

Abstract
Human immunodeficiency virus (HIV) infection of a host cell begins with fusion of the HIV and host cell membranes and is mediated by the gp41 protein, a single-pass integral membrane protein of HIV. The N-terminal 175 residues are the ectodomain which lies outside the virus. This paper describes production and characterization of an ectodomain construct containing the 154 N-terminal gp41 residues including fusion peptide (FP) that binds to target cell membranes. The Fgp41 sequence was derived from one of the African clade A strains of HIV-1 which have been less studied than European/North American clade B strains. Fgp41 expression at ~100 mg/L culture was evidenced by an approach that included amino acid type 13CO and 15N labeling of recombinant protein and solid-state NMR (SSNMR) spectroscopy of lyophilized whole cells. The approach did not require any protein solubilization or purification and may be a general approach for detection of recombinant protein. The purified Fgp41 yield was ~5 mg/L culture. SSNMR spectra of membrane-associated Fgp41 showed high helicity at residues C-terminal of the FP. This was consistent with “six-helix bundle” (SHB) structure which is the final gp41 state during membrane fusion. This observation plus negligible Fgp41-induced vesicle fusion supported a function for SHB gp41 of membrane stabilization and fusion arrest. SSNMR spectra of residues in the membrane-associated FP evidenced a mixture of molecular populations with either helical or β sheet FP conformation. These and earlier SSNMR data strongly support the existence of these populations in the SHB state of membrane-associated gp41.
Keywords: HIV, HIV-1, gp41, fusion peptide, solid-state NMR, secondary structure
The human immunodeficiency virus (HIV) is enveloped by a membrane obtained during budding from an infected host cell. An early step in HIV infection of a new cell is joining or “fusion” of the HIV and host cell membranes. This process is catalyzed by the ~350-residue HIV gp41 protein which is an integral membrane protein of the viral envelope (1). The ~175 N-terminal residues form the ectodomain which lies outside HIV; see Fig. 1a for most of the ectodomain sequence and identification of structural/functional regions. Prior to fusion, gp41 is non-covalently associated with the gp120 protein, Fig. 1b. Productive infection begins with binding of gp120 to receptor proteins in a target cell membrane and is followed by gp120 dissociation from gp41 (2). There are ensuing structural changes of gp41 and likely binding of the ~20-residue N-terminal “fusion peptide” (FP) region to target cell membranes with concurrent changes in the two membranes including mixing of lipids, formation of a single hemifusion diaphragm bilayer that separates the HIV and cell contents; and opening of the diaphragm to form a single membrane that encloses HIV and the cell (3). Although there are no high-resolution structures of full-length gp41, other structural and functional data support: (1) trimeric gp41; (2) an early-stage “pre-Hairpin intermediate” (PHI) state with a parallel trimer of fully extended ectodomains between the HIV membrane and the FP in the cell membrane; and (3) a final “six-helix bundle” (SHB) state with a gp41 trimer with each gp41 molecule having a N-helix-turn-C-helix Hairpin structure and parallel N-helices in the trimer interior and parallel C-helices on the trimer exterior, Fig. 1b (4–7). Studies of cell-cell fusion induced by gp120/gp41 complexes indicate that most membrane fusion steps with the exception of diaphragm opening occur prior to formation of the final SHB state (8).
Figure 1.

Panel a shows the Fgp41 amino acid sequence with regions encompassing the fusion peptide, N-helix, and C-helix in blue, red, and green, respectively. Panel b is a conceptual representation of gp41 structural states with time increasing from left to right and the same color coding as panel a. The sequence is from the HIV-1 clade A isolate, AY288087 or Q45D5. This is a primary isolate from a female patient in Kenya. The last eight residues of the sequence are non-native. The red and green regions were based on continuous helical regions in high-resolution structures of different soluble ectodomain gp41 constructs with Hairpin conformation (shown schematically in panel b). In these SHB structures, the red and green helices are antiparallel to one another.
The importance of the FP in fusion and infection has been highlighted by reduction in both functions with point mutations in the FP (9). Current understanding of gp41 is also based on smaller fragments of gp41 where fusogenic function has typically been assayed by fragment-induced perturbation/fusion of membrane vesicles. One such fragment is the HIV fusion peptide (HFP) which corresponds to the 20–30 N-terminal residues of gp41 and which has moderate fusogenicity (10). The functional significance of the PHI trimeric topology, Fig. 1b, has been supported by high fusogenicity of: (1) a cross-linked HFP trimer (HFPtr); and (2) “N70”, the 70 N-terminal residues of gp41 (10–13). The higher fusogenicity of N70 relative to HFP may also have a contribution from the N-helix residues that are C-terminal of the FP. Much larger ectodomain constructs have also been produced with N-helix and C-helix regions and form the thermostable SHB structure, Fig. 1b, which is the final gp41 state. Different approaches were used to obtain these FP-containing Hairpin constructs. In one approach, the FP and Hairpin regions were produced separately by chemical synthesis and bacterial expression, respectively, and “FP-Hairpin” was then made by native chemical ligation (11, 12). In another approach, a chimera was expressed in E. coli bacteria and contained a N-terminal molecular carrier protein (e.g. glutathione S-transferase) followed by the gp41 ectodomain (14–16). The carrier was cleaved during purification. There are conflicting results from different studies of the fusogenicity of such Hairpin constructs with reports of both very high and no fusion. There were some differences among the studies including: (1) deletion of loop residues between the N- and C-helices in some constructs; (2) lipid compositions of the vesicles including different fractions of negatively charged lipid; (3) use of smaller and less stable sonicated vesicles vs larger and more stable extruded vesicles; and (4) pHs that ranged between 3.0 and 7.5 (17).
Structural studies have also been carried out for some of the aforementioned fragments. A helical monomer HFP has been observed in detergent with one report of a continuous helix between residues 4 and 22 (18–22). However, to our knowledge, HFP does not induce fusion between detergent micelles. The structure of membrane-associated HFP has been probed mostly by solid-state nuclear magnetic resonance (SSNMR) spectroscopy with supporting data from other techniques such as infrared spectroscopy (23–25). SSNMR spectra of HFP associated with membranes lacking cholesterol show distinct populations of predominant β sheet and predominant α helical molecules while HFP associated with membranes with ~30 mol% cholesterol show only the β sheet conformation with antiparallel alignment of adjacent hydrogen bonded HFPs (10, 26–30). The biological relevance of membrane cholesterol is supported by the ~25 mol% cholesterol in host cell membranes and the ~45 mol% in HIV membranes (31). The FP structure appears to be similar in the highly fusogenic HFP trimer and in N70 whereas the FP-Hairpin construct with SHB structure showed approximately equal populations of molecules with either β sheet or helical FP structures (10, 12, 29). For membrane-associated N70, the N-helix residues appear to be predominantly helical and N70 is recognized by an antibody specific for trimeric coiled-coil N-helices (32). There are several high-resolution structures of Hairpin constructs without FP which show: (1) Hairpin structure of individual molecules; and (2) molecular trimers with SHB structure (4–7).
SSNMR requires production of multi-mg quantities of isotopically labeled protein and protein yields may be reduced by ligation and/or cleavage steps. This motivated one of the goals of the present study – expression of the FP-containing gp41 ectodomain (“Fgp41”) in bacteria without a chimera or ligation. This goal seemed reasonable because recently developed protocols yielded 20 mg protein/L culture for the full-length “FHA2” ectodomain of the influenza virus fusion protein (33–35). There is considerable diversity among HIV protein sequences in patient sera and in cell cultures. This motivated a second goal of the present study – functional and structural experiments on a gp41 ectodomain sequence that differed from the sequence of the earlier studies to address the generality of the functional and structural findings across strains of HIV. In these earlier studies, the sequence was from the HXB2 strain of HIV-1 which was first created in cell culture in 1984 and which is grouped with “clade B” HIV-1 prevalent in patients in North America and Europe (36). In some contrast, the gp41 sequence of the present study is from the primary HIV-1 isolate Q45D5 from the sera of a newly infected Kenyan woman (37). The Q45D5 isolate is grouped with clade A HIV-1 that is prevalent in Central and East Africa. The HXB2 and Q45D5 gp41 ectodomain sequences are provided in the Supporting Information. A third motivation for the present study was to provide comparative functional and SSNMR structural studies to the FP-Hairpin construct in which 46 contiguous residues including the native loop were replaced by the non-native SGGRGG sequence (11, 12, 17). FP-Hairpin did not induce vesicle fusion and inhibited fusion by constructs such as N70. The deletion of these residues in FP-Hairpin may be important as a 35-residue peptide which included the native loop region induced vesicle fusion under some conditions (38). The Fgp41 construct of the present study has the native sequence without deletion.
Materials and Methods
Materials
The Fgp41 plasmid was constructed within the pET24a(+) vector using the DNA sequence of the Q45D5 primary isolate which is grouped with clade A of HIV-1. The plasmid was transformed into BL21(DE3) chemically competent E. coli cells (Novagen, Gibbstown, NJ). The Fgp41 amino acid sequence is given in Fig. 1a and the correlative sequence from plasmid DNA extracted from the cells is provided in the Supporting Information. Other reagents and their sources include: Luria-Bertani Broth (LB) medium (Acumedia, Lansing, MI); n-octyl-β-D-thioglucopyranoside (bTOG) and isopropyl-β-D-thiogalactopyranoside (IPTG) (Anatrace, Maumee, OH); 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt) (POPG) (Avanti Polar Lipids, Alabaster, AL); isotopically labeled amino acids (Cambridge Isotope Laboratories, Andover, MA). Other materials were obtained from Sigma-Aldrich (St. Louis, MO).
Protein production and purification
The protocol was based on a previous protocol for the influenza virus fusion protein ectodomain (34). One key feature was initial bacterial growth in rich medium to high cell densities. Relative to initial growth in minimal medium, protein production was augmented by the cell densities and by the larger number of ribosomes per cell. Bacterial cell cultures were grown in media containing 15 mg/L kanamycin because the pET24a(+) vector contains a gene for kanamycin resistance. Bacterial cells in 1 mL of 80/20 (v/v) H2O/glycerol were added to two 2.8 L baffled fernbach flasks which each contained 1 L of LB and were capped with a foam plug. Bacterial growth to OD600 ≈ 4 occurred during overnight incubation at 37 °C with shaking at 140 rpm. The cell suspensions were centrifuged (10000g, 10 min) and the cell pellets were harvested and then resuspended in a single flask containing 1 L of fresh medium with M9 minimal salts, 2.0 mL of 1.0 M MgSO4, and 5.0 mL of 50% glycerol solution. Growth resumed after approximately one hour of incubation at 37 °C. At this time, 100 mg/L of 1-13C amino acid and 100 mg/L of 15N amino acid (or 100 mg/L of 1-13C, 15N amino acid) were added to the medium. IPTG was then added to a final concentration of 2 mM which induced expression of Fgp41 (6 hours, 23 °C). The cell pellet was harvested after centrifugation and stored at −80 °C. The wet cell mass was ~8 g.
A variant of this procedure was used to make whole cell NMR samples and included: (1) 50 mL culture volumes in 250 mL flasks; (2) overall reagent concentrations similar to those above including ~10 mg of labeled amino acid(s); and (3) in some cases, lyophilization of the cells.
The Fgp41 purification was based on the C-terminal His-tag and the following protocol is for ~3 g of wet cells. Most Fgp41 was in inclusion bodies so cells were lysed in a 40 mL solution containing 1% sodium dodecyl sulfate (SDS) as well as 50 mM sodium phosphate at pH 8.0, 300 mM NaCl, and 1 mM imidazole. Lysis was accomplished by sonication (4 × 1 min) with a tip sonifier (Branson, Danbury, CT) and the soluble and insoluble portions of the lysate were then separated by centrifugation (48000g, 20 min, 4 °C). The soluble lysate was mixed with 0.25 mL of cobalt His-Select resin (prepared using manufacturer’s instructions) and then agitated at ambient temperature for one hour. The resin with bound Fgp41 was separated from the lysate by flow through a fritted column. Purification was accomplished by flowing buffer (50 mM sodium phosphate at pH 8.0, 300 mM NaCl, and 0.5% SDS) through the resin with stepwise increases in [imidazole]. Weakly bound protein was first eluted with buffer containing [imidazole] = 1, 20, or 50 mM and tightly bound protein (predominantly Fgp41) was subsequently eluted with buffer containing [imidazole] = 250 mM. Excess SDS was precipitated by overnight incubation at 4°C and removed by centrifugation (16000g, 1 min). Fgp41 was typically dialyzed into “HEPES/MES” buffer (5 mM HEPES, 10 mM MES, pH 7.4). Fgp41 concentrations were quantified using ε280 = 46000 M−1·cm−1 which was estimated from the sum of the ε280s of individual tryptophans and tyrosines.
Circular dichroism (CD) spectroscopy
Spectra were obtained using a CD instrument (Chirascan, Applied Photophysics, Surrey, United Kingdom), 1 mm pathlength, a 260–200 nm spectral window, wavelength points separated by 0.5 nm, and 1 s signal averaging per point. Fgp41 samples were prepared by precipitation of excess SDS followed by overnight dialysis into HEPES/MES buffer at pH 7.4 with DTT added at two times the molar concentration of Fgp41 to prevent disulfide bond formation. Most spectra were obtained with [Fgp41] = 20 μM. For each sample, a reference spectrum was also taken of buffer without Fgp41 and the relevant Fgp41 spectrum was the difference between the Fgp41 + buffer and buffer only spectra.
Lipid mixing
One early step in fusion between the HIV and target cell membranes is mixing of lipids between the two membranes. This aspect of Fgp41 fusogenicity was probed by fluorescence-detected Fgp41-induced mixing of lipids between membrane vesicles. A set of vesicles was prepared that contained POPC:POPG lipids in 4:1 mol ratio and another set of “labeled” vesicles was prepared that contained an additional 2 mol % of the fluorescent lipid N-NBD-PE and 2 mol % of the quenching lipid N-Rh-PE. Large unilamellar vesicles (LUVs) were prepared by: (1) dissolving lipids in chloroform and then removing chloroform by nitrogen gas and overnight vacuum; (2) formation of pH 7.5 aqueous lipid dispersions with [total lipid] ≈ 5 mM and [HEPES] = 25 mM including five freeze-thaw cycles; and (3) ~20-fold extrusion through a polycarbonate filter with 0.1 μM diameter pores. The assay was done at 37 °C with continuous stirring in the HEPES buffer using a mixture of unlabeled vesicles ([total lipid] = 135 μM) and labeled vesicles ([total lipid] = 15 μM). After measuring the initial fluorescence F0, an aliquot of 30 μM Fgp41 in HEPES/MES buffer was added to the vesicle solution so that final [Fgp41] = 3 μM and Fgp41:total lipid = 0.02. Fgp41-induced fusion between labeled and unlabeled vesicles resulted in larger fluorophore-quencher distance and increased fluorescence. The fluorescence increase ΔFFgp41 was compared to the maximum fluorescence increase (ΔFmax) obtained after subsequent addition of Triton X-100 detergent which solubilized the vesicles. Assay parameters included: (1) fluorimeter (Photon Technology International); (2) excitation and emission wavelengths of 465 and 530 nm with 4 nm bandwidths; and (3) 1.8 mL of initial vesicle solution, 0.2 mL aliquot of Fgp41, and ~20 μL aliquot of 10% Triton X-100.
Membrane reconstitution
A homogeneous mixture of the POPC (28 mg) and POPG (7 mg) lipids and the bTOG (140 mg) detergent was made by: (1) dissolution in chloroform; (2) removal of chloroform by nitrogen gas and overnight vacuum; and (3) dissolution in HEPES/MES buffer. Fgp41 (~10 mg) was added to the solution and had been in affinity column eluents for which excess SDS had been removed. Dialysis of the bTOG/lipid/Fgp41 solution against HEPES/MES buffer removed bTOG with consequent liposome formation with bound Fgp41. Dialysis parameters included: (1) bTOG/lipid/Fgp41 solution in 10 kDa MWCO tubing (~15 mL initial volume); (2) 3L buffer volume; and (3) 3 day duration with one buffer change. The proteoliposome pellet was harvested after centrifugation (50000g, 3 hours) and unbound Fgp41 did not pellet under these conditions. The pellet was packed into a 4 mm diameter magic angle spinning (MAS) rotor with ~5 mg Fgp41 and ~20 mg total lipid in the 40 μL active sample volume.
Solid-state NMR spectroscopy
Data were obtained with a 9.4 T instrument (Agilent Infinity Plus) and a triple-resonance MAS probe whose rotor was cooled with nitrogen gas at −10 °C. Because of heating from MAS and rf radiation, we expect that water in the sample was liquid rather than solid. Experimental parameters included: (1) 8.0 kHz MAS frequency; (2) 5 μs 1H π/2 pulse and 2 ms cross-polarization time with 50 kHz 1H field and 70–80 kHz ramped 13C field; (3) 1 – 2 ms rotational-echo double-resonance (REDOR) dephasing time with a 9 μs 13C π pulse at the end of each rotor period except the last period and for some data, a 12 μs 15N π pulse at the center of each rotor period; and (4) 13C detection with 90 kHz two-pulse phase modulation 1H decoupling (which was also on during the dephasing time); and (5) 0.8 sec pulse delay (39). Data were acquired without (S0) and with (S1) 15N π pulses during the dephasing time and respectively represented the full 13C signal and the signal of 13Cs not directly bonded to 15N nuclei. The S0 – S1 (ΔS) difference signal was therefore dominated by the labeled 13COs in the sequential pairs targeted by the labeling. Spectra were externally referenced to the methylene carbon of adamantane at 40.5 ppm so that the 13CO shifts could be directly compared to those of soluble proteins (40).
Results
Detection of Fgp41 in whole cells by solid-state NMR
The bacterial growth and Fgp41 expression conditions were very similar to those used for FHA2, a construct corresponding to the full-length ectodomain (including fusion peptide) of the influenza virus HA2 fusion protein (34). Like Fgp41, FHA2 had a C-terminal hexahistidine tag and bacterial cell lysis and protein solubilization in buffer containing N-lauroylsarcosine detergent followed by affinity chromatography resulted in 10 mg purified FHA2/L culture. In some contrast, application of this protocol to cells induced to synthesize Fgp41 gave only 0.1 mg Fgp41/L culture. It was unclear whether the poor yield was due to low Fgp41 expression or to poor Fgp41 solubilization by the detergent.
The FHA2 solubilized by detergent was likely initially associated with the cell membrane. It was also shown that a much larger fraction of FHA2 was not solubilized by detergent and was likely constituted in inclusion bodies. The poor yield of Fgp41 might therefore be due to dominant incorporation in inclusion bodies. The molecular structure of FHA2 in inclusion bodies had been probed by: (1) adding specific 13CO and 15N labeled amino acids immediately prior to induction; and (2) recording REDOR SSNMR spectra of the whole cells after induction so that the filtered ΔS signal corresponded to the 13CO of a targeted residue in FHA2; and (3) correlation of the experimental peak 13CO shift to local conformation at this residue (41). A modified approach was applied to cells induced to express Fgp41 with the goal of assessing Fgp41 production. Addition of 10 mg of 1-13C, 15N Leu to 50 mL culture just prior to induction of expression targeted the 24 Leus and 6 LL repeats in the Fgp41 sequence, Fig. 1a. The isotropic 13CO regions of the REDOR S0, S1, and ΔS spectra of the cells are displayed in Fig. 2a,b. The Fig. 2a sample was an aliquot of the wet cell pellet obtained after the induction period and subsequent centrifugation and the Fig. 2b sample was an aliquot of this whole cell pellet that had been lyophilized. The spectra were similar for both wet and lyophilized cells with ~4 times greater signal-per-scan in the lyophilized cell sample because this sample had a higher fraction of non-aqueous cell mass. For either sample type, the intensity of the S1 spectrum was reduced relative to S0. This supported the presence of LL repeats in the protein produced during the induction period and correlated with the 6 LLs in the Fgp41 sequence. The ΔS spectra had prominent signals in the 13CO region and these were the only signals detectable above the noise (see Supporting Information for full spectra). Control cells were produced using unlabeled rather than labeled Leu. The resultant NMR spectra are displayed in Fig. 2c and had comparable S0 and S1 intensities with little 13CO ΔS signal. This provided further support that the 13CO ΔS signal from the labeled cells could be ascribed to LL repeats in protein produced during expression. Cells were also labeled with 1-13C Gly and 15N Phe which targeted the 11 Glys in the Fgp41 sequence and the single GF pair at G10F11. The resulting NMR spectra are displayed in Fig. 2d and included a prominent 13CO ΔS signal that was consistent with Fgp41 production.
Figure 2.

REDOR 13CO NMR spectra of whole bacterial cells produced by the sequential steps: (1) growth in rich medium; (2) growth in minimal medium; (3) addition of labeled or unlabeled amino acid(s); (4) induction of Fgp41 expression; and (5) centrifugation. The induction temperature and duration were either (a–c) 23 °C and ~2 hours or (d) 37 °C and ~5 hours. The left panels display S0 (blue) and S1 (red) spectra and the right panels display the ΔS ≡S0 – S1 spectra. The REDOR dephasing time was either (a–c) 1 ms or (d) 2 ms. For panels a,b,d, the dominant contribution to each ΔS spectrum was from residues with labeled 13COs that were directly bonded to labeled 15Ns. These residues were: (a, b) L33, L44, L54, L81, L134, and L149 of the LL sequential pairs of Fgp41; and (d) G10 of the G10/F11 unique sequential pair. Each S0 or S1 spectrum was processed with 100 Hz Gaussian line broadening and each ΔS spectrum was processed with either (a, d) 200 Hz or (b, c) 100 Hz line broadening. Polynomial baseline correction (typically 5th order) was applied to each spectrum. Each S0 or S1 spectrum was the sum of: (a) 100000; (b) 100000; (c) 127222; or (d) 48448 scans.
Both labeled and natural abundance 13COs contribute to the S0 and S1 NMR signals of the labeled whole cells. Fig. 3a(b) provides quantitative assessment of these two contributions and shows the full S0(S1) spectra of the Leu-labeled and unlabeled cells. In each panel, the two spectra were scaled to have equal intensity in the 0–90 ppm region because this region should be unaffected by labeling. The ratio of the unlabeled to labeled scaling factors was ~0.75 and matched the ratio of numbers of scans summed for the labeled vs unlabeled samples. This matching was expected because the signal intensities of individual scans were approximately equal to each other so the sum signal intensity increased linearly with number of scans. For panel a(b), the difference between the intensities in the 13CO region was the labeled Leu contribution to the S0(S1) signal. For these labeled Leu 13COs, there was smaller S1 intensity relative to S0. This is shown more clearly in Fig. 3c which displays the S0 and S1 spectra processed from labeled cell data – (0.75 × unlabeled cell data). For labeled Leu in the cells, the normalized experimental dephasing (ΔS/S0)exp = 0.13 ± 0.01 and was determined from the 13CO S0 and S1 intensities in panel c.
Figure 3.

REDOR 13C NMR spectra of lyophilized whole bacterial cells produced with either 1-13C, 15N Leu or unlabeled Leu. The cell production and NMR parameters are described in Fig. 2b, c caption. Panel a displays the S0 spectra of the (blue) labeled and (black) unlabeled cells with the relative intensities adjusted to obtain the best agreement in the 0–90 ppm region. Spectral intensity in this region should not be affected by the labeling. The incorporation of the labeled Leu into protein synthesized during the induction period is evidenced by the larger 13CO intensity for the labeled cell spectrum. Panel b displays the S1 spectra of the (red) labeled and (black) unlabeled cells. Panel c displays the S0 (blue) and S1 (red) spectra processed from the difference NMR data: (labeled cells) – (0.75 × unlabeled cells). The 0.75 factor reflected the ratio of the number of scans summed for the labeled cells relative to the number for the unlabeled cells and resulted in minimal signal in the 0–90 ppm region. The panel c spectra are representative of the 1-13C, 15N Leu incorporated into cellular protein.
The following model and analysis support that most of the labeled Leu was in Fgp41, i.e. Fgp41 was the dominant protein produced during expression. Consider the model: (1) The 24 Leus of Fgp41 are 13CO, 15N labeled; (2) the 13COs of the N-terminal Leus of the 6 LL repeats (directly bonded to 15N) have S1/S0 intensity ratio = 0.3; and (3) the other 18 Leu 13COs have S1/S0 = 1.0. Points (2) and (3) are based on earlier experiments and simulations (42). For the Fgp41 Leu 13COs, the (ΔS/S0)calc = [24 – 18 – (6)(0.3)]/24 = 0.17 which is close to (ΔS/S0)exp and supports dominant production of Fgp41 during the induction period. Additional evidence for Fgp41 production from SSNMR spectra is described later in the paper and resulted in an estimate of the ratio of mass Fgp41 to mass of lyophilized cells.
Optimization of Fgp41 solubilization
Initial cell lysis buffers contained either 8 M urea, 0.5% N-lauroylsarcosine, 0.5% Triton X-100, or 10% SDS – see Materials and Methods for other buffer components. The solubilization efficiency of the buffer was assessed using detection of a band at ~19 kDa (assigned to Fgp41) in the SDS-PAGE of the soluble lysate and then consideration of the absolute intensity of this band as well as its intensity relative to other bands in the gel lane. A dark Fgp41 band that was intense relative to other proteins was observed with lysis in buffer containing SDS, see Supporting Information for a representative gel. Bands that may be Fgp41 were also apparent for lyses in either urea or N-lauroylsarcosine but purifications of these lysates consistently yielded <1 mg Fgp41/L culture whereas purifications of SDS lysates yielded >1 mg Fgp41/L culture. Subsequent lyses were therefore done with SDS. The effect of SDS concentration on Fgp41 solubilization was further investigated by comparison of lysis in buffer containing either 0.5%, 1%, 3%, or 5% SDS. For 1%, a dark band that was intense relative to other proteins was observed, see Supporting Information. Subsequent lyses were done using 1% SDS. The effect of different sonication conditions during lysis on Fgp41 solubilization was also investigated. The darkest Fgp41 band was observed using four 1-minute cycles at 80% amplitude with 0.8 seconds on/0.2 seconds off. Increasing the number of cycles did not result in a darker band.
Optimization of Fgp41 expression
Experiments were carried out to investigate the effects on Fgp41 expression of: (1) [glycerol] in the expression medium; (2) [IPTG]; and (3) induction time. The protocol included: (1) overnight 37 °C cell growth from glycerol stock in 2 L of LB; (2) cell pelleting by centrifugation followed by resuspension in 1 L of LB; (3) growth at 37 °C for one hour; (4) transferring 100 mL aliquots of medium into separate flasks; (5) addition of glycerol and then IPTG with concomitant induction of expression at 23 °C; (6) cell pelleting by centrifugation followed by lysis in buffer with 1% SDS; and (7) SDS-PAGE of the soluble cell lysates with visual comparison of their Fgp41 band intensities. In general, only one parameter, e.g. [IPTG], was varied among a group of aliquots. Results included: (1) comparison between [IPTG] = 0.2 mM, 1.0, or 2.0 mM showed the darkest band at 2.0 mM; (2) comparison between [glycerol] = 0.1, 0.25, or 0.5% (v/v) showed the darkest bands for 0.1 and 0.25%; and (3) comparison between induction time = 2, 4, or 6 hours showed the darkest band for 6 hours. Subsequent experiments were done using [IPTG] = 2 mM, 0.25% glycerol, and 6 hour induction.
Optimization of Fgp41 purification
The basis for the development of the Fgp41 purification protocol was an earlier protocol developed in our lab for FHA2 (34). The Fgp41 band was observed with a modified protocol using buffers that contained 50 mM sodium phosphate at pH 8.0, 0.5% SDS, 300 mM NaCl, and imidazole with different concentrations. Relative to only washing with buffer containing [imidazole] = 20 mM, SDS-PAGE showed that sequential washes with buffers containing [imidazole] = 1, 20, and then 50 mM was more effective at washing non-Fgp41 proteins from the resin while leaving most Fgp41 bound to the resin. After the washes, the Fgp41 was eluted from the resin using buffer containing [imidazole] = 250 mM. The eluent was incubated overnight at 4 °C with consequent precipitation of excess SDS. Negligible Fgp41 precipitated as evidenced by very similar A280s for the eluent before and after incubation. SDS-PAGE showed that the eluent contained Fgp41 at high purity, Fig. 4a, and that Fgp41 could be membrane-reconstituted, Fig. 4b. The final purified yield of Fgp41 (as determined by A280) was ~5 mg/L culture. This yield was obtained using one hour initial mixing of the lysate and resin with similar yield obtained for two hour mixing and reduced 3 mg/L yield for four hour mixing. Increased proteolysis is one explanation for reduced yield with longer mixing time.
Figure 4.
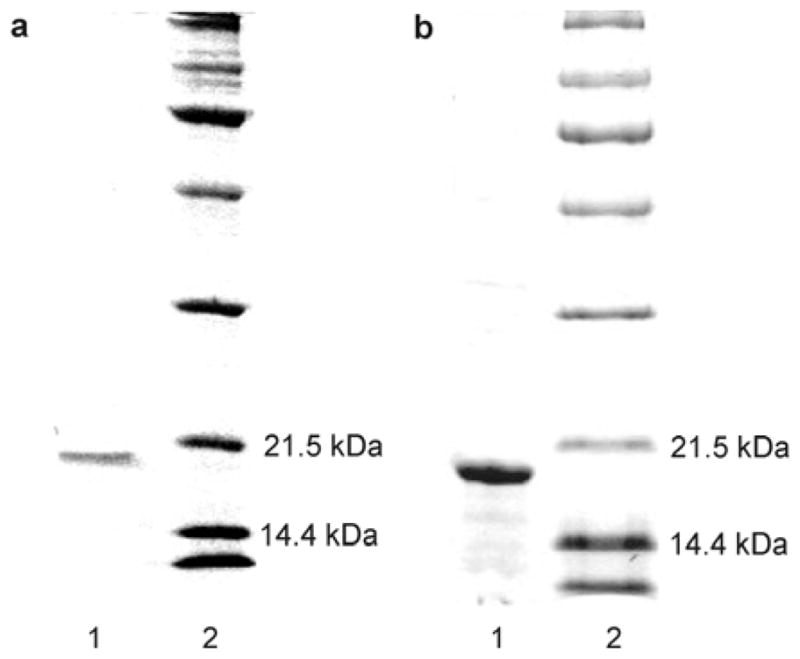
Panel a: SDS-PAGE gel of (lane 1) an aliquot of the purification eluent with [imidazole] = 250 mM and (lane 2) MW standards. Panel b: SDS-PAGE gel of (lane 1) an aliquot of the proteoliposome complexes formed during membrane reconstitution of Fgp41 and (lane 2), MW standards. The samples were boiled prior to loading on the gel.
CD spectroscopy
Fig. 5 (black trace) displays the CD spectrum of the purified Fgp41 after dialysis into HEPES/MES buffer at pH 7.4. Minima near 208 and 222 nm were diagnostic of α helical conformation as might be expected from the Hairpin structure, Fig. 1. The magnitude of θ222 showed a small linear decrease over the 25 – 100 °C range, Fig. 6, with | θ222| 100 C ≈ 0.8 × |θ222|25 C. The CD spectra at 25 °C were very similar before and after heating, Fig. 5, and showed that the temperature-dependent changes were reversible. This behavior was very similar to the temperature dependences of the CD spectra of the shorter Hairpin and FP-Hairpin constructs whose sequence was from the laboratory HXB2 strain of HIV-1 (11, 12). For these constructs, 46 contiguous residues including the native loop were replaced by 6 non-native residues. Subsequent differential scanning calorimetry experiments showed an unfolding transition centered at 110 °C for both constructs. Consideration of other CD measurements on this unfolded state indicate that for Fgp41, | θ222| unfolded ≈ 0.2 × | θ222| 25 C so even at 100 °C, Fgp41 appears to retain hyperthermostable Hairpin structure.
Figure 5.

CD spectra of Fgp41 at 25 °C. The black trace is for a sample which had not been heated and the red trace was obtained after heating to 100 °C with subsequent cooling to ambient temperature. Each trace is the difference between the CD spectrum of Fgp41 + buffer and the spectrum of buffer alone. The similarity of the red and black spectra support the reversibility of any thermal denaturation of Fgp41. Fgp41 samples were prepared by precipitation of excess SDS, subsequent dialysis in HEPES/MES buffer at pH 7.4, and addition of DTT at two times the molar concentration of Fgp41 to inhibit disulfide bond formation. For these spectra, [Fgp41] = 20 μM. Spectra for other Fgp41 samples were similar with minima near 208 and 222 nm that were diagnostic of α helical structure. In some spectra, the θ222 could be as low as –15000 degrees-cm2-dmol−1.
Figure 6.
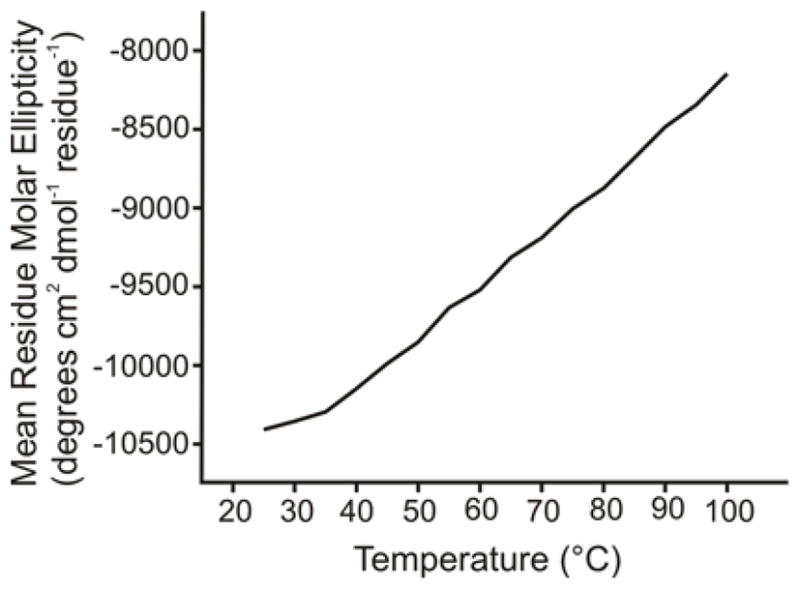
Plot of CD θ222 value vs temperature for Fgp41. No unfolding transition is apparent for temperatures up to 100 °C. Sample conditions were the same as in Figure 5.
Vesicle fusion
Fgp41 induced negligible intervesicle fusion at pH 7.5 as assayed by lipid mixing, Fig. 7. The fluorescence increase was ~2% of that observed for Triton X-100 detergent where Triton is commonly considered to induce 100% lipid mixing.
Figure 7.
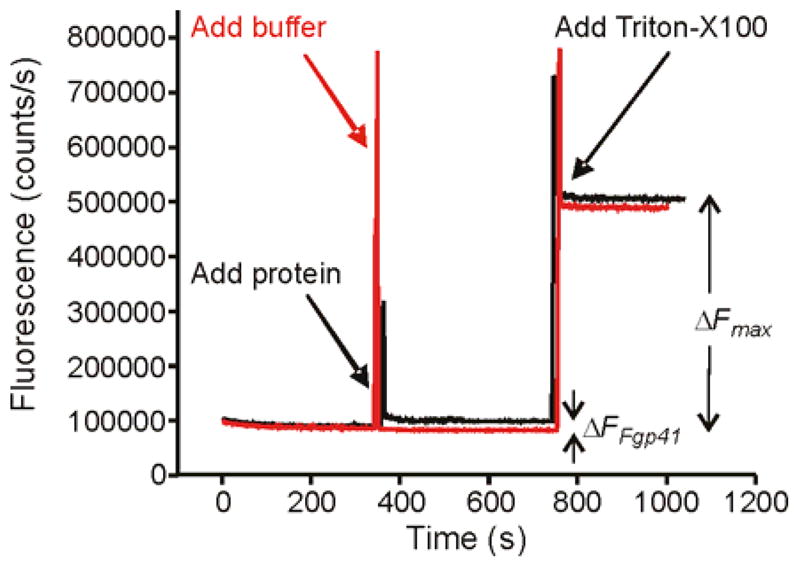
Vesicle fusion assayed by fluorescence. An aliquot of either Fgp41 + buffer (black trace) or buffer alone (red trace) was added to a vesicle solution at t = 350 s. Fgp41-induced vesicle fusion was evidenced by the fluorescence increase (ΔFFgp41) of the black trace. In either trace, Triton X-100 detergent was added at 750 s and solubilized the vesicles with resultant maximum fluorescence and fluorescence increase (ΔFmax). The spikes at 350 and 750 s were artifacts due to transient exposure to stray light. Assay parameters included vesicles with composition 4:1 POPC:POPG, Fgp41:total lipid mol ratio = 0.02, pH = 7.5, and T = 37 °C.
SSNMR of membrane-reconstituted Fgp41
Fig. 8 displays S0, S1, and ΔS REDOR SSNMR spectra of membrane-reconstituted Fgp41 labeled with different amino acids. Many of these spectra were deconvolved into a few Gaussian lineshapes, see Fig. 8 and Supporting Information. Table 1 presents the best-fit peak chemical shifts, linewidths, and integrated intensities of the individual lineshapes of the S0 spectra and Table 2 presents the lineshape parameters of the ΔS spectra. All fits were excellent as judged by the close agreement between the lineshape sum and the experimental intensity, see Supporting Information. These fittings were used to understand whether or not the N-helix and C-helix structures of the six-helix bundle were retained in the membrane-associated Fgp41 and to assess the distribution of conformations in the FP region.
Figure 8.

REDOR 13CO NMR spectra of Fgp41 reconstituted in membranes. The labeled amino acid(s) in the expression medium are shown. The left panels displays S0 (blue) and S1 (red) spectra, the middle panels display the best-fit Gaussian deconvolutions of the S0 spectra, and the right panels display the ΔS ≡ S 0 – S1 spectra. The REDOR dephasing time was either (a) 1 ms or (b–f) 2 ms and the dominant contribution to each ΔS spectrum was from residues with labeled 13COs which were directly bonded to labeled 15Ns. These residues were: (a) L33, L44, L54, L81, L134, and L149; (b) F8 and F11; (c) V7; (d) V2; (e) A15; and (f) G3. Each S0 or S1 spectrum was processed with 100 Hz Gaussian line broadening and each ΔS spectrum was processed with either (a, b) 100 Hz or (c–f) 200 Hz line broadening. Polynomial baseline correction (typically 5th order) was applied to each spectrum. Each S0 or S1 spectrum was the sum of: (a) 93424; (b) 115610; (c) 109504; (d) 110736; (e) 165216; or (f) 103717 scans.
Table 1.
Analysis and deconvolution of S0 and S1 solid-state NMR spectra of membrane-reconstituted Fgp41
| Fgp41 labeling | (ΔS/S0)exp (integrated)c |
S0 spectral deconvolution a |
Fraction of calculated S0 intensity b |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Peak shift (ppm) d | Peak width (ppm) e | Intensity (fraction of total) | Labeled | Nat. abund. Fgp41 | Nat. abund. lipid | Labeled in N- and C- helices | (ΔS/S0)calc,f | |||
| 1-13C, 15N Leu | 0.12 | 181.3 |
|
2.8 | 0.15 | |||||
| 178.5 | 2.8 | 0.60 | 0.86 | 0.07 | 0.07 | 0.68 | 0.15 | |||
| 175.3 | β | 3.7 | 0.25 | |||||||
| 1-13C Phe + 15N Leu | 0.15 | 183.2 |
|
3.0 | 0.02 | |||||
| 177.1 | 5.0 | 0.51 | 0.42 | 0.31 | 0.27 | 0 | 0.24 | |||
| 172.7 | β | 3.5 | 0.47 | |||||||
| 1-13C Val + 15N Phe | 0.07 | 182.1 |
|
2.7 | 0.02 | |||||
| 177.7 | 3.9 | 0.76 | 0.69 | 0.19 | 0.12 | 0.43 | 0.07 | |||
| 173.1 | β | 3.3 | 0.22 | |||||||
| 1-13C Val + 15N Gly | 0.08 | 178.6 |
|
2.9 | 0.37 | |||||
| 177.0 | 2.0 | 0.20 | 0.64 | 0.17 | 0.19 | 0.40 | 0.07 | |||
| 174.4 | β | 4.9 | 0.43 | |||||||
| 1-13C Ala + 15N Gly | 0.08 | 0.79 | 0.12 | 0.09 | 0.34 | 0.05 | ||||
| 1-13C Gly + 15N Leu | 0.11 | 0.77 | 0.15 | 0.08 | 0.14 | 0.06 | ||||
Spectral deconvolution was done with three Gaussian lineshapes whose peak shifts, linewidths, and intensities were independently varied until there was minimal difference between the sum of the lineshapes and the experimental lineshape. For all cases, there was excellent agreement between the best-fit deconvolution sum lineshape and experimental lineshape, see Supporting Information. Deconvolution was not meaningful for the 1-13C Ala and 1-13C Gly samples because the S0 spectra were broad and relatively featureless and the deconvolutions were dominated by a lineshape with ~7 ppm linewidth.
Spectral intensities were calculated with the following considerations: (1) 100% labeling of the Fgp41 residues corresponding to the labeled amino acid(s) with no scrambling to other amino acid types; (2) 1.0 relative intensity for each labeled 13CO; (3) 0.011 relative intensity for each natural abundance 13CO; (4) the Fgp41 natural abundance signal was the sum from backbone 13COs and Asn, Asp, Gln, and Glu sidechain 13COs; and (5) the lipid natural abundance signal was calculated using the experimental Fgp41:total lipid mol ratios. The specific ratio in each sample: 1-13C, 15N Leu, 0.011; 1-13C Phe + 15N Leu, 0.012; 1-13C Val + 15N Phe, 0.016; 1-13C Val + 15N Gly, 0.009; 1-13C Ala + 15N Gly, 0.013; and 1-13C Gly + 15N Leu, 0.019. The labeled 13CO fraction in N- and C- helices was based on the red and green regions in Fig. 1a.
The typical uncertainty of (ΔS/S0)exp was ±0.02 as determined from the standard deviation of integrals of regions of the S0 and S1 spectra which contained noise rather than signal.
The reasons for assignment of peaks to specific conformations are provided in the main text.
Full-width at half-maximum linewidth.
The (ΔS/S0)calc were based on: (1) the fraction of the S0 signal from labeled 13COs directly bonded to labeled 15Ns; and (2) a S1/S0 intensity ratio for these 13COs of 0.70 (1 ms dephasing time) or 0.85 (2 ms dephasing time). These ratios were based on experimental REDOR data of crystalline amino acid samples as well as simulations. The 1 ms dephasing time was used for the Fgp41 sample labeled with 1-13C, 15N Leu and the 2 ms dephasing time was used for the other Fgp41 samples.
Table 2.
Deconvolution of ΔS spectra of membrane-reconstituted Fgp41a
| Fgp41 labeling | Peak shift (ppm) b | Peak width (ppm) c | Intensity (fraction of total) | |
|---|---|---|---|---|
| 1-13C, 15N Leu | 182.1 |
|
1.4 | 0.10 |
| 178.3 | 3.1 | 0.82 | ||
| 174.6 | β | 1.5 | 0.08 | |
| 1-13C Phe + 15N Leu | 176.9 | helix | 4.9 | 0.37 |
| 172.6 | β | 4.3 | 0.63 | |
| 1-13C Val + 15N Phe | 181.8 |
|
3.3 | 0.12 |
| 177.4 | 3.9 | 0.57 | ||
| 172.2 | β | 4.2 | 0.31 | |
Spectral deconvolution was done with two or three Gaussian lineshapes whose peak shifts, linewidths, and intensities were independently varied until there was minimal difference between the sum of the lineshapes and the experimental lineshape. For all cases, there was excellent agreement between the best-fit deconvolution sum lineshape and experimental lineshape, see Supporting Information. Deconvolution was not meaningful for the other samples because the ΔS spectra were broad and relatively featureless.
The reasons for assignment of peaks to specific conformations are provided in the main text.
Full-width at half-maximum linewidth.
Fig. 8a displays the 13CO spectra of the 1-13C, 15N Leu-labeled sample. The S0 spectrum targeted the 24 Leus in the Fgp41 sequence and the ΔS spectrum targeted the L33, L44, L54, L81, L134, and L149 13COs which are the N-terminal Leus in LL repeats. The 13CO signal was the only discernible feature in the ΔS spectrum, see Supporting Information. Both the S0 and ΔS spectra had high signal-to-noise and were fitted well to the sum of three components. In both cases, the two higher shift components comprised >75% of the integrated intensity and were assigned to helical conformation because their peak shifts were much closer to the characteristic shifts of helical Leus (Gaussian distribution of 178.5 ± 1.3 ppm) than to β strand Leus (175.7 ± 1.5 ppm) (43). The 13CO S0 spectrum had contributions from the labeled Fgp41 Leus, as well as natural abundance sites in Fgp41 and lipids. Calculated relative fractional contributions are listed in Table 1 and show that the Fgp41 Leus dominate the spectrum. Using a S1/S0 intensity ratio of 0.3 for the N-terminal Leus of the LL pairs and a ratio of 1.0 for other 13COs (based on model compound studies and simulations), the (ΔS/S0)calc for the sample was 0.15 and correlated reasonably well with the (ΔS/S0)exp of 0.12 ± 0.02 (42).
If the SHB structure were retained in membrane-associated Fgp41, then the fractional contribution to the S0 13CO intensity of Leus in the N- and C-helices would be 0.68. This correlated well with the experimental fractional S0 intensity of 0.75 in helical conformation and supports retention of SHB structure upon membrane binding. Further support for this structure was the correlation between the experimental helical fractional intensity of 0.92 in the ΔS spectrum and the location of the six LL repeats in the N- and C-helices.
Spectra of the remaining labeled samples provided information about structure in the putative SHB region as well as in the FP. Fig. 8b displays spectra from a sample with 1-13C Phe and 15N Leu labeling. There are three Phes in the sequence: F8 and F11 in the FP, and F96 which would be in the loop region of a SHB structure. There was ~0.4 fractional contribution of the labeled Phe 13COs to the S0 spectrum and ~0.3 contributions each from natural abundance 13COs in Fgp41 and lipid. The S0 spectrum was well-fitted to the sum of three lineshapes. The two lineshapes with higher peak shifts comprised ~0.5 fractional contribution of the total intensity and the shifts were generally consistent with helical protein conformation. The peak shift of the other lineshape was consistent with β strand protein conformation and with lipid shifts. The labeled F8 and F11 13COs in the FP were directly bonded to labeled Leu 15Ns with S1/S0 of ~0.15 for 2 ms dephasing time (42). The other 13COs had S1/S0 of ~1. The (ΔS/S0)calc was close to (ΔS/S0)exp and the ΔS spectrum was dominated by the F8 and F11 13CO signals. The ΔS spectrum was well-fitted to two lineshapes with the higher (lower) peak shifts consistent with helical (β strand) Phe 13CO shift distributions of 177.1±1.4 (174.3±1.6) ppm (43). The lower ~173 experimental peak shift matched well with the 173 ppm peak shifts measured for F8 and F11 of the membrane-associated HFP fragment (26–28). This peptide has been shown to form small oligomers with antiparallel β sheet structure (30). For membrane-bound Fgp41, the ratio for F8 + F11 of helical to β strand/sheet intensities was ~1:2 and was consistent with two Fgp41 populations with different FP conformations.
Fig. 8c displays the spectra and analysis for a sample labeled with 1-13C Val and 15N Phe. The analysis approach was the same as in the previous paragraph. The eight labeled Val 13COs made a fractional contribution of ~0.7 to the S0 signal. The S0 spectrum was well-fitted to three lineshapes and the two higher shift lineshapes comprised ~0.8 fraction of the total intensity and had shifts that correlated with the helical rather than the β strand Val 13CO distribution (177.7±1.4 vs 174.8±1.4 ppm) (43). The lineshape with lowest peak shift correlated with β strand/sheet conformation. The high helical content was consistent with SHB structure for membrane-bound Fgp41. The (ΔS/S0)calc matched (ΔS/S0)exp. The ΔS spectrum was dominated by V7 and was well-fitted to three lineshapes which indicated a ratio of helical to β strand/sheet populations of ~2:1. This ΔS spectrum confirmed two Fgp41 populations with different FP conformations while the difference in population ratio relative to the Fig. 8b ΔS spectrum may reflect lower signal-to-noise of the Fig. 8c spectrum, sample-to-sample variation, and/or conformational differences between V7 and F8 + F11.
Fig. 8d displays the spectra and analysis for a sample labeled with 1-13C Val and 15N Gly. As with Fig. 8c, analysis of the S0 spectrum of Fig. 8d supported a dominant helical conformation consistent with six-helix bundle structure. Comparison of the two spectra provided insight into sample-to-sample variation and the robustness of the S0 deconvolution. The (ΔS/S0)calc matched (ΔS/S0)exp. The ΔS spectrum was dominated by V2 and extended broadly over 170–180 ppm region so that deconvolution was not meaningful. As noted in the previous paragraph, this shift range includes the helical and β strand/sheet shift distributions and the ΔS spectrum was therefore consistent with a mixture of Fgp41 populations with helical and β strand/sheet conformations at V2 in the FP. We note that the V2 13CO signal of the membrane-associated HFP was also broader than signals from residues 6–12 in the interior hydrophobic region (26).
Fig. 8e and f display spectra from samples that were labeled with 1-13C Ala + 15N Gly or 1-13C Gly + 15N Leu. The analyses are presented together because of the similar results. The S0 spectra were broad and featureless over the 170–185 ppm range so that deconvolution was not meaningful. This spectral breadth was understood by considering that although the fractional contribution of the labeled 13COs to the total S0 intensity was ~0.8, the labeled contribution from N- and C-helices in a SHB structure would be ~0.25. About half of the S0 intensity would be from labeled 13COs in the FP and loop regions. The earlier Fig. 8a–d analyses supported a mixture of helical and β strand/sheet shifts for FP 13COs and broad signals are also expected from 13COs in the less-ordered loop region. For the Fig. 8e, f spectra, there were relatively good agreements between (ΔS/S0)calc and (ΔS/S0)exp and the ΔS spectra were respectively dominated by the A15 and G3 13COs. These ΔS spectra extended over 170–180 ppm and as with the V2 ΔS spectrum, the breadth correlated with being near one end of the FP region and with the spectral breadth observed for the corresponding residues in the membrane-associated HFP (26).
Analysis of the SSNMR spectra of lyophilized whole cells
Comparison of the ΔS spectrum of 1-13C, 15N Leu labeled cells, Fig. 2b, to the ΔS spectrum of the unlabeled cells, Fig. 2c, shows a clear effect from using labeled Leu. The “labeled cell difference” S0 (S1) spectrum, Fig. 3c, is the difference between the S0 (S1) spectra of the labeled and unlabeled cells and shows only the contribution of the labeled Leu. Deconvolution was applied to the labeled cell difference S0 spectrum and to the labeled cell ΔS spectrum. Both spectra were well-fitted to the sum of three Gaussian lineshapes, see Table 3 and Supporting Information, and were dominated by the 1-13C, 15N Leu incorporated into cell protein produced during the expression period. In order to understand the fraction of Fgp41 in this protein, comparison was made between the deconvolutions of: (1) the ΔS spectrum of labeled cells and the ΔS spectrum of membrane-reconstituted Fgp41; and (2) the S0 spectrum of labeled cell difference and the S0 spectrum of membrane-reconstituted Fgp41. For either case, there were striking similarities in the deconvolutions including the peak chemical shifts and the large fraction of the total intensity in the two high shift peaks corresponding to helical conformation. These similarities as well as the detection of large ΔS signals provide additional strong evidence that Fgp41 is the predominant labeled protein in the cells. This result was used to conservatively estimate that there was at least 3 mg of Fgp41 in the lyophilized labeled cell NMR sample. Other inputs for this estimate were: (1) the mass Fgp41 in the membrane-reconstituted sample was ~5 mg; (2) the membrane and whole cell data were acquired on the same spectrometer and were the sums of about the same numbers of scans; and (3) for the membrane-reconstituted and whole cell samples, the integrated 13CO intensities of the ΔS spectra were within 20% agreement and there was similar agreement for the S0 spectra. There was ~50 mg total cell mass in the whole cell NMR sample so the ratio of mass Fgp41 to total dry cell mass was ~0.05. There was ~2 g dry cell mass/L culture so prior to solubilization and purification, there was ~100 mg Fgp41/L culture. The much smaller purified yield of ~5 mg Fgp41 /L culture points to solubilization and purification rather than expression as the limiting factors in Fgp41 production.
Table 3.
Deconvolution of spectra of lyophilized cells induced to produce Fgp41a
| Sample/ spectrum | Spectrum type | Peak shift (ppm) b | Peak width (ppm) c | Intensity (fraction of total) | |
|---|---|---|---|---|---|
| 1-13C, 15N Leu cells | ΔS | 182.1 |
|
2.2 | 0.03 |
| 177.4 | 4.5 | 0.82 | |||
| 173.1 | β | 3.2 | 0.15 | ||
| 1-13C, 15N Leu cells – (0.75 × unlabeled cells) | S0 | 180.8 |
|
8.9 | 0.18 |
| 177.6 | 5.6 | 0.69 | |||
| 172.1 | β | 3.1 | 0.13 | ||
Spectral deconvolution was done with three Gaussian lineshapes whose peak shifts, linewidths, and intensities were independently varied until there was minimal difference between the sum of the lineshapes and the experimental lineshape. For both cases, there was excellent agreement between the best-fit deconvolution sum lineshape and experimental lineshape, see Supporting Information.
The reasons for assignment of peaks to specific conformations are provided in the main text.
Full-width at half-maximum linewidth.
Because relatively harsh conditions were needed to solubilize Fgp41 in the cells, it seems likely that most Fgp41 was in inclusion bodies. Detection of predominant helical conformation for the Leus in Fgp41 in the lyophilized cells including those in the N- and C-helices of a putative SHB structure suggests that this structure is retained in inclusion bodies.
Discussion
To our knowledge, earlier reports of bacterial production of large quantities of the ectodomain of HIV gp41 including the FP have always included a N-terminal carrier protein like glutathione S-transferase that was subsequently cleaved. The present study describes production of Fgp41 which has a much shorter six-residue C-terminal histidine tag rather than a N-terminal carrier protein. The ~5 mg purified yield Fgp41 per liter culture is about half that of the highest reported yield with a carrier protein (16).
Initial attempts at solubilization and purification gave only 0.1 mg Fgp41/L culture and SSNMR was applied to detect whether this poor yield was due to very low expression. The successful approach included identifying an abundant amino acid in Fgp41 (24 Leus) that was the first amino acid of an abundant sequential pair (6 LLs). The procedure included: (1) inducing cells in minimal medium with either 1-13C, 15N Leu or unlabeled Leu; (2) cell pellet lyophilization; and (3) taking 13C REDOR SSNMR spectra of the lyophilized whole cells with short dephasing time. As expected, the spectra of the labeled and unlabeled cells were very similar in the aliphatic 13C shift region but the labeled cells had greater intensity in the 13CO region. The labeled cell – unlabeled cell difference spectra were therefore assigned to Leu 13COs incorporated into protein produced during the expression period. The (ΔS/S0)exp of the difference spectra was close to the (ΔS/S0)calc estimated for Fgp41 and provided strong evidence that Fgp41 was expressed in large quantities in the bacteria. Calibration with standard spectra led to the estimate of ~100 mg Fgp41/L culture. The harsh conditions needed to solubilize Fgp41 in the cells and the ~5 mg purified Fgp41/L culture evidenced that most Fgp41 is in inclusion bodies in the bacteria. This approach to detection of recombinant protein in whole cells by SSNMR has several strengths including: (1) small (~50 mL) culture volumes; (2) small (~10 mg) quantities of isotopically labeled amino acids; and (3) simple sample preparation protocol without protein solubilization or purification. The main drawback might be the few days of SSNMR spectrometer time. Future studies with other proteins are needed to test the generality of the approach.
Interpretation of the SSNMR spectra using this approach will likely not be greatly affected by some “scrambling”, i.e. conversion of the labeled amino acids into other amino acids. For example, transfer of the 15N from the labeled amino acid to other amino acids would likely result in a larger number of labeled 13CO-15N sequential pairs and therefore larger ΔS signal and more sensitive detection of the recombinant protein. Support for minimal scrambling of the Fgp41 sample labeled with 1-13C, 15N Leu included: (1) expression done at lower temperature for short 2h duration; (2) (ΔS/S0)exp for both the whole cell and membrane-reconstituted samples that were close to the values calculated using models without scrambling; and (3) deconvolutions of the S0 and ΔS 13CO spectra of these samples which agreed nearly quantitatively with the expected secondary structure distributions of the 24 Leu’s and the 6 N-terminal Leu’s in LL pairs, respectively, see Tables 1–3 (44).
The CD spectra and melting curves of purified Fgp41 support thermostable SHB structure and this structure was retained upon membrane binding as evidenced by a predominant sharp (3 ppm) helical 13CO feature in the ΔS spectrum of Fgp41 produced with 1-13C, 15N Leu. This feature was assigned to the sum of 13CO signals from six Leus which are in N- and C-helices in SHB structure. The SHB was also observed for the membrane-associated FP-Hairpin construct whose sequence was from a different HIV clade than Fgp41 and for which 46 contiguous residues including the native loop were replaced by a six non-native residues. By contrast, Fgp41 had the full native sequence of its clade. The similar results for Fgp41 and FP-Hairpin support the SHB as the final stable structure for membrane-associated gp41, Fig. 1b.
There are two Cys’s in the Fgp41 sequence that are separated by five residues. These Cys’s are likely on either side of the tip of the loop in the Hairpin structure and therefore positioned to form an intramolecular disulfide bond (4). For the laboratory strain HXB2 sequence, these Cys’s are replaced by Ala’s, see Supporting Information. The unfolding temperature of the HXB2 Hairpin structure is 105 °C which should be within a few degrees of that of Fgp41, Fig. 6 (12, 14). It is therefore unlikely that the disulfide bond of Fgp41 contributes appreciably to the thermostability of the Hairpin structure of Fgp41.
Fgp41 induced negligible inter-vesicle lipid mixing at pH 7.5 which correlated with the same result for FP-Hairpin. Gp41 in the final SHB state may therefore be fusion-inactive at least with respect to lipid mixing which occurs early in either fusion of membranes of HIV and host cells or in gp41-mediated cell-cell fusion. This view is supported by other fusion data showing that most membrane changes occur prior to formation of the final gp41 SHB state (8). For vesicles with negative charge, FP-Hairpin and related SHB gp41 constructs induce lipid mixing at pHs much lower than 7 (e.g. 4) and the pH-dependent functional difference has been correlated to changes in protein-membrane electrostatics (17). It is therefore likely that Fgp41 will also induce lipid mixing at these lower pHs. Over the past 25 years, there have been a series of experimental studies by different groups to determine whether HIV infects cells through direct fusion at the plasma membrane or through an endocytic mechanism (45, 46). In our view, the preponderance of data for either route support HIV-cell fusion at pH ≈ 7 where SHB gp41 is fusion-inactive. There may be some differences among enveloped viruses as there is significant evidence for fusion activity of the folded influenza virus fusion protein ectodomain FHA2 (33, 35).
Relative to the sharp 3 ppm ΔS 13CO signal from six Leu residues in the SHB, broader (4–10 ppm) ΔS 13CO signals were observed from (typically) one residue in the FP. These breadths indicate conformational heterogeneity in the FP (24, 25). This point was further supported by the ΔS spectra of V7, F8, and F11 which were reasonably deconvolved into helical and β-sheet signals and indicated two populations of Fgp41 with distinct FP conformations. Helical and β sheet FP signals were also observed for membrane-associated FP-Hairpin samples even though there were differences between the Fgp41 and FP-Hairpin samples including: (1) two of the sixteen FP residues were different; (2) lipids were ester-linked (Fgp41) vs ether-linked (FP-Hairpin); (3) membrane reconstitution was based on detergent dialysis (Fgp41) vs simple mixing of protein and vesicle solutions (FP-Hairpin); and (4) unfrozen Fgp41 vs frozen FP-Hairpin samples (12). Detection of helical and β sheet FP populations in both sample types strongly supports existence of these populations in membrane-associated gp41 in its final SHB state. In the future, it would be very interesting to study a larger gp41 construct that contains the transmembrane domain and for which there may be close contact between the FP and transmembrane domains.
For most non-bacterial proteins produced in bacteria, a large fraction of the protein in the cells is found in “inclusion bodies” which are macroscopic non-crystalline solid aggregates (41, 47, 48). Inclusion body formation appears to be largely independent of protein sequence. There are little data about the structure(s) of recombinant protein molecules in inclusion bodies. In the present study, deconvolutions of the S0 and ΔS spectra of the 1-13C, 15N Leu-labeled inclusion body Fgp41 in cells resulted in lineshapes with similar peak shifts and relative intensities as those of membrane-associated Fgp41 with folded SHB structure. It therefore seems likely that at least the SHB fold exists for most Fgp41 molecules in inclusion bodies. This is the second protein for which there is SSNMR evidence for a stable fold in inclusion bodies. This work highlights the potential of SSNMR for probing protein structure and aggregation in inclusion bodies.
MAS SSNMR structural studies of proteins are generally done by one of two approaches: (1) uniform 13C and 15N labeling, unambiguous assignment of most crosspeaks in multidimensional NMR spectra, and structural interpretation of the peak shifts and the crosspeak intensities of nuclei far apart in the sequence; or (2) specific (often residue or at least amino-acid type) labeling, and quantitative SSNMR measurements (e.g. shifts or dipolar couplings) to test specific structural models (49–51). The choice of approach for a particular protein depends on protein size and quantity as well as NMR linewidths. Approach (1) is more feasible for smaller proteins, high protein concentrations, and narrow (<1 ppm) linewidths. The present study is an example of approach (2) which was appropriate given the 162-residues, Fgp41:lipid ≈ 0.01 (with additional dilution of Fgp41 in the sample from water); the 3–10 ppm 13CO linewidths; and the possibility of FP conformational heterogeneity (shown to be true in this study). The approach considered a model based on the existing high-resolution SHB structures of gp41 fragments and the extensive residue-specific SSNMR data for membrane-associated HFP.
Supplementary Material
Acknowledgments
Dr. William Wedemeyer is acknowledged for the gp41 cDNA, Dr. Jun Sun is acknowledged for Fgp41 vector construction, Dr. Lisa Lapidus is acknowledged for the CD spectrometer and fluorimeter, and Dr. Kelly Sackett is acknowledged for assistance with the lipid mixing assay.
Abbreviations
- BOG
n-Octyl-β-D-glucopyranoside
- bTOG
n-Octyl-β-D-thioglucopyranoside
- C8E5
pentaethylene glycol monooctyl ether
- CD
circular dichroism
- C-helix
C-terminal helix
- ΔS
S0 – S1
- (ΔS/S0)calc
calculated (ΔS/S0)
- (ΔS/S0)exp
experimental (ΔS/S0)
- DTT
dithiothreitol
- FP
fusion peptide, HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HEPES/MES
5 mM HEPES, 10 mM MES, pH 7.4
- HFP
HIV fusion peptide
- HIV-1
human immunodeficiency virus type 1
- IPTG
isopropyl-β-D-thiogalactopyranoside
- LB
Luria-Bertani broth
- LUV
large unilamellar vesicle
- MAS
magic angle spinning
- MES
2-(N-morpholino)ethanesulfonic acid
- MW
molecular weight
- MWCO
molecular weight cut-off
- N-helix
N-terminal helix
- N-NBD-PE
N-(7-nitro-2,1,3-benzoxadiazol-4-yl)phosphatidylethanolamine
- N-Rh-PE
N-(lissamine Rhodamine B sulfonyl)phosphatidylethanolamine
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt)
- PHI
pre-Hairpin intermediate
- REDOR
rotational-echo double-resonance
- SDS
sodium dodecyl sulfate
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SHB
six-helix bundle
- SIV
simian imunodeficiency virus
- SSNMR
solid-state nuclear magnetic resonance
Footnotes
The research was supported by the National Institutes of Health grant A147153.
Supporting Information Available: Fgp41 amino acid and DNA sequences and comparison to the HXB2 amino acid sequence, additional SDS-PAGE gels, full ΔS spectra of 1-13C, 15N Leu labeled samples, and spectral deconvolutions. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.White JM, Delos SE, Brecher M, Schornberg K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit Rev Biochem Mol Biol. 2008;43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melikyan GB. Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm. Retrovirology. 2008;5:111. doi: 10.1186/1742-4690-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chernomordik LV, Zimmerberg J, Kozlov MM. Membranes of the world unite! J Cell Biol. 2006;175:201–207. doi: 10.1083/jcb.200607083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caffrey M, Cai M, Kaufman J, Stahl SJ, Wingfield PT, Covell DG, Gronenborn AM, Clore GM. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998;17:4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang ZN, Mueser TC, Kaufman J, Stahl SJ, Wingfield PT, Hyde CC. The crystal structure of the SIV gp41 ectodomain at 1.47 A resolution. J Struct Biol. 1999;126:131–144. doi: 10.1006/jsbi.1999.4116. [DOI] [PubMed] [Google Scholar]
- 6.Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 7.Buzon V, Natrajan G, Schibli D, Campelo F, Kozlov MM, Weissenhorn W. Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. Plos Pathogens. 2010;6:e1000880. doi: 10.1371/journal.ppat.1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Markosyan RM, Cohen FS, Melikyan GB. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol Biol Cell. 2003;14:926–938. doi: 10.1091/mbc.E02-09-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freed EO, Delwart EL, Buchschacher GL, Jr, Panganiban AT. A mutation in the human immunodeficiency virus type 1 transmembrane glycoprotein gp41 dominantly interferes with fusion and infectivity. Proc Natl Acad Sci USA. 1992;89:70–74. doi: 10.1073/pnas.89.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang R, Prorok M, Castellino FJ, Weliky DP. A trimeric HIV-1 fusion peptide construct which does not self-associate in aqueous solution and which has 15-fold higher membrane fusion rate. J Am Chem Soc. 2004;126:14722–14723. doi: 10.1021/ja045612o. [DOI] [PubMed] [Google Scholar]
- 11.Sackett K, Nethercott MJ, Shai Y, Weliky DP. Hairpin folding of HIV gp41 abrogates lipid mixing function at physiologic pH and inhibits lipid mixing by exposed gp41 constructs. Biochemistry. 2009;48:2714–2722. doi: 10.1021/bi8019492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sackett K, Nethercott MJ, Epand RF, Epand RM, Kindra DR, Shai Y, Weliky DP. Comparative analysis of membrane-associated fusion peptide secondary structure and lipid mixing function of HIV gp41 constructs that model the early Pre-Hairpin Intermediate and final Hairpin conformations. J Mol Biol. 2010;397:301–315. doi: 10.1016/j.jmb.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan JH, Lai CB, Scott WRP, Straus SK. Synthetic fusion peptides of tick-borne Encephalitis virus as models for membrane fusion. Biochemistry. 2010;49:287–296. doi: 10.1021/bi9017895. [DOI] [PubMed] [Google Scholar]
- 14.Lev N, Fridmann-Sirkis Y, Blank L, Bitler A, Epand RF, Epand RM, Shai Y. Conformational stability and membrane interaction of the full-length ectodomain of HIV-1 gp41: Implication for mode of action. Biochemistry. 2009;48:3166–3175. doi: 10.1021/bi802243j. [DOI] [PubMed] [Google Scholar]
- 15.Cheng SF, Chien MP, Lin CH, Chang CC, Lin CH, Liu YT, Chang DK. The fusion peptide domain is the primary membrane-inserted region and enhances membrane interaction of the ectodomain of HIV-1 gp41. Mol Membr Biol. 2010;27:31–44. doi: 10.3109/09687680903333847. [DOI] [PubMed] [Google Scholar]
- 16.Lin CH, Lin CH, Chang CC, Wei TS, Cheng SF, Chen SSL, Chang DK. An efficient production and characterization of HIV-1 gp41 ectodomain with fusion peptide in Escherichia coli system. J Biotech. 2011;153:48–55. doi: 10.1016/j.jbiotec.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Sackett K, TerBush A, Weliky DP. HIV gp41 six-helix bundle constructs induce rapid vesicle fusion at pH 3.5 and little fusion at pH 7.0: understanding pH dependence of protein aggregation, membrane binding, and electrostatics, and implications for HIV-host cell fusion. Eur Biophys J. 2011;40:489–502. doi: 10.1007/s00249-010-0662-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang DK, Cheng SF, Chien WJ. The amino-terminal fusion domain peptide of human immunodeficiency virus type 1 gp41 inserts into the sodium dodecyl sulfate micelle primarily as a helix with a conserved glycine at the micelle-water interface. J Virol. 1997;71:6593–6602. doi: 10.1128/jvi.71.9.6593-6602.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris KF, Gao XF, Wong TC. The interactions of the HIV gp41 fusion peptides with zwitterionic membrane mimics determined by NMR spectroscopy. Biochim Biophys Acta. 2004;1667:67–81. doi: 10.1016/j.bbamem.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 20.Jaroniec CP, Kaufman JD, Stahl SJ, Viard M, Blumenthal R, Wingfield PT, Bax A. Structure and dynamics of micelle-associated human immunodeficiency virus gp41 fusion domain. Biochemistry. 2005;44:16167–16180. doi: 10.1021/bi051672a. [DOI] [PubMed] [Google Scholar]
- 21.Li YL, Tamm LK. Structure and plasticity of the human immunodeficiency virus gp41 fusion domain in lipid micelles and bilayers. Biophys J. 2007;93:876–885. doi: 10.1529/biophysj.106.102335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabrys CM, Weliky DP. Chemical shift assignment and structural plasticity of a HIV fusion peptide derivative in dodecylphosphocholine micelles. Biochim Biophys Acta. 2007;1768:3225–3234. doi: 10.1016/j.bbamem.2007.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pereira FB, Goni FM, Muga A, Nieva JL. Permeabilization and fusion of uncharged lipid vesicles induced by the HIV-1 fusion peptide adopting an extended conformation: dose and sequence effects. Biophys J. 1997;73:1977–1986. doi: 10.1016/S0006-3495(97)78228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grasnick D, Sternberg U, Strandberg E, Wadhwani P, Ulrich AS. Irregular structure of the HIV fusion peptide in membranes demonstrated by solid-state NMR and MD simulations. Eur Biophys J. 2011;40:529–543. doi: 10.1007/s00249-011-0676-5. [DOI] [PubMed] [Google Scholar]
- 25.Tristram-Nagle S, Chan R, Kooijman E, Uppamoochikkal P, Qiang W, Weliky DP, Nagle JF. HIV fusion peptide penetrates, disorders, and softens T-cell membrane mimics. J Mol Biol. 2010;402:139–153. doi: 10.1016/j.jmb.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J, Gabrys CM, Weliky DP. Solid-state nuclear magnetic resonance evidence for an extended beta strand conformation of the membrane-bound HIV-1 fusion peptide. Biochemistry. 2001;40:8126–8137. doi: 10.1021/bi0100283. [DOI] [PubMed] [Google Scholar]
- 27.Zheng Z, Yang R, Bodner ML, Weliky DP. Conformational flexibility and strand arrangements of the membrane-associated HIV fusion peptide trimer probed by solid-state NMR spectroscopy. Biochemistry. 2006;45:12960–12975. doi: 10.1021/bi0615902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiang W, Bodner ML, Weliky DP. Solid-state NMR spectroscopy of human immunodeficiency virus fusion peptides associated with host-cell-like membranes: 2D correlation spectra and distance measurements support a fully extended conformation and models for specific antiparallel strand registries. J Am Chem Soc. 2008;130:5459–5471. doi: 10.1021/ja077302m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiang W, Weliky DP. HIV fusion peptide and its cross-linked oligomers: efficient syntheses, significance of the trimer in fusion activity, correlation of β strand conformation with membrane cholesterol, and proximity to lipid headgroups. Biochemistry. 2009;48:289–301. doi: 10.1021/bi8015668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmick SD, Weliky DP. Major antiparallel and minor parallel beta sheet populations detected in the membrane-associated Human Immunodeficiency Virus fusion peptide. Biochemistry. 2010;49:10623–10635. doi: 10.1021/bi101389r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krasslich HG. The HIV lipidome: A raft with an unusual composition. Proc Natl Acad Sci USA. 2006;103:2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sackett K, Wexler-Cohen Y, Shai Y. Characterization of the HIV N-terminal fusion peptide-containing region in context of key gp41 fusion conformations. J Biol Chem. 2006;281:21755–21762. doi: 10.1074/jbc.M603135200. [DOI] [PubMed] [Google Scholar]
- 33.Curtis-Fisk J, Preston C, Zheng ZX, Worden RM, Weliky DP. Solid-state NMR structural measurements on the membrane-associated influenza fusion protein ectodomain. J Am Chem Soc. 2007;129:11320–11321. doi: 10.1021/ja073644g. [DOI] [PubMed] [Google Scholar]
- 34.Curtis-Fisk J, Spencer RM, Weliky DP. Isotopically labeled expression in E. coli, purification, and refolding of the full ectodomain of the Influenza virus membrane fusion protein. Prot Expr Purif. 2008;61:212–219. doi: 10.1016/j.pep.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim CS, Epand RF, Leikina E, Epand RM, Chernomordik LV. The final conformation of the complete ectodomain of the HA2 subunit of Influenza Hemagglutinin can by itself drive low pH-dependent fusion. J Biol Chem. 2011;286:13226–13234. doi: 10.1074/jbc.M110.181297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K, Ivanoff L, Petteway SR, Pearson ML, Lautenberger JA, Papas TS, Ghrayeb J, Chang NT, Gallo RC, Wongstaal F. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature. 1985;313:277–284. doi: 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
- 37.Painter SL, Biek R, Holley DC, Poss M. Envelope variants from women recently infected with clade A human immunodeficiency virus type 1 confer distinct phenotypes that are discerned by competition and neutralization experiments. J Virol. 2003;77:8448–8461. doi: 10.1128/JVI.77.15.8448-8461.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pascual R, Moreno MR, Villalain J. A peptide pertaining to the loop segment of human immunodeficiency virus gp41 binds and interacts with model biomembranes: Implications for the fusion mechanism. J Virol. 2005;79:5142–5152. doi: 10.1128/JVI.79.8.5142-5152.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J Magn Reson. 1989;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Morcombe CR, Zilm KW. Chemical shift referencing in MAS solid state NMR. J Magn Reson. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 41.Curtis-Fisk J, Spencer RM, Weliky DP. Native conformation at specific residues in recombinant inclusion body protein in whole cells determined with solid-state NMR spectroscopy. J Am Chem Soc. 2008;130:12568–12569. doi: 10.1021/ja8039426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang J. Ph D Thesis. Michigan State University; East Lansing, MI: 2003. [Google Scholar]
- 43.Zhang HY, Neal S, Wishart DS. RefDB: A database of uniformly referenced protein chemical shifts. J Biomol NMR. 2003;25:173–195. doi: 10.1023/a:1022836027055. [DOI] [PubMed] [Google Scholar]
- 44.Tong KI, Yamamoto M, Tanaka T. A simple method for amino acid selective isotope labeling of recombinant proteins in E. coli. J Biomol NMR. 2008;42:59–67. doi: 10.1007/s10858-008-9264-0. [DOI] [PubMed] [Google Scholar]
- 45.Grewe C, Beck A, Gelderblom HR. HIV: early virus-cell interactions. J AIDS. 1990;3:965–74. [PubMed] [Google Scholar]
- 46.Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell. 2009;137:433–444. doi: 10.1016/j.cell.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang L. Towards revealing the structure of bacterial inclusion bodies. Prion. 2009;3:139–145. doi: 10.4161/pri.3.3.9922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gatti-Lafranconi P, Natalello A, Ami D, Doglia SM, Lotti M. Concepts and tools to exploit the potential of bacterial inclusion bodies in protein science and biotechnology. Febs J. 2011;278:2408–2418. doi: 10.1111/j.1742-4658.2011.08163.x. [DOI] [PubMed] [Google Scholar]
- 49.Tycko R. Molecular structure of amyloid fibrils: insights from solid-state NMR. Quart Rev Biophys. 2006;39:1–55. doi: 10.1017/S0033583506004173. [DOI] [PubMed] [Google Scholar]
- 50.McDermott A. Structure and dynamics of membrane proteins by magic angle spinning solid-state NMR. Ann Rev Biophys. 2009;38:385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 51.Fowler DJ, Weis RM, Thompson LK. Kinase-active signaling complexes of bacterial chemoreceptors do not contain proposed receptor-receptor contacts observed in crystal structures. Biochemistry. 2010;49:1425–1434. doi: 10.1021/bi901565k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.