

Abstract
Lynch syndrome (LS) is caused by germline mutations in DNA mismatch repair (MMR) genes. M M R recognizes and repairs DNA mismatches and small insertion/deletion loops. Carriers of MMR gene variants have a high risk of developing colorectal, endometrial, ovarian, and other extracolonic carcinomas. We report on an ovarian cancer patient who carries a germline MSH2 c.1A>C variant which alters the translation initiation codon. Mutations affecting the MSH2 start codon have been described previously for LS-related malignancies. However, the patients often lack a clear family history indicative of LS and their tumors often fail to display microsatellite instability, a hallmark feature of LS. Therefore, the pathogenicity of start codon variants remains undefined. Loss of the MSH2 start codon has been predicted to result in a truncated protein translated from a downstream in-frame AUG that would lack the first 25 amino acids. We therefore purified recombinant MSH2(NΔ25)-MSH6 and MSH2(NΔ25)-MSH3 to examine their DNA lesion recognition and adenosine nucleotide processing functions in vitro. We found that the MSH2(NΔ25) mutant confers distinct biochemical defects on MSH2-MSH6, but does not have a significant effect on MSH2-MSH3. We confirmed that expression of the MSH2 c.1A>C cDNA results in the production of multiple protein products in human cells that may include the truncated and full-length forms of MSH2. An in vivo MMR assay revealed a slight reduction in MMR efficiency in these cells. These data suggest that mutation of the MSH2 initiation codon, while not a strong, high-risk disease allele, may have a moderate impact on disease phenotype.
Keywords: Lynch syndrome, DNA mismatch repair, missense variants, MSH2-MSH6
INTRODUCTION
The familial disease Lynch syndrome (LS) is caused by a heterozygous germline mutation in the DNA mismatch repair (MMR) gene MSH2, MLH1, MSH6, or PMS2 [1]. MMR deficiency results in an elevated mutation rate and cancer risk [2]. LS is characterized by early development of colorectal cancer or cancer at extracolonic sites including the endometrium, ovary, stomach, and small intestine. LS tumors display rapid progression and resistance to certain chemotherapeutic agents. Mutations in MSH2 account for 36% of reported MMR germline variants [3]. Most of these variants are nonsense or frameshift mutations that result in loss of a stable protein product, however, 18% of MSH2 variants are missense variants that may affect only a single amino acid. The significance of these variants to pathogenicity is often uncertain, raising a dilemma for physicians and genetic counselors who must manage the disease and determine cancer risk. Several labs, including our own, have begun to functionally characterize different MMR gene variants in an attempt to understand their significance to disease [4,5].
In this study, we analyzed a truncated MSH2 protein product predicted to result from a germline mutation of the MSH2 initiation codon in an ovarian cancer patient. Although rare, MSH2 start codon mutations have been reported previously in cancer patients [6–8]. One report by Kets et al., was of a brother and sister who each had bi-allelic germline mutations of MSH2. In addition to an initiation codon mutation, they carried a second germline mutation that results in the pathogenic deletion of the first 6 exons of MSH2. Bi-allelic mutations of a MMR gene that result in constitutional loss of MMR display a distinct spectrum of tumors including hematological cancers that develop in childhood [9,10]. However, the brother and sister displayed a more typical LS tumor spectrum with early adult onset suggesting that the initiation codon variant is not a complete loss-of-function allele. Interestingly, though the tumors displayed MSI, they retained expression of all four MMR proteins by immunohistochemistry (IHC), including MSH2. These results led the authors to conclude that the initiation codon variant may result in a truncated protein product that retains some MMR function. It was speculated that a second in-frame initiation codon at position +76 might be used when the MSH2 start codon is mutated [7,8]. The use of this downstream AUG to start translation would result in a truncated MSH2 protein missing the first 25 amino acids, MSH2(NΔ25).
We wished to compare the biochemical activity of the truncated MSH2(NΔ25) protein to wild-type MSH2. We purified recombinant MSH2(NΔ25) protein in combination with its wild-type heterodimer partners MSH6 or MSH3. The MSH2-MSH6 heterodimer specifically binds to single-base mispairs or insertions while MSH2-MSH3 binds to larger insertion/deletion loops (IDLs) in DNA [11]. Binding occurs via paired N-terminal mismatch binding domains. Close coordination between DNA mismatch binding and adenosine nucleotide processing converts MSH2-MSH6 into a sliding clamp form on DNA that recruits the MLH1-PMS2 heterodimer to promote lesion repair, thereby contributing to genomic stability [12,13]. The mechanism by which MSH2-MSH3 directs repair is less well understood, though it also likely involves a DNA lesion-dependent interaction with MLH1-PMS2 [14]. Therefore, we analyzed the recombinant MSH2(NΔ25)-MSH6 and MSH2(NΔ25)-MSH3 complexes for DNA lesion recognition, binding and hydrolysis of adenosine nucleotides, and the ability to form sliding clamps in the presence of adenosine nucleotides. Our data indicate that the MSH2(NΔ25) truncation affects the function of MSH2-MSH6 but not MSH2-MSH3. To study the cellular effects of the MSH2 c.1A>C variant, we introduced the mutated cDNA into a cancer cell line that lacks endogenous MSH2. We observed expression of MSH2 from this mutant cDNA that corresponded to a slight MMR deficiency compared to cells complemented with wild-type MSH2. Together, the biochemical and cellular data suggest that a mutation in the MSH2 start codon may affect MSH2-MSH6 function and contribute subtly to pathogenicity.
MATERIALS AND METHODS
Mutagenesis and Protein Purification
The DNA coding sequence for the MSH2(NΔ25) mutant was generated by PCR amplification from wild-type MSH2 cDNA. The following primers amplified a large fragment of MSH2 lacking the first 75 nucleotides; 5′>3′ ATGCCGGAGAAG, and 5′>3′ TCACGTAGTAACTTTTATTCGTGAAATG. The mutant sequence was subcloned into the baculovirus expression vector pFastBacDual (Invitrogen; Carlsbad, CA) that also contained wild-type N-terminal His6-tagged MSH6 or MSH3 sequence under a separate promoter. Wild-type and mutant MSH2-MSH6 or MSH2-MSH3 heterodimers were overexpressed in Sf9 insect cells. MSH2-MSH6 and MSH2-MSH3 heterodimers were purified as described previously [15,16], except that MSH2-MSH3 peak fractions from a heparin-Sepharose column were loaded onto a MonoQ column (GE Healthcare; Uppsala, Sweden) as described previously for MSH2-MSH6 [16].
ATPase Activity
MSH2-MSH6 ATPase activity was measured by incubating 15–100 nM protein with 16.5 nM [γ-32P]ATP (PerkinElmer Life Sciences; Waltham, MA) in 20 μL of buffer containing 25 mM Hepes (pH 8.1), 100 mM NaCl, 10mM MgCl2, 1 mM DTT, 15% glycerol, and 20–240 μM unlabeled ATP. Reactions were carried out in the presence of 41-bp double-stranded DNA oligonucleotide with or without a central G-T mismatch. The fraction of hydrolyzed [γ-32P]ATP was determined by charcoal binding as described previously [17]. Analysis of DNA-independent ATP hydrolysis was performed as above but with 200 nM MSH2-MSH6 protein. MSH2-MSH3 ATPase activity was measured similarly, except that 15–50 nM protein was incubated with 0.5–32 μM unlabeled ATP in the presence of a 41-bp substrate containing a central (CA)4 insertion in one strand.
Surface Plasmon Resonance
Real time interaction of MSH2-MSH6 or MSH2-MSH3 with G-T mismatched DNA or (CA)4 IDL-containing DNA, respectively, was monitored by surface plasmon resonance (SPR) using a Biacore T100 system (GE Healthcare). Double-stranded biotinylated DNA with a central G-T mispair within an 81-bp duplex or a (CA)4 insertion loop within a 41-bp duplex was affixed to a streptavidin sensor chip to ~100 resonance units (RU). 0–120 nM MSH2-MSH6 or 0–8 nM MSH2-MSH3 were injected over the chip at a flow rate of 30 μL/min for 3 minutes in SPR buffer containing 25 mM Hepes (pH 8.1), 110 mM NaCl, 1 mM DTT, 5 mM MgCl2, and 2% glycerol. To assess sliding clamp formation and ATP-dependent dissociation, ~80 RUs of 60-bp homoduplex or G-T mispair containing substrates with or with or without a 5′ digoxigenin modification on the strand opposite the 5′ biotinylated strand were affixed to the streptavidin sensor chip. Anti-digoxigenin antibody (Roche; Basel, Switzerland) was applied to block the free ends of DNA prior to addition of 20 nM MSH2-MSH6. Following a brief buffer dissociation step with SPR buffer, 50 μM ATP was added. To examine loading of multiple sliding clamps, MSH2-MSH6 and 50 μM ATP were loaded simultaneously. Samples were kept at 10°C prior to injection and experiments were performed at 25°C. Data were analyzed with GraphPad Prism (GraphPad Software, Inc.; La Jolla, CA) using a 1:1 binding or a 1-phase exponential decay model.
ATP Binding
ATP crosslinking was performed with 0.2 μM MSH2-MSH6 or MSH2-MSH3 protein incubated with 0.25 μM [γ-32P]ATP for 20 minutes at room temperature in 20 μL of buffer containing 25 mM Hepes pH 8.1, 110 mM NaCl, 1 mM DTT, 5% glycerol, (+/−) 5 mM MgCl2, and (+/−) 1 mM EDTA. Reactions were then UV-crosslinked for 10 minutes using a Stratalinker 1800 (Agilent Technologies; Santa Clara, CA), and separated by 6% SDS-PAGE. Bands were visualized using a Cyclone Storage Phosphor System (Perkin Elmer Life Sciences) and OptiQuant software (Packard Instrument Co.; Meriden, CT). An equivalent amount of protein was loaded on a separate gel and visualized by Coomassie Blue stain as a control for protein loading. ATPγS filter binding experiments were carried out with 100 nM MSH2-MSH6 protein, 0.5μM [35S]ATPγS (PerkinElmer Life Sciences; Waltham, MA), and 0.5–20 μM unlabeled ATPγS as described previously [16]. Bound [35S]ATPγS was quantified in an LS 6500 scintillation counter (Beckman Coulter; Brea, CA).
ADP Binding and Exchange for ATP
ADP binding and exchange for ATP was measured using 60 nM MSH2-MSH6 protein pre-incubated with 3.9 μM [3H]ADP (PerkinElmer Life Sciences) in buffer containing 25 mM Hepes pH 8.1, 110 mM NaCl, 1 mM DTT, 75 μg/μL BSA, 5 mM MgCl2, and 15% glycerol. For each time point, the reaction was started by combining 15μL of pre-incubated protein with 15 μL of buffer supplemented with 1 mM ATP and 120 nM of 41-bp G-T DNA oligonucleotide. Reactions were quenched by adding 4 mL of cold stop buffer containing 25 mM Hepes pH 9.1, 110 mM NaCl, 5 mM MgCl2, 15% glycerol. The amount of ADP that remained bound to MSH2-MSH6 was quantified by filter retention. Exchange curves were fit to a 1-phase exponential decay model using GraphPad Prism software.
Partial Proteolysis
Increasing amounts of trypsin (0–160 ng) were incubated with 1 μg MSH2-MSH6 in 20 μL of buffer containing 50 mM ammonium bicarbonate, 25 mM Hepes pH 8.1, 100 mM NaCl, 1 mM DTT, 1 mM MgCl2, 0.1 mM EDTA, and 5% glycerol for 45 minutes at 37°C. Digestions were stopped by the addition of Laemmli sample buffer and boiling. 0.5 μg of protein were separated by 8% SDS-PAGE and visualized by silver stain. Western blot analysis was performed with polyclonal antibodies to MSH6 (A300-023A, Bethyl Laboratories; Montgomery, TX) and MSH2 (PC57, EMD4Biosciences; San Diego, CA).
Lentiviral Expression of MSH2
To generate a cell line carrying the MSH2 c.1A>C variant, MSH2 cDNA was PCR amplified introducing the c.1A>C start codon mutation using the following primers: 5′>3′ CTGGCGGTGCAG, and 5′>3′ CGTAGTAACTTTTATTCGTGAAATG. The PCR product was subcloned into the lentiviral expression vector pCDH-EF1-MCS-T2A-Puro (System Biosciences, Mountain View, CA). Hec59 cancer cells were transduced with lentivirus and kept under 0.5 μg/ml puromycin selection. Hec59 cells infected with the empty and wild-type MSH2 lentivirus were generated previously (Mastrocola, 2010). Hec59 cell lines were cultured in DMEM/F-12 medium supplemented with 10% fetal bovine serum (Invitrogen). Whole cell extracts were probed using the following antibodies at 1:1000 dilutions; anti-MSH2 (EMD4Biosciences), anti-MSH6 (Bethyl Laboratories), anti-MSH3 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Ku86 (Santa Cruz Biotechnology).
Mismatch Repair Assay
The MMR heteroduplex substrate was prepared as previously reported (Mastrocola, 2010). To measure repair efficiency, Hec59 cell lines carrying wild-type or mutant MSH2 were transfected with 1.5 μg of the heteroduplex plasmid and 1 μg of pDsRed2-N1 (Clonetech, Mountain View, CA) using Lipofectamine 2000 (Invitrogen). After 40 hours, cells were harvested and analyzed for fluorescence intensity by flow cytometetry and quantitated using FACSDiva software (BD Biosciences, Franklin Lakes, NJ). EGFP fluorescence was normalized to RFP fluorescence to control for transfection efficiency.
RESULTS
A Rare Germline MSH2 Mutation in an Atypical LS-Suspected Carcinoma
Diagnosis of LS relies primarily on family history, the presence of microsatellite instability in tumors, and negative IHC detection of MMR proteins in tumors [1]. We report on a suspected LS cancer patient with a germline MMR gene mutation whose tumor does not show microsatellite instability in a panel of five markers (BAT-25, BAT-26, MONO-27, NR21, NR24), and is positive for all four MMR proteins by IHC as performed by Genzyme Genetics. The patient has a family history of cancer and was diagnosed with a Stage IIIC moderate grade serous carcinoma of the ovary at age 53. A comprehensive pedigree was obtained (Figure 1A). Although family history does not entirely satisfy the Bethesda Criteria for LS diagnosis [18], three maternal cases of colon cancer prompted sequencing for mutations in MMR genes. A mutation was found within the start codon of MSH2 (c.1A>C) in the patient. No other mutations were detected in MSH2, MSH6 or MLH1. In addition, the breast cancer susceptibility genes BRCA1 and BRCA2 were negative for mutation. The MSH2 c.1A>C variant was also detected in an unaffected sister, who opted to have a prophylactic oophorectomy. Genetic testing confirmed that the mother is not a carrier of the variant, suggesting paternal inheritance. The father died at a young age and was therefore unavailable for testing.
Figure 1.

Pedigree analysis of a patient with the mutation hMSH2 c.1A>C and possible hMSH2 truncation mutation. (A) The proband is indicated by an arrow. Shaded boxes indicate a primary diagnosis of cancer according to the legend. Current age, age at diagnosis, and/or age at death is indicated for each individual. The sister of the proband is also a carrier of the hMSH2 c.1A>C mutation (*). (B) The coding sequence for the N-terminus of MSH2 reveals in-frame ATG codons at amino acids 76 and 199 that may be used as alternative start sites. (C) The deletion of 25 amino acids from the N-terminus of hMSH2 has been mapped to the hMSH2-hMSH6 crystal structure [23], as shown in red. The MSH2 and MSH6 monomers are indicated in green and blue respectively. Mismatched DNA is shown in yellow. Structural representations were created in PyMol (The PyMOL Molecular Graphics System, Schrödinger, LLC)
MSH2(NΔ25) Mutant Protein Retains Interaction with MSH6
Cancer-associated germline variants in the MSH2 initiation codon have been reported previously [6–8]. The tumors from these patients stained positively for MSH2 by IHC consistent with the production of a near-full length protein product from the variant allele [6,8]. It has been speculated that a second in-frame initiation codon at the +76 position (Figure 1B) could be used to generate an N-terminal truncation of MSH2, MSH2(NΔ25), in which the first 25 amino acids of the mismatch binding domain are missing (Figure 1C) [7,8]. In order to examine possible defects in protein function caused by this truncation, we examined the biochemical activities of recombinant MSH2(NΔ25)-MSH6 heterodimers in vitro.
The biological activity of MSH2 depends on formation of a heterodimer with either MSH6 or MSH3 [11]. MSH2-MSH6 is the major mismatch recognition complex in the cell. Strong interactions between MSH2 and MSH6 are formed via contacts between the C-terminal ATPase domains and between the mismatch binding and clamp domains of each monomer [19]. We expressed MSH2(NΔ25) and MSH6 proteins from a dual expression construct in Sf9 cells. Co-purification of MSH2(NΔ25) with His6-tagged MSH6 in an apparent 1:1 ratio indicates that the loss of amino acids 1–25 does not affect MSH2 interaction with MSH6 (Figure 4A, coomassie stained gel). Further, we analyzed MSH2(NΔ25)-MSH6 heterodimer stability by measuring the dimer to monomer ratio following urea denaturation. The N-terminal truncation of MSH2 does not significantly affect heterodimer stability (data not shown).
Figure 4.

Nucleotide binding and exchange properties of MSH2(NΔ25)-MSH6. (A) ATP binding activity. Wild-type MSH2-MSH6 and MSH2(NΔ25)-MSH6 proteins were UV- crosslinked to radiolabeled [γ-32P]ATP in the presence or absence of Mg2+ (to allow or inhibit hydrolysis). Radiolabeled ATP was visualized by phosphorimaging. To control for protein loading, a separate gel was equivalently loaded with each protein and Coomassie stained. (B) ATPγS binding. 100 nM of wild-type and mutant heterodimers were incubated with increasing concentrations of [35S]ATPγS. The amount of protein- bound nucleotide was determined by filter binding. (C) ADP binding and exchange for ATP. MSH2-MSH6 wild-type and mutant heterodimers were incubated with [3H]ADP. Exchange of ADP was initiated by adding mismatched DNA and excess ATP. Reactions were allowed to proceed for 0–90 seconds. The amount of ADP bound after each time point was determined by filter binding.
The MSH2(NΔ25)-MSH6 Heterodimer Has Decreased Steady-State ATPase Activity
Two highly-conserved composite ATP binding and hydrolysis motifs are located within the ATPase domains of MSH2 and MSH6 [20]. The MSH2-MSH6 heterodimer displays intrinsic low-level ATPase activity that is stimulated by mismatched DNA, and this adenosine nucleotide processing is essential for the MMR activity of MSH2-MSH6 [17,21,22]. Steady-state ATPase activity of MSH2(NΔ25)-MSH6 was determined in the absence of DNA, and in the presence of homoduplex (G-C) or mismatched (G-T) DNA using the Norit method to measure release of inorganic phosphate [17]. Like wild-type MSH2-MSH6, the mutant heterodimer displays weak ATPase activity in the absence of DNA (Figure 2). Similarly, ATPase activity is increased for both wild-type and mutant protein in the presence of homoduplex DNA, and is further stimulated by a G-T mismatch. However, at each condition the kcat of the mutant is decreased by nearly half when compared to wild-type (Figure 2, Table 1). This suggests that although MSH2(NΔ25)-MSH6 protein is able to undergo cycles of ATP hydrolysis, one or several steps in the reaction may be compromised.
Figure 2.
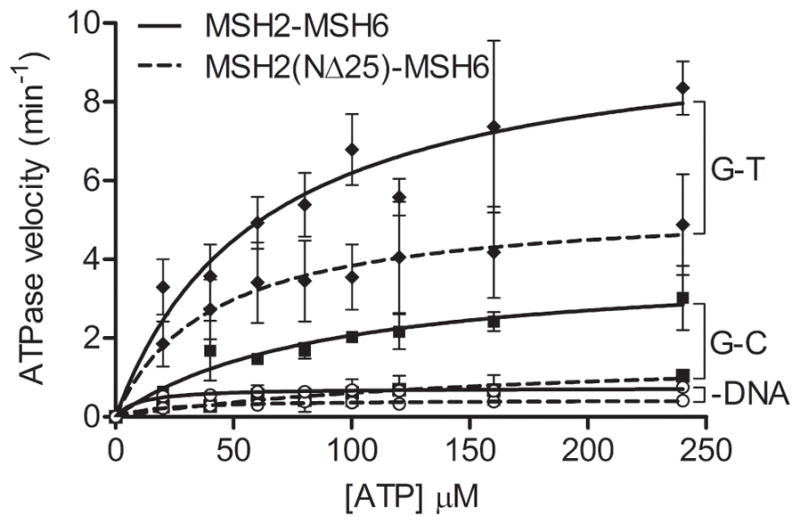
ATP hydrolysis activity of MSH2(NΔ25)-MSH6. Steady-state ATPase activity was determined by measuring the release of radiolabeled inorganic phosphate. Low- level ATPase in the absence of DNA was compared to G-C homoduplex- and G-T mismatch-stimulated activity for MSH2(NΔ25)-MSH6 and wild-type MSH2-MSH6 protein. Error bars represent standard deviation of three or more independent experiments.
Table 1.
ATP Hydrolysis and Mismatch Binding of MSH2(NΔ25)-MSH6.
| MSH2-MSH6 protein | ATPase Activity No DNA | ATPase Activity G/T DNA | ATPase Activity G/C DNA | DNA Binding | |||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| KM | kcat | KM | kcat | KM | kcat | KD(G/T) | |
|
| |||||||
| μM | min−1 | μM | min−1 | μM | min−1 | nM | |
| wild-type | 9.6 ± 2.1 | 0.7 ± 0.0 | 60.6 ± 17.4 | 10.0 ± 1.1 | 87.9 ± 25.6 | 3. 9 ± 0.5 | 5.98 ± 1.11 |
| NΔ25 | 19.9 ± 5.0 | 0.4 ± 0.0 | 40. 8 ± 5.9 | 5.4 ± 0.2 | 184.4 ± 81.8 | 1.7 ± 0.4 | 9.23 ±2.01 |
MSH2(NΔ25)-MSH6 Binds to Mismatched DNA
Mismatch recognition occurs through amino-terminal mismatch binding domains of both MSH2 and MSH6 [23]. The MSH2-MSH6 crystal structure reveals that the majority of protein-DNA interaction occurs via the MSH6 monomer, which makes extensive contacts with the DNA backbone and confers specificity for mismatched DNA. The mismatch binding domain of MSH2 only interacts with the DNA backbone via a lysine at position 6 which is deleted in the MSH2(NΔ25) protein [23], thus we wished to examine the ability of the MSH2(NΔ25) mutant heterodimer to bind to a mismatch. The kinetics of MSH2(NΔ25)-MSH6 binding to an 81-bp oligonucleotide containing a central G-T mispair was examined by surface plasmon resonance (SPR) (Figure 3). The KD(G/T) of the MSH2(NΔ25)-MSH6 mutant is modestly increased in comparison to wild-type protein suggesting slightly reduced affinity for heteroduplex DNA (Table 1).
Figure 3.
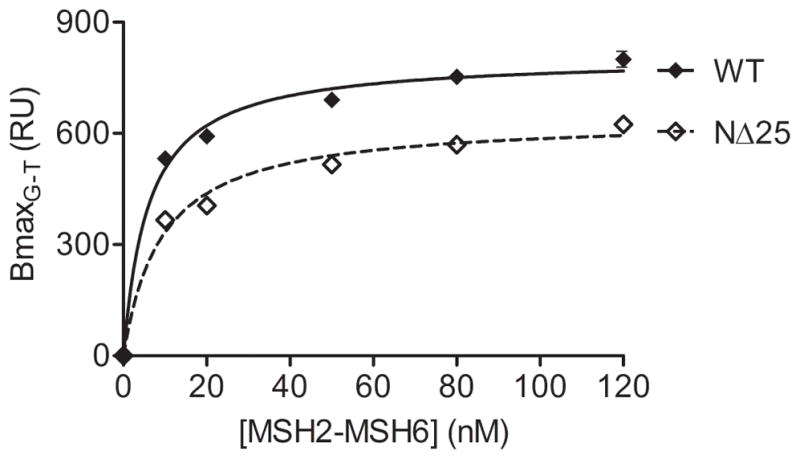
Mismatch binding properties of MSH2(NΔ25)-MSH6. G-T mismatch binding was measured by surface plasmon resonance (SPR). Maximum protein binding, measured in resonance units (RU), to an 81-bp G-T double-stranded DNA oligonucleotide was determined for increasing concentrations of MSH2-MSH6 protein. Curves were generated using a hyperbolic fit of the data.
Adenosine Nucleotide Affinity is Decreased in MSH2(NΔ25)-MSH6 Heterodimers
The ATPase cycle requires mismatch-induced exchange of ADP for ATP [24]. Although ATP binding is sufficient to induce dissociation of MSH2(NΔ25)-MSH6 from a mismatch, we wanted to measure nucleotide affinity, and exchange of ADP for ATP directly for this mutant. The MSH2 and MSH6 monomers have differential specificity for nucleotides [25]. MSH6 has a higher affinity for ATP while MSH2 has a higher affinity for ADP. We examined ATP binding by UV crosslinking of radiolabeled [γ-32P]ATP to MSH2(NΔ25)-MSH6 (Figure 4A). The ATPase activity of MSH2-MSH6 is magnesium- dependent [17], therefore the experiment was performed in the presence and absence of magnesium to allow or prohibit hydrolysis of the γ-labeled phosphate. Under magnesium-free conditions, the labeled ATP is bound to both subunits of the wild-type heterodimer with a much stronger binding to the MSH6 subunit. However, the binding of labeled ATP to the MSH6 subunit of the MSH2(NΔ25)-MSH6 is dramatically reduced. We also examined adenosine nucleotide binding by filter retention of the poorly hydrolyzable ATP analog, ATPγS (Figure 4B). Consistent with UV crosslinking results, the KD(ATPγS) for MSH2(NΔ25)-MSH6 heterodimer was reduced by approximately 40% compared to wild-type protein (Table 2).
Table 2.
Nucleotide binding, exchange, and ATP-induced dissociation properties of MSH2(NΔ25)-MSH6.
| MSH2-MSH6 protein | ATPγS binding | ADP→ATP exchange | ATP-induced dissociation* | |
|---|---|---|---|---|
| free end | blocked end | |||
|
| ||||
| KD(ATPγS) | T1/2 | koff 10−2 | koff 10−2 | |
|
| ||||
| μM | s | s−1 | s−1 | |
| wild-type | 0.59 ± 0.04 | 1.99 | 32.5± 0.9 | 0.22± 0.002 |
| NΔ25 | 0.98 ± 0.25 | 4.54 | 32.2± 1.0 | 0.16± 0.002 |
dissociation with 50 μM ATP
We also analyzed mismatch-provoked exchange of ADP for ATP by filter binding. Purified MSH2(NΔ25)-MSH6 was pre-bound to [3H]-ADP and the reaction initiated with a 41-bp G-T oligonucleotide and excess ATP. Initial binding of ADP was significantly lower in the mutant heterodimer, and the mismatch-provoked loss of ADP occurred more slowly in the mutant (Figure 4C, Table 2). This may be due to decreased affinity of MSH2(NΔ25) for ATP.
Altered Conformation of MSH2(NΔ25)-MSH6
Since MSH2(NΔ25)-MSH6 displays reduced affinity for ATP, we investigated whether ATP-induced conformational changes are affected. Partial digestion of MSH2(NΔ25)-MSH6 with increasing amounts of trypsin was used to examine conformational states in the absence of nucleotide and in the presence of either ATP or ATPγS. The nucleotide-free form of wild-type MSH2-MSH6 is particularly susceptible to trypsin digestion (Figure 5, silver stain panel). Examination of the proteolytic peptides by Western blot revealed that MSH6 is extensively digested and most of the fragments run off an 8% acrylamide gel (Figure 5, Western blot panel). The visible bands on the silver stained gel correspond to bands detected with an anti-MSH2 antibody suggesting that MSH2 is largely protected from proteolysis in the absence of nucleotide. In comparison, the MSH2(NΔ25)-MSH6 heterodimer is almost entirely digested in the absence of nucleotide. Limited amounts of MSH2(NΔ25) peptides are detected by Western blot with an anti-MSH2 antibody particularly at the 80 and 160 ng trypsin concentrations, suggesting a more trypsin-sensitive conformation for MSH2(NΔ25) compared to wild-type MSH2. We cannot rule out, however, that the anti-MSH2 and anti-MSH6 polyclonal antibodies fail to recognize all digested peptides.
Figure 5.

Partial proteolytic digestion of MSH2(NΔ25)-MSH6. Heterodimer conformational states were observed by trypsin proteolysis. 1 μg of MSH2-MSH6 wild- type or mutant heterodimer protein were incubated in the absence or presence of 1 mM adenosine nucleotide as indicated and increasing amounts of trypsin (0–180 ng). Protein fragmentation was visualized by silver-stained SDS-PAGE (left panel), and by Western blot with anti-hMSH6 (center panel) and anti-hMSH2 (right panel) antibodies.
Addition of adenosine nucleotide results in increased detection of proteolytic peptides for both wild-type and mutant MSH2-MSH6. Increased protection from trypsin access suggests that the heterodimers are in a more closed conformation in the ATP bound form. Though the overall pattern is similar, we do observe that hMSH2(NΔ25) is slightly more susceptible to digestion than wild-type protein in the presence of ATP and ATPγS. This is especially evident when examining the proteolysis fragments by Western blot with the polyclonal MSH2 antibody. These data indicate that the N-terminal truncation of MSH2 confers a modestly altered conformational state on the MSH2(NΔ25)-MSH6 heterodimer.
MSH2(NΔ25)-MSH6 forms sliding clamps on mismatched DNA
Addition of ATP to MSH2-MSH6 bound to a G-T mismatch results in a conformational change that releases the protein from the mismatch and allows it to diffuse along DNA as a sliding clamp [24]. In an SPR experiment, the MSH2-MSH6 sliding clamp rapidly slides off the free end of DNA not tethered to the sensor chip [26]. The free end can be blocked by modification of the oligo with digoxigenin and addition of an anti-digoxigenin antibody. Generation of double blocked end DNA resulted in a dramatic decrease in the ATP-induced koff of MSH2-MSH6 compared to free-end DNA (Figure 6A and Table 2) consistent with the trapping of sliding clamps on DNA. Simultaneous addition of ATP and MSH2-MSH6 protein reveals an increase in mass consistent with multiple sliding clamps on double blocked end DNA, whereas accumulation of sliding clamps fails to occur on free end DNA (Figure 6C). We observed similar formation of sliding clamps with the MSH2(NΔ25)-MSH6 protein indicating that the modest effects of the mutation on ATP-induced conformational changes do not eliminate sliding clamp formation (Figure 6B and D, Table 2).
Figure 6.

ATP-induced dissociation and sliding clamp formation of MSH2-MSH6 from mismatched DNA. The interaction of 20 nM MSH2-MSH6 with mismatched (G:T) or homoduplex (G:C) DNA, having either free or α-digoxigenin blocked ends, was monitored by SPR. Binding of MSH2-MSH6 (A, C) and MSH2(NΔ25)-MSH6 (B, D) to DNA was recorded in the absence of ATP followed by a brief dissociation phase in buffer. When indicated, buffer was supplemented with 50 μM ATP (A, B) or 50 μM ATP along with 20 nM MSH2-MSH6 (C, D).
The MSH2(NΔ25) Variant Does Not Affect Adenosine-Nucleotide Processing or Lesion Recognition of MSH2-MSH3
MSH2 forms functional complexes with either MSH6 or MSH3. Although MSH6 is the more abundant lesion recognition complex in the cell and mutations in MSH6 account for a significant percentage of LS cases, there is increasing evidence that MSH2-MSH3 has a role in cancer protection. Loss of MSH3 has been found to cause genomic instability in colorectal cancer [27], and a germline mutation in MSH3 was recently reported to be associated with familial hamartomatous polyposis and cancer [28]. To investigate whether carriers of the MSH2 start codon mutation may be predisposed to cancer via disruption of the MSH2-MSH3 heterodimer, we analyzed ATPase activity, ATP binding, and IDL interaction with recombinant MSH2(NΔ25)- MSH3.
Compared to wild-type MSH2-MSH3, the mutant heterodimer displays only a slight defect in IDL-stimulated ATPase activity (Figure 7A, Table 3). This slight defect is not due to decreased DNA binding since MSH2(NΔ25)-MSH3 heterodimers recognize a (CA)4 IDL as well as wild-type (Figure 7B, Table 3). Lastly, the MSH2(NΔ25) mutant protein does not affect ATP binding to either the MSH2 or MSH3 subunit as shown by UV crosslinking studies (Figure 7C). We conclude that the MSH2(NΔ25) truncation does not significantly alter the function of MSH2-MSH3 in vitro.
Figure 7.

Biochemical analysis of MSH2(NΔ25)-MSH3. (A) Steady-state ATPase activity was determined by measuring the release of radiolabeled inorganic phosphate in the presence of (CA)4 insertion-deletion loop for MSH2(NΔ25)-MSH3 and wild-type protein. Error bars represent standard deviation of three independent experiments. (B) DNA binding was measured by SPR. Maximum protein binding, measured in resonance units (RU) to an oligo containing a (CA)4 IDL was determined for increasing concentrations of MSH2-MSH3 protein. Curves were generated using a hyperbolic fit of the data. (C) ATP binding activity was measured by UV-crosslinking wild-type and mutant MSH2-MSH3 to radiolabeled [γ-32P]ATP in the presence or absence of Mg2+. Radiolabeled ATP was visualized by phosphorimaging. A coomassie stained gel indicates equivalent protein loading.
Table 3.
ATP Hydrolysis and Mismatch Binding of MSH2(NΔ25)-MSH3.
| MSH2-MSH3 protein | ATPase Activity (CA)4 DNA | DNA Binding | |
|---|---|---|---|
|
| |||
| KM | kcat | KD(CA)4 | |
|
| |||
| μM | min−1 | nM | |
| wild-type | 7.2 ± 1.4 | 3.3 ± 0.2 | 0.22 ± 0.05 |
| NΔ25 | 8.9 ± 1.8 | 2.7 ± 0.2 | 0.21 ± 0.03 |
Expression of the MSH2 c.1A>C Variant in Human Cells
We wished to determine whether the MSH2 c.1A>C variant results in expression of a truncated protein in human cells as predicted for MSH2 start codon mutations [8]. We utilized a lentiviral expression system to stably re-introduce wild-type MSH2 or the MSH2 c.1A>C variant into MSH2 null Hec59 endometrial cancer cells [29]. Expression of MSH2 c.1A>C restored MSH2 protein production in Hec59 cells compared to empty vector controls (Figure 8A). Recombinant MSH2(NΔ25) from Sf9 insect cells migrates slightly faster than full-length MSH2 on SDS-PAGE, however, the variant MSH2 expressed in Hec59 cells appears indistinguishable from wild-type. Interestingly, both the wild-type and variant MSH2 protein from Hec59 cells migrate more slowly than the Sf9 expressed recombinant MSH2 proteins suggesting that the proteins may be post-translationally modified in the human cells. These modifications may mask migration differences caused by a deletion 25 amino acids. Alternatively, full-length MSH2 may be produced by the variant cDNA in addition to the NΔ25 peptide. We observe three MSH2 peptides in the MSH2 c.1A>C expressing cells that may represent translation initiation from the mutant CUG codon as well as downstream AUG sites (see Figure 1B). Reports from in vitro translation experiments suggest that CTG can function as a very weak start codon in eukaryotic cells if surrounded by appropriate sequence and secondary structure [30,31].
Figure 8.

Expression of c.1A>C variant MSH2 in human cancer cells. (A) Whole cell extracts from Hec59 cells transduced with wild-type MSH2 (ATG) or MSH2 c.1A>C (CTG) lentiviral constructs. Extracts were analyzed by Western blotting with antibodies against MSH2, MSH6, and MSH3. Recombinant MSH2-MSH6 and MSH2(NΔ25)- MSH6 protein is shown for reference. Ku86 is a loading control. (B) In vivo repair of a mismatch-containing plasmid encoding EGFP was measured in wild-type MSH2 (ATG) and MSH2 c.1A>C (CTG) Hec59 cell lines by monitoring restoration of EGFP expression relative to a RFP transfection control. Three independent experiments were quantified by flow cytometry.
To determine whether the MSH2 protein produced from the c.1A>C variant affects MMR function, we performed an in vivo MMR assay to measure correction of a G-T mismatch in an EGFP reporter plasmid [32]. We observed that vector only control cells were incapable of restoring GFP fluorescence, while expression of wild-type MSH2 led to a dramatic increase in GFP-positive cells consistent with repair of the mismatch (Figure 8B). MSH2 c.1A>C cells also appeared to restore GFP fluorescence, however, we consistently saw modest, yet statistically significant (p<0.05), reductions in levels of repair.
DISCUSSION
Many MMR gene variants from cancer families have now been reported in the literature, and a significant proportion of these are missense variants where a full-length protein with potentially altered function is expressed. The pathogenicity of missense variants is questionable as many are likely not total loss-of-function mutations [4,5]. Classification of MMR gene missense variants is important for patient diagnosis, monitoring, and treatment, especially as increased testing leads to the discovery of more variant alleles. This problem becomes even more difficult in those patients with a limited family history of cancer that does not entirely fulfill the revised Amsterdam criteria for LS [18]. It is possible that missense variants that modestly alter MMR activity may function as weak disease alleles resulting in less penetrance and increased age of onset. Functional analysis of MMR variant-derived proteins can aid in understanding their relevance to cancer.
In the reported cases of missense mutations of the MSH2 start codon, family histories and disease phenotypes do not present as classical LS. Therefore, we chose to study the MSH2 start codon variant c.1A>C identified in one of our patients to evaluate any functional defects that may underlie disease. We studied the MSH2(NΔ25) protein which is suspected to be translated from an alternate downstream AUG. Recombinant MSH2(NΔ25) protein retains interaction with its binding partners MSH6 and MSH3. MSH2(NΔ25)-MSH6 has reduced ATPase activity, DNA binding activity, and adenosine nucleotide binding affinity in vitro as compared to the wild-type MSH2-MSH6. Further, the conformational state of MSH2(NΔ25)-MSH6 may be slightly altered, suggesting that loss of the N-terminal 25 amino acids of MSH2 could confer defects in protein folding that affects the heterodimer function. We conclude that MSH2(NΔ25) is not a complete loss-of-function mutation, but displays biochemical defects that may have significance.
We also asked whether the truncated form of MSH2 may affect MSH2-MSH3 function. Evidence from yeast studies suggests that the Msh2 mismatch binding domain has a critical role in the context of the Msh2-Msh3 heterodimer. A yeast strain harboring a deletion of the entire Msh2 mismatch binding domain retained function in Msh2-Msh6-directed repair assays, but was entirely defective in repair of larger insertion-deletion loops, specific to the repair activity of Msh2-Msh3 [33]. Our results suggest no effect of the NΔ25 deletion on MSH2-MSH3 IDL binding or IDL-stimulated ATPase, however, our deletion was significantly smaller than the portion of Msh2 deleted in the previous yeast study.
To examine the in vivo consequences of this variant, we expressed the MSH2 c.1A>C cDNA in MSH2-null human cancer cells. We determined that a protein product is produced from this mutant MSH2. Interestingly, we observed multiple MSH2 protein products including one that may represent full-length MSH2 as well as the predicted truncated protein product. A full length MSH2 protein may be produced from this allele if the resultant CUG codon is capable of being used as a start codon. In vitro evidence suggests that translation can initiate from a CUG codon in the proper sequence and structure context [30,31]. In vivo, CUG codons upstream of the initial AUG can serve as weak initiation codons for some mammalian genes, thus resulting in multiple isoforms of the proteins [34,35]. Expression of multiple MSH2 isoforms from the same allele may affect MMR function through reduced levels of wild-type MSH2 production. Additionally, production of an MSH2(NΔ25) protein that can bind mismatches, but not bind and hydrolyze ATP as efficiently may interfere with MMR by competing with the wild-type protein. Our in vivo MMR assay suggests that expression of the MSH2 c.1A>C variant results in a small, but statistically significant decrease in MMR. If the ratio of full-length MSH2 to truncated MSH2 were to decrease, any dominant negative effect would increase. Other MSH2 start codon variants reported in the literature include a c.1A>T variant [7] that likely does not produce an alternate start codon [30]. These patients may only produce truncated MSH2, potentially leading to a more dramatic functional defect.
In summary, this study has shown that a MSH2 c.1A>C variant from a non-classical LS patient results in the production of MSH2 protein that includes a peptide with an N-terminal deletion of 25 amino acids. This MSH2(NΔ25) protein has modest biochemical defects that may cause it to contribute to pathogenicity. The extent of the biochemical defect, combined with the possibility that it is produced in combination with full-length MSH2, suggest this would not be a strong cancer allele. The modest effect on MMR function may underlie the lack of MSI detected in the tumor from our patient. However, a weaker mutator phenotype that falls below the level of detection using traditional MSI testing may exist. Additionally, the variant may affect non-repair functions of the MMR pathway that could contribute to disease phenotype. The MMR proteins are involved in signaling to cell cycle checkpoint and apoptosis machinery in response to high levels of DNA damage [13], which may have an impact on their ability to function as tumor suppressors [36]. The molecular mechanism by which the MMR proteins function in this checkpoint response is not entirely clear. The functional analysis of missense variants may reveal single amino-acid alterations that can separate the repair and checkpoint response functions of MSH2-MSH6, while still contributing to disease progression [37,38].
Acknowledgments
This work was funded through National Cancer Institute grant CA115783 to C.D.H.
Abbreviations
- MSH
MutS homolog
- MMR
mismatch repair
- LS
Lynch syndrome
- IHC
immunohistochemistry
- SPR
surface plasmon resonance
- RU
resonance units
References
- 1.Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76(1):1–18. doi: 10.1111/j.1399-0004.2009.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duval A, Hamelin R. Genetic instability in human mismatch repair deficient cancers. Ann Genet. 2002;45(2):71–75. doi: 10.1016/s0003-3995(02)01115-2. [DOI] [PubMed] [Google Scholar]
- 3.Peltomaki P, Vasen H. Mutations associated with HNPCC predisposition--Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20(4–5):269–276. doi: 10.1155/2004/305058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ou J, Niessen RC, Lutzen A, et al. Functional analysis helps to clarify the clinical importance of unclassified variants in DNA mismatch repair genes. Hum Mutat. 2007;28(11):1047–1054. doi: 10.1002/humu.20580. [DOI] [PubMed] [Google Scholar]
- 5.Heinen CD. Genotype to phenotype: Analyzing the effects of inherited mutations in colorectal cancer families. Mutation Research/DNA Repair. 2010;693:32–45. doi: 10.1016/j.mrfmmm.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnetson RA, Cartwright N, van Vliet A, et al. Classification of ambiguous mutations in DNA mismatch repair genes identified in a population-based study of colorectal cancer. Hum Mutat. 2008;29(3):367–374. doi: 10.1002/humu.20635. [DOI] [PubMed] [Google Scholar]
- 7.Farrington SM, Lin-Goerke J, Ling J, et al. Systematic analysis of hMSH2 and hMLH1 in young colon cancer patients and controls. Am J Hum Genet. 1998;63(3):749–759. doi: 10.1086/301996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kets CM, Hoogerbrugge N, van Krieken JH, Goossens M, Brunner HG, Ligtenberg MJ. Compound heterozygosity for two MSH2 mutations suggests mild consequences of the initiation codon variant c. 1A>G of MSH2. Eur J Hum Genet. 2009;17(2):159–164. doi: 10.1038/ejhg.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Felton KE, Gilchrist DM, Andrew SE. Constitutive deficiency in DNA mismatch repair. Clin Genet. 2007;71(6):483–498. doi: 10.1111/j.1399-0004.2007.00803.x. [DOI] [PubMed] [Google Scholar]
- 10.Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet. 2008;124(2):105–122. doi: 10.1007/s00439-008-0542-4. [DOI] [PubMed] [Google Scholar]
- 11.Acharya S, Wilson T, Gradia S, et al. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc Natl Acad Sci U S A. 1996;93(24):13629–13634. doi: 10.1073/pnas.93.24.13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 13.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7(5):335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 14.Iyer RR, Pluciennik A, Genschel J, Tsai M-S, Beese LS, Modrich P. MutLalpha and Proliferating Cell Nuclear Antigen Share Binding Sites on MutSbeta. J Biol Chem. 2010;285(15):11730–11739. doi: 10.1074/jbc.M110.104125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson T, Guerrette S, Fishel R. Dissociation of mismatch recognition and ATPase activity by hMSH2-hMSH3. J Biol Chem. 1999;274(31):21659–21664. doi: 10.1074/jbc.274.31.21659. [DOI] [PubMed] [Google Scholar]
- 16.Cyr JL, Heinen CD. Hereditary cancer-associated missense mutations in hMSH6 uncouple ATP hydrolysis from DNA mismatch binding. J Biol Chem. 2008;283(46):31641–31648. doi: 10.1074/jbc.M806018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gradia S, Acharya S, Fishel R. The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell. 1997;91(7):995–1005. doi: 10.1016/s0092-8674(00)80490-0. [DOI] [PubMed] [Google Scholar]
- 18.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerrette S, Wilson T, Gradia S, Fishel R. Interactions of human hMSH2 with hMSH3 and hMSH2 with hMSH6: examination of mutations found in hereditary nonpolyposis colorectal cancer. Mol Cell Biol. 1998;18(11):6616–6623. doi: 10.1128/mcb.18.11.6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haber LT, Walker GC. Altering the conserved nucleotide binding motif in the Salmonella typhimurium MutS mismatch repair protein affects both its ATPase and mismatch binding activities. EMBO J. 1991;10(9):2707–2715. doi: 10.1002/j.1460-2075.1991.tb07815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blackwell LJ, Bjornson KP, Modrich P. DNA-dependent activation of the hMutSalpha ATPase. J Biol Chem. 1998;273(48):32049–32054. doi: 10.1074/jbc.273.48.32049. [DOI] [PubMed] [Google Scholar]
- 22.Studamire B, Quach T, Alani E. Saccharomyces cerevisiae Msh2p and Msh6p ATPase activities are both required during mismatch repair. Mol Cell Biol. 1998;18(12):7590–7601. doi: 10.1128/mcb.18.12.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell. 2007;26(4):579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 24.Gradia S, Subramanian D, Wilson T, et al. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol Cell. 1999;3(2):255–261. doi: 10.1016/s1097-2765(00)80316-0. [DOI] [PubMed] [Google Scholar]
- 25.Martik D, Baitinger C, Modrich P. Differential specificities and simultaneous occupancy of human MutSalpha nucleotide binding sites. J Biol Chem. 2004;279(27):28402–28410. doi: 10.1074/jbc.M312108200. [DOI] [PubMed] [Google Scholar]
- 26.Heinen CD, Wilson T, Mazurek A, Berardini M, Butz C, Fishel R. HNPCC mutations in hMSH2 result in reduced hMSH2-hMSH6 molecular switch functions. Cancer Cell. 2002;1(5):469–478. doi: 10.1016/s1535-6108(02)00073-9. [DOI] [PubMed] [Google Scholar]
- 27.Haugen AC, Goel A, Yamada K, et al. Genetic Instability Caused by Loss of MutS Homologue 3 in Human Colorectal Cancer. Cancer Res. 2008;68(20):8465–8472. doi: 10.1158/0008-5472.CAN-08-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang SC, Lee JK, Smith EJ, et al. Evidence for an hMSH3 defect in familial hamartomatous polyps. Cancer. 2011;117(3):492–500. doi: 10.1002/cncr.25445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe Y, Haugen-Strano A, Umar A, et al. Complementation of an hMSH2 defect in human colorectal carcinoma cells by human chromosome 2 transfer. Mol Carcinog. 2000;29(1):37–49. [PubMed] [Google Scholar]
- 30.Kozak M. Context effects and inefficient initiation at non-AUG codons in eucaryotic cell-free translation systems. Mol Cell Biol. 1989;9(11):5073–5080. doi: 10.1128/mcb.9.11.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozak M. Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotides in positions +5 and +6. EMBO J. 1997;16(9):2482–2492. doi: 10.1093/emboj/16.9.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou B, Huang C, Yang J, Lu J, Dong Q, Sun LZ. Preparation of heteroduplex EGFP plasmid for in vivo mismatch repair activity assay. Anal Biochem. 2009;1(388):167–169. doi: 10.1016/j.ab.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SD, Surtees JA, Alani E. Saccharomyces cerevisiae MSH2-MSH3 and MSH2-MSH6 complexes display distinct requirements for DNA binding domain I in mismatch recognition. J Mol Biol. 2007;366(1):53–66. doi: 10.1016/j.jmb.2006.10.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerashchenko MV, Su D, Gladyshev VN. CUG start codon generates thioredoxin/glutathione reductase isoforms in mouse testes. J Biol Chem. 2010;285(7):4595–4602. doi: 10.1074/jbc.M109.070532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tee MK, Jaffe RB. A precursor form of vascular endothelial growth factor arises by initiation from an upstream in-frame CUG codon. Biochem J. 2001;359:219–226. doi: 10.1042/0264-6021:3590219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heinen CD, Schmutte C, Fishel R. DNA repair and tumorigenesis: lessons from hereditary cancer syndromes. Cancer Biol Ther. 2002;1(5):477–485. doi: 10.4161/cbt.1.5.160. [DOI] [PubMed] [Google Scholar]
- 37.Lin DP, Wang Y, Scherer SJ, et al. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004;64(2):517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- 38.Yang G, Scherer SJ, Shell SS, et al. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004;6(2):139–150. doi: 10.1016/j.ccr.2004.06.024. [DOI] [PubMed] [Google Scholar]