

Background: Aromatic residues line glycoside hydrolase active sites mediating ligand binding.
Results: Binding affinity is significantly altered upon tryptophan to alanine mutation, although relative to the location in the active site.
Conclusion: Aromatic-carbohydrate interactions are employed in a variety of functionalities within the purview of ligand binding.
Significance: Understanding the functional role of aromatic residues in the active site is necessary for the rational design of new carbohydrate-active enzymes.
Keywords: Carbohydrate-binding Protein, Carbohydrate Processing, Enzyme Mutation, Molecular Modeling, Thermodynamics, Biomass Conversion, Carbohydrate-π Interactions, Free Energy Simulations, Ligand Binding, Thermodynamic Integration
Abstract
Proteins employ aromatic residues for carbohydrate binding in a wide range of biological functions. Glycoside hydrolases, which are ubiquitous in nature, typically exhibit tunnels, clefts, or pockets lined with aromatic residues for processing carbohydrates. Mutation of these aromatic residues often results in significant activity differences on insoluble and soluble substrates. However, the thermodynamic basis and molecular level role of these aromatic residues remain unknown. Here, we calculate the relative ligand binding free energy by mutating tryptophans in the Trichoderma reesei family 6 cellulase (Cel6A) to alanine. Removal of aromatic residues near the catalytic site has little impact on the ligand binding free energy, suggesting that aromatic residues immediately upstream of the active site are not directly involved in binding, but play a role in the glucopyranose ring distortion necessary for catalysis. Removal of aromatic residues at the entrance and exit of the Cel6A tunnel, however, dramatically impacts the binding affinity, suggesting that these residues play a role in chain acquisition and product stabilization, respectively. The roles suggested from differences in binding affinity are confirmed by molecular dynamics and normal mode analysis. Surprisingly, our results illustrate that aromatic-carbohydrate interactions vary dramatically depending on the position in the enzyme tunnel. As aromatic-carbohydrate interactions are present in all carbohydrate-active enzymes, these results have implications for understanding protein structure-function relationships in carbohydrate metabolism and recognition, carbon turnover in nature, and protein engineering strategies for biomass utilization. Generally, these results suggest that nature employs aromatic-carbohydrate interactions with a wide range of binding affinities for diverse functions.
Introduction
Protein-carbohydrate recognition is a ubiquitous phenomenon in biology that is important for a vast range of biochemical functions (1–7). A common means of selective protein binding to carbohydrates is accomplished by the aromatic amino acids, tryptophan, tyrosine, and phenylalanine, wherein aromatic rings can stack with planar faces of carbohydrate rings via carbohydrate-π interactions (8–22). To date, many studies have been conducted to determine the effect of aromatic-carbohydrate interactions in model (9–15) and real systems (16–22), with the aim of quantifying the strength and types of interactions for this important biological recognition pattern. A particularly dramatic example of the prevalence of aromatic-carbohydrate interactions is the ubiquity of these motifs observed throughout the 125 families of glycoside hydrolases (GHs),2 which catalyze hydrolysis of the glycosidic linkages in carbohydrates, and the related 63 families of carbohydrate-binding modules, which are responsible for increasing the binding affinity of GH enzymes to carbohydrate substrates by many orders of magnitude (8, 23). In each of the structurally characterized GH families, aromatic residues line the enzyme tunnels and clefts for binding, processing, and stabilization of carbohydrate ligands. In carbohydrate-binding modules, aromatic residues are commonly found on the substrate-binding faces, which form favorable electrostatic and hydrophobic interactions with carbohydrates (24, 25).
In nature, processive GHs provide the majority of direct hydrolytic potential in the conversion of insoluble carbohydrate polymers, such as cellulose and chitin, to soluble sugars for utilization as food sources (26–30). From crystal structures of processive chitinases and cellulases, it is known that these enzymes typically exhibit long tunnels or deep clefts lined with aromatic and polar residues that can thread single polymer chains to their active sites for hydrolysis (28–30). Processive GHs are generally hypothesized to bind to their insoluble, polymeric substrates, find a free chain end, thread the chain into the enzyme tunnel to form the catalytically active complex, conduct hydrolysis, expel the soluble product, and then repeat this catalytic cycle processively (31, 32) until the enzyme reaches the end of the polymer chain, becomes inhibited by obstacles on the surface, or detaches from the polymer chain (33). It should be noted, however, that processive cellulases have also been observed to exhibit endo-initiation activity (34, 35). The primary hypothesis in the literature to date is that aromatic residues in processive enzyme tunnels facilitate carbohydrate processivity, but the thermodynamic and kinetics of processivity have not been elucidated directly at the molecular level (28–30).
Several studies have examined the role of aromatic residues in cellulase and chitinase tunnels with respect to processivity and enzyme activity on substrates of varying accessibility (36–42). Watanabe et al. (41) conducted many of these original studies related to various chitinases. More recently, Horn et al. (36) mutated two tryptophans in the Serratia marcescens family 18 chitinase B (ChiB). They demonstrated that a single mutation (W97A) directly upstream of the catalytic site in ChiB significantly reduced the ability of the enzyme to degrade crystalline chitin, but increased the conversion rate by 29-fold on soluble chitosan. The authors also measured the degree of processivity between the wild type ChiB and the W97A mutant and demonstrated that the W97A mutation converted ChiB to a nonprocessive enzyme, even on soluble substrates (36). The same group later demonstrated similar trends for another S. marcescens chitinase, ChiA (38). Overall, this work suggests that the ability for an enzyme to be processive on insoluble substrates comes at a significant detriment to its substrate turnover frequency.
For aromatic-carbohydrate interactions in cellulases, a tryptophan residue (Trp-272) at the entrance to the tunnel of the processive family 6 cellulase, Cel6A, from Trichoderma reesei (an anamorph of Hypocrea jecorina), was mutated to alanine and aspartic acid (39). In both cases, the activity of the mutant on crystalline cellulose was significantly reduced, whereas the activity on amorphous cellulose was not affected (39). The authors interpret these results to suggest that the entrance tryptophan residue in Cel6A is crucial in binding to cellodextrin chains in crystalline substrates but plays little functional role when the substrate is more accessible. Similar observations have been made for Cel7A (43, 44). Additionally, Vuong and Wilson (37) mutated a tryptophan (Trp-464) in Thermobifida fusca Cel6B directly upstream of the catalytic site to alanine and tyrosine. For both mutations, they observed reduced activity on crystalline cellulose and increased activity on amorphous and soluble substrates, although the extent of processivity on filter paper was similar to the wild type. The authors concluded that Trp-464 helps to bind the cellulose chain to the active site tunnel, but based on increased activity on various accessible substrates, this function is not necessary for noncrystalline substrates. It is worthwhile to note that processivity has been measured in distinctly different ways for the cellulases and chitinases discussed above and thus may not be directly comparable.
We recently combined observations from the work of Horn et al. (36) on chitinases with a computational analysis of cellulose and chitin decrystallization to propose a microkinetic model of processive enzyme action (45–47). We hypothesize that the work that an enzyme must conduct to decrystallize a carbohydrate polymer from a crystalline substrate is directly offset by a favorable ligand binding free energy (31, 32, 45, 46). By removing aromatic residues in processive enzyme tunnels, the ligand binding free energy may be reduced sufficiently to switch the function from a processive enzyme to a nonprocessive enzyme; however, in the case of modified substrates that are either more accessible (48) or easier to decrystallize (45, 49), it is a direct extension from this work that engineered enzymes and enzyme mixtures significantly different from those found in nature may offer significant gains in conversion rates for industrial processes using modified cellulose. This, in turn, could lower production costs for the burgeoning renewable fuels and chemicals industry. Quantitative understanding of the effects of the aromatic-carbohydrate interactions in cellulase tunnels is thus a key factor in the rational design of new cellulases and other carbohydrate-active enzymes. Understanding the role that aromatic-carbohydrate interactions play in the structure and function of carbohydrate-active enzymes is similarly important. This remains an open question, for which molecular level simulations can offer unique insights.
EXPERIMENTAL PROCEDURES
We use thermodynamic integration (TI) (50, 51) to calculate the change in ligand binding free energy for removal of aromatic residues in the catalytic tunnels of the well characterized processive family 6 T. reesei Cel6A cellulase (29, 39, 52). Cel6A is a nonreducing end-specific enzyme with six binding sites numbered +4 (upstream of the catalytic site) to −2 (down the tunnel from the catalytic site), as shown in Fig. 1. The Cel6A catalytic domain tunnel exhibits tryptophan residues (Trp-135, Trp-269, Trp-272, and Trp-367), which we mutate individually to alanine.
FIGURE 1.

Aromatic-carbohydrate interactions in the processive Cel6A tunnel. A, surface representation of T. reesei Cel6A showing the catalytic domain with the cello-oligomer within the binding tunnel. The carbohydrate-binding module and linker are not shown. Tryptophans are shown in blue. The catalytic residue, aspartic acid 221 (D221), is shown in magenta. B, a detailed view of the Cel6A catalytic domain tunnel.
The TI calculations were performed using NAMD with dual topology methodology, wherein a hybrid residue containing atoms from both the tryptophan and alanine interacts with the system over a scaled coupling parameter during the transformation from reactant to product (50, 53, 55). The atoms of the hybrid residue do not interact with each other over the course of the simulation. In the TI simulations, the tryptophan is referred to as the reactant and the alanine as the product. The change in free energy was determined through numerical integration of sets of simulations probing the electrostatics and van der Waals interactions separately. The free energies, G, for each state were then combined according to Equation 1 to obtain the relative ligand binding free energy, ΔΔG, where the states are wild type (reactant) to mutant (product).
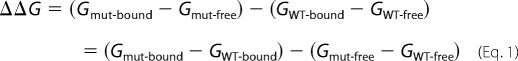 |
The alchemical cycle for measuring ΔΔG is shown under supplemental Figs. S1 and S2 along with error analysis details. From the ΔΔG for each mutation, we determined the binding affinity for all point mutations as in Equation 2,
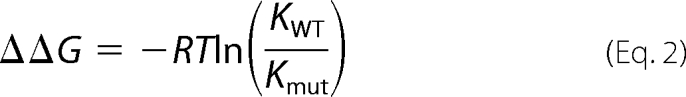 |
where ΔΔG is the relative binding free energy, R is the universal gas constant, T is absolute temperature, and Kmut and KWT are the partition coefficients for the mutant and wild type structures, respectively. All of the TI simulations were started using a snapshot at 18 ns from molecular dynamics (MD) simulations as described below. Simulations from which the TI data were collected were equilibrated for an additional 0.5 ns prior to collection of 14.5 ns of TI data.
In addition to the TI calculations, we use long MD simulations (250 ns) to quantify the energetics and fluctuations along the catalytic tunnels of the wild type and four mutant enzymes. These MD simulations provide insights into the role of the aromatic residues as a function of location in the tunnel in ligand acquisition and stabilization. CHARMM (56) was used to build, minimize, and equilibrate the protein structure from the wild type crystal structure (Protein Data Bank code 1QK2) (57). Final system sizes were on the order of 52,000 atoms, of which roughly 46,000 atoms were water. Subsequent MD simulations were performed using NAMD in the NVT ensemble at 300 K for 250 ns. The CHARMM27 force field with the CMAP correction (56, 58, 59) was used to describe the protein, whereas glycans and cellulose used the CHARMM35 carbohydrate force field (60–62). The system preparation and computational methods are described in more detail under supplemental data.
RESULTS
Table 1 lists the predicted change in relative binding free energy and binding affinity from the TI calculations. These results reveal several interesting observations. The ligand binding free energies of the tunnel entrance tryptophan, Trp-272, and the product side tryptophan, Trp-135, are quite large at 3.8 and 9.4 kcal/mol, respectively. Surprisingly, the results for Trp-367 and Trp-269, at the +1 and +2 sites, respectively, show only minor changes in relative ligand binding free energy. The relative binding affinity, Kwt/Kmut, was calculated from the ligand binding free energy as discussed under supplemental Table S1. The binding affinities of W135A and W272A indicate a clear preference for the wild type in these positions. The W367A and W269A mutations only slightly alter the relative binding affinity in comparison to W135A and W272A.
TABLE 1.
Relative ligand binding free energies (ΔΔG) and relative binding affinities (Kwt/Kmut) between the wild type and mutant enzyme states calculated from TI for Cel6A
| Mutation | Binding site | ΔΔG | ΔΔG error | Kwt/Kmut |
|---|---|---|---|---|
| kcal/mol | ||||
| W135A | −2 | 9.4 | 0.36 | 6.5E+06 |
| W367A | +1 | 1.6 | 0.41 | 1.5E+01 |
| W269A | +2 | −1.3 | 0.58 | 1.1E−01 |
| W272A | +4 | 3.8 | 0.41 | 5.5E+02 |
To further elucidate the role of the various aromatic residues in the Cel6A tunnel, we conducted five separate 250-ns MD simulations of the individual mutants and the wild type enzyme. From these simulations, we calculated the root mean square fluctuation (r.m.s. fluctuation) of the protein (Fig. 2) and of the ligand on a binding site basis (Fig. 3). The interaction energies of the protein with the ligand and the hydrogen bonds between the ligand and protein were also calculated and are provided under supplemental Fig. S3. Analyses of the glycosylation effects on the enzyme structure, which are minimal, are provided under supplemental Figs. S11–13. These analyses included comparisons of the r.m.s. fluctuation and r.m.s. deviation of the entire enzyme along with comparisons of ligand hydrogen bonding and interaction energies on a binding site basis.
FIGURE 2.

Root mean square fluctuation of the Cel6A enzyme by residue number from 250-ns MD simulations of the Cel6A wild type (WT) and 4 mutants: A, W135A; B, W269A; C, W272A; and D, W367A.
FIGURE 3.

Root mean square fluctuation of the cello-oligomer ligand labeled by the glucose binding site from 250-ns MD simulations of the Cel6A wild type and 4 mutants. Labels below the x axis indicate position of the tryptophan residues relative to the binding site. Error bars were obtained through 5-ns block averaging.
Evaluating the r.m.s. fluctuation of the protein over the course of the 250 ns MD simulations reveals interesting behavior in the loops forming the active site tunnel in some cases. The W135A mutant (Fig. 2A) exhibits increased fluctuations in both the 170–190 and 400–410 residue regions. Each of these residue segments correspond to tunnel-forming active site loops. The former loop, residues 172–182, has been previously linked to a conformational change as a function of active site mutations, increasing active site solvent accessibility although having no effect on ligand conformation (52). The W272A mutant (Fig. 2C) similarly exhibits increased fluctuation of the 400–410 active site loop region with minimal differences in the other loop region. This is likely a result of the increased interaction of the protein with the ligand as a result of the increased ligand flexibility in the +4 binding site, as will be shown below in Fig. 3. Only moderate effects on the fluctuation of the protein are observed in the W269A and W367A mutants with respect to wild type (Fig. 2, B and D).
The r.m.s. fluctuation of the ligand over the course of the MD simulations also lends insight into the molecular level aspects contributing to the relative ligand binding free energy. Comparing the MD simulations of the W272A mutant to that of the wild type Cel6A enzyme reveals that mutation of tryptophan to alanine in this binding site substantially alters the cellodextrin fluctuations, as shown in Fig. 3, which suggests that this residue is crucial for stabilizing the ligand at the entrance of the tunnel, as hypothesized by experimental studies (39). Fig. 3 also illustrates destabilization of the entire ligand in all six binding sites for the W269A and W367A mutants. It is particularly interesting that for these two mutants, the fluctuations in neighboring binding sites (e.g. the −1 and +2 sites for the W367A mutant) are affected, whereas for W135A and W272A the mutations to alanine have less of an impact on neighboring site fluctuations. The interaction energies of either wild type tryptophans or mutant alanines also exhibit this trend as illustrated under supplemental Fig. S4. The higher fluctuation of the −1 binding site in W269A and W367A than in all other mutations likely contributes entropically to the decreased relative binding free energy of these two mutants relative to W135A and W272A. All mutations result in increased fluctuations at the −2 binding site, and all reactant side (upstream of active site) mutations increase fluctuations at the +4 binding site.
The change in ligand binding free energy is most dramatic for W135A. Because it is located on the product side of the tunnel, it is directly involved in cellobiose binding, which is related both to stabilization of the product during catalysis and product inhibition (i.e. through competitive binding) (63). Although evaluation of the interaction energy of the entire protein with the individual binding sites (supplemental Fig. S3) yields little additional insight in terms of the relative ligand binding free energy, it does provide further evidence for the importance of the binding affinity of the product side residues, where the interaction energy of glucose in the −2 binding site for the wild type and the four mutants is substantially greater than any other site along the tunnel. This is the same general result that Mertz et al. (64) obtained using computational docking techniques. Reduction of this evaluation to either tryptophan or alanine with the glucose unit, as shown under supplemental Fig. S4, demonstrates that removal of any of the tryptophans in the tunnel has a significant local effect on interaction energy.
As mentioned above, the relative ligand binding free energies for W269A and W367A in Cel6A are small in comparison to W135A and W272A. These results suggest that tryptophans at the +1 and +2 sites in Cel6A are less important for direct binding to the ligand. Alternatively, these residues may create the necessary physical shape of the tunnel in the catalytically active complex, and play a role in the ring distortion at the −1 site wherein the glucose exhibits the 2SO conformation instead of the chair conformation, which is energetically favorable in solution (57). To examine this hypothesis, we calculated the Cremer-Pople ring amplitude (65) of the glucose residue in the −1 site from MD simulations for the wild type and four mutants as shown in Fig. 4. The 2SO conformation, corresponding to that of glucose in the −1 site of Cel6A, is defined by an amplitude of 0.73 Å. The chair conformation, 1C4 or 4C1, is defined by an amplitude of 0.57 Å (66). As the information presented in Fig. 4 was obtained from MD trajectories, the ring pucker amplitude fluctuates around these conformational definitions over time. In all cases except the W272A mutant, the conformation of the glucose ring in the −1 subsite relaxes to the chair conformation. Interestingly, the wild type −1 subsite ring cycles between chair and skew conformations, with skew as the preferential conformation. This suggests that the tryptophans local to the catalytic site, Trp-135, Trp-269, and Trp-367, are crucial for maintaining the ring conformation needed for catalysis, and that Trp-272 does not contribute to this function. Given the similarity of the tunnel region surrounding the catalytic site in W272A compared with wild type, we hypothesize that W272A MD simulation would likely exhibit similar −1 subsite conformational fluctuations as observed in the wild type upon extended sampling time.
FIGURE 4.

Cremer-Pople ring pucker amplitude of the −1 binding site glucose from 250-ns MD simulations of the Cel6A wild type and 4 mutants.
Detailed analyses of the hydrogen bonding of active site residues and general active site behavior can also provide insights into the results obtained from the relative ligand binding free energy and the long MD simulations. In the wild type MD simulation in the −1 binding site, we observed hydrogen bonding of residue Asp-221, the catalytic residue, to Asp-175 over the course of the simulation (supplemental Fig. S7). The hydrogen bonding of Asp-221 to another carboxylic acid residue allows for the sharing of a proton between the two residues. When this hydrogen bond is broken, Asp-221 is able to act as a catalytic acid pushing a proton onto a free electron pair on the glycosidic oxygen. Hydrogen bonding of these two residues does not occur in the W135A, W269A, and W367A MD simulations. For the wild type, Asp-221/Asp-175 hydrogen bonding is disrupted concomitantly with the conformational change of the −1 glucose ring. This crucial hydrogen bond is relatively stable over the course of the W272A MD simulation. In the wild type simulation, the distance of the carboxyl oxygen of Asp-221 from the glycosidic oxygen connecting the cellulose chain between the +1/−1 binding sites indicates hydrogen bonding between Asp-221 and Asp-175 ceases as Asp-221 moves away from Asp-175 and closer to the glycosidic oxygen. When the −1 glucopyranose ring changes from the skew to chair conformation in the wild type, this distance exhibits an average value of ∼4 Å. In the W135A, W269A, and W367A simulations, the Asp-221 residue quickly moves away from the protein toward the ligand preceding the glucopyranose ring conformational change, likely as a result of the increased volume of the active site tunnel near the catalytic acid. This movement of Asp-221 toward the glycosidic oxygen is consistent with the observations of Koivula et al. (52). We also observe hydrogen bond formation between the catalytic acid, Asp-221, and the primary alcohol group of the −1 site glucose following a conformational change in this ring. Hydrogen bonding of the glucose primary alcohol to the Tyr-169 residue is consistent regardless of glucose conformation.
Related to the increased fluctuations observed in the 170–190 residue region in Fig. 2A, we evaluated hydrogen bonding between residues Tyr-169/Arg-174 and Arg-174/Asp-175 and calculated the r.m.s. deviation of the 172–182 residue region. Koivula et al. (52) observed a conformational change in the 172–182 loop as part of their structural studies that resulted in the breakage of interactions between Asp-175/Asp-221 and Tyr-169/Arg-174 and a 6-Å conformational change of Ser-181. From the r.m.s. deviation of this loop (supplemental Fig. S5B), we did not observe a similar conformational change over the course of the 250-ns MD simulations, likely a function of the limited time scale for conformational sampling. We suspect, however, based on the r.m.s. fluctuation in Fig. 2A, that W135A is undergoing a conformational change of the 172–182 region. Decreased hydrogen bonding between Tyr-169 and Arg-174 is observed in the W135A simulation in comparison to wild type, although all of the mutants exhibit this to some extent (supplemental Fig. S9). Opposite to that observed by Koivula et al. (52), we find increased hydrogen bonding between Arg-174 and Asp-175 for W135A (supplemental Fig. S10). It is likely that this behavior, which we attribute to large conformational fluctuations, is a function of the mutation.
A final analysis of the wild type residue cross-correlation obtained from normal mode analysis (Fig. 5) shows strong positive correlations of residues Trp-135, Trp-269, and Trp-367 with the active site region, whereas Trp-272 is much less correlated. The positive correlation of Trp-135 with the active region, residues 163 to 178, includes a portion of the active site loop associated with increased fluctuations, as discussed above. Residue Trp-269 is correlated with section 221–231, and residue Trp-367 is correlated with section 399–403. The correlation value of each of these three residues, Trp-135, Trp-269, and Trp-367, is generally in the range of 0.6. Notably, the positive correlation of residue Trp-272 with any region of the active site is less than 0.4. Finally, whereas the MD simulation results indicate the dynamics of the 400–410 loop region are affected by the W135A and W272A mutations, normal mode analysis does not show a cross-correlation. This is likely a shortcoming of the capabilities of normal mode analysis, as it is well accepted that normal mode analysis may miss some loop activity, particularly in small sections such as the 10-residue loop here (67). The results from normal mode analysis further verify the roles of the aromatic residues as suggested by TI and MD simulations.
FIGURE 5.

Residue cross-correlation map of wild type Cel6A. A positive value denotes a positive correlation of motion, whereas negative values are indicative of negative correlation of motion. Zero represents a total lack of correlated modes. This map was calculated using the first 100 modes. Faint white lines running from left to right have been placed at locations on the map corresponding to Trp-135, Trp-269, Trp-272, and Trp-367.
DISCUSSION
Overall, this work presents several roles for aromatic-carbohydrate interactions in cellulase tunnels. First, residues near the entrance of the tunnels in Cel6A and Cel7A have been shown to be crucial for hydrolyzing insoluble substrates experimentally (39, 43, 44). Here we show that removal of this residue in Cel6A, Trp-272, leads to significantly reduced binding affinity and greater fluctuations of the oligomer chain in the tunnel. Further MD simulations, including that of the entire Cel6A complex on a crystalline cellulose microfibril, similar to work done on Cel7A (68), are warranted to examine how mutation of this tunnel entrance tryptophan affects the behavior and stability of the catalytically active complex on an insoluble substrate. Second, we have demonstrated that the aromatic residue on the product side of the catalytic domain tunnel in Cel6A plays a significant role in binding, as the removal of this residue dramatically affects the ligand binding free energy. Specifically, our MD simulations of the Cel6A wild type indicate that the interactions at the −2 site are much higher than anywhere else along the cellodextrin chain, suggesting that this aromatic residue plays a significant role in stabilization of the product state during catalysis and likely contributes to product inhibition via association of the product to the −1 and −2 sites.
Last, and most surprisingly, our results for the +1 and +2 site residues directly preceding the hydrolysis site suggest functionality not directly associated with tight binding. Rather, we propose the roles of Trp-367 and Trp-269 include structuring the active site tunnel appropriately for ideal positioning of the ligand for catalysis. Mutation of these residues, specifically Trp-367, would almost certainly result in a decrease in activity on crystalline cellulose substrates. Without the requisite distortion at the −1 binding site, the glycosidic oxygen is further from the catalytic residue, shown in Fig. 1A, and the anomeric carbon is in a less favorable position for nucleophilic attack, and therefore not appropriately preactivated for catalysis (69). Interestingly, an experimental study of T. fusca Cel6B, a family 6 GH similar to T. reesei Cel6A, showed that a mutation corresponding to W367A (W464A in T. fusca Cel6B) resulted in a significant decrease in both activity and binding to bacterial microcrystalline cellulose, with a more moderate reduction in activity on phosphoric acid-swollen cellulose (37). Processivity of the mutated T. fusca Cel6B remained similar to wild type. The T. fusca Cel6B experimental results combined with our results suggest that mutations of tryptophans in the +1 and +2 binding sites of family 6 cellulases may have little effect on processivity, which in this GH family is linked to low ligand binding free energies of aromatic residues near the catalytic site. This observation can be contrasted to the observed processivity differences for GH family 18 chitinases, further suggesting that, in general, the results from Horn et al. (36) are linked closely to higher ligand binding affinities. This will be examined in future work.
From the simulation results obtained in this study and previous experimental results (36, 37), we hypothesize that the thermodynamic nature of aromatic-carbohydrate interactions in glycoside hydrolases is a function of multiple factors, including the ligand conformation (both the glycosidic linkage conformations and the ring conformations), the ligand type, the hydrolytic mechanism (which dictates the position of the ring distortion), the nature of the binding site (i.e. a tunnel or a cleft with surrounding binding site residues), and the GH protein fold (i.e. the GH family). This hypothesis may suggest discontinuity in the strength of aromatic-carbohydrate interactions in the equivalent ligand binding sites across GH families. For example, evaluation of the potential energies of the glycosidic bond between the −1 and +1 sites from the β-1,4 φ/ϕ angles in the crystal structures of Cel6A and the family 18 chitinase ChiB indicates the potential energy of the ChiB ligand conformation is 1–2 kcal/mol higher than the cellodextrin chain in the Cel6A crystal structure, which may manifest itself as a higher ligand binding free energy in ChiB than in Cel6A (70, 71). Additionally, ChiB hydrolyzes the glycosidic linkage through a substrate-assisted retaining mechanism, wherein the substrate acts as a nucleophile (72). In contrast, Cel6A uses a single displacement inverting mechanism, where the nucleophilic attack is made by a water molecule and a catalytic base is required to abstract the proton from the water, increasing its nucleophilicity (73, 74). Each of these differences likely directly contributes to the residue functionality in active sites of GHs. It is possible that aromatic-carbohydrate functional roles may only be specific to GHs of sufficiently similar fold and hydrolysis mechanisms, which we will examine in future work.
There are several direct extensions to this work and related work (36, 38, 45, 46). In terms of converting processive enzymes to nonprocessive enzymes, as shown by Horn et al. (36), it would be of significant interest to determine the relative extents of processivity of a processive enzyme with the appropriate aromatic mutations (e.g. W97A for ChiB and possibly W367A for Cel6A) relative to a corresponding natural nonprocessive enzyme (i.e. an endoglucanase). Nonprocessive enzymes, such as endoglucanase Cel6B from Humicola insolens (75), would be ideal for such comparisons within this family. In a future study, we will examine the role of aromatic residues in corresponding processive and nonprocessive enzymes via a similar TI approach. Moreover, experimental measurements using techniques such as isothermal titration calorimetry (22, 76, 77), could potentially validate the trends observed in the changes in ligand binding free energy calculated here with TI. However, this would require substrates with modified linkages (e.g. with thioether bonds) such as those used in structural studies so that the catalytically active complex can be formed without hydrolytic activity. Calculation of the entire range of torsional parameters for glycosidic linkages of chitobiose and cellobiose, and similarly for ring distortion, using a consistent and sufficiently accurate model would also be another step toward identifying the energetic contributions to ligand binding free energies across the many GH families.
CONCLUSIONS
Free energy calculations, MD simulations, and normal mode analysis have been used to examine the functional roles of aromatic residues in the active site of the well characterized T. reesei processive cellulase, Cel6A. Residues associated with ligand acquisition and product stabilization/inhibition have the greatest influence on ligand binding free energy and thus are likely to have a large impact on enzyme activity and processivity. Unexpectedly, the +1 and +2 site tryptophan residues appear not to participate in the tight binding to the ligand, but rather in stabilization of the ligand for the formation of the catalytically active substrate conformation. Comparison of the results here for Cel6A to experimental results for family 18 chitinases as a function of binding site indicates the role of aromatic-carbohydrate interactions in processivity and ligand binding may not be reduced to a model simply based on tunnel position relative to the location of catalysis. As a result, protein engineering strategies focused on improved biomass utilization may not be as generalized as previously expected and may require tailoring specific to the enzyme family. Based on the results of this study, it is apparent that many factors contribute to the thermodynamic nature of aromatic-carbohydrate interactions in GH families, which suggests additional studies on the strength of aromatic-carbohydrate interactions in the context of the protein folds, hydrolytic mechanisms, and the substrate types. More generally, this study suggests that nature employs aromatic-carbohydrate interactions over a very large range of binding affinities for multiple functions.
Supplementary Material
Acknowledgments
Computer time was provided by the Texas Advanced Computing Center Ranger cluster and the National Institute for Computational Sciences Athena cluster under the National Science Foundation Teragrid Grant MCB090159 and the National Renewable Energy Laboratory Computational Sciences Center supported by the Department of Energy Office of Energy Efficiency and Renewable Energy under Contract DE-AC36–08GO28308. We thank Michelle Kuttel for helpful discussions regarding ring parameters, Deanne Sammond, Seonah Kim, and Stephen Chmely for help with figure preparation, and Alex MacKerell for providing updated force field parameters. We also thank Peter Ciesielski, Steve Decker, Mike Resch, and Deanne Sammond for helpful discussions and a critical reading of the manuscript. Fig. 1 was made with PyMOL (78). The glucose ring conformation and hydrogen bonding analyses were performed using VMD (54, 79, 80).
This work was supported by the Department of Energy Office of the Biomass Program.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S13.
- GH
- glycoside hydrolases
- TI
- thermodynamic integration
- MD
- molecular dynamics
- r.m.s.
- root mean square.
REFERENCES
- 1. Gagneux P., Varki A. (1999) Glycobiology 9, 747–755 [DOI] [PubMed] [Google Scholar]
- 2. Crocker P. R., Paulson J. C., Varki A. (2007) Nat. Rev. Immunol. 7, 255–266 [DOI] [PubMed] [Google Scholar]
- 3. Crocker P. R., Feizi T. (1996) Curr. Opin. Struct. Biol. 6, 679–691 [DOI] [PubMed] [Google Scholar]
- 4. Lis H., Sharon N. (1998) Chem. Rev. 98, 637–674 [DOI] [PubMed] [Google Scholar]
- 5. Gilbert H. J., Stålbrand H., Brumer H. (2008) Curr. Opin. Plant Biol. 11, 338–348 [DOI] [PubMed] [Google Scholar]
- 6. Brewer C. F., Miceli M. C., Baum L. G. (2002) Curr. Opin. Struct. Biol. 12, 616–623 [DOI] [PubMed] [Google Scholar]
- 7. Weis W. I., Drickamer K. (1996) Annu. Rev. Biochem. 65, 441–473 [DOI] [PubMed] [Google Scholar]
- 8. Boraston A. B., Bolam D. N., Gilbert H. J., Davies G. J. (2004) Biochem. J. 382, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kiehna S. E., Laughrey Z. R., Waters M. L. (2007) Chem. Commun. 39, 4026–4028 [DOI] [PubMed] [Google Scholar]
- 10. Laughrey Z. R., Kiehna S. E., Riemen A. J., Waters M. L. (2008) J. Am. Chem. Soc. 130, 14625–14633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ramírez-Gualito K., Alonso-Ríos R., Quiroz-García B., Rojas-Aguilar A., Díaz D., Jiménez-Barbero J., Cuevas G. (2009) J. Am. Chem. Soc. 131, 18129–18138 [DOI] [PubMed] [Google Scholar]
- 12. Terraneo G., Potenza D., Canales A., Jiménez-Barbero J., Baldridge K. K., Bernardi A. (2007) J. Am. Chem. Soc. 129, 2890–2900 [DOI] [PubMed] [Google Scholar]
- 13. Screen J., Stanca-Kaposta E. C., Gamblin D. P., Liu B., Macleod N. A., Snoek L. C., Davis B. G., Simons J. P. (2007) Angew. Chem. Int. Ed. Engl. 46, 3644–3648 [DOI] [PubMed] [Google Scholar]
- 14. Vandenbussche S., Diaz D., Fernandez-Alonso M. C., Pan W. D., Vincent S. P., Cuevas G., Canada F. J., Jimenez-Barbero J., Bartik K. (2008) Chem. Eur. J. 14, 7570–7578 [DOI] [PubMed] [Google Scholar]
- 15. Shental-Bechor D., Levy Y. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 8256–8261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Laurent N., Voglmeir J., Flitsch S. L. (2008) Chem. Commun. 37, 4400–4412 [DOI] [PubMed] [Google Scholar]
- 17. del Carmen Fernández-Alonso M., Cañada F. J., Jiménez-Barbero J., Cuevas G. (2005) J. Am. Chem. Soc. 127, 7379–7386 [DOI] [PubMed] [Google Scholar]
- 18. Paulson J. C., Blixt O., Collins B. E. (2006) Nat. Chem. Biol. 2, 238–248 [DOI] [PubMed] [Google Scholar]
- 19. Gabius H. J., Siebert H. C., André S., Jiménez-Barbero J., Rüdiger H. (2004) ChemBioChem 5, 740–764 [DOI] [PubMed] [Google Scholar]
- 20. Williams S. J., Davies G. J. (2001) Trends Biotechnol. 19, 356–362 [DOI] [PubMed] [Google Scholar]
- 21. Smith E. A., Thomas W. D., Kiessling L. L., Corn R. M. (2003) J. Am. Chem. Soc. 125, 6140–6148 [DOI] [PubMed] [Google Scholar]
- 22. Zolotnitsky G., Cogan U., Adir N., Solomon V., Shoham G., Shoham Y. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 11275–11280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) Nucleic Acids Res. 37, D233-D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beckham G. T., Matthews J. F., Bomble Y. J., Bu L., Adney W. S., Himmel M. E., Nimlos M. R., Crowley M. F. (2010) J. Phys. Chem. B 114, 1447–1453 [DOI] [PubMed] [Google Scholar]
- 25. Lehtiö J., Sugiyama J., Gustavsson M., Fransson L., Linder M., Teeri T. T. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 484–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Himmel M. E., Ding S. Y., Johnson D. K., Adney W. S., Nimlos M. R., Brady J. W., Foust T. D. (2007) Science 315, 804–807 [DOI] [PubMed] [Google Scholar]
- 27. Stern R., Jedrzejas M. J. (2008) Chem. Rev. 108, 5061–5085 [DOI] [PubMed] [Google Scholar]
- 28. Divne C., Ståhlberg J., Reinikainen T., Ruohonen L., Pettersson G., Knowles J. K., Teeri T. T., Jones T. A. (1994) Science 265, 524–528 [DOI] [PubMed] [Google Scholar]
- 29. Rouvinen J., Bergfors T., Teeri T., Knowles J. K., Jones T. A. (1990) Science 249, 380–386 [DOI] [PubMed] [Google Scholar]
- 30. van Aalten D. M., Synstad B., Brurberg M. B., Hough E., Riise B. W., Eijsink V. G., Wierenga R. K. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5842–5847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beckham G. T., Bomble Y. J., Bayer E. A., Himmel M. E., Crowley M. F. (2011) Curr. Opin. Biotechnol. 22, 231–238 [DOI] [PubMed] [Google Scholar]
- 32. Chundawat S. P. S., Beckham G. T., Himmel M. E., Dale B. E. (2011) Annu. Rev. Chem. Biomol. Eng. 2, 121–145 [DOI] [PubMed] [Google Scholar]
- 33. Jalak J., Väljamäe P. (2010) Biotechnol. Bioeng. 106, 871–883 [DOI] [PubMed] [Google Scholar]
- 34. Boisset C., Fraschini C., Schülein M., Henrissat B., Chanzy H. (2000) Appl. Environ. Microbiol. 66, 1444–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kurasin M., Väljamäe P. (2011) J. Biol. Chem. 286, 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Horn S. J., Sikorski P., Cederkvist J. B., Vaaje-Kolstad G., Sørlie M., Synstad B., Vriend G., Vårum K. M., Eijsink V. G. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 18089–18094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vuong T. V., Wilson D. B. (2009) Appl. Environ. Microbiol. 75, 6655–6661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zakariassen H., Aam B. B., Horn S. J., Vårum K. M., Sørlie M., Eijsink V. G. H. (2009) J. Biol. Chem. 284, 10610–10617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Koivula A., Kinnari T., Harjunpää V., Ruohonen L., Teleman A., Drakenberg T., Rouvinen J., Jones T. A., Teeri T. T. (1998) FEBS Lett. 429, 341–346 [DOI] [PubMed] [Google Scholar]
- 40. Katouno F., Taguchi M., Sakurai K., Uchiyama T., Nikaidou N., Nonaka T., Sugiyama J., Watanabe T. (2004) J. Biochem. 136, 163–168 [DOI] [PubMed] [Google Scholar]
- 41. Watanabe T., Ariga Y., Sato U., Toratani T., Hashimoto M., Nikaidou N., Kezuka Y., Nonaka T., Sugiyama J. (2003) Biochem. J. 376, 237–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Uchiyama T., Katouno F., Nikaidou N., Nonaka T., Sugiyama J., Watanabe T. (2001) J. Biol. Chem. 276, 41343–41349 [DOI] [PubMed] [Google Scholar]
- 43. Igarashi K., Koivula A., Wada M., Kimura S., Penttilä M., Samejima M. (2009) J. Biol. Chem. 284, 36186–36190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. von Ossowski I., Ståhlberg J., Koivula A., Piens K., Becker D., Boer H., Harle R., Harris M., Divne C., Mahdi S., Zhao Y., Driguez H., Claeyssens M., Sinnott M. L., Teeri T. T. (2003) J. Mol. Biol. 333, 817–829 [DOI] [PubMed] [Google Scholar]
- 45. Beckham G. T., Matthews J. F., Peters B., Bomble Y. J., Himmel M. E., Crowley M. F. (2011) J. Phys. Chem. B 115, 4118–4127 [DOI] [PubMed] [Google Scholar]
- 46. Beckham G. T., Crowley M. F. (2011) J. Phys. Chem. B 115, 4516–4522 [DOI] [PubMed] [Google Scholar]
- 47. Payne C. M., Himmel M. E., Crowley M. F., Beckham G. T. (2011) J. Phys. Chem. Lett. 2, 1546–1550 [Google Scholar]
- 48. Jeoh T., Ishizawa C. I., Davis M. F., Himmel M. E., Adney W. S., Johnson D. K. (2007) Biotechnol. Bioeng. 98, 112–122 [DOI] [PubMed] [Google Scholar]
- 49. Chundawat S. P., Bellesia G., Uppugundla N., da Costa Sousa L., Gao D., Cheh A. M., Agarwal U. P., Bianchetti C. M., Phillips G. N., Jr., Langan P., Balan V., Gnanakaran S., Dale B. E. (2011) J. Am. Chem. Soc. 133, 11163–11174 [DOI] [PubMed] [Google Scholar]
- 50. Kirkwood J. G. (1935) J. Chem. Phys. 3, 300–313 [Google Scholar]
- 51. Kollman P. (1993) Chem. Rev. 93, 2395–2417 [Google Scholar]
- 52. Koivula A., Ruohonen L., Wohlfahrt G., Reinikainen T., Teeri T. T., Piens K., Claeyssens M., Weber M., Vasella A., Becker D., Sinnott M. L., Zou J. Y., Kleywegt G. J., Szardenings M., Ståhlberg J., Jones T. A. (2002) J. Am. Chem. Soc. 124, 10015–10024 [DOI] [PubMed] [Google Scholar]
- 53. Frenkel D., Smit B. (2002) Understanding Molecular Simulations: From Algorithms to Applications, 2nd Ed., [Google Scholar]
- 54. Kuttel M., Gain J., Burger A., Eborn I. (2006) J. Mol. Graph. Model. 25, 380–388 [DOI] [PubMed] [Google Scholar]
- 55. Straatsma T. P., McCammon J. A. (1991) J. Chem. Phys. 95, 1175–1188 [Google Scholar]
- 56. Brooks B. R., Brooks C. L., 3rd, Mackerell A. D., Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M. (2009) J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zou J., Kleywegt G. J., Ståhlberg J., Driguez H., Nerinckx W., Claeyssens M., Koivula A., Teeri T. T., Jones T. A. (1999) Structure 7, 1035–1045 [DOI] [PubMed] [Google Scholar]
- 58. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiorkiewicz-Kuczera J., Yin D., Karplus M. (1998) J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 59. Mackerell A. D., Jr., Feig M., Brooks C. L., 3rd (2004) J. Comput. Chem. 25, 1400–1415 [DOI] [PubMed] [Google Scholar]
- 60. Guvench O., Hatcher E., Venable R. M., Pastor R. W., MacKerell A. D. (2009) J. Chem. Theor. Comp. 5, 2353–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guvench O., Greene S. N., Kamath G., Brady J. W., Venable R. M., Pastor R. W., Mackerell A. D., Jr. (2008) J. Comput. Chem. 29, 2543–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guvench O., Mallajosyula S. S., Raman E. P., Hatcher E. R., Vanommeslaeghe K., Foster T. J., Jamison F. W., MacKerell A. D. (2011) J. Chem. Theory Comput. 7, 3162–3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bu L., Beckham G. T., Shirts M. R., Nimlos M. R., Adney W. S., Himmel M. E., Crowley M. F. (2011) J. Biol. Chem. 286, 18161–18169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mertz B., Hill A. D., Mulakala C., Reilly P. J. (2007) Biopolymers 87, 249–260 [DOI] [PubMed] [Google Scholar]
- 65. Cremer D., Pople J. A. (1975) J. Am. Chem. Soc. 97, 1354–1358 [Google Scholar]
- 66. Hill A. D., Reilly P. J. (2007) J. Chem. Inf. Model. 47, 1031–1035 [DOI] [PubMed] [Google Scholar]
- 67. Brooks B., Karplus M. (1983) Proc. Natl. Acad. Sci. U.S.A. 80, 6571–6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhong L., Matthews J. F., Hansen P. I., Crowley M. F., Cleary J. M., Walker R. C., Nimlos M. R., Brooks C. L., 3rd, Adney W. S., Himmel M. E., Brady J. W. (2009) Carbohydr. Res. 344, 1984–1992 [DOI] [PubMed] [Google Scholar]
- 69. Biarnés X., Ardèvol A., Planas A., Rovira C., Laio A., Parrinello M. (2007) J. Am. Chem. Soc. 129, 10686–10693 [DOI] [PubMed] [Google Scholar]
- 70. Dowd M. K., French A. D., Reilly P. J. (1992) Carbohydr. Res. 233, 15–34 [DOI] [PubMed] [Google Scholar]
- 71. Yui T., Kobayashi H., Kitamura S., Imada K. (1994) Biopolymers 34, 203–208 [Google Scholar]
- 72. Jitonnom J., Lee V. S., Nimmanpipug P., Rowlands H. A., Mulholland A. J. (2011) Biochemistry 50, 4697–4711 [DOI] [PubMed] [Google Scholar]
- 73. Knowles J. K. C., Lentovaara P., Murray M., Sinnott M. L. (1988) J. Chem. Soc. Chem. Commun. 1401–1402 [Google Scholar]
- 74. Claeyssens M., Tomme P., Brewer C. F., Hehre E. J. (1990) FEBS Lett. 263, 89–92 [DOI] [PubMed] [Google Scholar]
- 75. Davies G. J., Brzozowski A. M., Dauter M., Varrot A., Schülein M. (2000) Biochem. J. 348, 201–207 [PMC free article] [PubMed] [Google Scholar]
- 76. Norberg A. L., Eijsink V. G., Sorlie M. (2010) Thermochim. Acta 511, 189–193 [Google Scholar]
- 77. Norberg A. L., Karlsen V., Hoell I. A., Bakke I., Eijsink V. G., Sørlie M. (2010) FEBS Lett. 584, 4581–4585 [DOI] [PubMed] [Google Scholar]
- 78. DeLano W. L. (2010) The PyMOL Molecular Graphics System, 1.3 Ed., Schrodinger, LLC, New York [Google Scholar]
- 79. Cross S., Kuttel M. M., Stone J. E., Gain J. E. (2009) J. Mol. Graph. Model. 28, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Humphrey W., Dalke A., Schulten K. (1996) J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.