

Abstract
Alkene insertion into Pd–N bonds is a key step in Pd-catalyzed oxidative amidation of alkenes. A series of well-defined Pd(II)-sulfonamidate complexes have been prepared and shown to react via insertion of a tethered alkene. The Pd–amidate and resulting Pd–alkyl species have been crystallographically characterized. The alkene insertion reaction is found to be reversible, but complete conversion to oxidative amination products is observed in the presence of O2. Electronic-effect studies reveal that alkene insertion into the Pd–N bond is favored kinetically and thermodynamically with electron-rich amidates.
The discovery of PdII-catalyzed oxidative coupling of ethylene and water (the Wacker Process) >50 years ago inspired extensive efforts to develop methods for the oxidative amination of alkenes (aza-Wacker reactions).1 Early studies showed that many alkyl- and arylamine nucleophiles coordinate strongly to PdII and inhibit catalytic turnover, and therefore much of this work focused on stoichiometric reactions of nitrogen nucleophiles with preformed PdII–alkene complexes.2 In 1982, Hegedus and McKearin demonstrated that p-toluenesulfonamides (tosylamides) could be used in catalytic intramolecular aza-Wacker reactions, with benzoquinone as the oxidant.3 More recently, amide-type nucleophiles have been used in aerobic oxidative amination reactions,4 including enantioselective5 and intermolecular6 applications. Mechanistic studies suggest that these aza-Wacker reactions often proceed via alkene insertion into the Pd– N bond of the amidate ligand, not attack of a nitrogen nucleophile onto a PdII–coordinated alkene.7,8 The first fundamental studies of alkene insertion into Pd–N bonds have been reported only recently by the groups of Wolfe and Hartwig with PdII–anilide complexes.9–11 Analogous reactions with Pd–amidates are unknown. Here, we describe well-defined PdII–sulfonamidate complexes that undergo alkene insertion into the Pd–N bond, and the reaction is shown to be reversible. The presence of O2 influences the fate of the resulting alkyl-PdII species. These observations, elaborated below, have important implications for catalytic reactions, including oxidative and non-oxidative transformations.
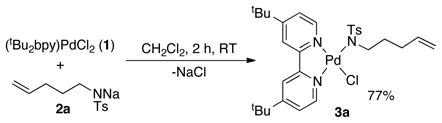
A well-defined PdII–sulfonamidate complex, suitable for fundamental investigation, was prepared by adding a solution of (tBu2bpy)PdCl2 (1, tBu2bpy = 4,4′-di-tert-butyl-2,2′-bipyridine) to a suspension of sodium tosylamidate (2a) in CH2Cl2 at room temperature. The air- stable Pd-amidate complex 3a was obtained in good yield (eq 1) and was characterized by 1H and 13C NMR spectroscopy, mass spectrometry and X-ray crystallography (Figure 1, left).12 Initial attempts to observe alkene insertion into the Pd–N bond of 3a were carried out in benzene, THF and dichloromethane, but no reaction was observed (Scheme 1). In chloroform, however, 3a reacted very slowly at room temperature, affording the alkyl-PdII amidopalladation (AP) product 4a in good yield (Figure 1, right). At higher temperatures, 4a underwent further reaction via β-hydride elimination and could not be obtained cleanly from the reaction mixture. The reaction proved to be much more efficient in dimethylsulfoxide (DMSO) as the solvent, proceeding in 84% yield in 12 h.
Figure 1.

X-ray crystal structures of 3a (left) and 4a (right) with thermal ellipsoids shown at 40% and 50% probability level, respectively. Most hydrogen atoms have been omitted for clarity.12
Scheme 1.

Amidopalladation of an Alkene
 |
(1) |
The beneficial effect of a polar solvent (DMSO) suggested that the reaction proceeds via an ionic intermediate. At least two reasonable ionic mechanisms can be considered: (1) dissociation of the amidate, followed by alkene coordination and nucleophilic attack of the pendant amidate on the alkene (trans-AP), or (2) chloride dissociation, followed by alkene coordination and insertion into the Pd–N bond (cis-AP), and reassociation of the chloride ligand to the PdII center. Two experimental observations provide support for the latter, cis-AP pathway. Addition of chloride to the reaction mixture strongly inhibits the reaction (Figure 2), consistent with a mechanism involving pre-equilibrium dissociation of chloride prior to the AP step. In addition, we prepared a Pd-sulfonamidate complex with a stereochemically defined, deuterium-labeled substrate probe (5, Scheme 2).13 The reaction of this complex under an atmosphere of O2 led to products 6 and 7, which arise from cis-AP of the alkene, followed by β-hydride elimination. No products arising from competing trans-AP of the alkene were observed.
Figure 2.
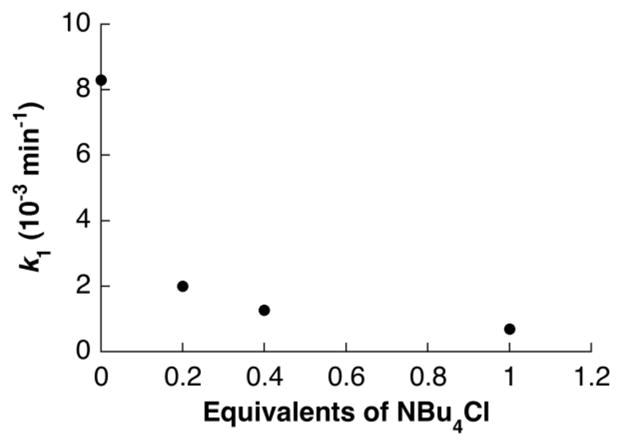
Chloride inhibition on amidopalladation for 3a
Scheme 2.

Isotopic Labeling Study Demonstrating that C–N Bond-Formation Proceeds via cis-Amidopalladation
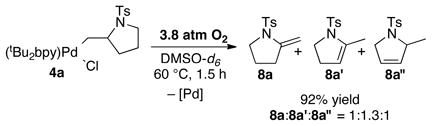
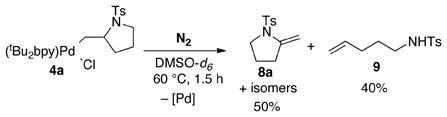
As expected from the reaction illustrated in Scheme 2, the alkyl-PdII complex 4a is susceptible to β-hydride elimination. Heating a DMSO solution of 4a to 60 °C under aerobic conditions led to a mixture of isomeric products 8a, 8a′ and 8a″ (eq 2).14 A different outcome was observed, however, when the reaction was performed under anaerobic conditions: the β-hydride elimination products 8a–8a″ were obtained in 50% yield, together with a 40% yield of 4-pentenyl tosylamide 9 (eq 3).
 |
(2) |
 |
(3) |
The formation of 9 in eq 3 was unexpected, but this result can be rationalized if alkene insertion into the Pd–N bond is reversible (i.e., 4a ⇆ 3a). According to the mechanism in Scheme 3, β-hydride elimination from 4a forms the enamide products 8a–8a″ together with a PdII–hydride species 10. In the absence of O2,15 HCl can form via reductive elimination from 10 and react with 3a to afford the alkenyl tosylamide 9. This proposal implies that HCl reacts much more rapidly with 3a than with 4a. In order to test this hypothesis, excess HCl (~60 equiv) was added to a DMSO-d6 solution of 4a at room temperature. Rapid and quantitative formation of 9 was observed in this reaction, together with (tBu2bpy)PdCl2 (1, eq 4); pyrrolidine 11, the product of protonolysis of the Pd–C bond of 4a, was not observed.
Scheme 3.

Proposed Mechanism for the Parallel Formation of 8 and 9 under Anaerobic Conditions
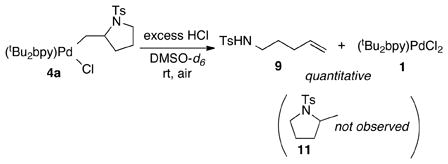 |
(4) |
Additional, more-direct evidence for reversible amidopalladation of the alkene was obtained in the investigation of a series of substituted PdII-sulfonamidate complexes (Figure 3). The reactions of four different para-substituted benzenesulfonamidate complexes [X = Me (3a), OMe (3b), Cl (3c) and NO2 (3d)] were monitored by 1H NMR spectroscopy at 30 °C in DMSO-d6 under aerobic conditions (Figure 3a). Each of the Pd complexes underwent clean amidopalladation of the alkene to afford an equilibrium mixture of complexes 3a–d and the corresponding alkyl–PdII species 4a–d (Figure 3b), together with slower concomitant formation of heterocycles 8–8″a–d via β-hydride elimination from 4a–d. The data were fit to a simplified kinetic model, 3 ⇆ 4 → 8–8″, that enabled quantitative comparison of kinetic and thermodynamic constants associated with the two observable steps (Figure 3c). Electronic effects on these parameters were probed via Hammett analysis (Figure 3d).16
Figure 3.

Kinetic studies of alkene insertion/β-hydride elimination reactions of four PdII–sulfonamidate complexes. Conditions: 3.68 mM 3, 3.8 atm O2, DMSO, 30 °C, 7–12 h. Note: Every fifth data point is shown to enhance clarity.
Alkene insertion into the Pd–N bond (i.e., cis-AP, k1) is favored for electron-rich amidates. This trend is similar to that observed previously for irrreversible alkene insertion into Pd–anilides, and it is consistent with an alkene insertion mechanism that formally corresponds to intramolecular nucleophilic attack of the amidate ligand onto the coordinated alkene.9 The reverse reaction, β-amidate elimination, (k−1) is favored for electron-deficient amidates (Figures 3c and 3d). Together, these trends cause the equilibrium constant to be largest for the p-Me derivatives 3/4a (K1 ~ 10) and smallest for the electron-deficient p-NO2 derivative 3/4d (K1 ~ 1) (Figure 3c). That K1 is largest for the p-Me, and not the p-OMe derivative, appears to reflect the lack of “resonance” electronic effects in the amidate elimination step k−1; the Hammett correlation for k−1 is best fit with the “inductive” Hammett parameter σI, rather than σp, which incorporates both resonance and inductive effects. The k2 values estimated from these fits show that β-hydride elimination is favored with more-electron-rich derivatives, consistent with a formal “hydride”-transfer mechanism in which electron-donating groups stabilize the build-up of positive charge on the adjacent carbon atom in the transition state. The combined electronic effects, K1·k2, reveal that the p-Me (tosylamidate) derivative 3a is the most reactive complex toward formation of the oxidative amidation products 8–8″.
Overall, these observations have important implications for catalysis. For example, the development of enantioselective Wacker-type oxidation reactions has been a long-standing challenge in the field of asymmetric catalysis.17 Alkene insertion into the Pd–N bond of 3a results in formation of a new stereogenic center (Scheme 1), and such steps provide the basis for enantioselective oxidative amination reactions (e.g., eq 5).5c Recent work has highlighted the importance of controlling the stereochemical course of the nucleopalladation step in enantioselective reactions (i.e., cis- vs. trans-nucleopalladation).1f The results reported here reveal that nucleopalladation could be reversible, in which case β-hydride elimination or another termination step will be the stereochemistry-determining step of the reaction. To our knowledge, this possibility has not been considered previously in Wacker-type reactions.
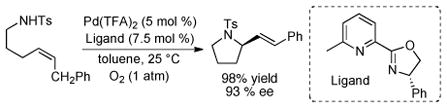 |
(5) |
Reversible C–N bond formation has been observed in reactions involving trans-AP of an alkene,18 but reversible insertion of an alkene into a Pd–N bond has not been observed previously. The latter observation, combined with the significantly more-facile protonolysis of Pd–N bonds relative to Pd–C bonds (cf. eq 4), represents a key challenge for the development of Pd-catalyzed hydroamination reactions that proceed via cis-AP pathways.
In summary, this study has led to key insights into reactions of PdII–sulfonamidates with alkenes, perhaps most notably demonstrating that alkene insertion into the Pd–N bonds of such species is facile and reversible. This work provides an important foundation for more-thorough characterization of cis-amidopalladation reactions relevant to important catalytic transformations.
Supplementary Material
Acknowledgments
We thank Dr. Ilia Guzei for performing X-ray structure determinations and Drs. Charlie Fry and Monika Ivancic for training and experimental assistance in the acquisition of NMR spectroscopic data. We are grateful for financial support from the NIH (R01 GM67163). Instrumentation was partially funded by the NSF (CHE-9974839, CHE-9629688, CHE-9629688, and CHE-8813550) and the NIH (1 S10 RR13866-010).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures, characterization data, kinetic data, and X-ray crystallographic data (CIF) for compound 3a and 4a. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews, see: Bäckvall JE. Acc Chem Res. 1983;16:335–342.Hegedus LS. Tetrahedron. 1984;40:2415–2434.Larock RC, Zeni G. Chem Rev. 2004;104:2285–2309. doi: 10.1021/cr020085h.Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem Rev. 2007;107:5318–5365. doi: 10.1021/cr068006f.Minatti A, Muñiz K. Chem Soc Rev. 2007;36:1142–1152. doi: 10.1039/b607474j.McDonald RI, Liu G, Stahl SS. Chem Rev. 2011;111:2981–3019. doi: 10.1021/cr100371y.
- 2.See refs. 1a,b and the follow representative primary references: Cope AC, Kliegman JM, Friedrich EC. J Am Chem Soc. 1967;89:287–291.Åkermark B, Bäckvall JE, Hegedus LS, Zetterberg K, Siiralah-Hansén K, Sjöberg K. J Organomet Chem. 1974;72:127–138.Bäckvall JE. Tetrahedron Lett. 1978:163–166.Hegedus LS, Allen GF, Bozell JJ, Waterman EL. J Am Chem Soc. 1978;100:5800–5807.Pugin B, Venanzi LM. J Organomet Chem. 1981;214:125–133.Åkermark B, Zetterberg K. J Am Chem Soc. 1984;106:5560–5561.Hegedus LS, Åkermark B, Zetterberg K, Olsson LF. J Am Chem Soc. 1984;106:7122–7126.
- 3.Hegedus LS, McKearin JM. J Am Chem Soc. 1982;104(9):2444–2451. [Google Scholar]
- 4.See, for example: van Benthem RATM, Hiemstra H, Longarela GR, Speckamp WN. Tetrahedron Letters. 1994;35:9281–9284.Rönn M, Bäckvall JE, Andersson PG. Tetrahedron Lett. 1995;36:7749–7752.Larock RC, Hightower TR, Hasvold LA, Peterson KP. J Org Chem. 1996;61:3584–3585. doi: 10.1021/jo952088i.Stahl SS, Fix SR, Brice JL. Angew Chem, Int Ed. 2002;41:164–166. doi: 10.1002/1521-3773(20020104)41:1<164::aid-anie164>3.0.co;2-b.Trend RM, Ramtohul YK, Ferreira EM, Stoltz BM. Angew Chem, Int Ed. 2003;42:2892–2895. doi: 10.1002/anie.200351196.
- 5.(a) Scarborough CC, Bergant A, Sazama GT, Guzei IA, Spencer LC, Stahl SS. Tetrahedron. 2009;65:5084–5092. doi: 10.1016/j.tet.2009.04.072. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He W, Yip KT, Zhu NY, Yang D. Org Lett. 2009;11:5626–5628. doi: 10.1021/ol902348t. [DOI] [PubMed] [Google Scholar]; (c) Jiang F, Wu ZX, Zhang WB. Tetrahedron Lett. 2010;51:5124–5126. [Google Scholar]; (d) McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. Org Lett. 2011;13:2830–2833. doi: 10.1021/ol200784y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Hosokawa T, Takano M, Kuroki Y, Murahashi SI. Tetrahedron Lett. 1992;33:6643–6646. [Google Scholar]; (b) Stahl SS, Timokhin VI, Anastasi NR. J Am Chem Soc. 2003;125:12996–12997. doi: 10.1021/ja0362149. [DOI] [PubMed] [Google Scholar]; (c) Brice JL, Harang JE, Timokhin VI, Anastasi NR, Stahl SS. J Am Chem Soc. 2005;127:2868–2869. doi: 10.1021/ja0433020. [DOI] [PubMed] [Google Scholar]; (d) Kim SK, Lee JM, Ahn DS, Jung DY, Lee J, Do Y, Chang SK. J Am Chem Soc. 2006;128:12954–12962. doi: 10.1021/ja0639315. [DOI] [PubMed] [Google Scholar]; (e) Rogers MM, Kotov V, Chatwichien J, Stahl SS. Org Lett. 2007;9:4331–4334. doi: 10.1021/ol701903r. [DOI] [PubMed] [Google Scholar]; (f) Obora Y, Shimizu Y, Ishii Y. Org Lett. 2009;11:5058–5061. doi: 10.1021/ol902052z. [DOI] [PubMed] [Google Scholar]; (g) Liu X, Hii KK. Eur J Org Chem. 2010;2010:5181–5189. [Google Scholar]
- 7.See ref. 1a and the following: Isomura K, Okada N, Saruwatari M, Yamasaki H, Taniguchi H. Chem Lett. 1985:385–388.Liu GS, Stahl SS. J Am Chem Soc. 2006;128:7179–7181. doi: 10.1021/ja061706h.Muñiz K, Hövelmann CH, Streuff J. J Am Chem Soc. 2007;130:763–773. doi: 10.1021/ja075041a.Liu GS, Stahl SS. J Am Chem Soc. 2007;129:6328–6335. doi: 10.1021/ja070424u.
- 8.For recent catalytic and fundamental studies of alkene amidation reactions with carbamate and carboxamide nucleophiles that proceed via trans-amidopalladation, see the following: Michael FE, Cochran BM. J Am Chem Soc. 2008;130:2786–2792. doi: 10.1021/ja0734997.Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J Am Chem Soc. 2009;131:15945–15951. doi: 10.1021/ja906915w.
- 9.(a) Neukom JD, Perch NS, Wolfe JP. J Am Chem Soc. 2010;132:6276. doi: 10.1021/ja9102259. [DOI] [PubMed] [Google Scholar]; (b) Klinkenberg JL, Hartwig JF. J Am Chem Soc. 2010;132:11830. doi: 10.1021/ja1023404. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Neukom JD, Perch NS, Wolfe JP. Organometallics. 2011;30:1269. [Google Scholar]; (d) Hanley PS, Hartwig JF. J Am Chem Soc. 2011;133:15661–15673. doi: 10.1021/ja205722f. [DOI] [PubMed] [Google Scholar]
- 10.Prior to fundamental studies of alkene insertion, Wolfe and coworkers provided stereochemical evidence for a cis-amidopalladation step: Wolfe JP, Ney JE. Angew Chem, Int Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060.Wolfe JP, Ney JE. J Am Chem Soc. 2005;127:8644–8651. doi: 10.1021/ja0430346.Wolfe JP, Nakhla JS, Kampf JW. J Am Chem Soc. 2006;128:2893–2901. doi: 10.1021/ja057489m.
- 11.For leading references to fundamental studies of alkene reactivity with other transition-metal amido complexes, see: Hong S, Marks TJ. Acc Chem Res. 2004;37:673–686. doi: 10.1021/ar040051r.Müller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem Rev. 2008;108:3795–3892. doi: 10.1021/cr0306788.Zhao P, Krug C, Hartwig JF. J Am Chem Soc. 2005;127:12066–12073. doi: 10.1021/ja052473h.Hoover JM, DiPasquale A, Mayer JM, Michael FE. J Am Chem Soc. 2010;132:5043–5053. doi: 10.1021/ja906563z.Leitch DC, Platel RH, Schafer LL. J Am Chem Soc. 2011;133 doi: 10.1021/ja202448b.
- 12.See Supporting Information for further details.
- 13.For the synthesis and previous application of this substrate probe, see ref. 7d.
- 14.This reaction was carried out in an NMR tube. Because gas-liquid mixing is slow in an NMR tube, an elevated pressure of O2 was used to ensure at least 1 equiv of O2 was dissolved in solution.
- 15.The reaction of O2 with (N-N)Pd(H)Cl complexes was the focus of a recent study: Decharin N, Popp BV, Stahl SS. J Am Chem Soc. 2011;133:13268–13271. doi: 10.1021/ja204989p.
- 16.Data acquisition focused on the alkene insertion/deinsertion steps (K)). Thus, the reliability of the k2 data is constrained by the limited data obtained for the β-hydride elimination step.
- 17.For recent progress in this area, see refs. 1f, 4d, 5 and the following additional leading references: Uozumi Y, Kyota H, Kato K, Ogasawara M, Hayashi T. J Org Chem. 1999;64:1620–1625. doi: 10.1021/jo982104m.Sasai H, Arai MA, Kuraishi M, Arai T. J Am Chem Soc. 2001;123:2907–2908. doi: 10.1021/ja005920w.
- 18.See, for example: Vitagliano A, Hahn C, Morvillo P. Eur J Inorg Chem. 2001:419–429.Michael FE, Cochran BM. J Am Chem Soc. 2008;130:2786–2792. doi: 10.1021/ja0734997.Timokhin VI, Stahl SS. J Am Chem Soc. 2005;127:17888–17893. doi: 10.1021/ja0562806.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.