

Abstract
Microglia are the resident mononuclear phagocytes of the CNS parenchyma and represent an initial line of defense against invading microorganisms. Microglia utilize Toll-like receptors (TLRs) for pathogen recognition and TLR2 specifically senses conserved motifs of Gram-positive bacteria including lipoproteins, lipoteichoic acids, and peptidoglycan (PGN) leading to cytokine/chemokine production. Interestingly, primary microglia derived from TLR2 knockout (KO) mice over-expressed numerous IL-12 family members, including IL-12p40, IL-12p70, and IL-27 in response to intact S. aureus, but not the less structurally complex TLR2 ligands Pam3CSK4 or PGN. The ability of intact bacteria to augment IL-12 family member expression was specific for Gram-positive organisms since numerous Gram-negative strains were unable to elicit exaggerated responses in TLR2 KO microglia. Inhibition of SYK or IRAK4 signaling did not impact heightened IL-12 family member production in S. aureus-treated TLR2 KO microglia, whereas PI3K, MAPK, and JNK inhibitors were all capable of restoring exaggerated cytokine expression to WT levels. Additionally, elevated IL-12 production in TLR2 KO microglia was ablated by a TLR9 antagonist, suggesting that TLR9 drives IL-12 family member production following exposure to intact bacteria that remains unchecked in the absence of TLR2 signaling. Collectively, these findings indicate crosstalk between TLR2 and TLR9 pathways to regulate IL-12 family member production by microglia. The summation of TLR signals must be tightly controlled to ensure the timely cessation and/or fine tuning of cytokine signaling to avoid non-specific bystander damage due to sustained IL-12 release.
Keywords: S. aureus, TLR2, TLR9, microglia, MAPK, JNK, PI3K
INTRODUCTION
Toll-like receptors (TLRs) are a group of germ-line encoded pattern recognition receptors (PRRs) expressed by cells of the innate and, more recently appreciated, adaptive immune systems (Blasius and Beutler 2010; Kawai and Akira 2010). TLRs mediate rapid cellular activation and proinflammatory mediator production in response to conserved pathogen-associated molecular patterns (PAMPs) found in bacteria, viruses, and fungi (Blasius and Beutler 2010; Kawai and Akira 2010). TLR2 plays an important role in recognizing a wide array of PAMPs, including lipoproteins, lipoteichoic acids, and peptidoglycan (PGN) found in Gram-positive bacteria such as S. aureus (Takeuchi et al. 2000; Takeuchi et al. 1999). Indeed, recent studies have established the importance of TLR2 during Gram-positive CNS infections (Echchannaoui et al. 2002; Kielian et al. 2005b; Stenzel et al. 2008; Takeuchi et al. 2000; Vidlak et al. 2010). However, intact Gram-positive bacteria are capable of activating multiple TLRs since they harbor a thick cell wall composed of several TLR2 ligands as well as unmethlyated CpG DNA, the latter of which activates TLR9 (Alexopoulou et al. 2001). To date, the number of studies investigating TLR pathways triggered by intact bacteria in various cell types is limited. Instead, most reports have utilized purified PAMPs, which is less representative of events elicited during infection. For this reason, we have examined the impact of TLR2 deficiency on microglial activation following exposure to intact bacteria.
Microglia are the resident phagocytes of the CNS parenchyma and provide an initial line of defense against bacterial colonization (Aloisi 2001; Colton 2009), in part, due to their constitutive expression of numerous TLRs including TLR2 (Kielian 2006; Okun et al. 2011). Previous work from our laboratory revealed that although TLR2 is pivotal for PGN recognition, inflammatory cytokine expression in response to intact S. aureus was qualitatively similar. However, one exception was IL-12p40, which was significantly elevated in TLR2 KO microglia following exposure to intact bacteria (Kielian et al. 2005a).
The IL-12 cytokine family includes members composed of a common p40 subunit that heterodimerizes with a unique subunit that identifies each cytokine (Goriely et al. 2008; Trinchieri 2003). IL-12p40 is produced in excess of the other IL-12 family subunits and can exert negative regulatory effects as a homodimer by competitively binding the IL-12 receptor; however, it can also act as a macrophage chemoattractant and promote dendritic cell migration in response to bacterial stimulation (Cooper and Khader 2007; Gillessen et al. 1995). IL-12p70 is composed of both heavy (p40) and light chain (p35) subunits (Gately et al. 1998) and plays a central role in Th1 development and NK cell activation (Jana et al. 2003; Kobayashi et al. 1989). IL-23 is formed by the combination of p40 and p19 subunits and both IL-12 and IL-23 are critical for anti-bacterial host defense and have also been linked to autoimmune diseases, including multiple sclerosis and arthritis (Gee et al. 2009; Goriely et al. 2008; McAleer and Kolls 2011). IL-27 is comprised of a p28 subunit and Epstein-Barr virus induced gene 3 (EBI3),(Ghilardi et al. 2004; Goriely et al. 2008; Lankford and Frucht 2003) and antagonizes the actions of IL-23 to inhibit Th17 development (Ivanov et al. 2007). TLR ligands, in particular CpG DNA engagement of TLR9, induce the expression of several IL-12 family subunits including p40, p35, and p19, and the crosstalk between TLRs and IFN-γ influences both p40 and p35 production (Bode et al. 2009; Ma et al. 1996; Re and Strominger 2001). Based on their wide array of functions, IL-12 family members are key players in dictating the nature of CNS inflammatory responses (Gran et al. 2004).
The objective of this study was to define the degree of crosstalk between TLR2 and TLR9 signaling in the context of intact bacteria and how these receptors regulate IL-12 family member production in microglia. Collectively, our results demonstrate that TLR9 triggers IL-12 family member production in response to intact Gram-positive bacteria, which becomes exaggerated in the absence of TLR2 signaling. These findings have identified a novel pathway of TLR crosstalk and highlight the importance of examining cellular responses to intact bacteria in addition to purified PAMPs, since the former is more reflective of events encountered during CNS bacterial infection in vivo.
MATERIALS AND METHODS
Primary microglial cultures
TLR2 knockout (KO) mice were generously provided by Dr. Shizuo Akira (Osaka University, Japan) and backcrossed with C57BL/6 animals for a minimum of eight generations prior to use in these studies, with C57BL/6 mice used as wild type (WT) controls (Charles River, Frederick, MD). Primary mixed glial cultures were prepared on the same day from the cerebral cortices of neonatal TLR2 KO and WT mice as previously described (Esen and Kielian 2007). Microglia were harvested from mixed glial cultures using a differential shaking technique with a purity of > 98%. The animal use protocol, approved by the University of Nebraska Medical Center Animal Care and Use Committee, is in accord with the National Institutes of Health guidelines for the use of rodents.
Bacterial strains, TLR ligands, and inhibitors
A USA300 community-acquired methicillin-resistant S. aureus (CA-MRSA) isolate recovered from a patient with a fatal brain abscess was kindly provided by Dr. Costi Sifri (University of Virginia School of Medicine, Charlottesville, VA)(Sifri et al. 2007). Citrobacter koseri strain 4036, originally isolated from the cerebrospinal fluid of an infant with meningitis, was kindly provided by Dr. J.G. Vallejo (Baylor College of Medicine, Houston, TX) (Liu and Kielian 2009). Other bacterial isolates including S. aureus Newman, Streptococcus pneumoniae, Streptococcus pyogenes, Staphylococcus epidermidis, Pseudomonas aeruginosa, and Escherichia coli were provided by Dr. Paul Dunman (University of Rochester Medical Center, Rochester, NY). Bacterial strains were heat-inactivated as previously described and used to stimulate microglia at 107 colony forming units (cfu)/well (Kielian et al. 2002). In some experiments, microglia were exposed to live bacteria at a multiplicity of infection (MOI) of 1:1 for 6 h to prevent bacterial overgrowth of microglial cultures. Microglia were also treated with purified TLR ligands, including stimulatory and inhibitory ODNs (ODNs 1826 and 2088, respectively), Pam3CSK4, and PGN-derived from S. aureus based on previously optimized concentrations (verified low endotoxin; all from Invivogen, San Diego, CA) (Gurley et al. 2008). Inhibitors for phosphatidylinositol 3-kinase (PI3K; wortmannin), MAP Kinase Kinase (MAPKK; PD98059), c-Jun N-terminal kinase (JNK; SP600125), and MAP Kinase Kinase 1/2 (MEK1/2; U0126) were purchased from Invivogen. Inhibitors for spleen tyrosine kinase (Syk) and IL-1R-associated kinase 1/4 (IRAK1/4) were purchased from Calbiochem (San Diego, CA).
Enzyme-linked immunosorbent assay (ELISA)
Comparisons in cytokine and chemokine expression between TLR2 WT and KO microglia were performed using standard sandwich ELISA kits according to the manufacturer's instructions (OptEIA mouse IL-12p40, IL-12p70, and CCL2/MCP-1, BD Biosciences, San Diego, CA; IL-23 and IL-27, eBiosciences, San Diego, CA).
Cell Viability Assays
To assess whether various inhibitors exhibited any toxic effects on microglia, standard MTT assays were performed as previously described based on the mitochondrial conversion of (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) into formazan crystals (Kielian et al. 2005a).
Illumina oligonucleotide microarray
Transcriptional profiling of S. aureus-stimulated TLR2 WT and KO microglia was performed as previously described using Illumina Sentrix MouseRef-8 Expression BeadChips containing 24,000 well-annotated RefSeq transcripts with approximately 30-fold redundancy (Garg et al. 2009). For statistical analysis, expression data were filtered to include only probes with a consistent signal on each chip; the probe original signal filter value was established at detecton p-value < 0.02. The resulting dataset was next analyzed with DIANE 6.0 (an overview of DIANE can be found online at http://www.grc.nia.nih.gov/branches/rrb/dna/diane_software.pdf). Using DIANE, the results were normalized with a Z-Score transformation (Cheadle et al. 2003) followed by principal component analysis (PCA). To determine gene expression changes caused by each specific RNA comparison, Z-Scores for paired treatment groups were compared using the Z-Ratio statistic (Cheadle et al. 2003):
Expression changes for individual genes were considered significant if they met four criteria: Z-Ratio above 1.5 or below -1.5; false detection rate (FDR) (Tusher et al. 2001) of less than 0.30; a p-value statistic for Z-Score replicability below 0.05; and mean background-corrected signal intensity greater than zero. A total of 1,700 differentially expressed genes were identified as significant (p < 0.05) based on Z-scores and organized into common pathways. Although numerous genes were differentially expressed, only a subset is presented (i.e. fold-change or Z-score > 2.0) to limit the size of tables. A complete listing of microarray results has been placed in the GEO repository (accession number GSE24935; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24935).
Microarray bioinformatics analysis
Gene expression values, provided as Z-scores, were taken from the microarray data and analyzed by the Parameterized Analysis of Gene Enrichment (PAGE) algorithm (Kim and Volsky 2005) to organize genes according to known biological pathways based on data sets provided by the Gene Ontology and MIT Broad Institutes. Pathways were identified if they encompassed a minimum of three differentially expressed genes in the microarray data set. A p-value ≤ 0.05 and FDR of ≤ 0.3 were used as cutoff criteria for the selection of significant pathways. In addition, a network of TLR2-regulated genes was constructed using Ingenuity Pathways Analysis IPA (Ingenuity Systems, Redwood City, CA) and/or Pathway Studio 6.1 to demonstrate various molecules that are both positively and negatively impacted following TLR2 loss.
Quantitative Real-Time RT-PCR (qRT-PCR)
Total RNA from TLR2 WT and KO microglia was isolated using the TriZol reagent and treated with DNase 1 (both from Invitrogen, Carlsbad, CA) before use in qRT-PCR studies. ABI Assays-on-Demand™ Taqman kits for TNF receptor associated factor 6 (TRAF6), IFN-β, and TGF-β were used to confirm gene expression levels identified by microarray analysis. Calculations were normalized against cycle thresholds for the housekeeping gene GAPDH and are presented as the fold-induction (2−ΔΔCt) value relative to unstimulated microglia.
Statistical Analysis
Significant differences in cytokine production between TLR2 KO versus WT microglia across multiple experimental groups were determined by one-way analysis of variance (ANOVA) followed by the Holm-Sidak method for multiple pair-wise comparisons using Sigma Stat (SPSS Science, Chicago, IL). For all analyses, a p-value of less than 0.05 was considered statistically significant.
RESULTS
TLR2 loss elevates IL-12 family cytokine production upon exposure to intact S. aureus
Previous work from our laboratory demonstrated that IL-12p40 production was significantly elevated in TLR2 KO microglia in response to intact S. aureus but not the TLR2 ligand PGN (Kielian et al. 2005a). To further investigate this relationship and determine whether it extended to additional IL-12 family members, TLR2 WT and KO microglia were exposed to intact heat-inactivated S. aureus, whereupon inflammatory cytokine production was assessed. TLR2 KO microglia produced significantly more IL-12p40, IL-12p70, and IL-27 in response to intact S. aureus compared to WT cells (Fig. 1A-C, respectively). In contrast, the production of other inflammatory mediators, such as CCL2, was equivalent between TLR2 KO and WT microglia (Fig. 1D), indicating that the absence of TLR2 signaling only affects the expression of a subset of inflammatory genes. IL-23 expression was also examined; however, it was consistently below detectable levels in all treatment groups (i.e. < 15.6 pg/ml; data not shown). Interestingly, the same trend of elevated IL-12 family member expression was also observed in TLR2 KO macrophages (Supplemental Figs. 1 and 2), suggesting a conserved regulatory response among these two mononuclear phagocyte populations.
Figure 1. TLR2 loss leads to elevated IL-12 family cytokine production in microglia upon exposure to intact S. aureus.

Primary TLR2 WT and KO microglia were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to 107 cfu heat-inactivated S. aureus for 24 h, whereupon IL-12p40 (A), IL-12p70 (B), IL-27 (C) and CCL2 (D) production was quantitated by ELISA. Significant differences between TLR2 KO versus WT microglia are indicated by asterisks (*, p < 0.001). Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across three separate experiments.
Exaggerated IL-12 family member expression by TLR2 KO microglia is selective for Gram-positive bacteria
To address the concept of specificity in tailoring immune responses to distinct pathogens (Gekara et al. 2009; Santos-Sierra et al. 2006; Vance et al. 2009), we next examined whether heightened IL-12 family member cytokine release in TLR2 KO microglia extended to other Gram-positive or Gram-negative bacteria. Interestingly, TLR2 KO microglia over-produced IL-12p40, p70, and IL-27 in response to several live Gram-positive bacterial strains, including multiple S. aureus isolates, Streptococcus pneumonia, and Streptococcus pyogenes (Fig. 2). Exaggerated IL-12 family production in the absence of TLR2 signaling was not influenced by a virulence factor produced by viable organisms since similar responses were also observed with heat-inactivated bacteria (Fig. 1 and data not shown). Microglia treated with live S. aureus produced less IL-12p40 compared to heat-killed bacteria (i.e. 0.5 vs. 10 ng/ml, respectively). This may be explained, in part, by the different intervals used for microglial exposure to live versus heat-killed organisms (i.e. 6 and 24 h, respectively) to prevent culture overgrowth with the former. This provided additional time for IL-12p40 accumulation in cultures exposed to heat-killed bacteria; therefore, it is difficult to make direct comparisons between each treatment paradigm. In contrast, TLR2 KO and WT microglia produced similar levels of IL-12p40, p70, and IL-27 upon exposure to various Gram-negative bacteria including Pseudomonas aeruginosa, Escherichia coli, and Citrobacter koseri, which are abundant in the TLR4 ligand LPS (Fig. 3). This selectivity was also observed in TLR2 KO macrophages (Supplemental Figs. 2 and 3), highlighting the fact that both microglia and macrophages are capable of pathogen discrimination and that TLR2 loss only impacts IL-12 family member production in response to Gram-positive but not Gram-negative bacteria.
Figure 2. Exaggerated IL-12 family member production by TLR2 KO microglia is conserved upon exposure to various streptococcal and staphylococcal species.

Primary TLR2 WT and KO microglia were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to 2 × 105 cfu/well of live Gram-positive bacteria for 6 h, whereupon IL-12p40, IL-12p70, and IL-27 production was quantitated by ELISA. Significant differences between TLR2 KO versus WT microglia are indicated by asterisks (*, p < 0.05). Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across three separate experiments.
Figure 3. Gram-negative bacteria do not augment IL-12 family member production in TLR2 KO microglia.

Primary TLR2 WT and KO microglia were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to 2 × 105 cfu/well of live Gram-negative bacteria for 6 h, whereupon IL-12p40, IL-12p70, and IL-27 production was quantitated by ELISA. Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across three separate experiments.
Identification of differentially expressed genes following TLR2 loss using microarray and pathway analysis
Due to the myriad of potential pathways that could be responsible for the increase in IL-12 family member expression in TLR2 KO microglia upon exposure to intact S. aureus, microarray analysis was performed to identify probable candidates that could be targeted in follow-up studies. Although we had previously performed microarray analysis on TLR2 WT and KO microglia utilizing a targeted array of approximately 240 genes, the microarray used in the current study encompassed the entire mouse genome. Microarray analyses, filtered to only include significant (p < 0.05) changes in gene expression, revealed roughly 1,700 differentially regulated genes between TLR2 WT and KO microglia, a subset of which were organized into functional groups to identify potential pathways that may be responsible for exaggerated IL-12 family member production (Table 1). This analysis revealed that TLR2 loss led to significant perturbations in several signaling pathways, including MAPK and interferon regulatory factors (IRFs), providing candidate signaling cascades that may be responsible for regulating IL-12 expression in microglia following exposure to intact Gram-positive bacteria. Similar changes in gene profiles between TLR2 WT and KO microglia were observed at both 6 and 12 h following S. aureus exposure (data not shown).
Table 1.
Analysis of differential gene expression in primary TLR2 KO versus WT microglia in response to intact S. aureus
| Gene Name* | Accession # | Z-score (TLR2 KO vs. TLR2 WT) |
|---|---|---|
| Signaling | ||
| SOX12 | NM_011438.2 | 4.65 |
| MAP2K1 | NM_008927.3 | 4.16 |
| RAB10 | NM_016676.4 | 3.93 |
| Jak1 | NM_146145.2 | 3.89 |
| c-Fos | NM_010234.2 | 3.78 |
| RAB9 | NM_019773.1 | 3.37 |
| REL | NM_009044.2 | 3.125 |
| RIN2 | NM_028724.3 | 3.11 |
| SOX21 | NM_177753.2 | 3.03 |
| Rgs-1 | NM_015811.1 | 2.79 |
| NFIB | NM_008687.2 | 2.65 |
| NFKBIB | NM_010908.3 | 2.64 |
| RhoB | NM_007483.2 | 2.46 |
| MAP4K4 | NM_008696.2 | 2.23 |
| VAV3 | NM_020505.2 | 2.23 |
| RelB | NM_009046.2 | 2.04 |
| ASAH1 | NM_019734.1 | -7.14 |
| UNC93B | NM_019449.1 | -5.66 |
| PIK3AP1 | NM_031376.3 | -4.93 |
| RAB7 | NM_009005.1 | -4.77 |
| PLD3 | NM_011116.1 | -7.77 |
| IRF1 | NM_008390.1 | -4.53 |
| SGK3 | NM_177547.3 | -4.42 |
| IRF9 | NM_008394.2 | -4.07 |
| IRAK3 | NM_028679.3 | -2 |
| Chemokine | ||
| CXCL9 | NM_008599.3 | 5.59 |
| CXCL4 | NM_019932.2 | 3.85 |
| CXCL1 | NM_008176.1 | -4.73 |
| Cytokine | ||
| IFN-β | NM_010510.1 | 1.52 |
| TNFRSF1B | NM_011610.1 | -8.3 |
| G-CSF | NM_009971.1 | -6.93 |
| IL-10 | NM_010548.1 | -4.53 |
| TGF-β1 | NM_009369.3 | -3.53 |
| Receptor | ||
| AIF1 | NM_019467.2 | 3.38 |
| CSF2RB2 | NM_007781.2 | 2.56 |
| IL-10RB | NM_008349.4 | 2.53 |
| TLR2 | NM_011905.2 | -18.7 |
| EMR1 | NM_010130.3 | -7.64 |
| P2RX4 | NM_011026.2 | -4.86 |
| LOX | NM_010728.1 | -4.12 |
| TLR8 | NM_133212.2 | -3.88 |
| Enzymes | ||
| PTGS2 | NM_011198.2 | 4.45 |
| PTGES3 | NM_019766.4 | 4.38 |
| CTSA | NM_001038492.1 | 3.85 |
| CTSS | NM_021281.1 | 3.83 |
| ADAM9 | NM_007404.1 | 3.74 |
| MMP3 | NM_010809.1 | 4.1 |
| CAT | NM_009804.1 | 2.0 |
| CTSZ | NM_022325.4 | 1.7 |
| NOS2 | NM_010927.3 | -6.36 |
| TIMP-1 | NM_011593.2 | -5.55 |
| MMP13 | NM_008607.1 | -4.97 |
| Miscellaneous | ||
| RTN1 | NM_153457.6 | 6.1 |
| VCAM-1 | NM_011693.2 | 2.2 |
MAP2K1, mitogen-activated protein kinase kinase 1; RAB10, Ras-related protein Rab-10; Jak1, Janus kinase 1; RAB9, Ras-related protein Rab-9; RIN2, Ras and Rab interactor 2; Rgs-1, regulator of G-protein signaling 1; NFIB, nuclear factor I/B; NFKBIB, NF-kappa-B inhibitor beta; RhoB, Ras homolog gene family, member B; MAP4K4, mitogen-activated protein kinase kinase kinase kinase 4; VAV3, guanine nucleotide exchange factor VAV3; ASAH1, acid ceramidase; UNC93B, transmembrane endoplasmic reticulum protein UNC-93B; PIK3AP1, Phosphoinositide 3-kinase adapter protein 1; RAB7, Ras-related protein Rab-7; PLD3, phospholipase D3; IRF1, interferon regulatory factor 1; SGK3, serum/glucocorticoid regulated kinase-like; IRF9, interferon regulatory factor 9; IRAK3, interleukin-1 receptor-associated kinase 3; CXCL9, monokine induced by IFN-γ (MIG); CXCL4, platelet factor-4 (PF4); CXCL1, keratinocyte-derived cytokine (KC); IFN-β, interferon-beta; TNFRSF1B, tumor necrosis factor receptor superfamily member 1B; G-CSF, granulocyte-colony stimulating factor; IL-10, interleukin-10; TGF-β1, transforming growth factor-β1; AIF1, allograft inflammatory factor 1; CSF2RB2, colony stimulating factor 2 receptor, beta 2; IL-10RB, interleukin-10 receptor beta; TLR2, Toll-like receptor 2; EMR1, EGF-like module-containing mucin-like hormone receptor-like 1; P2RX4, P2X purinoceptor 4; LOX, lectin-like oxidized LDL receptor; TLR8, Toll-like receptor 8; PTGS2, prostaglandin-endoperoxide synthase 2, cyclooxygenase-2 (COX-2); PTGES3, prostaglandin E synthase 3; CTSA, cathepsin A; CTSS, cathepsin S; ADAM9, disintegrin and metalloproteinase domain-containing protein 9; MMP3, matrix metalloproteinase-3; CAT, catalase; CTSZ, cathepsin Z; NOS2, inducible nitric oxide synthase; TIMP-1, tissue inhibitor of metalloproteinase-1; MMP-13, matrix metalloproteinase-13; RTN1, reticulon-1; VCAM-1, vascular cellular adhesion molecule-1.
Pathway analysis was also performed to identify cascades that were affected by TLR2 loss in response to intact bacteria. As expected, numerous pathways were augmented in TLR2 WT microglia following S. aureus exposure, many of which are involved in proinflammatory mediator signaling (Table 2). Although S. aureus does not possess LPS, pathways related to LPS signaling were increased, which was not surprising since several Gram-positive motifs, including PGN and lipoteichoic acids, can trigger similar proinflammatory pathways. The loss of TLR2 led to increased expression of negative regulatory signal transduction pathways as well as factors influencing myeloid dendritic cell differentiation (Table 2).
Table 2.
Pathway analysis of genes differentially expressed in primary TLR2 KO versus WT microglia in response to intact S. aureus
| Pathways increased in TLR2 WT microglia | |
|---|---|
| GO term | GO ID |
| Inflammatory response | GO:0006954 |
| Protein sumoylation | GO:0016925 |
| Immune response | GO:0006955 |
| GTPase activity | GO:0003924 |
| GTP binding | GO:0005525 |
| Cell motility | GO:0006928 |
| Positive regulation of apoptosis | GO:0043065 |
| Clathrin adaptor complex | GO:0030131 |
| Diacylglycerol binding | GO:0019992 |
| LPS-mediated signaling pathway | GO:0031663 |
| Response to LPS | GO:0032496 |
| Leukotriene biosynthetic process | GO:0019370 |
| Protein serine/threonine kinase activity | GO:0004674 |
| Clathrin coated vesicle | GO:0030136 |
| Pathways increased in TLR2 KO microglia | |
|---|---|
| GO term | GO ID |
| Negative regulation of signal transduction | GO:0009968 |
| Myeloid dendritic cell differentiation | GO:0002284 |
To visualize the cadre of molecular targets for TLR2 in microglia, an interaction map was formulated (Fig. 4). Interestingly, TLR2 signaling was shown to negatively impact several molecules including the transcription factor Rel, catalase (CAT), matrix metalloproteinase-3 (MMP-3), and vascular cellular adhesion molecule-1 (VCAM-1). Indeed, as shown by our microarray analysis, the loss of TLR2 signaling in TLR2 KO microglia led to increases in these same molecules (Table 1). The interaction map of molecules that are both positively and negatively regulated by TLR2 in microglia highlights the complexity whereby the receptor can impact multiple cellular responses (Fig. 4). Microarray analysis also revealed that the production of some cytokines unrelated to the IL-12 family was also altered in the absence of TLR2 signaling. For example, multiple interferon regulated genes were significantly changed, as well as genes involved in TGF-β expression (Table 1). These differences were confirmed by qRT-PCR, where IFN-β levels were significantly increased in TLR2 KO microglia, whereas TGF-β was reduced (Fig. 5). Collectively, microarray analysis identified candidate pathways that were targeted in subsequent studies to elucidate mechanisms contributing to exaggerated IL-12 family member production in TLR2 KO microglia.
Figure 4. Interaction network depicting molecules both positively and negatively impacted by TLR2.

Molecules that are regulated by TLR2 in microglia following S. aureus exposure are presented with their putative roles defined at the left. Molecules that are positively affected by TLR2 are delineated with arrows, whereas those that are negatively regulated by TLR2 are depicted by blunted lines. Molecules that were upregulated in the microarray analysis are depicted in red and those shown in purple were downregulated.
Figure 5. TLR2 differentially regulates inflammatory genes in microglia.

TLR2 WT and KO microglia were seeded at 2 × 106 cells per well in 6-well plates and incubated overnight. The following day, cells were stimulated with 107 heat-inactivated S. aureus/well for 6 or 12 h, whereupon total RNA was isolated and examined for IFN-β (A) or TGF-β (B) expression by qRT-PCR. Gene expression levels were calculated after normalizing cytokine signals against GAPDH and are presented as the fold-induction (2−ΔΔCt) value relative to unstimulated microglia (mean ± SD from three independent experiments). Significant differences between TLR2 KO versus WT microglia are indicated by asterisks (*, p < 0.05).
Loss of TLR2-dependent signaling leads to unchecked TLR9 activation and heightened IL-12 production in microglia
Since CpG DNA in intact bacteria can activate TLR9, a potent inducer of IL-12p40, its role in regulating IL-12 family member production was next examined. Similar to what was observed with intact S. aureus, triggering TLR9 activation with CpG ODN treatment also led to exaggerated IL-12p40 expression in TLR2 KO microglia (Fig. 6). As expected, IL-12p40 release in TLR2 KO microglia was completely attenuated in response to Pam3CSK4 (Fig. 6), in agreement with our previous studies with PGN, since both represent TLR2 ligands. Similar findings were obtained for IL-12p70 and IL-27 (data not shown).
Figure 6. IL-12p40 expression is exaggerated in TLR2 KO microglia following exposure to intact S. aureus and the TLR9 ligand ODN.
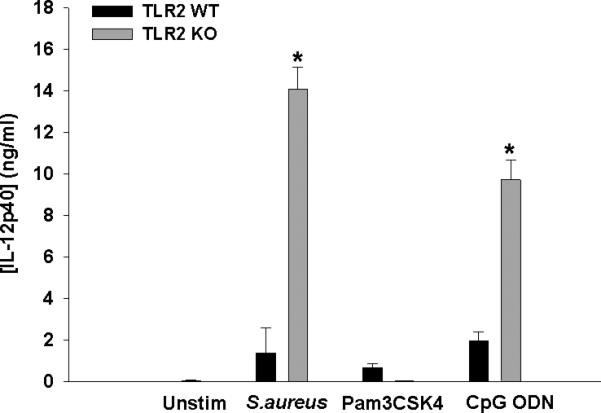
Primary TLR2 WT and KO microglia were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to heat-inactivated S. aureus (107 cfu/well), Pam3CSK4 (1 μg/ml), or CpG ODN (0.1 μM) for 24 h, whereupon IL-12p40 production was quantitated by ELISA. Significant differences between TLR2 KO versus WT microglia are indicated by asterisks (*, p < 0.001). Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across three separate experiments.
To further demonstrate the involvement of TLR9, microglia were exposed to an inhibitory ODN that blocks TLR9 signaling (Krieg et al. 1998; Stunz et al. 2002). Importantly, inhibitory ODN treatment of TLR2 KO microglia prevented exaggerated IL-12 production typically elicited by intact S. aureus with minimal effect on WT cells (Fig. 7). Further evidence to suggest TLR2-TLR9 crosstalk in regulating microglial IL-12 family member production was demonstrated by the finding that cytokine secretion was equivalent in TLR9 KO and WT microglia following S. aureus exposure, where TLR2 signaling remained intact (Fig. 8). Similar findings were obtained with TLR2 KO macrophages, where inhibitory ODN treatment was also capable of restoring IL-12 production to WT levels (Supplemental Fig. 4). Collectively, these findings indicate that TLR2 normally regulates TLR9-dependent signaling to control IL-12 production by an, as of yet, unidentified mechanism.
Figure 7. Inhibitory ODN ablates exaggerated IL-12p40 expression in TLR2KO microglia exposed to intact S. aureus.
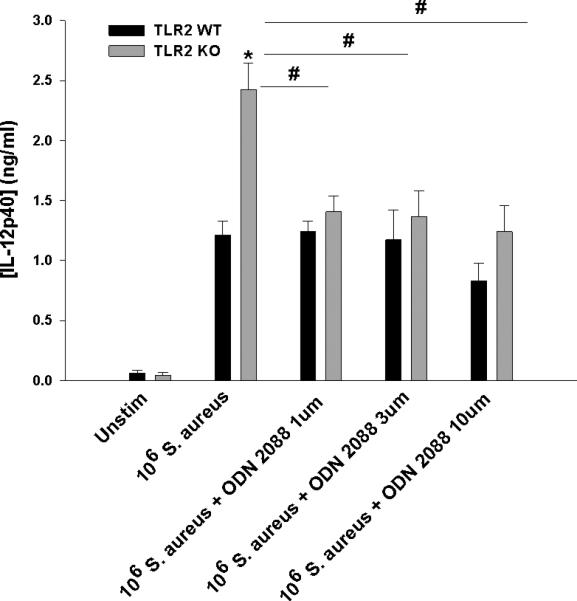
Primary TLR2 WT and KO microglia were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to heat-inactivated S. aureus (106 cfu/well) ± the indicated concentrations of the inhibitory CpG ODN 2088 for 24 h, whereupon IL-12p40 production was quantitated by ELISA. Significant differences between TLR2 KO versus WT microglia are indicated by asterisks (*, p < 0.05), whereas differences between TLR2 KO microglia treated with S. aureus only versus S. aureus + ODN 2088 are indicated by hatched signs (#, p < 0.05). Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across two separate experiments.
Figure 8. IL-12 family member production is not exaggerated in TLR9 KO microglia.

Primary WT, TLR2 KO, and TLR9 KO microglia were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to heat-inactivated S. aureus (107 cfu/well) ± the inhibitory CpG ODN 2088 for 24 h, whereupon IL-12p40 and IL-12p70 production was quantitated by ELISA. Significant differences between TLR2 KO versus WT and TLR9 KO microglia are indicated by asterisks (*, p < 0.05), whereas differences between TLR2 KO microglia treated with S. aureus only versus S. aureus + inhibitor are indicated by hatched signs (#, p < 0.05). Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across two separate experiments.
Heightened IL-12p40 production by TLR2 KO microglia is influenced by PI3K but not SYK or IRAK4 signaling pathways
After establishing that excessive IL-12 family member production occurred in the context of intact TLR9 but absence of TLR2 signaling, we next investigated signal transduction pathway(s) that may impact this phenomenon. Four signaling cascades can be triggered following TLR engagement, namely NF-κB, MAPK, PI3K, and members of the IFN regulatory factor (IRF) family (Kawai and Akira 2010). Of the four signaling pathways, our microarray analysis identified increases in members of the PI3K and MAPK family (Table 1). Based on this, we elected to first inhibit PI3K, a major signaling molecule for TLR2 activation and IRAK4, which is further upstream in the pathway. SYK was chosen to examine possible inflammasome involvement due to reported SYK activation of NLRP3-containing inflammasomes and downstream inflammatory cytokine production (Gross et al. 2009). Inhibition of PI3K signaling by wortmannin attenuated IL-12p40 production in TLR2 KO microglia in a dose-dependent manner and returned expression to nearly WT levels at the highest dose examined without any evidence of toxicity (Fig. 9A and B). In contrast, although SYK and IRAK1/4 inhibitors attenuated IL-12p40 production at various doses in both TLR2 KO and WT microglia, neither was able to return cytokine production in the former to WT levels (Fig. 9C and E). In addition to IL-12p40, IL-12p70 and IL-27 production was similarly affected by the inhibitors tested (data not shown). Taken together, these findings indicate that PI3K signaling contributes to the exaggerated production of IL-12 family cytokines in the absence of TLR2.
Figure 9. The ability of intact S. aureus to augment IL-12p40 production in TLR2 KO microglia is influenced by PI3K but not SYK or IRAK4 signaling pathways.

Primary TLR2 WT and KO microglia were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were pre-treated for 1 h with various concentrations of wortmannin (A and B), SYK (C and D), or IRAK4 (E and F) inhibitors. Following the pre-treatment period, microglia were exposed to 107 heat-inactivated S. aureus/well for 24 h, whereupon IL-12p40 production was quantitated by ELISA. Cell viability was evaluated by MTT assays where the mean OD570 values are reported for each inhibitor tested (B, D, and F). Significant differences between TLR2 WT and KO microglia are denoted by asterisks (*, p < 0.05), whereas differences between TLR2 KO microglia treated with S. aureus only versus S. aureus + inhibitors are indicated by hatched signs (#, p < 0.05). Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across three separate experiments.
MAPK/JNK signaling impacts IL-12 expression in the context of TLR2 loss
Our microarray studies revealed significant increases in molecules belonging to the MAPK/JNK kinase family in TLR2 KO microglia (Table 1). Therefore, this pathway was also targeted to evaluate its role in regulating excessive IL-12 production in response to S. aureus. All three MAPK inhibitors tested (MAPKKK, JNK, and MEK1/2) restored IL-12p40 production in TLR2 KO microglia to levels observed in WT cells in a dose-dependent manner without any evidence of toxicity (Fig. 10). In addition to IL-12p40, IL-12p70 and IL-27 production was similarly affected by the inhibitors tested (data not shown). Collectively, these findings indicate that MAPK/JNK pathways also contribute to exaggerated IL-12 family member production in the absence of TLR2 signaling.
Figure 10. TLR2 delivers a regulatory signal to attenuate IL-12 production in microglia via a MAPK/JNK pathway.

Primary TLR2 WT and KO microglia were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were pre-treated for 1 h with JNK (A and B), MEK1/2 (C and D), or MAPKK (E and F) inhibitors. Following the pre-treatment period, microglia were exposed to 107 heat-inactivated S. aureus for 24 h, whereupon IL-12p40 production was quantitated by ELISA. Significant differences between TLR2 WT and KO microglia are denoted by asterisks (*, p < 0.05), whereas differences between TLR2 KO microglia treated with S. aureus only versus S. aureus + inhibitors are indicated by hatched signs (#, p < 0.05). Cell viability was evaluated by MTT assays where the mean OD570 values are reported for each inhibitor tested (B, D, and F). Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across three separate experiments.
DISCUSSION
Microglial TLR2 plays an important role in sensing S. aureus- derived PAMPs (Kielian et al. 2005a); however, TLR2 loss also leads to increased IL-12 family cytokine production in response to intact bacteria, suggesting that TLR2 may also deliver a regulatory signal(s) to attenuate cytokine release. Our results indicate that TLR9 triggers microglial IL-12 family member production in response to intact Gram-positive bacteria and that the lack of TLR2 signaling leads to exaggerated cytokine release. This conclusion was formulated by several key findings. First, we showed that IL-12 production is similar in TLR9 WT and KO microglia, which both possess intact TLR2. Second, we demonstrated that treatment of TLR2 KO microglia with inhibitory ODN, which blocks TLR9 signaling, prevents excessive IL-12 family mediator release. This data strongly suggests that TLR2-TLR9 crosstalk regulates IL-12 family mediator expression in microglia. Both MAPK and PI3K inhibitors inhibited IL-12 family member production in TLR2 KO microglia, and to a lesser extent, in WT cells. This led us to conclude that MAPK and PI3K signaling are major pathways responsible for IL-12 family member release in the absence of TLR2 signaling. In contrast, we suggest that TLR2 engagement induces comparatively less MAPK and PI3K signaling in WT microglia because of TLR9 crosstalk; therefore, WT cells were only slightly affected by inhibitor treatment. We propose that the mechanism of action leading to exaggerated IL-12 family member production in TLR2 KO microglia is primarily mediated by the inability to regulate ongoing TLR9 signaling in the absence of TLR2 (Fig. 11).
Figure 11. Relationship between TLR2 and signaling pathways regulating IL-12 family member expression in microglia.
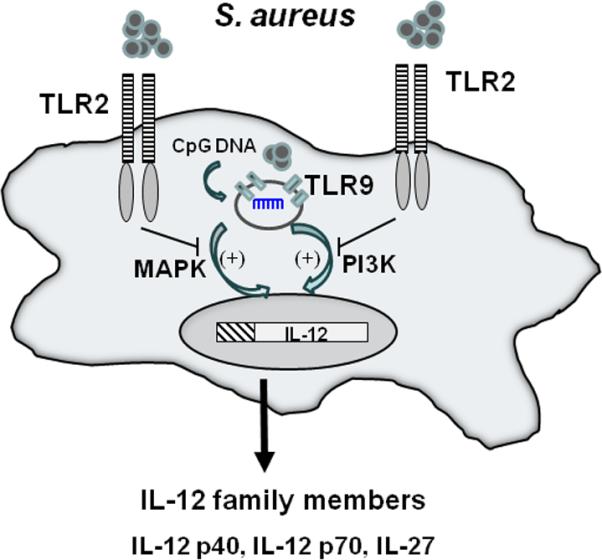
Intact Gram-positive bacteria trigger TLR9 activation via CpG DNA, which stimulates IL-12 family member production. In the absence of TLR2, TLR9 activation remains unchecked, resulting in exaggerated cytokine release. Both PI3K and MAPK pathways influence IL-12 expression in response to intact bacteria.
To the best of our knowledge, this report is a novel example of TLR crosstalk and, by extension, implies that under normal circumstances TLR2 holds TLR9 signaling in check to control IL-12 and IL-27 production. Indeed, several reports have identified molecules that attenuate TLR signaling including Tollip, SARM, TANK, and SHP-1 (Kawai and Akira 2010; O'Neill and Bowie 2007). However, it is important to note that we cannot definitively conclude that TLR2 acts directly on TLR9 to modulate IL-12 production. Indeed, since only intact Gram-positive bacteria elicited heightened IL-12 release and whole bacteria harbor numerous PAMPs, it is likely that multiple receptors are engaged at the microglial surface and/or intracellularly. In this case, the integrated balance of these receptor signaling pathways would dictate IL-12 production and the regulatory effect of TLR2 on TLR9 signaling could be indirect. Indeed, S. aureus has been shown to engage numerous surface receptors including CD36 and mannose receptors that are expressed on microglia (Lefkowitz et al. 1997; Stuart et al. 2005; Zimmer et al. 2003). In addition, Shida et al. demonstrated that TLR2 KO macrophages exhibited increased IL-12 production when exposed to a combination of the Gram-positive bacterium L. casei and PGN, the latter of which would normally trigger TLR2 signaling (Shida et al. 2009). Although not examined by these investigators, TLR9 engagement by bacterial DNA may be responsible for augmenting IL-12 release, as demonstrated in the current study. Additionally, the cytoplasmic pattern recognition receptor NOD2 has been reported to negatively impact IL-12 production (Watanabe et al. 2008) and antagonize TLR2-dependent signaling (Tsai et al. 2011; Watanabe et al. 2004; Watanabe et al. 2006). PARP-1 has also been shown to effect TLR-mediated signaling (Farez et al. 2009); however, the potential impact of these alternative inhibitory mechanisms is beyond the scope of the current study. The finding that Gram-positive but not Gram-negative bacteria augmented IL-12 production in TLR2 KO microglia, yet both harbor CpG DNA, can be explained by a recent study by Bode et al. where TLR4 was shown to negatively regulate IL-12 production (Bode et al. 2009). Since TLR4-mediated signaling remains intact in TLR2 KO microglia, LPS present in the outer cell wall of Gram-negative bacteria is able to trigger TLR4 resulting in controlled IL-12 release, which was observed in the present study.
An intriguing aspect of the current report was the relative selectivity of intact Gram-positive bacteria to augment the expression of IL-12 family cytokines but not other inflammatory mediators. CpG DNA has been shown to induce high levels of IL-12 by trigging epigenetic modifications within the IL-12p40 promoter, including acetylation and nucleosome remodeling as well as transcriptional effects via NF-κB (Bode et al. 2009; Heeg et al. 2008). Furthermore, c-Rel is involved in the induction of p40, p35, and p19 in response to TLR stimulation (Goriely et al. 2008; Sanjabi et al. 2000). Our microarray analysis identified increases in c-Rel (3-fold) and RelB (2-fold) in TLR2 KO microglia exposed to intact S. aureus. These NF-κB family members may play important roles in IL-12 transcriptional regulation in microglia, which appears plausible based on a recent study demonstrating that CpG DNA stimulation resulted in prolonged RelA activity at the IL-12p40 promoter. Conversely, TLR2 ligands were capable of inducing S536 phosphorylation of RelA, which has been implicated in early NF-κB termination (Bode et al. 2009) and may be partially responsible for downregulating IL-12p40 production. By extension, RelA-dependent activation of the IL-12 promoter by CpG DNA in the absence of TLR2 would be expected to proceed unchecked, resulting in exaggerated cytokine expression as observed in the current study. However, this possibility remains speculative at the present time.
The concept of TLR crosstalk, as shown here by the ability of TLR2 and TLR9 to impact IL-12 production, is in agreement with other recent reports. For example, Butchi et al. demonstrated that the synthetic TLR7 agonist, imiqimod, could inhibit TLR9-induced cytokine/chemokine secretion in microglia and astrocytes (Butchi et al. 2010) and TLR7-deficient microglia displayed enhanced cytokine responses to CpG ODN stimulation (Butchi et al. 2010). However, an important distinction between these studies and the current report is our use of intact bacteria, which is expected to more accurately mimic responses encountered during infection in vivo. Another recent study has revealed that PGN can exert negative effects on IL-12p40 production in human monocytes following exposure to intact S. aureus (Chau et al. 2009). The ability of PGN to inhibit cytokine production was attributed to IL-10 and PI3K signaling, although the role of TLR2 was not examined. We did not find any evidence for autocrine/paracrine IL-10 in regulating IL-12p40 production by S. aureus stimulated microglia in this study, since treatment of WT microglia with an IL-10 neutralizing antibody could not mimic the exaggerated IL-12p40 response observed in TLR2 KO microglia (data not shown). Another possible negative regulator is TGF-β, which waits examination in future studies.
In summary, we have demonstrated that TLR2 deficiency results in aberrant IL-12 family member expression by microglia when exposed to intact S. aureus or other Gram-positive species. We propose that the ability of inhibitory ODN to ablate heightened IL-12 production in TLR2 KO microglia is due to the failure to deliver negative regulatory signals to TLR9 in the context of TLR2 deficiency. It is currently unclear whether this is a direct or indirect effect stemming from TLR2 loss. The regulatory signals altered following TLR2 loss discriminate between Gram-positive and –negative bacteria, likely based upon the disparate ligands contained within their cell walls. To the best of our knowledge, this study is the first to report a regulatory effect of TLR2 activation on IL-12 family member production in response to intact Gram-positive bacteria and delineates the specificity of TLR signaling and subsequent inflammatory cytokine expression. This point is particularly relevant given the fact that natural polymorphisms exist in the human population that result in TLR2 inactivation (Kang and Chae 2001; Kang et al. 2002).
Supplementary Material
Supplemental Figure 1. TLR2 loss leads to elevated IL-12 family cytokine production in macrophages upon exposure to intact S. aureus. Thioglycollate-elicited peritoneal macrophages from TLR2 WT and KO mice were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to 107 cfu heat-inactivated S. aureus for 24 h, whereupon IL-12p40 (A), IL-12p70 (B), IL-27 (C) and CCL2 (D) production was quantitated by ELISA. Significant differences between TLR2 KO versus WT macrophages are indicated by asterisks (*, p < 0.001). Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across three separate experiments.
Supplemental Figure 2. Exaggerated IL-12p40 production by TLR2 KO macrophages is conserved upon exposure to various streptococcal and staphylococcal species. Thioglycollate-elicited peritoneal macrophages from TLR2 WT and KO mice were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to 2 × 105 cfu/well of live Gram-positive bacteria for 6 h, whereupon IL-12p40 production was quantitated by ELISA. Significant differences between TLR2 KO versus WT macrophages are indicated by asterisks (*, p < 0.05). Results are reported as the mean ± SD of two independent wells for each experimental treatment and were identical across two separate experiments.
Supplemental Figure 3. Gram-negative bacteria do not augment IL-12p40 production in TLR2 KO macrophages. Thioglycollate-elicited peritoneal macrophages from TLR2 WT and KO mice were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to 2 × 105 cfu/well of live Gram-negative bacteria for 6 h, whereupon IL-12p40 production was quantitated by ELISA. Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across two separate experiments.
Supplemental Figure 4. TLR9 blockade prevents exaggerated IL-12p40 expression in TLR2 KO macrophages. Thioglycollate-elicited peritoneal macrophages from TLR2 WT and KO mice were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to heat-inactivated S. aureus (107 cfu/well) ± the inhibitory CpG ODN 2088 for 24 h, whereupon IL-12p40 production was quantitated by ELISA. Significant differences between TLR2 KO versus WT microglia are indicated by asterisks (*, p < 0.05), whereas differences between TLR2 KO microglia treated with S. aureus only versus S. aureus + ODN 2088 are indicated by hatched signs (#, p < 0.05). Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across two separate experiments.
ACKNOWLEDGEMENTS
This work was supported by the NIH National Institute of Neurological Disorders and Stroke (RO1 NS055385) to T.K. and, in part, by the Intramural Research Program of the NIH, National Institute on Aging. The authors thank Dr. Shizuo Akira for the use of TLR2 KO mice, Drs. Costi Sifri and Paul Dunman for various bacterial isolates, and Ms. Kari Nelson for proofreading the manuscript.
REFERENCES
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Aloisi F. Immune function of microglia. Glia. 2001;36:165–79. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–15. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Bode KA, Schmitz F, Vargas L, Heeg K, Dalpke AH. Kinetic of RelA activation controls magnitude of TLR-mediated IL-12p40 induction. J Immunol. 2009;182:2176–84. doi: 10.4049/jimmunol.0802560. [DOI] [PubMed] [Google Scholar]
- Butchi NB, Du M, Peterson KE. Interactions between TLR7 and TLR9 agonists and receptors regulate innate immune responses by astrocytes and microglia. Glia. 2010;58:650–64. doi: 10.1002/glia.20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau TA, McCully ML, Brintnell W, An G, Kasper KJ, Vines ED, Kubes P, Haeryfar SM, McCormick JK, Cairns E. Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nat Med. 2009;15:641–8. doi: 10.1038/nm.1965. others. [DOI] [PubMed] [Google Scholar]
- Cheadle C, Vawter MP, Freed WJ, Becker KG. Analysis of microarray data using Z score transformation. J Mol Diagn. 2003;5:73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AM, Khader SA. IL-12p40: an inherently agonistic cytokine. Trends Immunol. 2007;28:33–8. doi: 10.1016/j.it.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, Landmann R. Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis. 2002;186:798–806. doi: 10.1086/342845. [DOI] [PubMed] [Google Scholar]
- Esen N, Kielian T. Effects of low dose GM-CSF on microglial inflammatory profiles to diverse pathogen-associated molecular patterns (PAMPs). J Neuroinflammation. 2007;4:10. doi: 10.1186/1742-2094-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farez MF, Quintana FJ, Gandhi R, Izquierdo G, Lucas M, Weiner HL. Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat Immunol. 2009;10:958–64. doi: 10.1038/ni.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Nichols JR, Esen N, Liu S, Phulwani NK, Syed MM, Wood WH, Zhang Y, Becker KG, Aldrich A. MyD88 expression by CNS-resident cells is pivotal for eliciting protective immunity in brain abscesses. ASN Neuro. 2009;1 doi: 10.1042/AN20090004. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gately MK, Renzetti LM, Magram J, Stern AS, Adorini L, Gubler U, Presky DH. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol. 1998;16:495–521. doi: 10.1146/annurev.immunol.16.1.495. [DOI] [PubMed] [Google Scholar]
- Gee K, Guzzo C, Che Mat NF, Ma W, Kumar A. The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm Allergy Drug Targets. 2009;8:40–52. doi: 10.2174/187152809787582507. [DOI] [PubMed] [Google Scholar]
- Gekara NO, Dietrich N, Lyszkiewicz M, Lienenklaus S, Weiss S. Signals triggered by a bacterial pore-forming toxin contribute to toll-like receptor redundancy in gram-positive bacterial recognition. J Infect Dis. 2009;199:124–33. doi: 10.1086/595562. [DOI] [PubMed] [Google Scholar]
- Ghilardi N, Kljavin N, Chen Q, Lucas S, Gurney AL, De Sauvage FJ. Compromised humoral and delayed-type hypersensitivity responses in IL-23-deficient mice. J Immunol. 2004;172:2827–33. doi: 10.4049/jimmunol.172.5.2827. [DOI] [PubMed] [Google Scholar]
- Gillessen S, Carvajal D, Ling P, Podlaski FJ, Stremlo DL, Familletti PC, Gubler U, Presky DH, Stern AS, Gately MK. Mouse interleukin-12 (IL-12) p40 homodimer: a potent IL-12 antagonist. Eur J Immunol. 1995;25:200–6. doi: 10.1002/eji.1830250133. [DOI] [PubMed] [Google Scholar]
- Goriely S, Neurath MF, Goldman M. How microorganisms tip the balance between interleukin-12 family members. Nat Rev Immunol. 2008;8:81–6. doi: 10.1038/nri2225. [DOI] [PubMed] [Google Scholar]
- Gran B, Zhang GX, Rostami A. Role of the IL-12/IL-23 system in the regulation of T-cell responses in central nervous system inflammatory demyelination. Crit Rev Immunol. 2004;24:111–28. doi: 10.1615/critrevimmunol.v24.i2.20. [DOI] [PubMed] [Google Scholar]
- Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–6. doi: 10.1038/nature07965. others. [DOI] [PubMed] [Google Scholar]
- Gurley C, Nichols J, Liu S, Phulwani NK, Esen N, Kielian T. Microglia and Astrocyte Activation by Toll-Like Receptor Ligands: Modulation by PPAR-gamma Agonists. PPAR Res. 2008;2008:453120. doi: 10.1155/2008/453120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeg K, Dalpke A, Peter M, Zimmermann S. Structural requirements for uptake and recognition of CpG oligonucleotides. Int J Med Microbiol. 2008;298:33–8. doi: 10.1016/j.ijmm.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19:409–17. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana M, Dasgupta S, Saha RN, Liu X, Pahan K. Induction of tumor necrosis factor-alpha (TNF-alpha) by interleukin-12 p40 monomer and homodimer in microglia and macrophages. J Neurochem. 2003;86:519–28. doi: 10.1046/j.1471-4159.2003.01864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ME, Draghi DC, Karlowsky JA, Sahm DF, Bradley JS. Prevalence of antimicrobial resistance in bacteria isolated from central nervous system specimens as reported by U.S. hospital laboratories from 2000 to 2002. Ann Clin Microbiol Antimicrob. 2004;3:3. doi: 10.1186/1476-0711-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TJ, Chae GT. Detection of Toll-like receptor 2 (TLR2) mutation in the lepromatous leprosy patients. FEMS Immunol Med Microbiol. 2001;31:53–8. doi: 10.1111/j.1574-695X.2001.tb01586.x. [DOI] [PubMed] [Google Scholar]
- Kang TJ, Lee SB, Chae GT. A polymorphism in the toll-like receptor 2 is associated with IL-12 production from monocyte in lepromatous leprosy. Cytokine. 2002;20:56–62. doi: 10.1006/cyto.2002.1982. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kielian T. Immunopathogenesis of brain abscess. J Neuroinflammation. 2004;1:16. doi: 10.1186/1742-2094-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–30. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia. 2005a;49:567–76. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Haney A, Mayes PM, Garg S, Esen N. Toll-like receptor 2 modulates the proinflammatory milieu in Staphylococcus aureus-induced brain abscess. Infect Immun. 2005b;73:7428–35. doi: 10.1128/IAI.73.11.7428-7435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Mayes P, Kielian M. Characterization of microglial responses to Staphylococcus aureus: effects on cytokine, costimulatory molecule, and Toll-like receptor expression. J Neuroimmunol. 2002;130:86–99. doi: 10.1016/s0165-5728(02)00216-3. [DOI] [PubMed] [Google Scholar]
- Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med. 1989;170:827–45. doi: 10.1084/jem.170.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg AM, Wu T, Weeratna R, Efler SM, Love-Homan L, Yang L, Yi AK, Short D, Davis HL. Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs. Proc Natl Acad Sci U S A. 1998;95:12631–6. doi: 10.1073/pnas.95.21.12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankford CS, Frucht DM. A unique role for IL-23 in promoting cellular immunity. J Leukoc Biol. 2003;73:49–56. doi: 10.1189/jlb.0602326. [DOI] [PubMed] [Google Scholar]
- Lefkowitz DL, Lincoln JA, Lefkowitz SS, Bollen A, Moguilevsky N. Enhancement of macrophage-mediated bactericidal activity by macrophage-mannose receptor-ligand interaction. Immunol Cell Biol. 1997;75:136–41. doi: 10.1038/icb.1997.18. [DOI] [PubMed] [Google Scholar]
- Liu S, Kielian T. Microglial activation by Citrobacter koseri is mediated by TLR4- and MyD88-dependent pathways. J Immunol. 2009;183:5537–47. doi: 10.4049/jimmunol.0900083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Chow JM, Gri G, Carra G, Gerosa F, Wolf SF, Dzialo R, Trinchieri G. The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells. J Exp Med. 1996;183:147–57. doi: 10.1084/jem.183.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAleer JP, Kolls JK. Mechanisms controlling Th17 cytokine expression and host defense. J Leukoc Biol. 2011 doi: 10.1189/jlb.0211099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naesens R, Ronsyn M, Druwe P, Denis O, Ieven M, Jeurissen A. Central nervous system invasion by community-acquired meticillin-resistant Staphylococcus aureus. J Med Microbiol. 2009;58:1247–51. doi: 10.1099/jmm.0.011130-0. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–64. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011;34:269–81. doi: 10.1016/j.tins.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re F, Strominger JL. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J Biol Chem. 2001;276:37692–9. doi: 10.1074/jbc.M105927200. [DOI] [PubMed] [Google Scholar]
- Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST. Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc Natl Acad Sci U S A. 2000;97:12705–10. doi: 10.1073/pnas.230436397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Sierra S, Golenbock DT, Henneke P. Toll-like receptor-dependent discrimination of streptococci. J Endotoxin Res. 2006;12:307–12. doi: 10.1179/096805106X118762. [DOI] [PubMed] [Google Scholar]
- Shida K, Kiyoshima-Shibata J, Kaji R, Nagaoka M, Nanno M. Peptidoglycan from lactobacilli inhibits interleukin-12 production by macrophages induced by Lactobacillus casei through Toll-like receptor 2-dependent and independent mechanisms. Immunology. 2009;128:e858–69. doi: 10.1111/j.1365-2567.2009.03095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifri CD, Park J, Helm GA, Stemper ME, Shukla SK. Fatal brain abscess due to community-associated methicillin-resistant Staphylococcus aureus strain USA300. Clin Infect Dis. 2007;45:e113–7. doi: 10.1086/522171. [DOI] [PubMed] [Google Scholar]
- Stenzel W, Soltek S, Sanchez-Ruiz M, Akira S, Miletic H, Schluter D, Deckert M. Both TLR2 and TLR4 are required for the effective immune response in Staphylococcus aureus-induced experimental murine brain abscess. Am J Pathol. 2008;172:132–45. doi: 10.2353/ajpath.2008.070567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart LM, Deng J, Silver JM, Takahashi K, Tseng AA, Hennessy EJ, Ezekowitz RA, Moore KJ. Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J Cell Biol. 2005;170:477–85. doi: 10.1083/jcb.200501113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stunz LL, Lenert P, Peckham D, Yi AK, Haxhinasto S, Chang M, Krieg AM, Ashman RF. Inhibitory oligonucleotides specifically block effects of stimulatory CpG oligonucleotides in B cells. Eur J Immunol. 2002;32:1212–22. doi: 10.1002/1521-4141(200205)32:5<1212::AID-IMMU1212>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–6. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–51. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- Tsai WH, Huang DY, Yu YH, Chen CY, Lin WW. Dual roles of NOD2 in TLR4-mediated signal transduction and -induced inflammatory gene expression in macrophages. Cell Microbiol. 2011;13:717–30. doi: 10.1111/j.1462-5822.2010.01567.x. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance RE, Isberg RR, Portnoy DA. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe. 2009;6:10–21. doi: 10.1016/j.chom.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidlak D, Mariani MM, Aldrich A, Liu S, Kielian T. Roles of Toll-like receptor 2 (TLR2) and superantigens on adaptive immune responses during CNS staphylococcal infection. Brain Behav Immun. 2010 doi: 10.1016/j.bbi.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, Fuss IJ, Kitani A, Strober W. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118:545–59. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–8. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kitani A, Murray PJ, Wakatsuki Y, Fuss IJ, Strober W. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. 2006;25:473–85. doi: 10.1016/j.immuni.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Zimmer H, Riese S, Regnier-Vigouroux A. Functional characterization of mannose receptor expressed by immunocompetent mouse microglia. Glia. 2003;42:89–100. doi: 10.1002/glia.10196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. TLR2 loss leads to elevated IL-12 family cytokine production in macrophages upon exposure to intact S. aureus. Thioglycollate-elicited peritoneal macrophages from TLR2 WT and KO mice were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to 107 cfu heat-inactivated S. aureus for 24 h, whereupon IL-12p40 (A), IL-12p70 (B), IL-27 (C) and CCL2 (D) production was quantitated by ELISA. Significant differences between TLR2 KO versus WT macrophages are indicated by asterisks (*, p < 0.001). Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across three separate experiments.
Supplemental Figure 2. Exaggerated IL-12p40 production by TLR2 KO macrophages is conserved upon exposure to various streptococcal and staphylococcal species. Thioglycollate-elicited peritoneal macrophages from TLR2 WT and KO mice were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to 2 × 105 cfu/well of live Gram-positive bacteria for 6 h, whereupon IL-12p40 production was quantitated by ELISA. Significant differences between TLR2 KO versus WT macrophages are indicated by asterisks (*, p < 0.05). Results are reported as the mean ± SD of two independent wells for each experimental treatment and were identical across two separate experiments.
Supplemental Figure 3. Gram-negative bacteria do not augment IL-12p40 production in TLR2 KO macrophages. Thioglycollate-elicited peritoneal macrophages from TLR2 WT and KO mice were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to 2 × 105 cfu/well of live Gram-negative bacteria for 6 h, whereupon IL-12p40 production was quantitated by ELISA. Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across two separate experiments.
Supplemental Figure 4. TLR9 blockade prevents exaggerated IL-12p40 expression in TLR2 KO macrophages. Thioglycollate-elicited peritoneal macrophages from TLR2 WT and KO mice were seeded at 2 × 105 cells per well in 96-well plates and incubated overnight. After 24 h, cells were exposed to heat-inactivated S. aureus (107 cfu/well) ± the inhibitory CpG ODN 2088 for 24 h, whereupon IL-12p40 production was quantitated by ELISA. Significant differences between TLR2 KO versus WT microglia are indicated by asterisks (*, p < 0.05), whereas differences between TLR2 KO microglia treated with S. aureus only versus S. aureus + ODN 2088 are indicated by hatched signs (#, p < 0.05). Results are reported as the mean ± SD of three independent wells for each experimental treatment and were identical across two separate experiments.