

Abstract
Detection of biologically interesting, low-abundance proteins in complex proteomes such as serum typically requires extensive fractionation and high-performance mass spectrometers. Processing of the resulting large datasets involves trade-offs between confidence of identification and depth of protein coverage, that is, higher stringency filters preferentially reduce the number of low-abundance proteins identified. In the current study, alternative database search and results filtering strategies were evaluated using test samples ranging from purified proteins to ovarian tumor secretomes and human serum in order to maximize peptide and protein coverage. Full and partial tryptic searches were compared because substantial numbers of partial tryptic peptides were observed in all samples, and the proportion of partial tryptic peptides was particularly high for serum. When data filters that yielded similar false discovery rates (FDR) were used, full tryptic searches detected far fewer peptides than partial tryptic searches. In contrast to the common practice of using full tryptic specificity and a narrow precursor mass tolerance, more proteins and peptides could be confidently identified using a partial tryptic database search with a 100 ppm precursor mass tolerance followed by filtering of results using 10 ppm mass error and full tryptic boundaries.
Keywords: Database search, data filtering, trypsin, partial tryptic peptide, mass accuracy, peptide false discovery rate (FDR), search time, complex proteome
Introduction
The publication of the first database search engine, SEQUEST,1 set the stage for the later, rapid application of mass-spectrometer-based analyses of proteomes. Additional search algorithms, such as Mascot,2 X!Tandem,3 and MyriMatch4 were developed, which have similar capacities to automatically correlate thousands to millions of peptide fragmentation spectra with protein sequence databases to identify the most likely peptide sequences. However, in a typical proteomics study, only a modest percentage of the resulting peptide-spectrum matches (PSMs) are correct, for multiple reasons that have been outlined in a recent review.5 To remove incorrect PSMs, different strategies can be used to filter the search result, including: applying a cutoff filter on PSM matching scores such as Xcorr for SEQUEST;6 using a statistical model to estimate distributions of correct and random PSMs and then setting a desired cutoff filter (discriminant score in PeptideProphet™)7; combining multiple match scores and setting separate thresholds for each sub population (IDPicker)8; or employing machine learning to distinguish correct versus random matches based on multiple PSM properties (Percolator)9. For all these approaches, a target-decoy search strategy10–12 can be adapted to estimate a peptide false discovery rate (FDR). In this strategy, either a reverse sequence of true proteins or computer-generated random sequences (“decoy”) are used as surrogates to estimate the level of matches due to random chance. That is, if the forward and decoy databases are the same size and share similar peptide lengths and compositions, the assumption is that similar numbers of sequences will hit the forward and reverse sequence databases by random chance. Therefore, the percentage of the decoy matches in the final dataset after filtering out poor scoring hits is an objective indicator of the overall quality and specificity of the search result. However, a tradeoff between sensitivity and specificity exists here; i.e., stringent filters usually will yield low FDR but will result in fewer peptide and protein identifications, while a relaxed filter yields more peptide and protein identifications with higher uncertainty.
The major goal of most proteomics studies is to identify as many proteins as possible in biological samples, especially those proteins or protein changes most critical to biological functions. Identification of the most abundant proteins is rarely a challenge because peptides from these proteins have strong signals and are selected more frequently for analysis in data-dependent acquisition methods. Low-abundance proteins usually are the most biologically interesting because they frequently include the proteins that drive or regulate biological processes, but reliable detection of low-abundance proteins in complex samples is more challenging because peptide signals are weaker and MS/MS spectra have more noise from chemical background, interfering peptides, etc. To enhance detection of low-abundance proteins, particularly in samples with a wide dynamic range of abundance such as serum or plasma, extensive sample pre-fractionation and extended LC-MS/MS analyses usually are required.6, 13 This results in large datasets that, in some cases, may contain millions of MS2 spectra. Since large datasets are more prone to false protein identifications,14 efficient processing requires a strategy optimized both for analysis speed and confidence of the identification.
Advances in mass spectrometer design and performance have contributed to the size and complexity of MS/MS datasets. For example, hybrid instruments such as the LTQ-FT and LTQ-Orbitrap feature both fast scan cycles and high mass accuracy, which can be improved further through either internal calibration15 or post-acquisition recalibration,16 laying the foundation for an era of “precision proteomics.”17 For data-dependent discovery studies, these hybrid instruments provide the best sensitivity when performing survey scans in the FT analyzer and MS2 scans in the ion trap,18 because the main contributor to depth of analysis is the number of MS2 scans, which can be performed more rapidly in the ion trap. At the same time, highly accurate precursor ion masses can generate more high-confidence peptide identifications than low-mass-accuracy data, because the mass accuracy of the PSM is an effective search result filter that can remove most erroneous random matches.18, 19 The benefit of high mass accuracy is particularly dramatic for low-abundance, phosphopeptide identifications.20
In addition to being used as a post-search data filter, high-mass-accuracy precursor ion data enable the potential use of a narrow precursor mass tolerance during the database search, thereby shortening the computational time. But recent studies using Mascot21 and SEQUEST22 showed that use of tight precursor tolerances during the database search step resulted in fewer peptide identifications. Both studies suggested that a wide precursor tolerance for the database search, combined with a narrow mass accuracy filtering of search results, was more sensitive due to more effective filtering of false positive matches. Interestingly when we scanned proteomics studies in the Journal of Proteome Research that utilized high-mass-accuracy instruments, most database searches were performed using very narrow precursor tolerances.
Furthermore, most published database search methods only considered sequences with dual-tryptic termini (full tryptic). This typical usage of full tryptic boundaries for searches also is of interest because substantial numbers of partial tryptic peptides can be detected in MS2 scans, even though these peptides typically are present in much lower concentrations than their full-tryptic counterparts.23 The impact on conventional data analyses strategies of partial tryptic peptide spectra has not been fully addressed and is of particular interest for specimens where substantial biological proteolytic activity is likely to occur, such as in cell secretomes and serum. In this study, we analyzed samples ranging from purified proteins to complex samples, including human serum and tumor secretomes using 1D-SDS gel fractionation followed by LC–MS/MS analysis (GeLC–MS/MS). The proportion of MS/MS spectra that were due to partial tryptic sequences was substantial for all samples types and was particularly high for human serum. The effects of full and partial tryptic database searches for serum proteomes were evaluated, together with alternative post-search filtering strategies. These results indicated that the relatively large number of partial tryptic spectra in these datasets had a substantial negative impact on the accuracy of peptide identifications when full tryptic specificity was used for the database search. In addition, partial tryptic searches identified more peptides and proteins for a fixed peptide FDR. An optimized combination of precursor mass tolerance and subsequent mass accuracy filter was determined for partial tryptic database search results.
Materials and Methods
Materials
Human albumin (A8763) was purchased from Sigma-Aldrich (St. Louis, MO). Sequencing grade modified trypsin was from Promega Corporation (Madison, WI). Recombinant human Peroxiredoxin 6 (Prdx6) protein was purified in the laboratory. Amicon® Ultra centrifugal filters (10,000 Da cut off) were from Millipore (Billerica, MA). Ultra-pure urea and dithiothreitol (DTT) were from GE Healthcare, Ltd. (Giles, U.K.). All other reagents were from Sigma-Aldrich.
In-solution digestion of purified proteins
Five micrograms of lyophilized protein were solubilized in 10 μL of denature solution (100 mM ammonium bicarbonate, 10 M urea). Proteins were reduced at 37 °C for 30 min with 7 mM DTT. Free cysteine residues were alkylated with 20 mM iodoacetamide at 37 °C for 60 min. The reaction was stopped by quenching iodoacetamide with additional DTT at 37 °C for 15 min. Ammonium bicarbonate (25 mM) was added to the solution to dilute urea to 2 M. Proteolytic digestion was carried out overnight with a trypsin-to-protein ratio of 1:100. The reaction was stopped by adjusting the sample to pH 2–3 with formic acid. The sample was diluted to 20 fmole/μL with 0.1% formic acid in H2O and typically 1 μL was analyzed by LC-MS/MS.
Secretome sample preparation
Residual fresh human ovarian tumor tissue was used with informed consent under institutional review board approved protocols. Fresh tumor was cut into small pieces about 1 mm3 and the tissue was washed three times with 1 mL serum-free medium for 1 min each followed by incubation in 1 mL serum-free medium for 4 hr in 5% CO2, 95% air at 37 °C. After centrifugation, the supernatant was stored frozen at −80°C until needed. For proteome analysis, 500 μL aliquots were thawed, filtered through a 0.1 μm membrane, and concentrated to 25 μL by ultra-filtration. A 20 μL aliquot of the concentrated sample was separated for 2 cm by SDS-PAGE. The gel lane was sliced into 20 × 1-mm slices. Each gel slice was reduced using 20 mM Tris(2-carboxyethyl)phosphine (TCEP) for 30 min at 37 °C and alkylated using 40 mM iodoacetamide for 60 min at 37 °C. After lyophilization, gel pieces were digested overnight with 0.4 μg trypsin at 37 °C.
Serum sample preparation
Human serum was collected from a healthy donor with informed consent under an institutional review board approved protocol. Major proteins were depleted using a ProteoPrep® 20 Plasma Immunodepletion LC Column (Sigma-Aldrich). The depleted serum was separated 4 cm by SDS-PAGE, gel lanes were cut into uniform 1-mm pieces and were alkyated and digested with trypsin, as described above.
In-gel digestion of individual proteins
One μg aliquots of the proteins of interest were separated by SDS-PAGE by running the tracking dye to within 0.5 cm of the gel bottom, followed by staining with colloidal Coomassie. The majority of the stained band of interest was excised (4 mm × 1mm × 1mm), alkylated and digested by trypsin, as described above. The digestion was diluted to 20 fmole/μL with 0.1% formic acid in H2O, and 1 μL was typically analyzed by LC–MS/MS.
NanoLC–MS/MS
A nanoACQUITY HPLC (Waters, Milford, MA), interfaced with an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA), was used. Trypsin digestions were separated using a 75 μm i.d. × 25 cm PicoFrit (New Objective, Inc., Woburn, MA) column packed with 3 μm MAGIC C18-AQ resin. Peptides were eluted using a gradient formed by solvent A (0.1 % formic acid in H2O) and solvent B (0.1 % formic acid in acetonitrile) as follows: 3–28% B over 42 min, 28–50% B over 25.5 min, 50–80% B over 5 min, and constant 80% B over 5 min. A 25-min blank gradient was run between sample injections to minimize carryover. Full scans were performed at 60,000 R in the Orbitrap with simultaneous data-dependant MS2 in the LTQ on the six most intense ions. Monoisotopic peak selection (MIPS) was enabled, singly charged ions were rejected for MS2 and the Lock Mass function was not used. Dynamic exclusion was enabled and analyzed precursors were excluded for 45 sec. The LTQ Orbitrap XL mass spectrometer control software was version 2.4 SP1.
Data Processing
MS2 data were extracted and searched using the SEQUEST algorithm (Ver. 28, rev. 13, University of Washington, Seattle, WA) in BioWorks (Ver. 3.3.1, Thermo Fisher Scientific). The FASTA database (human UniRef 100, Ver. May, 2009) was downloaded from Protein Information Resource (PIR), Georgetown University, Washington, D.C. A decoy database was generated by reversing the amino acid sequence of each protein in UniRef 100, and it was then appended to the forward database. The combined database was indexed using either full or partial tryptic specificity with up to two missed cleavages. SEQUEST search parameters were set to the same trypsin specificity (full or partial tryptic, up to two missed cleavages) used to index the database. Other search parameters included either a 1.1 Da or 100 ppm precursor mass tolerance, a 1 Da fragment ion mass tolerance, variable methionine oxidization (+15.9949), and static cysteine carboxamidomethylation of cysteine (+57.0215 Da). Search results were stored in SRF files and transferred into SQT files by mspire (mass spectrometry proteomics in Ruby). 24 Consensus protein lists were built by DTASelect (Ver 2.0, licensed from Scripps Research Institute, La Jolla, CA) after applying filters within DTASelect for mass accuracy, Xcorr score, ΔCN score, minimum peptide per protein, and tryptic status (y =1 for partial tryptic or 2 for full tryptic peptides). For each data filter, the FDR was estimated from the ratio of decoy database peptide or protein counts divided by forward database peptide or protein counts, expressed as a percentage. Redundant or non-redundant peptide counts described in results were taken directly from DTASelect, which counted different charge states and variable modifications such as methionine oxidation as separate peptides. Unique peptide counts were obtained using an in-house script that collapsed different charge states and variable modifications of a unique sequence into a single count. To determine actual mass accuracy of each dataset, forward PSM were used that passed the following stringent filter: Xcorr [+1] ≥ 1.9, Xcorr [+2] ≥ 2.5, or Xcorr [+3] ≥ 4, and ΔCN ≥ 0.12, and at least three peptides per protein. Proteins and PSMs that were common to both the 1.1 Da search and 100 ppm searches and unique peptide sequences were determined using in-house Ruby scripts.
Result and Discussion
Partial tryptic peptides are detected with high frequency in trypsin-digested samples
Trypsin is the most widely used protease in proteomics studies due to its high specificity, robustness, and optimal average size of tryptic peptides. Because the enzyme is considered to be highly specific, database searches typically are conducted using full tryptic boundaries. The most common rule for full tryptic peptides is “cleavage after arginine or lysine, but not before proline.” However, a recent bioinformatics study observed substantial trypsin cleavages before proline and suggested that the proline restriction should be removed.25 This is consistent with our experimental observations and, in this study, the proline restriction was not used for either the full or partial tryptic searches. When using full tryptic boundaries, any spectra that result from partial tryptic peptides will yield incorrect identifications. To determine the extent of partial tryptic peptides in representative specimens, we performed in-gel and in-solution trypsin digestion of purified human albumin and Prdx6. Digestion products from 20 fmole of protein digests were analyzed by LC–MS/MS, and raw data were searched with a partial tryptic setting and a mass tolerance of 1.1 Da. PSMs were filtered using mass accuracy ≤ 10 ppm and ΔCN ≥0.05 in DTASelect. Under this default filter, the redundant peptide FDR was approximately 1% for albumin and 2–3% for Prdx6. Corresponding full tryptic and partial tryptic peptides for each condition are listed in Supplemental Table 1. Peptides with different charge states and modifications were consolidated into unique, unmodified peptides. Substantial numbers of partial tryptic peptides were identified in all four samples (Figure 1A). The in-solution digested samples contained more incompletely cleaved peptides (internal Arg and/or Lys residuals) than the in-gel samples, which resulted in more observed unique peptides in the in-solution digestion and indicated that the in-solution digestion was somewhat less efficient than the in-gel method, presumably due to the much lower enzyme-to-substrate ratio commonly used for in-solution digests. In contrast, the portion of unique observed peptides that had partial tryptic boundaries was lower for the in-gel digests. This is probably because the SDS gel separates the intact protein from fragments caused by prior non-tryptic proteolysis events.
Figure 1. Partial tryptic peptides are observed at substantial levels for diverse types of samples.

(A) Unique full and partial tryptic peptides for individual purified proteins digested with trypsin either in-solution or in-gel. Peptides that passed a ΔCN ≥=0.05 and mass error ≤ 10 ppm filter were consolidated into a minimum list of unique peptides by collapsing different charge states and variable modifications (Met oxidation) into single entries. (B) Unique full and partial tryptic peptides identified in human serum and an ovarian tumor secretome using the same data filter as in panel A are shown. (C) Percentage of all observed unique peptides that have partial tryptic boundaries.
We performed similar GeLC-MS/MS analyses on human serum (40 fractions) and human tumor secretomes (20 fractions) as representatives of complex biological samples. Unique peptides that passed the mass accuracy ≤ 10 ppm and ΔCN ≥0.05 filter were sorted into full or partial tryptic peptides (Figure 1B). A total of 8,943 unique peptides with a redundant peptide FDR of 0.3% were identified in the serum sample, and 27,855 unique peptides with a redundant peptide FDR of 0.2% were identified in the tumor secretome. This large difference in identified peptides was due primarily to the well-known, much greater dynamic range of protein concentrations in serum compared with tissue secretomes. The percentages of all detected unique peptides that were due to partial trypsin cleavage peptides are shown in Figure 1C for albumin, serum, and the tumor secretome. Interestingly, more than 50% of all identified peptides in the serum sample had partial tryptic boundaries.
To evaluate the cleavage mechanisms responsible for the observed partial tryptic peptides, we inspected all 16 partial tryptic peptides from Prdx6 (Table 1). For all partial tryptic peptides, the corresponding full tryptic peptides also were detected in the sample. Fifteen partial tryptic peptides were due to N-terminal truncation. Four of these partial tryptic peptides were detected at the same retention time as the corresponding full tryptic peptides in both the in-gel digestion sample and the in-solution digestion sample, indicating these observed partial tryptic peptides were most likely due to in-source fragmentation in the mass spectrometer. This in-source decay could be reduced by decreasing the tube-lens voltage on the ion source, but some peptides still maintained fragility even with low tube-lens voltage, and lower tube-lens voltages typically reduced the total number of identified full tryptic peptides. Five partial tryptic peptides eluted at different retention times than the corresponding full tryptic peptides and were observed in both in-solution and in-gel digests. Cleavage in these cases occurred on the C-terminal side of W, F, L, and M, indicating cleavage due to a chymotryptic activity. Chymotrypsin is a common contaminant in trypsin preparations from pancreatic extracts. In addition, trypsin undergoes autolysis, and the autolysis product, pseudotrypsin, has a chymotrypsin-like activity.26 Although methylation of trypsin helps to reduce autolysis, pseudotrypsin activity may not be eliminated. 27, 28 The last group of partial tryptic peptides were observed only in the in-solution digest and extracted ion chromatograms of these precursor ions did not show any peaks at the expected retention times in the in-gel digests. This indicates that these partial tryptic sequences were derived from proteomic fragments present in the original protein sample, which were separated from the intact protein on the SDS gel. The presence of these large non-tryptic fragments, presumably from partial proteolysis during protein purification, appears to be the major reason that the in-gel digestions generated slightly less partial tryptic peptides for the individual proteins (Figure 1A, 1C). Of course, when the entire gel lane is analyzed, as in the case of the serum and secretome samples, partial tryptic fragments from pre-analytical proteolysis will not be removed.
Table 1.
List of 16 partial tryptic peptides and their full tryptic counterparts from purified recombinant human Prdx6
| Full/partial tryptic peptidesa | Cleavage mechanismb | RT match | Only found in in-solution digestion |
|---|---|---|---|
| VVFVFGPDKK | in-source | yes | |
| LPFPIIDDR | in-source | yes | |
| DFTPVCTTELGR | in-source | yes | |
| LIALSIDSVEDHLAWSK | in-source | yes | |
| LIALSIDSVEDHLAWSK | chymotrypsin | ||
| DFTPVCTTELGR | chymotrypsin | ||
| LSILYPATTGR | chymotrypsin | ||
| DGDSVMVLPTIPEEEAK | chymotrypsin | ||
| VATPVDWKDGDSVMVLPTIPEEEAK | chymotrypsin | ||
| PGGLLLGDVAPNFEANTTVGR | sample degradation | yes | |
| DINAYNCEEPTEKLPFPIIDDR | sample degradation | yes | |
| DINAYNCEEPTEKLPFPIIDDR | sample degradation | yes | |
| DINAYNCEEPTEKLPFPIIDDR | sample degradation | yes | |
| DINAYNCEEPTEKLPFPIIDDR | sample degradation | yes | |
| VATPVDWKDGDSVMVLPTIPEEEAK | sample degradation | yes | |
| VATPVDWKDGDSVMVLPTIPEEEAK | sample degradation | yes |
The expected full tryptic sequence is shown with the observed partial tryptic sequence in bold and underlined
Likely cleavage mechanisms responsible for the observed partial tryptic peptides are based on retention time (RT) match to the full tryptic peptide, cleavage site, and sample specificity.
The above analyses show that in-source decay, chymotryptic activity during trypsin digestion, and pre-existing proteolytic fragments in the original sample contribute to the total group of observed partial tryptic sequences. As it would be very difficult to eliminate the contribution from any of these mechanisms for most proteomics studies, the effects of these sequences should be addressed during data analysis. The extent of detected partial tryptic peptides in LC–MS/MS experiments will be dependent on multiple factors such as depth of analysis, sample characteristics and whether entire proteomes or single gel bands are analyzed. It is not surprising that in complex samples such as serum where substantial proteolysis can occur both in vivo and during sample processing, the number of observed partial tryptic peptides can exceed the number of complete tryptic peptides. Another reason why we detected a larger proportion of peptides as partial tryptic peptides in serum compared with the tumor secretome is the larger dynamic range of serum and the fact that more fractions were analyzed. In serum, low-yield, partial tryptic peptides from high- and medium-abundance proteins often be more abundant than high-yield, complete tryptic peptides from lower-abundance proteins. In contrast, the tumor secretome had far fewer partial tryptic peptides detected because: 1) it contained a large number of proteins present at similar levels and the capacity of the mass spectrometer was primarily devoted to acquiring data from the higher-abundant full tryptic peptides for these proteins; 2) only half as many fractions were analyzed compared to serum due to this reduced dynamic range, and 3) there is probably less physiological proteolysis during a four-hour acquisition of the tumor secretome compared with in vivo proteolysis in the blood and proteolysis during blood clotting. As the mechanisms of partial tryptic peptide production suggest, we routinely observe that the abundance levels of partial tryptic peptides are one to three orders-of-magnitude less abundant than the corresponding full tryptic peptides. These observations are consistent with those of a previous report.23 Considering the big differences in the relative abundances of partial tryptic peptides and corresponding full tryptic peptides, it is not surprising that experiments with limited depth of analysis, such as a single LC–MS analysis on a bacterial proteome, may only detect full tryptic peptides.29
Spectra from partial tryptic peptides result in random mismatches with good scores when using full tryptic searches
Since most datasets from in-depth analyses will have a proportion of spectra from partial tryptic sequences, the common practice of using full trypsin specificity will result in misidentifications for these spectra. In large datasets, some of these mismatches are likely to yield good apparent matches by random chance. To further explore this possibility, we searched the serum dataset described above with SEQUEST using either full or partial tryptic settings with all other search conditions held constant. The results from the partial tryptic search were subsequently filtered using DTASelect with mass accuracy ≤ 10 ppm, ΔCN ≥0.05, and full tryptic peptide boundaries (our default filter). The full tryptic search result was filtered with three different filters: 1) a filter equivalent to our default partial tryptic search filter (mass accuracy ≤ 10 ppm, ΔCN ≥0.05); 2) a SEQUEST parameters filter commonly used in prior publications 30, 31 (Xcorr [+1] ≥ 1.8, Xcorr [+2] ≥ 2.5, Xcorr [+3] ≥ 3.5, ΔCN ≥ 0.08); and 3) a more stringent filter including mass accuracy (mass accuracy ≤ 10 ppm, Xcorr (+1) ≥ 2, Xcorr (+2) ≥ 2.8, Xcorr (+3) ≥ 3.7, ΔCN ≥ 0.12). Figure 2A shows the redundant peptide counts and FDR for the four conditions. The partial tryptic search combined with our default partial tryptic filter produced the second-highest number of peptide IDs with the lowest FDR (0.3%). A full tryptic search with the same post-search filter generated a slightly higher number of peptide IDs but with a much higher FDR (4.1%). The stringent filter achieved a similar peptide FDR to our default filter but with ~17,000 less peptide identifications. Similar trends are observed when non-redundant peptide counts and FDRs are considered (Figure 2B). That is, when a highly stringent filter is applied to the full tryptic search results, a FDR similar to the partial tryptic dataset is obtained but with 1,682 less non-redundant peptides identified. Furthermore, the FDR at the non-redundant peptide level is unacceptably high for the full tryptic searches with the less stringent filters. Comparisons of the redundant and non-redundant peptide data (Figure 2A, 2B) show that identified peptides were repetitively sampled an average of about 10 times, which means that the most abundant peptides were sampled far more than 10 times. This is a problem that is more severe when working with serum or plasma than in cell lysates32 due to the very wide dynamic range that persists even after depletion of the 20 most abundant proteins and subsequent separation into 40 fractions. Highly repetitive sampling of the most abundant peptides results in dramatically lower FDRs estimated for redundant peptide matches because the many repetitive matches from abundant peptides are usually accurately identified, while single copy, low-intensity PSMs are less likely to be correctly identified due to a lower signal to noise ratio.
Figure 2. Comparisons of database search and data filtering strategies.

A 40-fraction serum proteome dataset was searched using SEQUEST with either full or partial tryptic boundaries. The resulting identifications were filtered using three alternative conditions (see text). (A) Redundant peptide counts and peptide FDRs. (B) Non-redundant peptide counts and FDRs. (C) Target database and decoy hits for single peptide proteins only. (D) Target database and decoy hits for proteins identified by more than one peptide. These data show that partial tryptic database searches combined with subsequent full tryptic filtering provide both good depth of analysis and high-confidence identifications.
In published reports using decoy databases to estimate FDR, it often is unclear whether FDR was calculated using redundant or non-redundant peptide counts. But this is a critical parameter that should be reported, especially for serum and plasma, because using redundant PSM counts to calculate peptide FDR is likely to result in over-confidence in the quality of results, as indicated by the 3.9% (redundant) vs. 24.1% (non-redundant) FDRs for the full tryptic search/commonly used filter or the 0.3% (redundant) vs. 1.8% (non-redundant) FDR for the partial tryptic search/default filter (Figure 2A, 2B). Using non-redundant PSM counts is more appropriate, particularly for serum or plasma proteomes, as it largely removes the bias caused by repetitive sampling of abundant peptides. This bias results in an approximately six-fold-higher FDR with the non-redundant peptide calculation method compared to the redundant calculation method for all data analysis strategies summarized in Figure 2.
As the data in Figure 2 illustrate, it is not appropriate to use the same filter on full and partial tryptic search data, in part because Xcorr and ΔCN values are affected by the larger search space in the partial tryptic search. There are certainly many database search programs and post-search filtering strategies that could be used. In this study, we used data filtering strategies within DTASelect that achieved similar FDRs to compensate for the differing search spaces of full and partial tryptic database searches. FDRs generated from the target-decoy search were still valid even with the enlarged search space of partial tryptic searches because decoy and target sequences were equal regardless of changes in search space. Thus, FDR-orientated filtering enabled us to perform a fair comparison between two conditions: the “default” filter with the partial tryptic search and the “stringent” filter with the full tryptic search. In addition to the two filters, we also used the “default” filter on the full tryptic search as a control for the filtering step. The “common” filter was used as an example of a commonly used full tryptic search/filtering strategy.
Interestingly, at the protein level, the two less stringent filters each identify more than 2,000 proteins (sum of values in Figures 2C and 2D), but most of these proteins are single hit proteins and nearly all of these single hit proteins are expected to be incorrect identifications (Figure 2C). The two stringent filters have far less single hit proteins and slightly less than half of these single hit proteins are likely to be incorrect identifications. Furthermore, less than 1% of the protein identifications based on two or more peptides are incorrect for the two stringent filters, while 27% and 24% of the proteins are incorrect for the less stringent filters (Figure 2D). Hence, while the two less stringent conditions produced the greatest numbers of target protein identifications, this advantage were negated by the impractically large number of apparent false forward database protein identifications.
Most importantly, the partial tryptic search/default filter analysis identified 438 proteins based on two or more peptides, compared with only 368 proteins for the full tryptic search/stringent filter analysis (Figure 2D), resulting in the best compromise between highly accurate protein identifications and comprehensiveness of proteome coverage. Database searches using partial trypsin specificity ensure that most high-quality spectra arising from partial tryptic sequences will be matched to the correct sequence. At the same time, subsequent elimination of partial sequences from the final dataset by restricting the final results to full tryptic boundaries is not detrimental to proteome coverage because most partial tryptic sequences are present at much lower yield than the corresponding full tryptic sequence. Therefore, most identified partial tryptic sequences will be from abundant proteins that are unambiguously identified by multiple full tryptic peptides. Filtering out the partial tryptic peptides has a negligible cost on protein coverage while greatly reducing FDR. A summary of the serum proteins identified by two or more peptides in the partial tryptic search using the default filter is shown in Supplemental Table 2A.
Evaluation of spectra that match partial tryptic sequences when they are forced to match full tryptic sequences
We extracted the 23,824 spectra that matched to partial tryptic peptides in the partial tryptic search result with mass accuracy ≤ 10 ppm and ΔCN ≥0.05, which is the same as our default filter with the exception of the normal requirement for full tryptic peptide boundaries. When the fate of these spectra in the full tryptic search was evaluated, the majority of spectra did not match any full tryptic sequences with high scores, as expected. However, 1,404 spectra matched full tryptic sequences with ≤10 ppm mass error and ΔCN ≥0.05, with 694 matched to decoy sequences and 710 matched to target full tryptic sequences. These 710 false matches typically match partial tryptic peptides from abundant serum proteins with higher scores (Table 2). Minimizing false positive hits in full tryptic database searches would require the elimination of most of these 1,404 spectra, as the partial tryptic search shows that these full tryptic identifications are incorrect. When a very stringent filter (mass accuracy ≤ 10 ppm, Xcorr [+1] ≥ 2, Xcorr [+2] ≥ 2.8, Xcorr [+3] ≥ 3.7, ΔCN ≥ 0.12) was used, only 13 PSMs passed the filter, nine hit decoy sequences, and four hit target sequences. But this very stringent filter severely suppressed depth of analysis compared to the partial tryptic search/default filter, as shown in Figures 2C and 2D. For an independent approach, the full tryptic data were processed using the Percolator algorithm, which has shown great power in distinguishing between correct and random matches based on multiple PSM properties. A redundant peptide FDR of 0.28% was selected to match the FDR obtained for the two more stringent filters shown in Figure 2A. The Percolator algorithm identified 66,093 redundant and 6,301 non-redundant peptides, which is much better than the 50,375 redundant and 4,975 non-redundant peptides obtained for the full tryptic search/stringent filter, but somewhat less than the 67,465 redundant and 6,657 non-redundant peptides obtained for the partial tryptic search/default filter results (Figure 2).
Table 2.
Representative false positive peptide identifications in a full tryptic search and the corresponding partial tryptic peptides from abundant serum proteins
| Full Tryptic Search | Partial Tryptic Search | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spectrum | Xcorr | dCN | Δ Mass | Peptide Sequence | Protein ID | Protein Name | Xcorr | dCn | ΔMass | Peptide Sequence | Protein ID | Protein Name |
| 1.1799 | 2.07 | 0.30 | −7.9 | RALGDPATPTEGPRR | UPI0000D61BBA | UPI0000D61BBA UniRef100 entry | 3.13 | 0.34 | −0.8 | QVDAVPANGQTPIQR | Q60FE4 | Fibronectin 1 |
| 2.3203 | 2.07 | 0.07 | −1 | MTILQTYFR | O75348 | Vacuolar ATP synthase subunit G 1 | 2.72 | 0.14 | −2.5 | TMEQLTPELK | UPI0000D820D0 | apolipoprotein B precursor |
| 4.5842 | 1.87 | 0.06 | −7.8 | MGPLSPARTLRLWGPR | Q96RP7 | Galactose-3-O-sulfotransferase 4 | 4.31 | 0.46 | 1 | GLTPGVEYVYTIQVLR | Q60FE4 | Fibronectin 1 |
| 6.4234 | 1.69 | 0.12 | −3.4 | GTLAAQAPHLCPR | Q59H20 | Hypothetical protein | 3.45 | 0.49 | 0 | AVTADGNAFIGDIK | UPI000014D030 | inter-alpha (globulin) inhibitor H1 |
| 8.7814 | 1.75 | 0.10 | 4.2 | QQLGWEAWLQYSFPLQLEPSAQTWGPGTLR | O00334 | Phosphatidylinositol 3-kinase delta catalytic subunit | 2.28 | 0.07 | 3.1 | SVVPVFYVFHYLETGNHWNIFHSDPLIEK | Q27I61 | Complement component 5 |
| 10.2892 | 2.43 | 0.11 | −6.2 | FSNDNIKHSQNMR | Q96M29 | Tektin-5 | 4.01 | 0.39 | 0.8 | SLCSDQQSHLEFR | UPI000014279E | protein S (alpha) |
| 12.4906 | 1.53 | 0.14 | −3.3 | VDGTRGRGGPAWR | UPI0000DD7E44 | PREDICTED: hypothetical protein | 3.13 | 0.47 | 2.8 | TLTGGNVFEYGVK | Q2TAZ5 | Complement factor H |
| 18.2465 | 2.05 | 0.15 | 0.5 | RDTFNHLTTWLEDAR | P61019 | Ras-related protein Rab-2A | 4.59 | 0.45 | 2.9 | SIPVCGQDQVTVAM*TPR | Q4QZ40 | Prothrombin |
| 19.2373 | 2.56 | 0.07 | −2.8 | SGNRDRTVQSPQSK | Q15695 | U2 small nuclear ribonucleoprotein auxiliary factor 35 kDa subunit | 3.63 | 0.21 | −0.4 | RPQGSCSLEGVEIK | P00751 | Complement factor B precursor |
| 23.4572 | 3.15 | 0.15 | 5.9 | QDMIIDKLNGNSTPEK | Q9NY46 | Sodium channel protein type 3 subunit alpha | 5.77 | 0.45 | −2.6 | PNFNGNTLDNDIMLIK | Promega sequencing grade modified trypsin | |
| 27.1682 | 2.40 | 0.10 | 5.6 | IDEKSLDESKNPAR | Q7RTM1 | Otopetrin-1 | 4.04 | 0.41 | −1.4 | ALSSVQESQVAQQAR | P02656 | Apolipoprotein C-III precursor |
| 27.3208 | 2.55 | 0.14 | 5.5 | EKDEQIRGLMEEGEK | Q59GK0 | TATA element modulatory factor 1 variant | 3.43 | 0.22 | −0.7 | LIHGPNLYCYSDVEK | P02790 | Hemopexin precursor |
| 28.4748 | 2.20 | 0.09 | −2.4 | PGTMMPEAFLQEAQIMKK | UPI0000456B50 | Proto-oncogene tyrosine- protein kinase Yes | 5.03 | 0.46 | −1.9 | LGEHNIDVLEGNEQFINAA | Promega sequencing grade modified trypsin | |
| 29.5703 | 2.36 | 0.09 | −2.8 | KPTEWPLKLVPQHR | Q9P1K3 | PRO1163 | 5.14 | 0.52 | −2 | VLNLPSGVTVLEFNVK | Q27I61 | Complement component 5 |
| 33.3622 | 2.40 | 0.08 | 3.9 | LNESDEQHQENEGTNQLVMGIQK | Q15545 | Transcription initiation factor TFIID subunit 7 | 5.64 | 0.39 | −1.4 | EPTM*YGEILSPNYPQAYPSEVEK | P09871 | Complement C1s subcomponent precursor |
| 39.4095 | 2.40 | 0.07 | 3.6 | GFTVKMHCYMNSASGNVSWLWK | Q53FS2 | CD79B antigen isoform 1 variant | 4.98 | 0.32 | 0.2 | EVSADQVATVM*WDYFSQLSNNAK | P06727 | Apolipoprotein A-IV precursor |
| 36.2075 | 2.86 | 0.07 | 4.1 | SIENDSDEVEERAENFPR | Q8N1H7 | Protein SIX6OS1 | 5.35 | 0.36 | −1.1 | HPNSPLDEENLTQENQDR | P01011 | Alpha-1-antichymotrypsin precursor |
The vast majority of the 710 spectra that incorrectly match target sequences in the forward full tryptic search show better matches to partial tryptic sequences than to the full tryptic sequences based upon Xcorr, ΔCN, and mass accuracy (Figure 3A). Furthermore, the distributions of Xcorr (+2), Xcorr (+3), and ΔCN scores are shifted toward higher values for partial tryptic matches compared with full tryptic matches (Figures 3B–D). These score distributions are consistent with the thresholds we used for the stringent filter that was applied to full tryptic search results; that is, Xcorr (+2) ≥ 2.8 and Xcorr (+3) ≥ 3.7 will exclude most of these incorrect, high-scoring matches to full tryptic sequences.
Figure 3. Comparisons of scores for partial and full tryptic searches.

Scores for the 710 spectra in a human serum data set that generated high-quality peptide matches in both the full or partial tryptic searches were compared. (A) The highest-scoring search method is indicated for each of the individual parameters analyzed (Xcorr, ΔCN, and mass accuracy). (B) Xcorr scores for doubly charged spectra. (C) Xcorr scores for triply charged spectra. (D) Distribution of ΔCN scores.
The similar good fit of some spectra to both full tryptic and partial tryptic peptides is further illustrated by annotation of one of these 710 spectra using the results from the full tryptic and partial tryptic searches (Figure 4). Although both sequence assignments fit the data reasonably well with most major peaks assigned, the partial tryptic sequence is clearly a better fit, consistent with the higher Xcorr and ΔCN scores. Taken together, these data show that the most efficient strategy for preventing misidentifications of these 710 spectra is to use a partial tryptic rather than full tryptic specificity during the database search.
Figure 4. Comparison of peptide matches from a full tryptic search (A) and a partial tryptic search (B) to a representative MS2 spectrum.

One argument against relaxing the tryptic termini restriction is that partial tryptic sequences dramatically increase the search space. The indexed database for the full tryptic search had 5,145,680 unique sequences, while the partial tryptic search database had 69,330,268 unique sequences. This 14-fold increase of search space was expected to shift the global distribution of PSM’s Xcorr scores to higher values, as previously reported33. Thus more random matches should pass the filter if the same thresholds would be used for both full and partial tryptic searches. However, such effects can be controlled by adjusting post-search data filters to achieve similar FDR as previously discussed. For example, in our serum partial tryptic search data, the non-redundant peptide FDR for spectra that matched full tryptic peptides was only 1.8% (Figure 2B), while the peptide FDR for spectra in the full tryptic search using the same filter was 22.6%, indicating a partial tryptic search coupled with filtering for full tryptic boundaries dramatically decreased random matches in the partial tryptic search data. Within the results from the partial tryptic search, the FDR was higher for the partial tryptic peptide matches than for the full tryptic peptide matches. One solution to this problem is to have a more stringent threshold for the partial matches separately as used in IDPicker.8 Another solution is to integrate tryptic status into a single “final score,” such as in Percolator9 and Peptide Prophet.7 But under these programs, the tryptic status of PSM is not readily apparent in filtered results and the relative weight of tryptic status may vary depends on the algorithm and data. A simple alternative strategy is to exclude all partial tryptic peptides from the final dataset. The validity of this approach is supported by the fact that the longer, full tryptic peptides were almost invariably detected for observed partial tryptic peptides (Table 1 and data not shown). Importantly, this indicates that including partial tryptic peptide matches in the final data will minimally contribute to detection of more proteins or higher sequence coverage.
A related potential concern is that random partial tryptic sequence matches could replace some true positive full tryptic matches due to the far larger number of partial tryptic candidates compared with full tryptic candidates. To evaluate this possibility, we took all spectra corresponding to the 50,375 sequences from the full tryptic search/stringent filtering result (Figure 2A) and mapped them back to the partial tryptic search result. Greater than 99% of these spectra had the same full tryptic sequence hit in the partial tryptic search result, indicating that the approximately 14-fold increased search space of a partial tryptic search had only a very minor negative effect on matching spectra from full tryptic peptides to the correct full tryptic sequence. This minor effect is greatly outweighed by the other factors discussed above.
Optimization of precursor mass tolerance in partial tryptic searches
Another potential drawback of partial tryptic searches is the greatly increased computational time required for the search due to the increased search space. To test the effects of search time, a human ovarian tumor secretome consisting of 20 gel slices from a GeLC-MS/MS experiment was used. An approximate five-fold increase in search time was required for a partial tryptic search compared with a full tryptic search when the same sequence database, computer and SEQUEST version were used with a 1.1 Da precursor mass tolerance (Figure 5A). But the total search time of the partial tryptic search could be dramatically reduced by reducing the mass tolerance to 100 ppm. Surprisingly, there were only minor further improvements on search time as the mass tolerance was further tightened. Of course, search time will be influenced by the specific search algorithm, codes that execute the algorithm, hardware factors, and specific databases and datasets that are used. While performance of other search algorithms may vary from that observed here for SEQUEST, it is expected that partial tryptic searches and wider precursor mass tolerances will usually increase computational time. Nonetheless, the improved depth of analysis of the partial tryptic search using intermediate mass tolerances such as 100 ppm, followed by filtering on full tryptic peptides and tighter mass tolerance, should be worth the increased computational time for datasets where maximizing depth of analysis is important. To further assess the effects of using narrow precursor tolerances, we compared the search result from the 1.1 Da precursor mass tolerance with that of the 100 ppm precursor tolerance at both the PSM and protein levels using a partial tryptic specificity and our default data filter (Figure 5B). Filtered non-redundant spectra, 27,705 from a 1.1 Da search and 28,939 from a 100 ppm search, were compared to each other. A total of 27,091 spectra were common to both data sets and 27,085 were matched to the same peptide sequence, while six spectra matched different peptides in the two searches. There also was extensive overlap at the protein level for the two data sets with greater than 97% of the protein identifications common to both datasets. Most of the minor observed variation at the protein level appeared to be due to database redundancy as has been previously observed.32 These data suggest that, for SEQUEST, switching to a moderately narrow precursor tolerance of 100 ppm can reduce partial tryptic search times to near minimal levels without significantly changing search results. A summary of the proteins identified in the tumor secretome using the 100 ppm partial tryptic search and the default filter is shown in Supplemental Table 2B.
Figure 5. Comparisons of precursor mass tolerance effects on search time and protein identifications.

An ovarian tumor secretome dataset was searched using SEQUEST as described in “Materials and Methods.” (A) Search time using different precursor mass tolerances. (B) Overlap of PSM and protein identifications between the 1.1 Da and 100 ppm precursor mass tolerance searches. Numbers of PSMs and proteins unique to each search condition are listed below the precursor mass tolerance. Filtered non-redundant peptide identifications were used.
An optimal combination of precursor mass tolerance and post-search mass accuracy filter yields high-quality protein identifications with reduced search times
Mass accuracy has been shown to be an effective filter for rejecting random matches and thus enhancing detection sensitivity for low-abundance peptides.18–20 Of course the appropriate final mass tolerance used to filter the data should be matched to a statistically valid actual mass accuracy for the instrument used to acquire the data.34 The data used in this study were generated on an LTQ-Orbitrap with monoisotopic peak selection enabled, where the final precursor mass value for creation of the DTA file is extracted from the full scan at 60,000 R by Bioworks. To estimate the actual mass accuracy of a given instrument, a pool of representative true peptides with good diversity should be used. For this purpose, we filtered partial tryptic search data with a special filter (Xcorr [+1] ≥ 1.9, Xcorr [+2] ≥ 2.5, Xcorr [+3] ≥ 4, ΔCN ≥ 0.12) and required that at least three peptides match each protein, which resulted in a non-redundant peptide FDR of 0.1% without any mass filter. The mass accuracy histogram of these PSMs exhibited an approximately normal distribution centered near zero (mean = 0.18 ppm, σ=2.5) (Figure 6A). This mass accuracy was similar to that recently reported using Lock Mass on the same type of instrument.35 These data justified the use of ±10 ppm as the mass accuracy filter as this is approximately ±4σ, which ensures that >99.99% of true hits will not be rejected on the basis of mass error, whereas a tighter filter, such as ±5 ppm, would result in the loss of about 5% of correct matches, which is substantial when dealing with large datasets. Furthermore, the peptide data used for calculating these mass error statistics is expected to preferentially represent more abundant peptides with good signal-to-noise due to the stringent criteria used to select these PSMs. But it is likely that the low-abundance peptides will exhibit somewhat larger variations in mass error.34
Figure 6. Determining optimal precursor mass tolerance and post-search mass accuracy filter.

An ovarian tumor secretome dataset was used to test different precursor mass tolerances and mass accuracy filter combinations. (A) Distribution of PSM mass accuracy for very high confident matches (solid line) with a superimposed normal distribution (dashed line). (B) Relationship between precursor mass tolerance and peptide FDR using a constant post-search filter. (C) Comparison of peptide FDR using different post-search filters with full tryptic/10 ppm, or partial tryptic/10 ppm, or partial tryptic/100 ppm searches.
The effects of using different mass tolerances with partial tryptic specificity were further evaluated by searching our ovarian tumor secretome with precursor tolerances ranging from 1.1 Da to 10 ppm. After applying the default filter, we found that tighter precursor tolerances led to higher peptide FDR (Figure 6B). These data show that using a precursor tolerance at 10 ppm results in a much higher FDR compared with 100 ppm while providing only a very minor saving in computational time (Figure 5A). Interestingly, the FDR determined using redundant peptides was only two to three times higher than for non-redundant peptide at all precursor mass tolerances in the ovarian tumor secretome dataset (Figure 6B). In contrast, for serum, the difference in FDR calculated using non-redundant peptides compared with redundant peptides was about six-fold higher for all database search and data filtering strategies (Figures 2A and 2B). This is apparently due to the lower redundancy of peptide identifications in the ovarian tumor secretome because it has a narrower range of protein abundances compared with serum.
To further evaluate effects of full and partial tryptic search parameters we searched our secretome data using three different conditions: a full tryptic search at 10 ppm, a partial tryptic search at 10 ppm, and a partial tryptic search at 100 ppm precursor tolerance. These results were subsequently processed using different filters and non-redundant peptide FDRs were calculated (Figure 6C). Of course for the full tryptic/10 ppm mass tolerance search, neither the mass accuracy nor the full tryptic post-search filters could reduce FDR because they were already incorporated into the search parameters. The observed nearly 50% FDR showed that this unfiltered data was not much better than random matches. Of the filters used here, only ΔCN moderately decreased the FDR. For the data generated by a 10 ppm/partial tryptic search, the mass filter alone could not be effective, and a FDR of 43.7% was observed. In contrast, the full tryptic filter alone dramatically decreased the FDR to 7.8% and it further improved with the addition of the ΔCN cutoff. For the result from the 100 ppm/partial tryptic search, using the mass accuracy filter at 10 ppm and the full tryptic filter in combination reduced the FDR below 2%, and when ΔCN was included the FDR was less than 1%. In contrast, conventional SEQUEST Xcorr filters were less effective, analogous to the observations described above for the serum proteome. For example, use of Xcorr [+1] ≥ 2, Xcorr [+2] ≥ 2.8, Xcorr [+3] ≥ 3.7, ΔCN ≥ 0.12) yielded a FDR of 5.05% on the full tryptic search at 10 ppm tolerance.
These analyses show database searches with 100 ppm mass tolerance and partial tryptic specificity are still superior to full tryptic searches with tight mass tolerance for samples such as the tumor secretome, which contain far smaller percentages of partial tryptic peptides compared with serum. A major advantage is that the search results subsequently can be filtered using a tighter mass tolerance and full tryptic boundaries, thereby greatly reducing the FDR with minimal loss of true positive identifications (Figure 2). Interestingly, filters based on mass accuracy, tryptic boundary status, and SEQUEST scores are relatively independent of each other and can be combined to achieve the very low non-redundant peptide FDRs demanded by large-scale, in-depth analysis of complex proteomes. Many newer algorithms for processing database search results allow automatic setting of desired FDR, but due to the large proportion of partial tryptic peptide spectra in most datasets, the use of partial tryptic searches is still advisable. In addition, one limitation of some programs is the lack of a convenient option for removing all partial tryptic peptides prior to setting the FDR since, as shown above, including the partial tryptic peptides in the final results has minimal value on proteome coverage.
Effects of including asparagine deamidation and other low frequency modifications in database searches
As shown above, it is advantageous to consider partial tryptic peptides in database searches primarily because a substantial proportion of the total spectra arise from such peptides. Similarly, methionine oxidation is routinely used in nearly all database search strategies because the majority of methionine containing peptides are typically detected in both the unoxidized and oxidized forms. To determine the impact of including lower frequency modifications in database searches, we conducted a series of parallel database searches of a serum proteome dataset where one variable modification at a time was considered in addition to methionine oxidation to methionine sulfoxide. The modifications considered included: asparagines deamidation, glutamine deamidation, tryptophan oxidation (+1 or +2 oxygen), methionine sulfone, pyroglutamate, carbamylation of lysine, and N-terminal carboxyaminomethylation. Among these modifications, asparagine deamidation was most commonly observed, with a frequency that was more than twice the next most common modification. This is not surprising as Asn deamidation has been reported to be among the most frequent chemical modifications of amino acids36, and occurs spontaneously when proteins or peptides are in aqueous solution.37 To evaluate the effects of considering Asn deamidation, we searched both the ovarian tumor secretome and human serum datasets with a 100 ppm precursor mass tolerance, both with and without variable Asn deamidation. The search time was nearly doubled with variable Asn deamidation enabled, but it was still much shorter than performing the search with a 1.1 Da precursor mass tolerance. Our default filters resulted in an increased number of non-redundant peptide identifications when enabling Asn deamidation in both samples, with a larger percent increase for serum (Figure 7). This higher deamidation level is presumably due to the long half-life of many abundant plasma proteins in the blood. However, considering deamidation did not increase the number of proteins identified and most deamidated peptides also were identified in the unmodified form. The non-redundant peptide FDR for the secretome data was 0.70% in the normal search result and 0.64% in the Asn deamidation search. In the serum sample, the Asn deamidation search generated a slightly higher FDR (2.4%) than that without the modification (1.8%). Overall, these results suggest that in contrast to considering the high frequency partial tryptic sequences, considering low frequency modifications such as Asn deamidation does not positively affect depth of analysis for large in-depth datasets.
Figure 7. Effects of considering Asn deamidation in database searches.
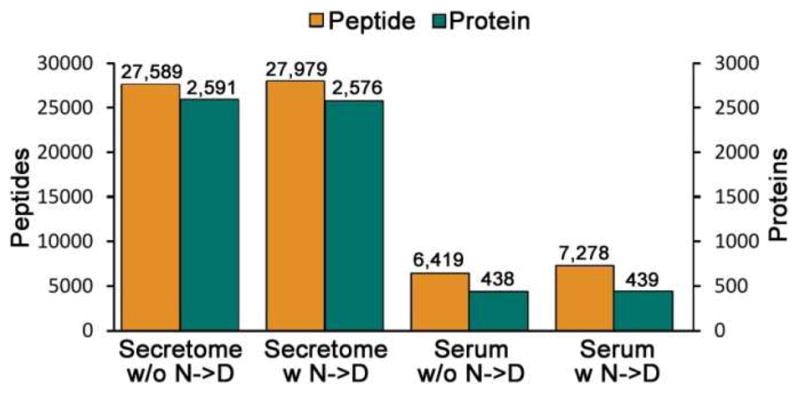
The ovarian tumor secretome and the human serum datasets were searched both with and without variable Asn deamidation. Non-redundant peptides and proteins with at least two peptide identifications are shown.
Conclusions
Use of extensive proteome fractionation with modern mass spectrometers can produce very large datasets of MS/MS spectra in which a substantial proportion are from partial tryptic peptides. These partial tryptic peptides arise from multiple sources including prior proteolysis in biological samples, non-tryptic cleavages during trypsin digestion and fragmentation in the mass spectrometer ion source. The proportion of spectra from partial tryptic peptides is particularly high for serum, and probably other biological fluids, apparently due to the very wide dynamic range of protein abundance coupled with substantial levels of intrinsic proteolytic activity. The common practice of using full tryptic specificity and narrow mass tolerances for database searches is detrimental to maximizing proteome coverage with a low FDR as some spectra from partial tryptic peptides will match full tryptic sequences by random chance. Use of very stringent filters to reduce the FDR of full tryptic search identifications will result in decreased depth of analysis because many low abundance true positive identifications will be simultaneously removed. In contrast, partial tryptic specificity and moderate mass tolerances such as 100 ppm for SEQUEST, allow the subsequent filtering of database search results using full tryptic boundaries and a tighter mass error filter that is matched to instrument performance. This strategy results in a superior depth of analysis with FDRs at the non-redundant peptide level that are typically less than 1%. Although use of partial tryptic database search specificity is highly advantageous, considering lower frequency events, such as asparagine deamidation in the database search, does not further improve depth of analysis.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants, CA120393 and CA131582 to D.W.S, as well as institutional grants to the Wistar Institute, including an NCI Cancer Core Grant, CA010815 and grants from the Pennsylvania Department of Health. HW was supported by NCI training grant CA009171. We thank the Wistar Proteomics Core Facility for their assistance, Mr. Peter Hembach for providing purified recombinant Prdx6 protein, Ms. Lynn Beer for human serum immunodepletion, and David J. Powell, Jr.. Ph.D., and George Coukos, M.D., Ph.D., for providing human ovarian tumor secretomes. We thank Ms. Mea Fuller for her assistance in preparing the figures and manuscript.
Footnotes
Supporting Information Available: Lists of full tryptic and partial tryptic peptides identified in purified protein digestions, human serum proteins identified using a partial tryptic search, and proteins identified in a ovarian tumor secretome using a partial tryptic search. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Eng JK, McCormack AL, Yates Iii JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5(11):976–989. doi: 10.1016/1044-0305(94)80016-2. %U http://www.sciencedirect.com/science/article/B6TH2-44FNFDS-4/2/5d823c970daabc608065d6cfdcaea001. [DOI] [PubMed]
- 2.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. %U http://www.ncbi.nlm.nih.gov/pubmed/10612281. [DOI] [PubMed]
- 3.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics (Oxford, England) 2004;20(9):1466–1467. doi: 10.1093/bioinformatics/bth092. %U http://www.ncbi.nlm.nih.gov/pubmed/14976030. [DOI] [PubMed]
- 4.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. Journal of Proteome Research. 2007;6(2):654–661. doi: 10.1021/pr0604054. %U http://www.ncbi.nlm.nih.gov/pubmed/17269722. [DOI] [PMC free article] [PubMed]
- 5.Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. Journal of Proteomics. 2010;73(11):2092–2123. doi: 10.1016/j.jprot.2010.08.009. %U http://www.ncbi.nlm.nih.gov/pubmed/20816881. [DOI] [PMC free article] [PubMed]
- 6.Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nature Biotechnology. 2001;19(3):242–247. doi: 10.1038/85686. %U http://www.sciencedirect.com/science/article/B6WVB-45DMMYK-1N/2/381a38f52aef0de9c49525b6344bb8c9. [DOI] [PubMed]
- 7.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical Chemistry. 2002;74(20):5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 8.Ma Z-Q, Dasari S, Chambers MC, Litton MD, Sobecki SM, Zimmerman LJ, Halvey PJ, Schilling B, Drake PM, Gibson BW, Tabb DL. IDPicker 2.0: Improved Protein Assembly with High Discrimination Peptide Identification Filtering. Journal of Proteome Research. 2009;8(8):3872–3881. doi: 10.1021/pr900360j. %U http://dx.doi.org/10.1021/pr900360j. [DOI] [PMC free article] [PubMed]
- 9.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nature Methods. 2007;4(11):923–925. doi: 10.1038/nmeth1113. %U http://www.ncbi.nlm.nih.gov/pubmed/17952086. [DOI] [PubMed]
- 10.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods. 2007;4(3):207–214. doi: 10.1038/nmeth1019. %U http://www.ncbi.nlm.nih.gov/pubmed/17327847. [DOI] [PubMed]
- 11.Moore RE, Young MK, Lee TD. Qscore: an algorithm for evaluating SEQUEST database search results. J Am Soc Mass Spectrom. 2002;13(4):378–386. doi: 10.1016/S1044-0305(02)00352-5. [DOI] [PubMed] [Google Scholar]
- 12.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res. 2003;2(1):43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 13.Tang HY, Ali-Khan N, Echan LA, Levenkova N, Rux JJ, Speicher DW. A novel fourdimensional strategy combining protein and peptide separation methods enables detection of lowabundance proteins in human plasma and serum proteomes. Proteomics. 2005;5(13):3329–3342. doi: 10.1002/pmic.200401275. [DOI] [PubMed] [Google Scholar]
- 14.Reiter L, Claassen M, Schrimpf SP, Jovanovic M, Schmidt A, Buhmann JM, Hengartner MO, Aebersold R. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Molecular & Cellular Proteomics: MCP. 2009;8(11):2405–2417. doi: 10.1074/mcp.M900317-MCP200. %U http://www.ncbi.nlm.nih.gov/pubmed/19608599. [DOI] [PMC free article] [PubMed]
- 15.Olsen JV, de Godoy LMF, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Molecular & Cellular Proteomics: MCP. 2005;4(12):2010–2021. doi: 10.1074/mcp.T500030-MCP200. %U http://www.ncbi.nlm.nih.gov/pubmed/16249172. [DOI] [PubMed]
- 16.Jung H-J, Purvine SO, Kim H, Petyuk VA, Hyung S-W, Monroe ME, Mun D-G, Kim K-C, Park J-M, Kim S-J, Tolic N, Slysz GW, Moore RJ, Zhao R, Adkins JN, Anderson GA, Lee H, Camp DG, Yu M-H, Smith RD, Lee S-W. Integrated Post-Experiment Monoisotopic Mass Refinement: An Integrated Approach to Accurately Assign Monoisotopic Precursor Masses to Tandem Mass Spectrometric Data. Analytical Chemistry. 2010 doi: 10.1021/ac101388b. %U http://www.ncbi.nlm.nih.gov/pubmed/20863060. [DOI] [PMC free article] [PubMed]
- 17.Mann M, Kelleher NL. Precision proteomics: The case for high resolution and high mass accuracy. Proceedings of the National Academy of Sciences. 2008;105(47):18132–18138. doi: 10.1073/pnas.0800788105. %U http://www.pnas.org/content/105/47/18132.abstract. [DOI] [PMC free article] [PubMed]
- 18.Haas W, Faherty BK, Gerber SA, Elias JE, Beausoleil SA, Bakalarski CE, Li X, Villen J, Gygi SP. Optimization and Use of Peptide Mass Measurement Accuracy in Shotgun Proteomics. Molecular Cellular Proteomics. 2006;5(7):1326–1337. doi: 10.1074/mcp.M500339-MCP200. %U http://www.mcponline.org/cgi/content/abstract/5/7/1326. [DOI] [PubMed]
- 19.Dieguez-Acuna FJ, Gerber SA, Kodama S, Elias JE, Beausoleil SA, Faustman D, Gygi SP. Characterization of mouse spleen cells by subtractive proteomics. Molecular Cellular Proteomics. 2005:M500137–MMCP200. doi: 10.1074/mcp.M500137-MCP200. %U http://www.mcponline.org/cgi/content/abstract/M500137-MCP200v1. [DOI] [PubMed]
- 20.Bakalarski CE, Haas W, Dephoure NE, Gygi SP. The effects of mass accuracy, data acquisition speed, and search algorithm choice on peptide identification rates in phosphoproteomics. Analytical and Bioanalytical Chemistry. 2007;389(5):1409–1419. doi: 10.1007/s00216-007-1563-x. %U http://www.ncbi.nlm.nih.gov/pubmed/17874083. [DOI] [PubMed]
- 21.Brosch M, Swamy S, Hubbard T, Choudhary J. Comparison of Mascot and X!Tandem performance for low and high accuracy mass spectrometry and the development of an adjusted Mascot threshold. Molecular & Cellular Proteomics: MCP. 2008;7(5):962–970. doi: 10.1074/mcp.M700293-MCP200. %U http://www.ncbi.nlm.nih.gov/pubmed/18216375. [DOI] [PMC free article] [PubMed]
- 22.Hsieh EJ, Hoopmann MR, MacLean B, MacCoss MJ. Comparison of database search strategies for high precursor mass accuracy MS/MS data. Journal of Proteome Research. 2010;9(2):1138–1143. doi: 10.1021/pr900816a. %U http://www.ncbi.nlm.nih.gov/pubmed/19938873. [DOI] [PMC free article] [PubMed]
- 23.Picotti P, Aebersold R, Domon B. The Implications of Proteolytic Background for Shotgun Proteomics. Mol Cell Proteomics. 2007;6(9):1589–1598. doi: 10.1074/mcp.M700029-MCP200. %U http://www.mcponline.org/cgi/content/abstract/6/9/1589. [DOI] [PubMed]
- 24.Prince JT, Marcotte EM. mspire: mass spectrometry proteomics in Ruby. Bioinformatics (Oxford, England) 2008;24(23):2796–2797. doi: 10.1093/bioinformatics/btn513. %U http://www.ncbi.nlm.nih.gov/pubmed/18930952. [DOI] [PMC free article] [PubMed]
- 25.Rodriguez J, Gupta N, Smith RD, Pevzner PA. Does Trypsin Cut Before Proline? Journal of Proteome Research. 2008;7(1):300–305. doi: 10.1021/pr0705035. %U http://dx.doi.org/10.1021/pr0705035. [DOI] [PubMed]
- 26.Keil-Dlouha V, Zylber N, Imhoff JM, Tong NT, Keil B. Proteolytic activity of pseudotrypsin. FEBS Letters. 1971;16(4):291–295. doi: 10.1016/0014-5793(71)80373-3. %U http://www.ncbi.nlm.nih.gov/pubmed/11945964. [DOI] [PubMed]
- 27.Rice RH, Means GE, Brown WD. Stabilization of bovine trypsin by reductive methylation. Biochimica Et Biophysica Acta. 1977;492(2):316–321. doi: 10.1016/0005-2795(77)90082-4. %U http://www.ncbi.nlm.nih.gov/pubmed/560214. [DOI] [PubMed]
- 28.Finehout EJ, Cantor JR, Lee KH. Kinetic characterization of sequencing grade modified trypsin. PROTEOMICS. 2005;5(9):2319–2321. doi: 10.1002/pmic.200401268. %U http://www.ncbi.nlm.nih.gov/pubmed/15880790. [DOI] [PubMed]
- 29.Olsen JV, Ong SE, Mann M. Trypsin Cleaves Exclusively C-terminal to Arginine and Lysine Residues. Molecular Cellular Proteomics. 2004;3(6):608–614. doi: 10.1074/mcp.T400003-MCP200. %U http://www.mcponline.org/cgi/content/abstract/3/6/608. [DOI] [PubMed]
- 30.Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, Green WN, Yates JR, Davis NG, El-Husseini A. Neural palmitoylproteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456(7224):904–909. doi: 10.1038/nature07605. %U http://www.ncbi.nlm.nih.gov/pubmed/19092927. [DOI] [PMC free article] [PubMed]
- 31.Ramya TNC, Weerapana E, Liao L, Zeng Y, Tateno H, Liao L, Yates JR, Cravatt BF, Paulson JC. In situ trans ligands of CD22 identified by glycan-protein photo-cross-linking enabled proteomics. Molecular & Cellular Proteomics: MCP. 2010;9(6):1339–1351. doi: 10.1074/mcp.M900461-MCP200. %U http://www.ncbi.nlm.nih.gov/pubmed/20172905. [DOI] [PMC free article] [PubMed]
- 32.Wang H, Chang-Wong T, Tang H-Y, Speicher DW. Comparison of Extensive Protein Fractionation and Repetitive LC-MS/MS Analyses on Depth of Analysis for Complex Proteomes. Journal of Proteome Research. 2010;9(2):1032–1040. doi: 10.1021/pr900927y. %U http://dx.doi.org/10.1021/pr900927y. [DOI] [PMC free article] [PubMed]
- 33.Resing KA, Meyer-Arendt K, Mendoza AM, Aveline-Wolf LD, Jonscher KR, Pierce KG, Old WM, Cheung HT, Russell S, Wattawa JL, Goehle GR, Knight RD, Ahn NG. Improving reproducibility and sensitivity in identifying human proteins by shotgun proteomics. Analytical Chemistry. 2004;76(13):3556–3568. doi: 10.1021/ac035229m. %U http://www.ncbi.nlm.nih.gov/pubmed/15228325. [DOI] [PubMed]
- 34.Zubarev R, Mann M. On the proper use of mass accuracy in proteomics. Molecular & Cellular Proteomics: MCP. 2007;6(3):377–381. doi: 10.1074/mcp.M600380-MCP200. %U http://www.ncbi.nlm.nih.gov/pubmed/17164402. [DOI] [PubMed]
- 35.Lee KA, Farnsworth C, Yu W, Bonilla LE. 24-Hour Lock Mass Protection. Journal of Proteome Research. 2011;10(2):880–885. doi: 10.1021/pr100780b. %U http://dx.doi.org/10.1021/pr100780b. [DOI] [PubMed]
- 36.Yang H, Zubarev RA. Mass spectrometric analysis of asparagine deamidation and aspartate isomerization in polypeptides. Electrophoresis. 2010;31(11):1764–1772. doi: 10.1002/elps.201000027. %U http://www.ncbi.nlm.nih.gov/pubmed/20446295. [DOI] [PMC free article] [PubMed]
- 37.Geiger T, Clarke S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. The Journal of Biological Chemistry. 1987;262(2):785–794. %U http://www.ncbi.nlm.nih.gov/pubmed/3805008. [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.