hand, GPR1 expression but not CCRL2, was detected in the islets, suggesting a potential role of GPR-1 as chemerin receptor in β-cell function.
Several groups have shown that chemerin is an adipokine11,12,13,14 that regulates adipocyte differentiation and lipolysis in autocrine and paracrine fashion11,13. Chemerin expression and secretion increase dramatically during adipogenesis, and loss of chemerin or ChemR23 expression in pre-adipocytes impairs their differentiation into mature adipocytes. In the present study, we did not detect significant difference in the body weight or the weight of epididymal fat pad at least until 12 weeks of age. Interestingly, serum NEFA levels were lower in chemerin (−/−) mice. These results are in line with previous findings that the absence of chemerin results in reduced basal and IBMX-stimulated lipolysis rate in 3T3L1 adipocytes in vitro11. It is noteworthy that, although the expression of adipocyte differentiation markers was unchanged in chemerin (−/−) mice, the expression of macrophage markers, CD68 and F4/80, and the accumulation of macrophages in adipose tissue were reduced in chemerin (−/−) mice. Given that chemerin is a chemoattractant like other chemokines for DCs and macrophages6,23, these data suggest that chemerin plays an important role in the accumulation of macrophages in adipose tissue. A recent study has shown that inflammation of adipose tissue caused by an accumulation of immune cells, including macrophages, plays an important role in obesity-induced metabolic dysfunction24. In chemerin (−/−) mice, irrespective of the reduced macrophage infiltration, glucose tolerance deteriorated, indicating that the effect of adipose tissue on the glucose intolerance may be small under this condition.
With respect to the role of chemerin in insulin signaling, in vitro studies have provided conflicting results; one study reported a decrease in insulin-stimulated glucose uptake in long incubation time25, whereas another study showed an increase in insulin-stimulated glucose uptake and insulin receptor substrate-1 tyrosine phosphorylation in shorter incubation time with chemerin treatment in adipocytes. In primary human skeletal muscle cells, chemerin treatment induces insulin resistance15. In db/db mice, chemerin treatment exacerbates glucose intolerance, by decreasing serum insulin levels, reducing adipose tissue glucose uptake and decreasing in liver and total tissue glucose uptake; however, these effects were not observed in normal mice16. In this study using glucose clamp studies, we have clearly shown that chemerin (−/−) mice demonstrate enhanced insulin sensitivity in the muscle and increased glucose production in the liver. Total insulin sensitivity, demonstrated by glucose infusion rate, was comparable in chemerin (−/−) and (+/+) mice suggesting that the different modes of action in the muscle and liver counterbalance each other at least under this condition. The muscle results of glucose clamp studies and of insulin-induced Akt phosphorylation are in line with previous report showing that chemerin treatment induces insulin resistance in vitro in human skeletal muscle15. The adipose tissue study showing that a decrease in insulin-induced Akt phosphorylation is compatible with previous report that chemerin treatment enhances insulin sensitivity in 3T3L1 adipocytes14. Taken together, chemerin plays different roles in the muscle, adipose tissue, and liver in the context of insulin action.
In summary, although further investigation is required to clarify the precise role of chemerin in the pathogenesis of diabetes, the findings herein present a physiological function of chemerin in glucose homeostasis in mice. The present study also demonstrated that chemerin plays an important role in the regulation of β-cell function.
Methods
Mice and in vivo experiments
Mouse experiments were performed according to the guidelines of the Animal Ethics Committee of Kobe University Graduate School of Medicine. Targeting strategy for disruption of the chemerin gene locus is depicted in Supplementary information. Chemerin transgenic mice were generated using the human chemerin cDNA under the control of liver-specific SAP promoter, which was a generous gift from Dr Miyazaki (Osaka University). Mice were maintained on a 12h/12h light/dark cycle and ad libitum access to food and water. The results presented here are of studies conducted on mice backcrossed at least for six generations with C57BL6 background and 7-15-week-old age-matched mice, unless specified. For HFD studies, 8-week-old male C57BL/6J mice were fed with a diet containing 45% fat, 10% protein, and 35% carbohydrates (Oriental Bioservice, Japan) for 6 weeks.
Glucose measurements and intraperitoneal glucose tolerance test
For the intraperitoneal glucose tolerance test, 50% glucose (1.5g/kg body weight) was intraperitoneally injected after 15-h fasting. Blood was collected from the tail vein at 0, 30, 60, 90, and 120 min after the injection.
Euglycemic hyperinsulinemic clamp Studies
Euglycemic hyperinsulinemic clamp studies were performed as previously described26. Details are presented in Supplementary information.
Islet morphometry, immunohistochemistry, and immunofluorescence
The pancreas was isolated, fixed in 4% paraformaldehyde and embedded in paraffin. Consecutive 4 μm sections were cut and mounted on slides. The percent islet area was quantified using Image J software, as described previously27,28. Immunohistochemistry and immunofluorescence were performed as previously described29. Anti-swine insulin antibody was obtained from DakoCytomation (Denmark). Anti-ChemR23, GPR1, CXCL2, and Mac3 antibodies were from SantaCruz (CA, USA), anti-MafA antibody was from Bethyl (Tx, USA).
Quantitative real time PCR and immunoblotting
Quantitative real-time PCR (QRPCR) was performed as previously described26,30, on a LightCycler system using the FastStart DNA Master SYBR Green I kit (Roche Diagnostics, Switzerland). Each value was normalized to 36B4 or β- actin expression. The primer sequences are provided in Supplementary information. Immunoblotting analysis was performed as previously described31. Anti-chemerin and ChemR23 antibodies have been described previously14. Anti-Akt, p-Akt (Ser473) antibodies were from Cell Signaling Tech (MA, USA).
Islet isolation and batch incubation
Mouse pancreatic islet isolation and batch incubation were performed as previously described32.
Perfusion of mouse pancreata
Overnight (12–16 h)-fasted mice at 8–15 weeks of age were used in perfusion experiments as previously described33. Details are presented in Supplementary information.
MIN6 cells and knockdown of chemerin and ChemR23
The mouse β-cell line, MIN6 was cultured in Dulbecco's modified Eagle's medium (DMEM) containing 25 mM glucose and 15% (v/v) heat-inactivated fetal calf serum under humidified conditions of 5% CO2, 95% air at 37 °C34. For chemerin and ChemR23 knockdowns of in MIN6 cells, the shRNA expression vector, pcPURm6i35 containing the target sequence for mouse chemerin or ChemR23 was transfected into MIN6 cells by reverse transfection using Lipofectamine 2000 reagent (Invitrogen) as previously described36,37.
Infection of the islets and MIN6 cells with Adenovirus
Mouse MafA cDNA (obtained by RT-PCR) was cloned into pENTR vector (Invitrogen). Adenoviruses were generated using ViraPower Adenoviral Expression System (Invitrogen), according to the manufacturer's instructions. Cells were infected with the adenovirus for 48 h. Adenovirus expressing lacZ was used as a negative control. The multiplicity of infection (MOI) for the islets and MIN6 cells were 125 and 8, respectively. The expression of MafA was verified in mouse embryonic fibroblasts (MEF) and MIN6 cells by immunoblotting (Supplementary Fig. 4e).
Statistics and data analysis
Results are presented as mean±SEM from at least 3 independent, unless stated otherwise. The data were analyzed using unpaired two-tailed Student's t test, analysis of variance (ANOVA), or Mann-Whitney U-test as appropriate. A P value of less than 0.05 was considered significant.
Detailed experimental procedures are provided in Supplementary methods.
Author Contributions
YT designed the project. YT, YO, GI, KC, MK and MT designed experiments. MT, HN, MY, KS, RK, SK, WF, KT, FZ, KH, HK and TA performed a significant amount of the experimental work. MT, RK, and KT performed most of the data collection and data analysis. YT directed the study and wrote the main manuscript text and prepared figures and tables. All authors reviewed the manuscript.
Supplementary Material
Supplementary information
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research from Japanese Ministry of Education Science, Sports and Culture 22591012 and 23659477 and a grant for 21st Century COE Program, “Center of Excellence for Signal Transduction Disease: Diabetes Mellitus as Model” from Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank C Ogata, K Imura, S Matsuda, and N Sakamoto for excellent technical assistance, Drs S Seino, K Minami, T Miki, S Tateya, W Ogawa, N Takahashi, H Kasai, Y Shigeyoshi, T Uchida, S Kadowaki, T Nomura, and H Iwakabe for excellent suggestions and support.
References
- Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol 115, 911–919; quiz 920 (2005). [DOI] [PubMed] [Google Scholar]
- Kershaw E. E. & Flier J. S. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 89, 2548–2556 (2004). [DOI] [PubMed] [Google Scholar]
- Charo I. F. & Taubman M. B. Chemokines in the pathogenesis of vascular disease. Circ Res 95, 858–866 (2004). [DOI] [PubMed] [Google Scholar]
- Rossi D. & Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol 18, 217–242 (2000). [DOI] [PubMed] [Google Scholar]
- Nagpal S., et al.. Tazarotene-induced gene 2 (TIG2), a novel retinoid-responsive gene in skin. J Invest Dermatol 109, 91–95 (1997). [DOI] [PubMed] [Google Scholar]
- Wittamer V., et al.. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. The Journal of experimental medicine 198, 977–985 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luangsay S., et al.. Mouse ChemR23 is expressed in dendritic cell subsets and macrophages, and mediates an anti-inflammatory activity of chemerin in a lung disease model. J Immunol 183, 6489–6499 (2009). [DOI] [PubMed] [Google Scholar]
- Ernst M. C. & Sinal C. J. Chemerin: at the crossroads of inflammation and obesity. Trends Endocrinol Metab (2010). [DOI] [PubMed] [Google Scholar]
- Zabel B. A., et al.. Mast cell-expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. The Journal of experimental medicine 205, 2207–2220 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnea G., et al.. The genetic design of signaling cascades to record receptor activation. Proc Natl Acad Sci U S A 105, 64–69 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goralski K. B., et al.. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. The Journal of biological chemistry 282, 28175–28188 (2007). [DOI] [PubMed] [Google Scholar]
- Bozaoglu K., et al.. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology 148, 4687–4694 (2007). [DOI] [PubMed] [Google Scholar]
- Roh S. G., et al.. Chemerin—a new adipokine that modulates adipogenesis via its own receptor. Biochem Biophys Res Commun 362, 1013–1018 (2007). [DOI] [PubMed] [Google Scholar]
- Takahashi M., et al.. Chemerin enhances insulin signaling and potentiates insulin-stimulated glucose uptake in 3T3-L1 adipocytes. FEBS Lett 582, 573–578 (2008). [DOI] [PubMed] [Google Scholar]
- Sell H., et al.. Chemerin is a novel adipocyte-derived factor inducing insulin resistance in primary human skeletal muscle cells. Diabetes 58, 2731–2740 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst M. C., Issa M., Goralski K. B. & Sinal C. J. Chemerin exacerbates glucose intolerance in mouse models of obesity and diabetes. Endocrinology 151, 1998–2007 (2010). [DOI] [PubMed] [Google Scholar]
- Ernst M. C. & Sinal C. J. Chemerin: at the crossroads of inflammation and obesity. Trends Endocrinol Metab 21, 660–667 (2010). [DOI] [PubMed] [Google Scholar]
- Zhou Q., Brown J., Kanarek A., Rajagopal J. & Melton D. A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455, 627–632 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., et al.. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol 25, 4969–4976 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramata S., Han S. I. & Kataoka K. Roles and regulation of transcription factor MafA in islet beta-cells. Endocrine journal 54, 659–666 (2007). [DOI] [PubMed] [Google Scholar]
- Harmon J. S., Stein R. & Robertson R. P. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. The Journal of biological chemistry 280, 11107–11113 (2005). [DOI] [PubMed] [Google Scholar]
- Kitamura Y. I., et al.. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell metabolism 2, 153–163 (2005). [DOI] [PubMed] [Google Scholar]
- Wittamer V., et al.. Neutrophil-mediated maturation of chemerin: a link between innate and adaptive immunity. J Immunol 175, 487–493 (2005). [DOI] [PubMed] [Google Scholar]
- Ouchi N., Parker J. L., Lugus J. J. & Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol 11, 85–97 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralisch S., et al.. Interleukin-1beta induces the novel adipokine chemerin in adipocytes in vitro. Regul Pept 154, 102–106 (2009). [DOI] [PubMed] [Google Scholar]
- Tateya S., Tamori Y., Kawaguchi T., Kanda H. & Kasuga M. An increase in the circulating concentration of monocyte chemoattractant protein-1 elicits systemic insulin resistance irrespective of adipose tissue inflammation in mice. Endocrinology 151, 971–979 (2010). [DOI] [PubMed] [Google Scholar]
- Bernal-Mizrachi E., Wen W., Stahlhut S., Welling C. M. & Permutt M. A. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. The Journal of clinical investigation 108, 1631–1638 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynihan K. A., et al.. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell metabolism 2, 105–117 (2005). [DOI] [PubMed] [Google Scholar]
- Kishimoto K., Kitazawa R., Kurosaka M., Maeda S. & Kitazawa S. Expression profile of genes related to osteoclastogenesis in mouse growth plate and articular cartilage. Histochem Cell Biol 125, 593–602 (2006). [DOI] [PubMed] [Google Scholar]
- Takahashi M., et al.. CXCL14 enhances insulin-dependent glucose uptake in adipocytes and is related to high-fat diet-induced obesity. Biochem Biophys Res Commun 364, 1037–1042 (2007). [DOI] [PubMed] [Google Scholar]
- Takahashi Y., et al.. Biologically inactive growth hormone caused by an amino acid substitution. The Journal of clinical investigation 100, 1159–1165 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki T., et al.. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci U S A 104, 19333–19338 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T., et al.. Distinct effects of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 on insulin secretion and gut motility. Diabetes 54, 1056–1063 (2005). [DOI] [PubMed] [Google Scholar]
- Miyazaki J., et al.. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132 (1990). [DOI] [PubMed] [Google Scholar]
- Jazag A., et al.. Single small-interfering RNA expression vector for silencing multiple transforming growth factor-beta pathway components. Nucleic Acids Res 33, e131 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morioka T., et al.. Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. The Journal of clinical investigation 117, 2860–2868 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunton J. E., et al.. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell 122, 337–349 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information