

Abstract
Cellular aging is linked to deficiencies in efficient repair of DNA double strand breaks and authentic genome maintenance at the chromatin level. Aging poses a significant threat to adult stem cell function by triggering persistent DNA damage and ultimately cellular senescence. Senescence is often considered to be an irreversible process. Moreover, critical genomic regions engaged in persistent DNA damage accumulation are unknown. Here we report that 65% of naturally occurring repairable DNA damage in self-renewing adult stem cells occurs within transposable elements. Upregulation of Alu retrotransposon transcription upon ex vivo aging causes nuclear cytotoxicity associated with the formation of persistent DNA damage foci and loss of efficient DNA repair in pericentric chromatin. This occurs due to a failure to recruit of condensin I and cohesin complexes. Our results demonstrate that the cytotoxicity of induced Alu repeats is functionally relevant for the human adult stem cell aging. Stable suppression of Alu transcription can reverse the senescent phenotype, reinstating the cells' self-renewing properties and increasing their plasticity by altering so-called “master” pluripotency regulators.
Key words: adult stem cells, senescence, SINE/Alu transposons, DNA damage, H2AX, ChIP-seq, cohesin, condensin, PML body, induced pluripotency
Introduction
The stem cell hypothesis of aging1,2 postulates that the gradual and coordinated age-related loss of DNA damage repair capacity results in DNA damage accumulation over time; by triggering the persistent DNA damage response (DDR), cellular senescence and ultimately, stem cell aging. Although senescence was previously reported to be an irreversible process,3 several line of evidence indicate that senescence can be suppressed pharmacologically.4,5 Recent investigations of the distribution of the DNA damagespecific chromatin modification (γH2AX) in the genome of cycling Saccharomyces cerevisiae cells revealed “fragile” genomic locations.6 This suggests that mapping sites of γH2AX enrichment could be fruitful to pinpoint at-risk genomic elements in other genomes, perhaps indicating that specific genomic regions or elements exist where DNA damage is less efficiently repaired in aged cells. These functional elements have not been previously identified in the context of human adult stem cell aging. Here, we report that Alu retrortansposal RNA drives persistent DDR through the alteration of chromatin structure and that this event is functionally important as an integral component to human adult stem cell senescence ex vivo.
Results
Human adipose-derived stem cells undergo senescence upon ex vivo expansion.
Here, we isolated adult adipose derived mesenchymal stem cells (hADSCs) (Fig. S1A), and investigated the mechanisms leading to their aging upon ex vivo expansion. As previously reported, upon isolation, hADSCs exhibit consistent self-renewing (SR) capacity until population doubling (PD)17, after which they displayed characteristic aging phenotypes7–9 (Fig. S1B). By PD37 (SR), hADSC cultures manifested a dramatic downregulation of the genes encoding cell cycle progression functions (Table 1) and accumulated non-dividing giant cells expressing the enzyme lysosomal pH 6 senescence-associated β-galactosidase (SA-β-Gal)10 (Fig. 1A). Cells self-renewed poorly, as determined by incorporation of 3[H] thymidine and bromodeoxyuridine (BrdU) into DNA (Fig. S1C and D). As hADSCs approached senescence, both mediators of DDR, phosphorylated form of histone variant H2AX (γH2AX),11 and p53 binding protein-1 (53BP1),12 form characteristic persistent DNA damage foci (Fig. 1B and C).11,13,14 The presence of these foci drastically increased from very rare in self-renewing ADSCs, to almost 90% in hADSCs approaching senescence (SEN hADSCs) (Fig. 1C).
Table 1.
Transcriptional changes in the cell cycle genes in senescent hADSC
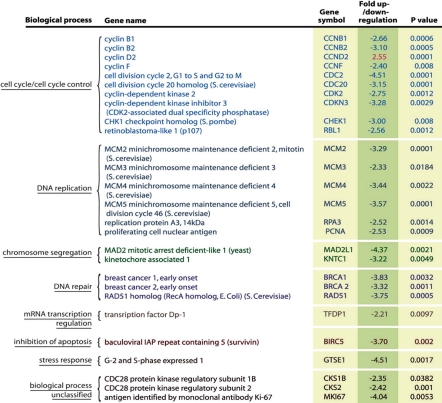 |
The list represents only the genes with statistically significant change of p < 0.005. The human cell cycle QPCR array was used for the profiling of 96 cell cycle related genes. Three independent experiments were performed for profiling senescent and self-renewing hADSC samples.
Figure 1.

Ex vivo aging of hADSCs is associated with formation of transcriptionally active persistent DNA damage foci and upregulation of transcriptional activity from Alu retrotransposons. (A) Immunohistochemical detection of senescence-associated β-galactosidase (SA-β-Gal) activity. Examples of hADSCs' morphological changes (10x magnification) shown in inserts. Bar graphs correspond to percentage of SA-β-Gal positive cells with progressive ex-vivo hADSC expansion, based on three independent experiments. Error bars are standard deviations from the mean. (B) DNA damage response (DDR) in senescent hADSCs. Representative immunostaining for the persistent γH2AX (green)/53BP1 (red) foci formation upon senescence of hADSCs. (C) Quantification of accumulation of persistent DNA damage foci with ex-vivo passaging of hADSCs. γH2AX was stained with affinity-purified rabbit polyclonal antibody. Histogram indicates the percentage of the cells with 1, 2, 3 or more than 3 foci. Representative examples are shown below. Foci formation was scored in self-renewing, SR (population doubling less than 17), pre-senescent, preSEN (population doubling more than 29, but less than 38) and senescent, SEN (population doubling greater than 39) hADSCs cultures. The growth curve of ADSC in shown in the Supplemental 1B. n = total number of nuclei counted in all 3 independent experiments. (D) Alu expression in SR and SEN hADSCs. Northern hybridization of self-renewing (SR) and senescent (SEN) hADSCs with Alu oligonucleotide probe. Alu and 7SL are indicated. Total RNA of 2 µg per lane was loaded as described in Materials and Methods. (E) Ribosomal small RNAs can be seen in the ethidium bromide stained gel for loading comparison. The ssRNA ladder sizes are indicated on the right. (F) Persistent γH2AX/53BP1 foci in senescent hADSC are associated with active transcription. Senescent hADSCs were incubated with hallogenated precursor FUr for 10 min in vivo to label nuclear RNA. After fixation, cells were immunolabelled with anti-BrdU antibody (red) to detect FUr incorporation sites in combination with anti-γH2AX (green). Arrows point to co-localization of the persistent DNA damage sites upon senescence with regions of high transcriptional activity. (G) Association of DNA damage foci with transription was further verified by confocal microscopy in co-immunostaing of DNA damage foci depicted by 53BP1 antibodies (green) with PML bodies (blue) and nascent RNA (red). Representative image of a single nucleus is shown. Spatial relationship between FUr incorporation sites, 53BP1 and PML bodies in a single 5 µm confocal section is shown. Image was analyzed by Imaris software and z1, z2 and z3 planes are shown. Cartoon demonstrates the orientations of z1, z2 and z3 planes within single z-section. Confocal sectioning confirms the tight association of nascent transcripts with persistent DNA damage sites in senescent hADSCs.
Senescence of hADSCs is associated with activation of Alu retrotransposons.
In concert with the suggested association between genotoxic stress-induced DDR and retrotransposon activation,15,16 we observed a dramatic increase in Alu transcriptional activity in senescent hADSC (Fig. 1D). We did not observe a generalized upregulation of other major Pol III dependent genes, such as 7SL (Fig. 1D) or rRNA genes (Fig. 1E). Thus, this robust Pol III-dependent transcriptional activation upon senescence of hADSCs appears to specific to Alu retrotransposons.
Persistent DNA damage 53BP-1/γH2AX foci apparently were transcriptionally active in SEN hADSC (Fig. 1F) as indicated by in vivo nascent transcript labeling with FUr17,18 as described in Materials and Methods, and their co-localization with nuclear PML bodies implicated many cellular processes in DNA repair and transcription19–21 (Fig. 1G). These observations are consistent with previous reports indicating a possible involvement of RNA component(s) in 53BP1 foci formation after IR-induced damage in NIH3T3 and HeLa cells.22 The transcriptional activity within persistent DNA damage foci seems to be PolIII dependent, because treatment with the inhibitor of Pol-III, tagetin, abrogates FUr-incorporation in the transcripts and impairs 53BP1 focus-forming capacity in SEN hADSCs (Fig. 2). These results suggest a possible connection between transcriptional activity of Alu retrotransposons and genomic locations engaged in persistent DNA damage foci formation.
Figure 2.

Persistent γH2AX/53BP1 foci in senescent hADSCs are associated with Pol III transcription. Senescent hADSCs were either cultured in the presence of 10 µM inhibitor of Pol III transcriptional activity, tagetin, for 2 h at 37°C (+tagetin) or in the absence of the inhibitor treatment (-tagetin). Nuclear RNA was labeled by addition of 2 mM FUr to the culture for 10 min at 37°C. After fixation, cells were immunolabelled with anti-BrdU antibody (red) to detect FUr incorporation sites in combination with anti-53BP1 (green). Double labeling experiment revealed FUr incorporation sites exclusively localized with persistent DNA damage sites throughout entire depth of z-stack images. Tagetin inhibition of Pol III dependent transcription results in complete disappearance of FUr incorporation, and loss of compaction of the DNA damage sites as detected by more defuse 53BP1 staining. Fifteen optical sections are shown for confocal analysis of immunostained samples for each of the experimental conditions. Bottom images show the merged signals with DAPI staining.
Genome-wide ChIP-seq location analysis of persistent DNA damage in hADSCs.
To identify genomic loci directly engaged in DDR in SEN hADSCs, we performed genome-wide profiling of hADSC chromatin using chromatin immunoprecipitation with an antibody against the γH2AX modified histone, followed by next generation sequencing (ChIP-seq) on the ABI SOLiD platform (Fig. 3A and Sup. Material). Asynchronous SR samples of hADSCs were used in similar ChIP-seq experiments to establish an overall representation of γH2AX modified chromatin and to define the locations of repairable DNA damage that does not affect the SR properties of the cells. Four replicate ChIPseq experiments each were performed for SR and SEN cells, and the resulting sequence tags were mapped to the human genome reference sequence (Table 2 and Sup. Material). Genomic maps of γH2AX sequence tags were subject to outlier removal, noise reduction, data merging from replicate experiments and clustering into contiguous mono-nucleosomal, midsize and large clusters (Sup. Material). The resulting genomic distributions of γH2AX modified nucleosomes and clusters were compared for SR and SEN cells (Fig. 3A) in order to assess the positional and quantitative changes in γH2AX-modified chromatin associated with these phenotypes.
Figure 3.

Genome-wide location analysis of γH2AX. (A) Relative chromosomal distributions of γH2AX tags in self-renewing and senescent cells illustrated for chromosomes 10 and 21. γH2AX tag enrichment levels, calculated as the log2 ratio of position-specific tag counts normalized by the genomic background, are shown for self-renewing (blue) and senescent (red) cells. Relative differences in γH2AX tag enrichment levels between cells, calculated as the absolute values of the differences in cell stage specific enrichment levels, are shown below the individual cell tracks. Below the difference tracks, the chromosomal locations of large clusters of γH2AX modified sites are shown for the self-renewing (blue) and senescent (red) cell types. (B) Differences in the relative γH2AX enrichment levels for self-renewing (SR) vs. senescent (SEN) cells across various genomic features. The absolute values of the normalized differences in γH2AX tag counts between cell types are shown on the y-axis. Blue bars show genomic features that have higher fractions in SR cells, and red bars show genomic features that have higher fractions in SEN cells. Error bars show the standard errors for the 4 replicates based on binomial distributions. (C) γH2AX enrichment levels in peritelomeric regions for self-renewing (SR-blue) and senescent (SEN-red) cell lines. Chromosome ends (telomeres) are shown at the origin of the x-axis, which then extends into the chromosome arms. Average γH2AX enrichment levels are calculated as the log2 ratio of the position-specific tag counts normalized to the genomic background averaged over all chromosome arms. (D) γH2AX enrichment levels in pericentric regions for self-renewing (SR-blue) and senescent (SEN-red) cell lines. Centromeres are shown as a gap centered on the x-axis, which extends into the chromosome arms in either direction. Average γH2AX enrichment levels are calculated as the log2 ratio of the position-specific tag counts normalized to the genomic background averaged over all chromosomes. (E) Fractional differences in the numbers of γH2AX sites within large clusters in pericentric regions between senescent vs. self-renewing cells. The fractional differences for γH2AX sites within large clusters between cell phenotypes are shown on the y-axis. Blue bars show chromosomes that have higher fractions of γH2AX sites within large clusters in SR cells, and red bars show chromosomes that have higher fractions of γH2AX sites within large clusters in SEN cells. Error bars show the standard errors for the four replicates based on binomial distributions.
Table 2.
γH2AX ChIP-seq tag and mapping data
| Experiment | Total tags | Mapped tags | % Mapped tags |
| SR1 | 25650863 | 6427476 | 25.058% |
| SR2 | 22864646 | 6740098 | 29.478% |
| SR3 | 23172888 | 7153067 | 30.868% |
| SR4 | 20554101 | 6610682 | 32.162% |
| SEN1 | 32114970 | 7915256 | 24.647% |
| SEN2 | 28469416 | 7569634 | 26.589% |
| SEN3 | 26694829 | 4911085 | 18.397% |
| SEN4 | 23942531 | 4780551 | 19.967% |
Our mapping analysis revealed that γH2AX-modified chromatin was distributed non-randomly, with the majority of γH2AX modifications (SR: 65.4%, SEN: 65.2%) mapping to transposable elements (TEs), most of which were Alu (SINE), L1 (LINE) or LTR retrotransposons (Table S1). Alu elements were the most enriched for γH2AX-modified nucleosomes in SR cells compared with SEN cells, whereas L1s had the greatest relative increase of γH2AX-modified nucleosomes in SEN cells (Fig. 3B). The majority of the mono-nucleosomal γH2AX sites (67.3% in SR and 65.8% in SEN) were associated with the retrotransposal portion of the genome, with SINEs contributing 34.9% and 30.0% and LINEs accounting for 32.4% and 35.8% of these sites in SR and SEN hADSCs, respectively (Table S1). Because the retrotransposon portion of the genome is known to impede the progression of replication machinery,23–25 a portion of γH2AX detected in asynchronously dividing cells (the SR sample) might be caused by replication-fork pausing or collapse as described previously in yeast.23,26–28 Our data also indicates that the majority of damage in SEN cells is not related to unrepaired damage accumulation caused by the collision of replication forks and transcriptional complexes in SR cells, since gene density and γH2AX tag density were significantly positively correlated in SR samples and negatively correlated in SEN samples (Table S2).
We also observed substantial differences between the SR and SEN samples in the amount of (as well as the genomic features occupied by) large contiguous γH2AX clusters. The total amount of large γH2AX clusters dropped more than 3-fold comparing SR (37,467,892 bp) and SEN (11,243,429 bp) samples (Table S1). In SEN non-replicating samples, large γH2AX clusters were shifted to intergenic regions, with a decrease in intragenic sequences from 47.9% in SEN samples to 35.2% in SR (Table S1). Together, these data suggested a quantitative and qualitative drift in DNA damage susceptibility occurring upon human adult stem cell ex vivo aging.
Pericentric chromatin is a source of the persistent DNA damage in senescent hADSCs.
Bioinformatic analysis allowed us to pinpoint genomic locations responsible for the formation of persistent DNA damage foci. Further analysis of ChIP-seq tags that bore a specific telomeric repeat sequence motif (TTA GGG/AAT CCC) among all sequencing tags (mapped uniquely combined with unmapped due to their repetitive nature) revealed that SR and SEN cell samples had similar numbers of γH2AX telomeric tags (1.66% vs. 1.63%). Similarly, peri-telomeric regions showed 1.5-fold (SR) and 2.1-fold (SEN) depletion in γH2AX-modified nucleosomes (Figs. 3C, S2 and S3), suggesting that in contrast to senescence of somatic cells driven by telomeric erosion-associated DNA damage accumulation,29–31 global telomeric and peri-telomeric DNA damage was an unlikely causal factor for hADSCs senescence.
Our data revealed that the distribution of γH2AX modified chromatin at pericentric regions was markedly different from those seen for peri-telomeres. There was a substantial over-representation of γH2AX modified chromatin in pericentromeres (Figs. 3D and S2), and a number of individual chromosomes exhibited highly enriched levels of pericentromeric γH2AX accumulation (Fig. 3E). Overall, pericentromeres exhibited 2.0- and 2.1-fold enrichments in γH2AX accumulation for SR and SEN cells, respectively (Fig. S3), thus indicating intrinsic susceptibility to DNA damage (i.e., “fragility”) of these chromosomal regions. Pericentromeres were not only enriched for γH2AX, but they were also one of the few genomic features that showed relatively greater γH2AX presence in SEN cells (Fig. 3B and D). Chromosomes 6, 10, 2 and 19 demonstrated the most significant enrichment of large γH2AX clusters in SEN cells (Fig. 3E). An increase in pericentric γH2AX accumulation on a subset of human chromosomes in SEN hADSCs could be indicative of: (1) defects in the assembly of kinetochores that provoke spindle-generated DNA damage, resulting in a compromised spindle-assembly checkpoint (SAC),32 and/or, (2) unresolved DSB, possibly due to changes in the characteristics of their pericentric heterochromatin and the molecular machinery responsible for its repair. Both of these events, acting separately or synergistically, could potentially trigger senescence of human adult stem cells.
We investigated both of these possibilities. We tested whether or not centromeric histone H3 variant (CENP-A), which is required to recruit many other centromere and kinetochore proteins,33 colocalizes with persistent DNA damage foci upon senescence. By using confocal fluorescent microscopy, we observed that CENP-A co-localized with persistent γH2AX/53BP1 foci in nearly 75% of the cases upon senescence of hADSCs (Fig. 4B and C), indicating that centromeric regions are embedded within persistent γH2AX/53BP1 DNA damage foci. Similar overlapping immunostaining patterns were observed when human anti-kinetochore serum (α-CREST) was prepped with γH2AX (Fig. S4A) or anti-53BP1 (Fig. S4B) antibodies. No detectable co-localization of centromeric regions with either 53BP1 (Fig. 4A) or γH2AX (data not shown) was observed in interphase of SR hADSCs, consistent with the fact that no prolonged cell cycle arrest was triggered in these cells (Fig. S1B–D), nor were γH2AX/53BP1 foci formed (Fig. 1B and C). These data further support the notion that the pericentric chromatin, rather then centromeres, per se, might drive DNA damage susceptibility, since no defects in the incorporation of CENP-A in centromeric chromatin or inner kinetochore assembly were observed.
Figure 4.

Centromeric regions are associated with persistent DNA damage foci in senescent hADSCs. (A) Immunofluorescent labeling of self-renewing hADSCs. Cells were seeded on coverslips and co-stained with anti-CENP-A (green) and anti-53BP1 (red) antibodies. DAPI staining is shown in blue. Confocal image of representative interphase nucleus is shown as separate channels and as a merged image. Four µm z-slice was analyzed by Imaris software and z1, z2 and z3 projections are shown. Self-renewing hADSCs show no focal damage associated sites. Centromeric areas are clearly visible. (B) Persistent DNA damage is associated with centromeres. Senescent hADSCs were seeded on coverslips and as in (A) and immunostaining was performed. An arrow depicts the co-localization of a centromeric region with persistent, senescence-associated γH2AX/53BP1 damage foci. Scale bar, 4 µm. (C) Quantification of CENP-A and 53BP1 co-localization in senescence. Senescent hADSCs were stained with antibodies against CENP-A (green) and 53BP1 (red) and DAPI (blue). Total of 200 cells were scored from three independent experiments. Error bars represent ± SAM. Example of higher magnification of the image is shown. Scale bar 1 µm. Images were analyzed by IMARIS software with optical sections representation as depicted on the left. Single 5 mm confocal section is shown. Image was analyzed by Imaris software and z1, z2 and z3 planes are shown. Cartoon demonstrates the orientations of z1, z2 and z3 planes within single z-section.
Increased transcriptional activity of Alu reptotransposons interferes with cohesin and condensin I recruitment at the damage sites.
The pericentric regions of the chromosomes shown to accumulate large clusters of γH2AX upon senescence (Figs. 3D, E, S3 and Table S1) are known to be enriched for Alu retrotransposal repeats.34 In multiple organisms, it has been shown that transcription from within pericentric heterochromatin is under tight control during the cell cycle and is required for the formation and maintenance of heterochromatin and sister chromatid cohesion.35,36 Because the persistent DNA damage foci associated with pericentric regions (Figs. 4B, C and S4) are actively transcribed (Fig. 1F and G) and sensitive to Pol-III inhibition (Fig. 2), and because we observed significant upregulation of transcriptional activity of Alu repeats upon hADSCs ex vivo aging (Fig. 1D), we next investigated if increased transcriptional activity of Alu retrotransposons could be recorded from the pericentric regions of the chromosomes susceptible for DNA damage accumulation in SEN hADSCs (Fig. 3E).
Chromosome 10 showed an increase of pericentric γH2AX tag accumulation in the SEN sample, including an excess of large clusters, whereas chromosome 21 had more pericentric γH2AX in the SR sample (Fig. 3E). RT-PCR was used to evaluate the transcriptional activity of individual Alu and MIR SINE sequences co-located with large γH2AX clusters in the pericentromeric regions of chromosome 10 (Fig. 5A) and chromosome 21 (Fig. S5). No transcriptional activity from MIR on ch10 was observed in either SR or SEN samples, whereas the transcription of Alus on both chromosomes were recorded in both cases. The chromosome 10 Alu sequence associated with a large cluster of γH2AX in the SEN sample was significantly upregulated in SEN vs. SR cells (Fig. 5A). On the other hand, AluJb and AluSx on chromosome 21 did not demonstrate significant differences in their transcriptional activity between SEN vs. SR cells (Fig. S5). Thus, the upregulation of Alu transcriptionl activity correlates with the presence of persistent DNA damage in SEN samples. Together, these data along with multiple reports from yeast and mammals,37–41 suggest a general role for the Pol-III transcription in genome organization, and prompted us to consider the hypothesis that, similar to centromeric tRNA genes in yeast,42 pericentric Alu retrotransposons in human adult stem cells might be integral to centromere function in self-renewing hADSCs, and an increase in the rate/level of their transcriptional activity upon ex vivo aging of hADSCs might be toxic to the chromatin environment and cause senescence.
Figure 5.

Pericentromere-associated persistent DNA damage in senescent hADSCs correlates with transcriptional upregulation of Alu retrotransposons and defects in recruitment of cohesin and condensin I complexes. (A) On the left: pericentric region of human chromosome 10. Red box on the chromosome ideogram is depicting the region (Chr10: 41,800,000–42,050,000) given in higher magnification. Annotation for the repeats in this location is given under the tracks according to the UCSC genome browser. Tracks for distribution of γH2AX nucleosomes are shown in blue for self-renewing, and in red for senescent hADSCs. Large γH2AX clusters are shown as solid boxes. On the right: schematic diagram showing the relative positions of Alu retrotransposons to the large senescence-associated, persistent γH2AX cluster from this genomic location. Grey and black boxes are indicative of the positions of MIR and Alu repeats. Representative example of transcriptional activity of these repeats as assessed by strand-specific RT-PCR is shown. Graph represents the quantitation of the RT-PCR results from seven independent experiments. Upregulation of transcriptional activity from Alu repeat in senescent cells correlates with formation of persistent DNA damage cluster upon senescence. Data shown are mean ± SEM **p = 0.00.014 (B) Cartoons are representing the composition of cohesin, condensin I and Eco1-dependent conversion of cohesin to cohesive state. (C) Loss of cohesin and condensin I in the pericentric location of persistent DNA damage in senescent hADSC. ChIP analysis of the pericentric repeats on chromosome 10 in self-renewing (blue bars) and senescent (red bars) hADSCs. Same repeats as in (A) were assessed as locations for recruitment of TFIIIC, Eco1 as well as components of cohesin (Rad21) and condensin I (CAP-H) complexes (n = 3, ±SEM). Schematic representation of the subunits of the cohesin and condensin I complexes, as well as a cartoon of previously reported function of Eco1, are shown. *p < 0.02, **p < 0.2.
Transcriptional activity of SINE/Alu retrotransposons is driven by the Pol-III transcriptional complex,43 and its general transcription factor TFIIIC, reportedly involved in the maintenance of high-order chromatin structure37,44–45 through structural organization of chromatin cohesion and condensation in yeast.42 We observed stable recruitment of TFIIIC to the analyzed retrotransposal repeats on chromosome 10 in both SR and SEN hADSC. Surprisingly, we observed a senescence-associated increase in the recruitment of TFIIIC to the MIR retrotransposal element located within the large γH2AX cluster even in the absence recorded MIR transcription (Fig. 5C). The specific association of cohesin with Alu elements has been reported in human cells,46 and multiple lines of evidence point to the critical role of the cohesin complex in DNA repair by homologous recombination47 as well as in spindle assembly and chromosome segregation during mitosis.34,47,48 Both cohesin and condensin complexes are indispensable for sister chromatid cohesion49 and DNA repair by homologous recombination (HR),50,51 while their deficiency has been shown to be responsible for triggering G1 and G2-M DNA damage checkpoints.52 Also, spindle assembly checkpoint (SAC) acts semi-redundantly with the DNA damage checkpoint to enact long-term cell cycle arrest in the presence of unresolved pericentric DSB, thereby preventing the segregation of the damaged chromatin.53–56
Therefore, we elected to investigate whether the reported toxicity of Alu RNA is related to an inability to repair pericentric DNA damage due to deficiencies in recruitment of the key complexes to the chromatin within or in direct proximity to DNA damage clusters. We investigated the recruitment of components of the cohesin complex Scc1/Rad21/Mcd1, the condensin I complex (Cap-H), and the histone acetylase Eco1, a regulator of Scc1/Rad21/Mcd1 cohesive state57–59 (Fig. 5B), to the Alu repeats in the vicinity of a persistent pericentric γH2AX cluster on chromosome 10. Data obtained by conventional ChIP analysis demonstrated a statistically significant loss in the recruitment of Scc1/Rad21/Mcd1 and Cap-H to the MIR and Alu transcription units during senescence of hADSC (SEN red bars in Fig. 5C) when compared with the status of the same genomic locations in SR cells (SR blue bar in Fig. 5C). Surprisingly, the recruitment of the histone acetylase Eco1 was largely correlated with the formation of persistent γH2AX clusters in senescence, and appeared to be Scc1/Rad21/Mcd1-independent. This suggests another, not yet identified, role for the Eco1 protein in DDR, different from its previously reported participation in the modulation of cohesiveness of Scc1/Rad21/Mcd1.46,57–59 Together these data further support the correlation between an increase in Alu transcription, persistent DNA damage and defects in pericentric recruitments of condensin I (Cap-H) and cohesin (Scc1/Rad21/Mcd1), and suggest an experimental approach to evaluate the functional significance of Alu toxicity in establishing the irreversible3 senescence of hADSCs.
Lentivirus-mediated depletion of the senescence-associated Alu transcripts re-instates the self-renewal of human ADSCs.
In order to evaluate the functional significance of Alu transcription in establishing and/or mediating cellular senescence upon ex vivo aging, we took advantage of the common sequence feature of Alu retrotransposons to generate a number of lenti-virus-delivered shRNA constructs carrying GFP for assessment of transduction efficiency (Fig. 6A and Materials and Methods). hADSCs transduced with lentiGFP expressing sh-132Alu RNA (Fig. 6B) demonstrated a near complete knockdown of the generic Alu transcript (Fig. 6C). hADSCs transduced with lentiGFP (control) or lentiGFP sh-193Alu exhibited little or no change at the Alu transcription level. Surprisingly, SEN hADSCs lines stably expressing sh-132Alu RNA exhibited a dramatically altered morphology and exhibited an increase in proliferation as detected in 3[H] thymidine uptake DNA synthesis experiments (Fig. 6D). In contrast, SEN hADSCs transduced with lentiGFP (control), or treated with polybrene alone (PB), did not show any significant changes in their proliferation rate when compared with wild type (wt) SEN hADSCs (Fig. 6D). These results suggest that forced suppression of Alu transcription was sufficient to overcome persistent DDR in senescent hADSCs. sh-132Alu expressing cells were uniformly positive for proliferation specific antigen Ki67, expression of which is strictly associated with cell proliferation (Fig. 6E). Loss of senescence-associated β-galactosidase activity and disassembly of persistent DNA damage foci (γH2AX) was recorded in sh-132Alu expressing cells. Infected and uninfected cell populations are shown in the same field (Fig. 6F). Faithful re-establishment of DNA synthesis upon knockdown of the Alu transcript was further recorded 96 h after transduction and is sustainable as sh-132Alu expressing cells demonstrate proliferation upon culturing for 40 d or longer (Fig. S6 and data not shown). Unexpectedly, when compared with wt SEN hADSCs, prior SEN hADSCs lines stably expressing sh-132Alu RNA demonstrated a statistically significant upregulation of pluripotency factors Nanogand Oct4 as detected by qPCR analysis (Fig. 6G). The levels of Nanog and Oct4 in wt SEN hADSCs were below the threshold of the qPCR method.
Figure 6.

Stable knockdown of generic Alu transcript in senescent human adult stem cells restores cell's proliferative properties and induces iPS-like phenotype. (A) Model of Alu retrotransposon. Secondary structure of generic Alu RNA. Regions for shRNA design are shown in blue. (B) Representative example of the efficiency of lentiviral transduction of hADSCs depicted by GFP. (C) Nothern blot hybridization of the RNA recovered from hADSCs cells stably expressing sh-RNA against Alu. Senescent hADSCs were infected with lentiGFP sh-193Alu, lentiGFP sh-132Alu or control no sh-RNA insert lentiGFP. RNA was isolated after 24 hrs post transduction and northern hybridization was performed with a Alu specific oligonucleotide. Senescent hADSCs stably expressing sh-132Alu show near complete knockdown of the Alu transcripts. (D) Proliferative properties of senescent hADSCs were reinstated in the cells upon stable knockdown of Alu transcripts. 3[H] thymidine uptake is shown. Senescent cells (wt) or senescent cells transduced with lentiGFP (control) or lentiGFP sh-132Alu were pulse-labeled with 1 µCi of 3[H] thymidine for 24 hrs either 24 or 96 h post infection. Data shown are mean ± SEM for triplicate measurements. (E) Immunostaining of prio senescent lentiGFP sh-132Alu ADSCs with proliferation marker Ki67. (F) Senescent ADSC cultures infected with lentiGFP sh-132Alu demonstrate a significant loss in senescence-associated SA-B-Gal activity and persistent DNA damage foci, indicated by gH2AX staining in GFP-expressing cells. (G) Expression of pluripotency markers Nanog and Oct4 was measured by qPCR analysis in senescent hADSCs (wt) and in hADSC with reversed-senescent phenotype upon stable knockdown of Alu transcription (lentiGFP sh-132Alu). RNA was isolated from the cells 96 h post infection. Results are expressed as relative quantity (DCt). Samples were normalized against β--actin. Data are shown as mean ± SEM (n = 3) ***p = 6.98e-05, *p = 0.03. (H) Model of Alu transcriptional toxicity upon ex-vivo human ADSCs aging.
Discussion
In conclusion, our data support the observation that a retrotransposal portion of the genome is particularly sensitive to DSBs, a type of DNA breakage recently linked directly to genomic rearrangements in cancer.15,60–63 Unexpectedly, our results imply the importance of a fine-turned balance between beneficial (allowed) rate of Alu retrotransposons transcription, critical for mediating pericentromeric integrity during cell cycle progression and the toxicity of elevated transcriptional activity from the Alu retrotransposons, ultimately leading to human adult stem cell aging ex vivo. Our data indicate that such toxicity of Alu transcription is associated with loss of the recruitment cohesin and condensin I complexes to pericentromeric regions, thus blocking efficient repair of pericentric regions, leading to formation of persistent DNA damage foci (Fig. 6H). This impairment of condensin I loading is likely due to an absence in the recruitment of condensin complex, rather than a deficiency in Eco1, consistent with previous reports demonstrating that condensin is indispensible for cohesin loading onto the chromosome64 and that depletion of the Cap-H subunit of the condensin I complex results in defects associated with alterations in the structural integrity of centromere-proximal heterochromatin in Drosophila.65 Our data further linking together physiological impairment in recruitment of cohesin/condensin I complexes and Alu toxicity to persistent DDR upon adult stem cell ex vivo aging, suggests that such regulation might be an integral part of cellular aging or age-associated disease.66 Our results, in concert with previously published data,4,5 also challenge the notion that cellular senescence and ultimately cellular aging, is always an irreversible process.
Materials and Methods
Antibodies.
Primary antibodies used were: 53BP1 (Bethyl Laboratories #A300-273A), BrdU (BD, 7580), CD31 (Invitrogen #HMCD3101), CD44 (Invitrogen #HMCD4401), CD45 (Invitrogen #HMCD4501), CD105 (Invitrogen #HMCD10520), Phospho-Chk1 (Ser345) (CST #2341), Phospho-Chk2 (Thr68) (CST #2661), CENP-A (Abcam #ab13939), human anti-centromeric autoantibody, α-CREST (Antibodies Inc., #15-235), γH2AX (Millipore #05-636). Secondary antibodies were AlexaFlour® conjugated donkey antibodies (Invitrogen).
Human ADSC isolation and expansion.
Human adipose derived stem cells were isolated from human subcutaneous white adipose tissue collected during liposuction procedures. The lipoaspirate was suspended in Hank's Buffered Salt Solution (HBSS), 3.5% Bovine Serum Albumin (BSA), 1% Collagenase, type II (Sigma) in 1:3 w/v ratio and shaken at 37°C for 50 min. The cells were filtered through a 70 µm mesh cell strainer (BD Falcon #352350), treated with Red Blood Cell Lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.3), and expanded ex vivo in DMEM/F12 complete medium (DMEM/F12, 10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, 2.5 µg/ml amphotericin B; Invitrogen) in 10% CO2 at 37°C and passaged at 80% confluency, changing medium every 72–96 h. Cumulative population doublings were calculated by summing the population doublings (PD = log(N/N0) × 3.33, where N0 is the number of cells plated in the flask and N is the number of cells harvested at this passage) across multiple passages as a function of the number of days it was grown in culture.
Surface marker characterization.
Five × 105 cells each were incubated for 30 min on ice in the dark with fluorochrome-conjugated antibodies (CD31, CD44, CD45 and CD105; Invitrogen) in PBS with 1% BSA (Sigma), washed and analyzed in a Guava EasyCyte Mini System (Guava Technologies, Millipore). Data analysis was done with FlowJo software (Tree Star, Ashland, OR).
Assessment of cellular senescence.
Senescence-associated β-galactosidase activity assay was done as described in manufacturer's kit (BioVision).
Immunofluorescence.
1–3 × 104 cells/well in 4-well slides were fixed with 4% paraformaldehyde and permeabilized with PBS, 0.5% Triton X-100. The blocking and antibody incubations were performed in 4% normal donkey serum (NDS; Jackson Immunochemicals) in PBS. The nuclei were counter-stained with 100 ng/ml 4′, 6-diamidino-2-phenylindole (DAPI; Sigma), and the slides were mounted in ProLong® Gold antifade aqueous mounting medium (Invitrogen). Analysis was performed as described in details in Supplemental Materials and Methods.
Expression profiling.
Human Cell Cycle qRCR Array (SABiosciences #PAHS-020C-2) was used for the profiling of self-renewing (SR) and senescent (SEN) hADSCs. Details of the experimental protocol and data analysis are given in Supplemental Materials and Methods online.
ChIP-Seq: nucleosomal ChIP and SoLiD library preparation.
Nucleosomal ChIP was performed as described in reference 31, and in Supplemental Materials and Methods online. Samples were sequenced on SOLiD™ System 2.0 (Applied Biosystems) according to manufacturer's protocol.
5-Fluorouridine labeling for RNA transcript detection in situ.
Senescent hADSCs, 1 × 104 cells/well in 4-well slides, were treated with 2 mM 5-Fluorouridine (FUr; Sigma) for 10 min. The nuclei were exposed with ice-cold CSK buffer (100 mM KCl, 300 mM sucrose, 10 mM Pipes pH 6.8, 3 mM MgCl2, 1 mM EGTA, 1.2 mM phenylmethylsulfonyl fluoride) with 0.5% Triton X-100 and fixed with 4% paraformaldehyde. Immunostaining was performed as described above for immunoflourescence.
RT PCR analysis of retrotransposal transcription and qPCR.
Genomic coordinates of the Alu elements tested in RT-PCR were from the March 2006 Assembly (NCBI36/hg18) of the Human Genome Browser at UCSC (http://genome.ucsc.edu); MIR: chr10: 41922815–41922906, Alu: chr10:41928992–41929118, AluJb: chr21:10141132–10141429 and AluSx: chr21:10145344–10145644. 100 ng of total RNA was used with the RT2 First Strand Kit (SABiosciences) per reaction. The primers for first strand synthesis are at locations outside of the Alu element sequences (external or reverse primers, Table S3) and forward primers within the Alu element sequence (internal forward primers, Table S3). RPL13A was used as a positive control.
Lentiviral shRNA constructs.
Lentiviral shRNA constructs to knockdown genetic Alu transcript were designed as follows: oligonucleotides Lenti sh-132 Alu RNA 5′-GAT CCC CCC ACC ACG CCC GGC TAA TTT TCA AGA GAA ATT AGC CGG GCG TGG TGG TTT TTG GAA A-3′ and Lenti sh-193 Alu RNA 5′-GAT CCC CCC CGG GTT CAA GCG ATT CTT TCA AGA GAA GAA TCG CTT GAA CCC GGG TTT TTG GAA A-3′, were annealed with equal amounts of their complementary strands, creating restriction site specific overhangs for cloning, and ligated into HindIII and BglII digested, gel purified pENTR/pTER+ vector.67 The constructs were confirmed by sequencing (sense strand sequence is shown above). Equal amounts of each constructs was mixed with pLenti-CMV-GFP DEST vector67 in LR Clonase reaction to recombine cloned shRNA production elements into a destination vector according to manufacturer's instructions (Invitrogen). The produced lentiviral plasmid was transformed into E. coli Stbl3 cells (Invitrogen) for amplification.
Lentiviral production and transduction.
293T cells were grown in DMEM complete medium (DMEM, high glucose, 10% FBS, 0.1 M nonessential amino acid, 6 mM L-glutamine, 1 mM pyruvate, 100 U/ml penicillin, 100 µg/ml streptomycin) for lentiviral production and transfected for 12 h with pLenti sh-132Alu or pLenti sh-193Alu and pCgpV, pRSV-Rev and pCMVVSV-G helper plasmids (Allele Biotechnology) in 2:1:1:1 molar ratio using Lipofectamine 2000 (Invitrogen) according to standard protocol.67 Medium was collected at 48 and 72 h and filtered (0.45 µm). Virus was precipitated with PEG and frozen in aliquots (−80°C). Lentiviral transductions were done in complete medium with 5 µg/ml Polybrene (Santa Cruz Biotechnology) for 12 h. Viral titers were determined by comparing GFP positive cells counts to total population.
Proliferation index.
For each condition in 3[H]-thymidine uptake assay, 10,000 cells were treated with 1 µCi 3[H]-thymidine (Perkin-Elmer, Boston, MA) in DMEM/F12 complete for 24 h. DNA was isolated from harvested cells and quantified with NanoDrop (ND-1000; NanoDrop Technologies Inc.). 3[H]-thymidine uptake into cellular DNA was measured with liquid scintillation counter (LS 6500; Beckman Instruments). Lentiviral transduction was done as above, and untreated and polybrene treated controls were performed in parallel.
Northern hybridization.
RNA was isolated from SR and SEN hADSCs (with or without lentiviral transduction) with mir-VANA (Ambion, Invitrogen) kit and 2 mg of total RNA/lane was run on a 7 M Urea, 6% polyacrylamide, TBE. Gel was stained with ethidium bromide, photographed and electroblotted onto Hybond™-N+ (Amersham, GE Healthcare). Hybridizations were performed in 6x SSC, 4x Denhardts', 0.1% SDS at 37°C. Oligonucleotide probes were labeled with Biotin-16-dUTP (Roche) by terminal transferase (NEB). Northern was visualized with streptavidin-HRP (Invitrogen) using ECL Plus western Blotting Detection Reagent (Amersham, GE Healthcare) and Amersham Hyperfilm (GE Healthcare). Oligonucleotide used as probes were:
Alu 132: 5′-CCA CCA CGC CCG GCT AAT TT-3′ and Alu 90: 5′-CGC GCG CCA CCA CGC CCG GCT AAT TTT TGT ATT TTT AGT AGA GAC GGG GTT TCA CCA TGT TGG CC-3′.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Jeri Jenkins for help with manuscript preparation, Max Sarrazin for help with figures, and ABI Life Technologies for reagents and technical assistance for SOLiD instrumentation. For critical reading of the manuscript we are grateful to Drs. B. Kennedy, G. Lithgow and M. Classon. We thank Dr. Denise Muñoz and Justin Winstead for assistance with library preparation and sequencing runs. This work was supported by National Institutes of Health pilot projects on UL1 DE019608, Buck Institute Trust Fund to V.V.L. and by the Alfred P. Sloan Foundation (BR-4839) to I.K.J. V.V.L. designed the experiments; J.W. and I.K.J. developed and applied bioinformatics method for data analysis; G.G., S.L.H., M.A., B.B. and E.L.L. performed the experiments; P.J.C. and M.D. provided the primary cells; B.P. and E.H. mapped the SOLiD data, G.S. provided technical assistance for SOLiD instrumentation; I.K.J., M.G.R. and V.V.L. discussed the results and wrote the paper.
Supplementary Material
References
- 1.Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520–528. doi: 10.1038/nature08982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirkwood TB. Intrinsic ageing of gut epithelial stem cells. Mech Ageing Dev. 2004;125:911–915. doi: 10.1016/j.mad.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 3.d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–522. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 4.Demidenko ZN, Blagosklonny MV. Quantifying pharmacologic suppression of cellular senescence: prevention of cellular hypertrophy versus preservation of proliferative potential. Aging. 2009;1:1008–1016. doi: 10.18632/aging.100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demidenko ZN, Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle. 2009;8:1901–1904. doi: 10.4161/cc.8.12.8810. [DOI] [PubMed] [Google Scholar]
- 6.Szilard RK, Jacques PE, Laramee L, Cheng B, Galicia S, Bataille AR, et al. Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nat Struct Mol Biol. 2010;17:299–305. doi: 10.1038/nsmb.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonab MM, Alimoghaddam K, Talebian F, Ghaffari SH, Ghavamzadeh A, Nikbin B. Aging of mesenchymal stem cell in vitro. BMC Cell Biol. 2006;7:14. doi: 10.1186/1471-2121-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kern S, Eichler H, Stoeve J, Kluter H, Bieback K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood or adipose tissue. Stem Cells. 2006;24:1294–1301. doi: 10.1634/stemcells.2005-0342. [DOI] [PubMed] [Google Scholar]
- 9.Fehrer C, Brunauer R, Laschober G, Unterluggauer H, Reitinger S, Kloss F, et al. Reduced oxygen tension attenuates differentiation capacity of human mesenchymal stem cells and prolongs their lifespan. Aging Cell. 2007;6:745–757. doi: 10.1111/j.1474-9726.2007.00336.x. [DOI] [PubMed] [Google Scholar]
- 10.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 12.Aguilera A, Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204–217. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 13.Stewart GS. Solving the RIDDLE of 53BP1 recruitment to sites of damage. Cell Cycle. 2009;8:1532–1538. doi: 10.4161/cc.8.10.8351. [DOI] [PubMed] [Google Scholar]
- 14.Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudin CM, Thompson CB. Transcriptional activation of short interspersed elements by DNA-damaging agents. Genes Chromosomes Cancer. 2001;30:64–71. doi: 10.1002/1098-2264(2000)9999:9999<::AID-GCC1066>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 16.Hagan CR, Sheffield RF, Rudin CM. Human Alu element retrotransposition induced by genotoxic stress. Nat Genet. 2003;35:219–220. doi: 10.1038/ng1259. [DOI] [PubMed] [Google Scholar]
- 17.Wang C, Politz JC, Pederson T, Huang S. RNA polymerase III transcripts and the PTB protein are essential for the integrity of the perinucleolar compartment. Mol Biol Cell. 2003;14:2425–2435. doi: 10.1091/mbc.E02-12-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen TA, Von Kaenel S, Goodrich JA, Kugel JF. The SINE-encoded mouse B2 RNA represses mRNA transcription in response to heat shock. Nat Struct Mol Biol. 2004;11:816–821. doi: 10.1038/nsmb813. [DOI] [PubMed] [Google Scholar]
- 19.Carbone R, Pearson M, Minucci S, Pelicci PG. PML NBs associate with the hMre11 complex and p53 at sites of irradiation induced DNA damage. Oncogene. 2002;21:1633–1640. doi: 10.1038/sj.onc.1205227. [DOI] [PubMed] [Google Scholar]
- 20.Dellaire G, Ching RW, Dehghani H, Ren Y, Bazett-Jones DP. The number of PML nuclear bodies increases in early S phase by a fission mechanism. J Cell Sci. 2006;119:1026–1033. doi: 10.1242/jcs.02816. [DOI] [PubMed] [Google Scholar]
- 21.Dellaire G, Kepkay R, Bazett-Jones DP. High resolution imaging of changes in the structure and spatial organization of chromatin, gamma-H2A.X and the MRN complex within etoposide-induced DNA repair foci. Cell Cycle. 2009;8:3750–3769. doi: 10.4161/cc.8.22.10065. [DOI] [PubMed] [Google Scholar]
- 22.Pryde F, Khalili S, Robertson K, Selfridge J, Ritchie AM, Melton DW, et al. 53BP1 exchanges slowly at the sites of DNA damage and appears to require RNA for its association with chromatin. J Cell Sci. 2005;118:2043–2055. doi: 10.1242/jcs.02336. [DOI] [PubMed] [Google Scholar]
- 23.de la Loza MC, Wellinger RE, Aguilera A. Stimulation of direct-repeat recombination by RNA polymerase III transcription. DNA Repair (Amst) 2009;8:620–626. doi: 10.1016/j.dnarep.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 24.Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci USA. 2008;105:9936–9941. doi: 10.1073/pnas.0804510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheifele LZ, Cost GJ, Zupancic ML, Caputo EM, Boeke JD. Retrotransposon overdose and genome integrity. Proc Natl Acad Sci USA. 2009;106:13927–13932. doi: 10.1073/pnas.0906552106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Admire A, Shanks L, Danzl N, Wang M, Weier U, Stevens W, et al. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev. 2006;20:159–173. doi: 10.1101/gad.1392506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deshpande AM, Newlon CS. DNA replication fork pause sites dependent on transcription. Science. 1996;272:1030–1033. doi: 10.1126/science.272.5264.1030. [DOI] [PubMed] [Google Scholar]
- 28.Cha RS, Kleckner N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science. 2002;297:602–606. doi: 10.1126/science.1071398. [DOI] [PubMed] [Google Scholar]
- 29.d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 30.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/S0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 31.Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol. 2005;7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- 32.Guerrero AA, Gamero MC, Trachana V, Futterer A, Pacios-Bras C, Diaz-Concha NP, et al. Centromere-localized breaks indicate the generation of DNA damage by the mitotic spindle. Proc Natl Acad Sci USA. 2010;107:4159–4164. doi: 10.1073/pnas.0912143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McClelland SE, Borusu S, Amaro AC, Winter JR, Belwal M, McAinsh AD, et al. The CENP-A NAC/CAD kinetochore complex controls chromosome congression and spindle bipolarity. EMBO J. 2007;26:5033–5047. doi: 10.1038/sj.emboj.7601927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schueler MG, Sullivan BA. Structural and functional dynamics of human centromeric chromatin. Annu Rev Genomics Hum Genet. 2006;7:301–313. doi: 10.1146/annurev.genom.7.080505.115613. [DOI] [PubMed] [Google Scholar]
- 35.Kato H, Goto DB, Martienssen RA, Urano T, Furukawa K, Murakami Y. RNA polymerase II is required for RNAi-dependent heterochromatin assembly. Science. 2005;309:467–469. doi: 10.1126/science.1114955. [DOI] [PubMed] [Google Scholar]
- 36.Grewal SI, Elgin SC. Transcription and RNA interference in the formation of heterochromatin. Nature. 2007;447:399–406. doi: 10.1038/nature05914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noma K, Cam HP, Maraia RJ, Grewal SI. A role for TFIIIC transcription factor complex in genome organization. Cell. 2006;125:859–872. doi: 10.1016/j.cell.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 38.Oki M, Kamakaka RT. Barrier function at HMR. Mol Cell. 2005;19:707–716. doi: 10.1016/j.molcel.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 39.Scott KC, Merrett SL, Willard HF. A heterochromatin barrier partitions the fission yeast centromere into discrete chromatin domains. Current biology: CB. 2006;16:119–129. doi: 10.1016/j.cub.2005.11.065. [DOI] [PubMed] [Google Scholar]
- 40.Lunyak VV, Prefontaine GG, Nunez E, Cramer T, Ju BG, Ohgi KA, et al. Developmentally regulated activation of a SINE B2 repeat as a domain boundary in organogenesis. Science. 2007;317:248–251. doi: 10.1126/science.1140871. [DOI] [PubMed] [Google Scholar]
- 41.Willoughby DA, Vilalta A, Oshima RG. An Alu element from the K18 gene confers position-independent expression in transgenic mice. J Biol Chem. 2000;275:759–768. doi: 10.1074/jbc.275.2.759. [DOI] [PubMed] [Google Scholar]
- 42.Iwasaki O, Noma KI. Global genome organization mediated by RNA polymerase III-transcribed genes in fission yeast. Gene. 2010 doi: 10.1016/j.gene.2010.12.011. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slagel VK, Deininger PL. In vivo transcription of a cloned prosimian primate SINE sequence. Nucleic Acids Res. 1989;17:8669–8682. doi: 10.1093/nar/17.21.8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Young LS, Dunstan HM, Witte PR, Smith TP, Ottonello S, Sprague KU. A class III transcription factor composed of RNA. Science. 1991;252:542–546. doi: 10.1126/science.1708526. [DOI] [PubMed] [Google Scholar]
- 45.Kundu TK, Wang Z, Roeder RG. Human TFIIIC relieves chromatin-mediated repression of RNA polymerase III transcription and contains an intrinsic histone acetyltransferase activity. Mol Cell Biol. 1999;19:1605–1615. doi: 10.1128/mcb.19.2.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hakimi MA, Bochar DA, Schmiesing JA, Dong Y, Barak OG, Speicher DW, et al. A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature. 2002;418:994–998. doi: 10.1038/nature01024. [DOI] [PubMed] [Google Scholar]
- 47.Blower MD, Karpen GH. The role of Drosophila CID in kinetochore formation, cell cycle progression and heterochromatin interactions. Nat Cell Biol. 2001;3:730–739. doi: 10.1038/35087045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernard P, Maure JF, Partridge JF, Genier S, Javerzat JP, Allshire RC. Requirement of heterochromatin for cohesion at centromeres. Science. 2001;294:2539–2542. doi: 10.1126/science.1064027. [DOI] [PubMed] [Google Scholar]
- 49.Sonoda E, Matsusaka T, Morrison C, Vagnarelli P, Hoshi O, Ushiki T, et al. Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells. Dev Cell. 2001;1:759–770. doi: 10.1016/S1534-5807(01)00088-0. [DOI] [PubMed] [Google Scholar]
- 50.Potts PR, Porteus MH, Yu H. Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 2006;25:3377–3388. doi: 10.1038/sj.emboj.7601218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jessberger R. Cohesin's dual role in the DNA damage response: repair and checkpoint activation. EMBO J. 2009;28:2491–2493. doi: 10.1038/emboj.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 54.Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet. 2006;40:209–235. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- 55.Lim HH, Zhang T, Surana U. Regulation of centrosome separation in yeast and vertebrates: common threads. Trends Cell Biol. 2009;19:325–333. doi: 10.1016/j.tcb.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 56.Dotiwala F, Harrison JC, Jain S, Sugawara N, Haber JE. Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Current biology. 2010;20:328–332. doi: 10.1016/j.cub.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ström L, Lindroos HB, Shirahige K, Sjogren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell. 2004;16:1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 58.Ünal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, et al. DNA damage response pathway uses histone modification to assemble a doublestrand break-specific cohesin domain. Mol Cell. 2004;16:991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 59.Unal E, Heidinger-Pauli JM, Koshland D. DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7) Science. 2007;317:245–248. doi: 10.1126/science.1140637. [DOI] [PubMed] [Google Scholar]
- 60.Weinstock DM, Richardson CA, Elliott B, Jasin M. Modeling oncogenic translocations: distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair (Amst) 2006;5:1065–1074. doi: 10.1016/j.dnarep.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 61.Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–1083. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eickbush TH. Repair by retrotransposition. Nat Genet. 2002;31:126–127. doi: 10.1038/ng897. [DOI] [PubMed] [Google Scholar]
- 63.Gosselin K, Martien S, Pourtier A, Vercamer C, Ostoich P, Morat L, et al. Senescence-associated oxidative DNA damage promotes the generation of neoplastic cells. Cancer Res. 2009;69:7917–7925. doi: 10.1158/0008-5472.CAN-08-2510. [DOI] [PubMed] [Google Scholar]
- 64.Samoshkin A, Arnaoutov A, Jansen LE, Ouspenski I, Dye L, Karpova T, et al. Human condensin function is essential for centromeric chromatin assembly and proper sister kinetochore orientation. PLoS ONE. 2009;4:6831. doi: 10.1371/journal.pone.0006831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oliveira RA, Coelho PA, Sunkel CE. The condensin I subunit Barren/CAP-H is essential for the structural integrity of centromeric heterochromatin during mitosis. Mol Cell Biol. 2005;25:8971–8984. doi: 10.1128/MCB.25.20.8971-84.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471:325–330. doi: 10.1038/nature09830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE. 2009;4:6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.