

Okilactomycin (1a) is a structurally interesting antitumor antibiotic that was isolated from Streptomyces griseoflavus in 1987.[1] In vitro studies have demonstrated that 1a exhibits significant antitumor and antiproliferative activity against both lymphoid leukemia L1210 cells and P388 leukemia cells with IC50 values of 216 nM and 89 nM, respectively.[1b] A closely related compound, chrolactomycin (1b), differs only in structure by the replacement of a methyl group with a methoxy moiety at the pyranone/lactone ring fusion and displays promising telomerase inhibition.[1c,d] In addition to their potent biological activity, these compounds posses a compact and intriguing architecture. The tricyclic core is characterized by a unique 6,5-fused tetrahydropyranone γ-lactone bicycle with a spiro fusion to a highly substituted cyclohexene. A strained deoxygenated dipropionate segment spans this unusual tricycle to generate a highly rigid tetracyclic topology. Despite the biological activity and structural complexity, there have been only limited reports on the synthesis of okilactomycin (1a) over the last two decades, namely from the laboratories of Takeda, Paquette, and Smith.[2] These synthetic efforts culminated in a total synthesis of unnatural enantiomer (−)-1 a and determination of the absolute configuration of the natural product by Smith et al. in 2007.[2d,e] There are no syntheses of chrolactomycin (1b) reported to date. We disclose herein a convergent synthesis of (−)-1 a utilizing a Prins-type Maitland–Japp cyclization strategy of two advanced fragments.
Our retrosynthetic plan is outlined in Scheme 1. Given the electrophilic nature of the exomethylene unit, we elected for a late-stage installation of this moiety. We envisioned that the key tetracyclic precursor could be accessed from the lactonization of seco-ester 2, and subsequent ring-closing metathesis (RCM) to install the 11-membered macrocycle. The tetrahydropyranone heart of the molecule would be formed from a convergent union of the corresponding α-hydroxy aldehyde 3 and β-hydroxy dioxinone 4 through a Prins cyclization.[3] The cyclohexenyl aldehyde could be accessed using an asymmetric Diels–Alder reaction in conjunction with functional group manipulation and Rubottom oxidation. The β-hydroxy dioxinone motif could be constructed using a vinylogous aldol reaction of an acetoacetate equivalent.
Scheme 1.

Retrosynthetic strategy. TBDPS =tert-butyldiphenylsilyl.
The synthesis of aldehyde 3 centered on an endo-selective Diels–Alder reaction to install the necessary substitution pattern. The requisite diene for this [4+2] strategy was constructed starting with the hydrozirconation/iodination reaction of benzyl-protected alkyne 5 (Scheme 2).[4] A lithium–halogen exchange of vinyl iodide 6 with nBuLi and subsequent treatment of the resulting vinyllithium species with Weinreb amide 7 afforded the desired enone in 65% yield as a >20:1 mixture of E/Z isomers. A selective Wittig olefination with ethyltriphenylphosphonium bromide provided diene 8 with >20:1 E/Z selectivity.[5,6] In the first key step of the synthesis, the core cyclohexene was formed in 86% yield with 20:1 diastereoselectivity for the endo product through the Diels–Alder reaction of diene 8 (1 equiv) with acrylimide 9 (1.1 equiv).[7,8] Early attempts using dialkyl aluminum halides or alternative Lewis acids for this cyclo-addition with realistic levels of diene (i.e. <10 equiv) resulted in poor conversion owing to rapid decomposition of acryl-amide 9. Ultimately, we found that AgPF6 was a crucial additive to achieve high yields of the desired product (10) when employing practical quantities and ratios of both reacting partners (ca. 1 equiv each). Studies are underway to understand and expand this practical improvement. The reductive cleavage of the Evans oxazolidinone auxiliary with LiBH4 and MeOH generated the alcohol in 83% yield. The oxidation of the primary alcohol with Dess–Martin period-inane provided aldehyde 11 (90% yield). Lastly, the installation of the α-hydroxyl group through a Rubottom oxidation produced α-hydroxy aldehyde 3 in 88% yield and 13:1 diastereoselectivity favoring the desired stereoisomer.[9]
Scheme 2.

Synthesis of cyclohexene 3. Reagents and conditions: a) Cp2Zr(H)Cl, NIS, THF, 84 % b) nBuLi, THF, −78 °C; then 7, 23°C, 65%. c) KHMDS, ethyltriphenylphosphonium bromide, THF, −78 to 0°C, 84%. d) Et2AlCl, AgPF6, CH2Cl2, −78 °C, 86 %, >20:1 d.r. 1e) LiBH4, MeOH, Et2O, 0 °C, 83%. f) Dess–Martin periodinane, CH2Cl2, 90%. g) TBSOTf, NEt3, CH2Cl2. h) DMDO, CH2Cl2, 0 °C; then acidic work-up, 88% over two steps. Bn =benzyl, Cp =cyclopenta-dienyl, DMDO=dimethyldioxirane, HMSD=hexamethyldisilazide, NIS =N-iodosuccinimide, TBS =tert-butyldimethylsilyl, THF =tetrahy-drofuran.
The corresponding β-hydroxy dioxinone fragment was assembled beginning with known aldehyde 13[10,2d,e] (derived in six steps from pseudoephedrine propionamide and TBDPS-protected iodoethanol). Using Kr4ger and Carreira3s copper-catalyzed vinylogous aldol reaction conditions with silyloxy diene 14 and aldehyde 13, β-hydroxy dioxinone 15 was formed in 70% yield and 10:1 diastereomeric ratio favoring the desired anti adduct (Scheme 3).[11] The resulting secondary alcohol was protected as a TBS ether, and the primary TBDPS group was selectively removed upon treatment with NH4F to afford the primary alcohol.[12] In anticipation of the ring-closing metathesis later in the route, the alcohol was converted into terminal olefin 16 with a Grieco elimination sequence.[13] The removal of the secondary TBS group with HF·py afforded the requisite β-hydroxy dioxinone 4 in 93% yield.
Scheme 3.

Synthesis of intermediates 4 and 17. Reagents and conditions: a) Cu(OTf)2, (R)-tol-binap, TBAT, THF, −50 °C, 70 %, 10:1 d.r. b) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 92%. c) NH4F, MeOH, 40 °C, 92%. d) 1. o-nitrophenyl selenocyanate, PBu3, THF; 2. H2O2, THF, 76%. e) HF·py, THF, 93 %. f) KOEt, CH2Cl2, 88%. g) HF·py, THF 94%. py =pyridine, TBAT =tetrabutylammonium difluorotriphenylsilicate, tol-binap=2,2′-Bis(di-p-tolylphosphino)-1,1′-binaphthyl. OTf =trifluoro-methanesulfononate.
With efficient routes to access both α-hydroxy aldehyde 3 and β-hydroxy dioxinone 4 in multigram quantities, we proceeded to investigate conditions for the key Prins-type fragment assembly reaction. Employing the Lewis acid conditions previously developed in our laboratory with scandium(III),[14] the condensation/Prins cyclization reaction of 3 and 4 was examined. Unfortunately, we found that β-hydroxy dioxinone 4 was unreactive with α-hydroxy aldehyde 3, and no cyclization took place. We decided to convert the dioxinone moiety into the corresponding ketoester based on reports by Clarke et al.[15a–c] indicating that a δ-hydroxy-β-ketoester could undergo a modified Maitland–Japp reaction[15d] with aldehydes to afford 2,6-cis-tetrahydropyran-4-ones. Thus, treatment of dioxinone 16 with KOEt smoothly provided a β-ketoester, where the protecting group was removed with HF·py to afford δ-hydroxy β-ketoester 17 without any observed lactonization (Scheme 3). With the fragments to pursue this modified approach in hand, the Lewis acid mediated coupling of ketoester 17 with α-hydroxy aldehyde 3 furnished trioxabicyclo[3.2.1]octane 18 in 35% yield instead of the desired tetrahydropyran (Scheme 4).[16]
Scheme 4.

Proposed pathway for the formation of 18. M.S. =molecular sieves.
The proposed pathway that leads to this product is shown in Scheme 4. The condensation of δ-hydroxy-β-ketoester 17 with aldehyde 3 forms oxocarbenium ion 19. Instead of enol cyclization by C2, O cyclization occurs to afford a second oxocarbenium ion 20. Trapping of the oxocarbenium ion with the tertiary hydroxy group affords trioxabicyclo[3.2.1]octane 18. The formation of this product is intriguing and suggests that the mechanism for the modified Maitland–Japp reaction under these conditions is similar to the condensation/Prins-type cyclization pathway found with a β-hydroxy dioxinone instead of a Knoevenagel/oxo-conjugate addition reaction.[15a,d]
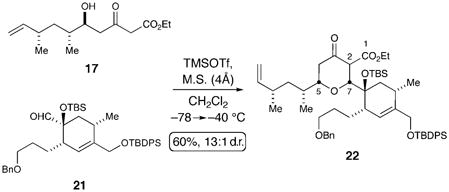
Despite the unexpected formation of tricycle 18, we were encouraged that δ-hydroxy-β-ketoester 17 was a competent coupling partner for the sterically demanding α-hydroxy aldehyde 3. A revised strategy was pursued involving the protection of the tertiary alcohol in 3 to facilitate the desired cyclization pathway. After extensive experimentation with various protecting groups, only TBS ether 21 survived the Lewis acid mediated conditions. Ultimately, the optimal conditions for the stereoselective coupling of 21 and 17 were TMSOTf in CH2Cl2, and led to the desired tetrahydropyranone 22 in a 60% yield and as a 13:1 mixture of diastereomers favoring the desired 2,6-cis isomer [Eq. (1)].
 |
(1) |
The union of these two fragments establishes the key C–O and C–C bonds of the central 6,5-fused pyranone/lactone in a single operation. In addition, this process relays the configuration of β-ketoester 17 to the new stereogenic center at C7 with high level of fidelity and constructs the full carbon skeleton of the natural product.
The global removal of the silyl groups of pyranone 22 was accomplished with aqueous HF and set the stage for completion of the synthesis. A lactonization between the unmasked hindered tertiary alcohol and pendant ethyl ester with KOtBu and subsequent stereoselective C methylation produced the key tricycle 23 in a 58% yield with only one purification over the three-step sequence (Scheme 5). Allylic alcohol 23 was reprotected as the TBDPS ether and the benzyl ether was cleaved under oxidative conditions with DDQ.[17] The resultant primary alcohol was converted into the terminal olefin without interference from the additional double bond in the molecule by using the Grieco protocol.[13]
Scheme 5.

Completion of the synthesis of (−)-okilactomycin. Reagents and conditions: a) aq HF, CH3CN. b) KOtBu, CH2Cl2. c) K2CO3, MeI, CH3CN, 70 °C, 58% over three steps). d) TBDPSCl, imidazole, CH2Cl2, 90%. e) DDQ (30 equiv), CH2Cl2, 61%. f) 1. o-nitrophenyl-selenocyanate, PBu3, THF; 2. H2O2, THF, 60%. g) Grubbs second-generation catalyst (40 mol%), CH2Cl2, 40 °C. h) H2, PtO2, EtOAc, 65 % over two steps. i) HF·py, THF, >99% j) LiHMDS, dimethylmethylideneammonium iodide, THF 95%. k) Dess–Martin periodinane, CH2Cl2, 83% l) NaClO2, NaH2PO4, 2-methyl-2-butene, tBuOH/THF/H2O (4:4:1), 50%. DDQ =2,3-dichloro-5,6-dicyanobenzoquinone.
At this stage, we pursued the planned ring-closing metathesis of 24 to form the macrocycle and complete the core of (−)-okilactomycin. This approach was originally proposed by Paquette[2b,c] and subsequently realized in the Smith synthesis.[2d] However, unique to our system was the presence of different functionality, most notably the olefin already installed in the cyclohexene ring. This internal alkene ultimately facilitates smooth installation of the α,β-unsaturated acid of the natural product (see below) and could have potentially compromised a metathesis approach. Pleasingly, the exposure of bis(olefin) 24 to Grubbs second-generation catalyst and subsequent hydrogenation furnished the tetracycle 25 in 65% yield over the two steps.[18] For our end-game approach, we were concerned that the exomethylene unit might be incompatible with conditions used to remove the silyl group, so we first removed the silyl unit and attempted to install the exocyclic olefin in the presence of the free allylic alcohol. Fortunately, silyl ether 25 was treated with HF·pyr to yield the allylic alcohol, and treatment with LiHMDS and dimethylmethylideneammonium iodide (Eschenmoser3s salt) cleanly installed the exocyclic olefin.[19] Finally, an oxidation to enal 26 with Dess–Martin periodinane (83% yield) followed by a Pinnick oxidation[20] afforded (−)-okilactomycin, which possessed identical characterization data (1H NMR, 13C NMR, HRMS, IR spectra) to the natural material.[21] The observed was −20 degcm3g−1dm−1 (MeOH, c =0.04 gcm−3) which is opposite in sign compared to isolated (+)-okilactomycin (+34 degcm3g−1dm−1, c = 1.0 gcm−3, MeOH).[2b,22]
In summary, the total synthesis of (−)-okilactomycin (1 a) has been achieved in 1.0% overall yield over 26 steps as the longest linear sequence (via 17). Stereoselective alkylation and Diels–Alder routes facilitated quick access to the δ-hydroxy β-ketoester and α-silyloxy aldehyde fragments, respectfully. A Lewis acid promoted Maitland–Japp reaction established the full carbon core with a high degree of diastereoselectivity for the 2,6-cis tetrahydropyrans core. This Prin-type transformation is one of the most advanced to date in terms of size and functionality of the reactants and further defines the potential of this approach for late-state unions of complex intermediates. The installation of the exocyclic olefin at the end of the synthesis and convergent nature of this route makes this synthesis amenable to the production of analogues and structure-activity relationship studies. The synthesis of these related compounds, which are intended for biological investigations, based on our complex fragment assembly Prins/Maitland–Japp route described here are ongoing in our laboratory.
Supplementary Material
Footnotes
Financial support for this work was provided by the NCI (R01 CA126827), the American Cancer Society (Research Scholar Grant), the Elsa Pardee Foundation, Amgen, and GlaxoSmithKline. Marianne Lalonde provided assistance with X-ray crystallographic analysis. W.J.M. thanks Abbott Laboratories for a graduate fellowship. We thank FMCLithium and Wacker Chemical for providing reagents, Dr. Chris Holmquist for assistance with chromatography, and Astellas Pharma for providing the spectroscopic data of natural (+)-okilactomycin.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201102037.
References
- 1.a) Imai HS, Kaniwa H, Tokunaga T, Fujita S, Furuya T, Matsumoto H, Shimizu M. J Antibiot. 1987;40:1483–1489. doi: 10.7164/antibiotics.40.1483. [DOI] [PubMed] [Google Scholar]; b) Imai HS, Suzuki KI, Morioka M, Numasaki Y, Kadota S, Nagai K, Sato T, Iwanami M, Saito T. J Antibiot. 1987;40:1475–1482. doi: 10.7164/antibiotics.40.1475. [DOI] [PubMed] [Google Scholar]; c) Nakai R, Kakita S, Asai A, Chiba S, Akinaga S, Mizukami T, Yamashita Y. J Antibiot. 2001;54:836–838. doi: 10.7164/antibiotics.54.836. [DOI] [PubMed] [Google Scholar]; d) Nakai R, Ishida H, Asai A, Ogawa H, Yamamoto Y, Kawasaki H, Akinaga S, Mizukami T, Yamashita Y. Chem Biol. 2006;13:183–189. doi: 10.1016/j.chembiol.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 2.a) Takeda K, Shimotani A, Yoshii E, Yamaguchi K. Heterocycles. 1992;34:2259–2261. [Google Scholar]; b) Boulet SL, Paquette LA. Synthesis. 2002:895–900. [Google Scholar]; c) Paquette LA, Boulet SL. Synthesis. 2002:888–894. [Google Scholar]; d) Smith AB, III, Basu K, Bosanac T. J Am Chem Soc. 2007;129:14872–14874. doi: 10.1021/ja077569l. [DOI] [PubMed] [Google Scholar]; e) Smith AB, III, Bosanac T, Basu K. J Am Chem Soc. 2009;131:2348–2358. doi: 10.1021/ja8084669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Cloninger MJ, Overman LE. J Am Chem Soc. 1999;121:1092–1093. [Google Scholar]; b) Zhang WC, Viswanathan GS, Li CJ. Chem Commun. 1999:291–292. [Google Scholar]; c) Crosby SR, Harding JR, King CD, Parker GD, Willis CL. Org Lett. 2002;4:3407–3410. doi: 10.1021/ol020127o. [DOI] [PubMed] [Google Scholar]; d) Jasti R, Vitale J, Rychnovsky SD. J Am Chem Soc. 2004;126:9904–9905. doi: 10.1021/ja046972e. [DOI] [PubMed] [Google Scholar]; e) Chan KP, Loh TP. Org Lett. 2005;7:4491–4494. doi: 10.1021/ol051951q. [DOI] [PubMed] [Google Scholar]; f) Chan KP, Ling YH, Loh TP. Chem Commun. 2007:939–941. doi: 10.1039/b616558c. [DOI] [PubMed] [Google Scholar]; g) Liu F, Loh T-P. Org Lett. 2007;9:2063–2066. doi: 10.1021/ol070506n. [DOI] [PubMed] [Google Scholar]; h) Pastor IM, Yus M. Curr Org Chem. 2007;11:925–957. [Google Scholar]; i) Van Orden LJ, Patterson BD, Rychnovsky SD. J Org Chem. 2007;72:5784–5793. doi: 10.1021/jo070901r. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Bahnck KB, Rychnovsky SD. J Am Chem Soc. 2008;130:13177–13181. doi: 10.1021/ja805187p. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Elsworth JD, Willis CL. Chem Commun. 2008:1587–1589. doi: 10.1039/b717078e. [DOI] [PubMed] [Google Scholar]
- 4.a) Hart DW, Schwartz J. J Am Chem Soc. 1974;96:8115–8116. [Google Scholar]; b) Schwartz J, Labinger JA. Angew Chem. 1976;88:402–409. [Google Scholar]; Angew Chem Int Ed Engl. 1976;15:333–340. [Google Scholar]
- 5.Maryanoff BE, Reitz AB. Chem Rev. 1989;89:863–927. [Google Scholar]
- 6.The E/Z ratio was determined by nOe interactions. See the Supporting Information for details.
- 7.a) Evans DA, Chapman KT, Bisaha J. J Am Chem Soc. 1984;106:4261–4263. [Google Scholar]; b) Evans DA, Chapman KT, Bisaha J. J Am Chem Soc. 1988;110:1238–1256. [Google Scholar]; c) Ho G, Mathre D. J Org Chem. 1995;60:2271–2273. [Google Scholar]
- 8.The absolute and relative configuration of 10 was determined by X-ray crystallographic analysis. CCDC 816131 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 9.a) Roush W, Barda D. J Am Chem Soc. 1997;119:7402–7403. [Google Scholar]; b) Roush W, Barda D, Limberakis C, Kunz R. Tetrahedron. 2002;58:6433–6454. [Google Scholar]; c) Roush WR, Limberakis C, Kunz RK, Barda DA. Org Lett. 2002;4:1543–1546. doi: 10.1021/ol025772+. [DOI] [PubMed] [Google Scholar]
- 10.a) Hanessian S, Murray PJ. Can J Chem. 1986;64:2232–2234. [Google Scholar]; b) Blasdel LK, Myers AG. Org Lett. 2005;7:4281–4283. doi: 10.1021/ol051785m. [DOI] [PubMed] [Google Scholar]
- 11.Kr4ger J, Carreira EM. J Am Chem Soc. 1998;120:837–838. [Google Scholar]
- 12.Zhang W, Robins MJ. Tetrahedron Lett. 1992;33:1177–1180. [Google Scholar]
- 13.Grieco PA, Gilman S, Nishizawa M. J Org Chem. 1976;41:1485–1486. [Google Scholar]
- 14.Morris WJ, Custar DW, Scheidt KA. Org Lett. 2005;7:1113–1116. doi: 10.1021/ol050093v. [DOI] [PubMed] [Google Scholar]
- 15.a) Clarke PA, Martin WHC. Org Lett. 2002;4:4527–4529. doi: 10.1021/ol027081j. [DOI] [PubMed] [Google Scholar]; b) Clarke PA, Martin WHC, Hargreaves JM, Wilson C, Blake AJ. Chem Commun. 2005:1061–1063. doi: 10.1039/b416247a. [DOI] [PubMed] [Google Scholar]; c) Clarke PA, Martin WHC, Hargreaves JM, Wilson C, Blake AJ. Org Biomol Chem. 2005;3:3551–3563. doi: 10.1039/b508252h. [DOI] [PubMed] [Google Scholar]; d) Japp FR, Maitland W. J Chem Soc Trans. 1904;85:1473–1489. [Google Scholar]
- 16.The configuration was assigned by an nOe interactions.
- 17.Ikemoto N, Schreiber SL. J Am Chem Soc. 1992;114:2524–2536. [Google Scholar]
- 18.For a related ring-closing metathesis strategy and execution, see references. [2d] and [2e].
- 19.a) Schreiber J, Maag H, Hashimoto N, Eschenmoser A. Angew Chem. 1971;83:355–357. [Google Scholar]; Angew Chem Int Ed Engl. 1971;10:330–331. [Google Scholar]; b) Nicolaou KC, Rutjes FPJT, Theodorakis EA, Tiebes J, Sato M, Untersteller E. J Am Chem Soc. 1995;117:1173–1174. [Google Scholar]
- 20.a) Lindgren BO, Nilsson T. Acta Chem Scand. 1973;27:888–890. [Google Scholar]; b) Kraus GA, Taschner MJ. J Org Chem. 1980;45:1175–1176. [Google Scholar]
- 21.We thank Astellas Pharma for providing spectroscopic data of (+)-okilactomycin for comparison.
- 22.With regard to this synthesis of the unnatural enantiomer of 1a, (−)-okilactomycin, since our intermediates up to and including 25 possessed (+) rotation values, we anticipated that the route would produce the natural (+) enantiomer. Comparing our structures to the previous synthesis by Smith (Ref. [2f]) was complicated since the absolute structure of (+)-1a in Figure 1 of Ref. [2f] should be the enantiomer. Importantly, all other structures and assignments in Ref. [2f] are correct (Prof. Amos Smith, private communication). Interestingly, both natural chrolactomycin and the recently isolated congeners of okilactomycin (okilactomycin A–D) have negative optical rotation values. Efforts are underway in our laboratory to produce the natural enantiomer along with structure analogues for our biological investigations.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.