

Here the authors develop a statistical mechanical model to predict structure and folding stability of RNA/RNA kissing-loop complex. They employ a recently developed virtual bond-based RNA folding model (Vfold model) to evaluate the entropy parameters for the different types of kissing loops. A benchmark test against the experiments suggests that the entropy calculation is reliable. As an application of the model, they applied the model to investigate the structure and folding thermodynamics for the kissing complex of HIV-1 dimerization initiation signal. The described method provides a new method to study RNA/RNA kissing complexes.
Keywords: RNA/RNA kissing complex, HIV dimerization, structural predictions, folding thermodynamics, energy landscape, three-dimensional structure (3D)
Abstract
We develop a statistical mechanical model to predict the structure and folding stability of the RNA/RNA kissing-loop complex. One of the key ingredients of the theory is the conformational entropy for the RNA/RNA kissing complex. We employ the recently developed virtual bond-based RNA folding model (Vfold model) to evaluate the entropy parameters for the different types of kissing loops. A benchmark test against experiments suggests that the entropy calculation is reliable. As an application of the model, we apply the model to investigate the structure and folding thermodynamics for the kissing complex of the HIV-1 dimerization initiation signal. With the physics-based energetic parameters, we compute the free energy landscape for the HIV-1 dimer. From the energy landscape, we identify two minimal free energy structures, which correspond to the kissing-loop dimer and the extended-duplex dimer, respectively. The results support the two-step dimerization process for the HIV-1 replication cycle. Furthermore, based on the Vfold model and energy minimization, the theory can predict the native structure as well as the local minima in the free energy landscape. The root-mean-square deviations (RMSDs) for the predicted kissing-loop dimer and extended-duplex dimer are ∼3.0 Å. The method developed here provides a new method to study the RNA/RNA kissing complex.
INTRODUCTION
RNA function is not solely determined by a single native structure; the alternative structures are also functionally important (Schultes and Bartel 2000; Nagel and Pleij 2002; Tucker and Breaker 2005). Predicting RNA structure and conformational changes requires a model for the folding free energy landscape. The development of a predictive model for the structure and energy landscapes of RNA–RNA complexes is strongly motivated by the widespread biological applications from mRNA splicing to microRNA-target recognition (Madhani and Guthrie 1994; Brunel et al. 2002; Lai 2003; Bartel 2004). During the mRNA splicing process, RNA–RNA complexes formed by small nuclear RNAs undergo multiple structural rearrangements in the different steps of splicing (Madhani and Guthrie 1992; Sashital et al. 2004; Cao and Chen 2006a; Sashital et al. 2007; Mitrovich and Guthrie 2007; Valadlkhan 2007). The importance of understanding and predicting RNA–RNA binding is also highlighted by the rapidly growing research on microRNA functions in post-transcriptional gene regulation. In microRNA-mediated gene regulation, short RNA molecules (microRNAs) bind to gene targets (at 3′ untranslated regions of target mRNA transcripts) to regulate gene expression. Emerging evidence suggests that microRNA–mRNA target recognition is determined not only by the local sequence complementarity at the binding site but also by the global (nonlocal) interplay between intermolecular and intramolecular base pairing. Incorporating the intermolecular and intramolecular competition in the model can lead to improvement in the predictions for microRNA activity (Didiano and Hobert 2006; Long et al. 2007). In addition, RNA–RNA dimerization has been found to play an important role in viral replication. For example, two copies of a genomic sequence have been proposed to play a critical role in the initiation of HIV-1 viral replication. Many RNA–RNA dimers are stabilized by tertiary interactions such as kissing-loop interactions and pseudoknotted interactions between the RNAs (Paillart et al. 1996, 2004; Jossinet et al. 1999; Kolb et al. 2000a,b, 2001a,b; Russell et al. 2004). The RNA–RNA interactions mentioned in the above biological processes demonstrate the need to have a model that can treat (1) conformational changes, (2) complex interplay between intermolecular and intramolecular base pairing, and (3) kissing interactions in RNA–RNA complexes.
Motivated by the biological significance of RNA–RNA interactions, several computational methods have been developed to predict the structures and stabilities of RNA/RNA complexes (Mathews et al. 1999; Lewis et al. 2003; Dimitrov and Zuker 2004; Rehmsmeier et al. 2004; Andronescu et al. 2005; Bernhart et al. 2006; Dirks et al. 2007). Similar predictive tools for DNA/DNA hybridization can be found in the DNA software package (SantaLucia and Hicks 2004). A number of these methods can treat intermolecular and intramolecular competitions (Andronescu et al. 2005; Bernhart et al. 2006; Cao and Chen 2006a). These models enable predictions of two-dimensional structures (base pairs) for the binding between small nuclear RNAs, between ribozyme and substrates, and between microRNAs and the targets. However, these methods are restricted to treat only RNA secondary structures (Lewis et al. 2003; Dimitrov and Zuker 2004; Rehmsmeier et al. 2004; Andronescu et al. 2005; Bernhart et al. 2006; Dirks et al. 2007) and cannot treat pseudoknotted structures such as the tertiary folds formed by loop–loop kissing interactions in the dimerization of human immunodeficiency virus type 1 (HIV-1) genomes (Skripkin et al. 1994; Laughrea and Jetté 1994; Li et al. 2006, 2008). We note that a recently developed model based on partition function calculations can account for complex kissing interactions (Huang et al. 2009). The importance of including the kissing interactions underscores the need to develop a rigorous free energy model for the formation of such structural motifs. Kissing loops can cause cross-linkage between different helices and between helices and loops. As a result of the cross-linkage, the folding free energy of the system becomes nonadditive; i.e., the total stability of the structure is not the simple additive sum of the stability of each structural subunit (Dill 1990). To account for the nonadditive free energy, especially the entropy, we need a physical model. Such physical entropy models have been shown to give an improved prediction for simple H-type pseudoknots (Cao and Chen 2006b, 2009; Andronescu et al. 2010; Sperschneider and Datta 2010; Sperschneider et al. 2011).
The evaluation of the conformational entropy is effectively a problem of counting the three-dimensional (3D) structures. In a previous study, we used a virtual bond-based coarse-grained RNA folding model (Vfold model) (Cao and Chen 2005) to evaluate the entropies and the free energies for RNA–RNA complexes at the level of secondary structures (Cao and Chen 2006a). The model was able to calculate the free energy landscape for secondary structures, which led to several predictions for the structures and conformational switches. Applications of the model to the yeast U2-U6 spliceosomal RNA complex showed two energetically favorable structures competing with each other. Moreover, the competition between inter- and intramolecular interactions causes conformational switches between the alternative structures. The predicted conformational switches might be related to the catalytic functions of the different stages of mRNA splicing.
In the present study, inspired by the biological significance of tertiary structural folds of RNA–RNA complexes, we apply the Vfold model to treat RNA–RNA kissing complexes. We evaluate the entropy parameters for the different structural motifs with the different (kissing) loop–loop contacts. With the calculated entropy parameters, we develop a model to predict the structure and folding thermodynamics for RNA–RNA complexes. As an application of the model, we will study the energy landscape of the HIV-1 dimerization initiation signal (DIS), which shows the kissing-loop dimer and the extended-duplex dimer coexisting in thermal equilibrium. The theoretical predictions are consistent with the two forms of RNA–RNA complexes observed in crystal and NMR structural measurements (Mujeeb et al. 1998, 1999; Ennifar et al. 1999, 2001; Takahashi et al. 2005; Ulyanov et al. 2006).
Our studies show that the kissing-loop dimer is stabilized by the coaxial stacking of two stems. Experiments find that protein NCp7 can activate the transition from the kissing-loop dimer to the extended-duplex dimer (Muriaux et al. 1996a). We propose that NCp7-binding can destabilize the kissing-loop dimer by inhibiting the coaxial stacking. In addition, we find that the extended-duplex dimer becomes energetically more favorable as the temperature increases, which is also consistent with the experiment (Muriaux et al. 1996b; Takahashi et al. 2000).
MATERIALS AND METHODS
Energetic parameters
For an RNA/RNA complex, while the free energies of base pairs and base stacks can be estimated from the empirical parameters (Turner rules), the evaluation of the loop free energy for a kissing complex requires a theory. Assuming the loop stability is dominated by the entropic component (instead of interaction energies), we can estimate the loop free energy as 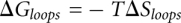 , where the loop entropy
, where the loop entropy 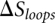 is determined by the statistics of 3D conformations:
is determined by the statistics of 3D conformations: 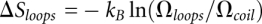 , where
, where 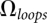 is the total number of conformations of the loops and
is the total number of conformations of the loops and 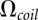 is the number of conformations of the coil state. The present form of the theory assumes weak loop–helix tertiary interactions, which may contribute a nonzero loop enthalpy to the free energy. For the loop–loop and intraloop interactions, we consider canonical base stacks as well as mismatched base stacks. Here a mismatched stack is formed by a non-Watson-Crick base pair stacked on a Watson-Crick base pair. The energetic parameters for a mismatched base stack is given by the Turner rules. The formation of the loop–loop and intraloop contacts can cause a large reduction in the conformational entropy. Our statistical mechanical model (Vfold) can calculate such conformational entropy parameters through a direct conformational count. In the following, we use a hairpin kissing-loop system to illustrate the method of entropy calculation.
is the number of conformations of the coil state. The present form of the theory assumes weak loop–helix tertiary interactions, which may contribute a nonzero loop enthalpy to the free energy. For the loop–loop and intraloop interactions, we consider canonical base stacks as well as mismatched base stacks. Here a mismatched stack is formed by a non-Watson-Crick base pair stacked on a Watson-Crick base pair. The energetic parameters for a mismatched base stack is given by the Turner rules. The formation of the loop–loop and intraloop contacts can cause a large reduction in the conformational entropy. Our statistical mechanical model (Vfold) can calculate such conformational entropy parameters through a direct conformational count. In the following, we use a hairpin kissing-loop system to illustrate the method of entropy calculation.
Structural model
The kissing complex consists of three stems and four loops (Fig. 1A). Usually, loop L2 and L4 are short, with ∼1 nucleotide (nt) (Ennifar et al. 2001). A short loop favors the formation of coaxial stacking interaction between stems H1 and H2 and between stems H2 and H3, which in turn can stabilize the kissing complex. In order to accurately predict the folding thermodynamics of kissing complex, we first need to estimate the entropy parameter for the formation of the kissing complex.
FIGURE 1.

(A) A schematic diagram for a kissing complex structure. Stems H1, H2, and H3 are coaxially stacked. Loops L1 and L3 span across stem H2. The lengths of loops L2 and L3 are usually ≤1 nt. (B) The virtual bond representation of the kissing complex structure.
We model stems H1, H2, and H3 as A-form helices. We use the atomic coordinates of the A-form helix to configure the helices (Arnott and Hukins 1972). The coordinates (r, θ, z) for P, C4, and N1 (or N9) atoms in the helix are (8.71 Å, 70.5 + 32.7i, −3.75 + 2.81i), (9.68 Å, 46.9 + 32.7i, −3.10 + 2.81i), and (7.12 Å, 37.2 + 32.7i, −1.39 + 2.81i) (i = 0, 1, 2, …) (Arnott and Hukins 1972). For the other strand, we negate θ and z. We assemble stems H1, H2, and H3 according to the coordinates of 8 nt (ai, a′i, aj, a′j, bi, b′i, bj, and b′j) in the junction. The coordinates of the 8 nt are adopted from the known NMR structure (Ennifar et al. 2001).
The bonds that connect the P, C4, and N1 (or N9) atoms are called virtual bonds. Each nucleotide is represented by three virtual bonds: P-C4, C4-N1 (or N9), and C4-P. We use the above three-vector virtual bond model (Vfold) to describe loop conformations. In the Vfold model, the conformational of each nucleotide is described by three virtual bonds: two bonds for the nucleotide backbone and a third bond for the sugar pucker orientation. A survey of the known RNA structures shows discrete distributions of the (pseudo)torsional angles for the virtual bonds (Olson 1980; Duarte and Pyle 1998; Cao and Chen 2005), and the discrete distribution of the torsional angles can be approximately represented in a diamond lattice. Therefore, we can model loop conformations as self-avoiding walks of the virtual bonds on diamond lattice.
We can also reduce the all-atom structures for the helices using the virtual bonds. Figure 1B shows the virtual bond representation of the assembled stems H1, H2, and H3. The connection between the A-form helix and the discrete loop conformations is realized through an iterative optimized algorithm (Ferro and Hermans 1971) for the coordinates of the four loop–helix interfacial nucleotides (ai, aj, bi, and bj) in the junctions. Figure 1B shows a conformation of loops L1 and L3. Both loops L1 and L3 span across the major groove of stem H2.
A key issue in the conformational count (conformational entropy) is the excluded volume interaction between loop and helix and between the different loops. Loop–helix excluded volume effect requires an accurate description of the helical structure. For example, for a loop (L1 or L3) that spans across a helix H2, the helix structure causes a nonmonotonic behavior of the loop conformation: the end–end distance of the loop, defined as the distance between the P atoms at the junction ai and at the junction aj, decreases with the length of helix H2 until H2 = 5 and then increases (Fig. 2A). In general, the volume exclusion between a loop and the helix that the loop spans across is highly significant and must be accounted for in the calculation of conformational entropy. For example, for loop L3, the excluded volume interaction from helix H3 is overwhelmingly stronger than that from helices H1 and H2 (Fig. 2C). Moreover, for kissing complexes, loops (such as L1 and L3) could be in a close proximity, causing excluded volume-induced coupling between loop conformations (Fig. 2B). In conclusion, the evaluation of loop entropy requires consideration of the loop conformations in the context of the global fold instead of individual, isolated loops.
FIGURE 2.

(A) The P-P end-end distance of loop L1 or L3 as a function of the length of helix (H2). (B) The calculated loop entropy as a function of loop length (L3). In the calculation, we fix (H1, H2, H3) = (7, 6, 7) bp. The lengths of loops L2 and L4 are fixed at 1 nt, and the length of L1 is 2 nt. For multiple short loops configured in a crowded spatial region, loop–loop volume exclusion can significantly reduce the number of the loop conformations. (C) The dependence of the entropy parameter on the length of stem H1 or H3.
Kissing-loop entropy
We calculate the kissing-loop entropy using exact enumeration method (Cao and Chen 2005, 2006b); for the calculated entropy as a function of the lengths of stem H2 and loops L1 and L3 with fixed loop length of 1 nt for L2 and L4 (Table 1). Here the loop and stems lengths are chosen according to experiments (Mujeeb et al. 1998).
TABLE 1.
In the table, we label the calculated conformational entropies [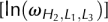 ] of the kissing complex at different stem lengths and different loop lengths
] of the kissing complex at different stem lengths and different loop lengths

The computational time for the exact enumeration increases exponentially as the loop length. In order to efficiently enumerate the loop conformations, we restrict the lengths of loops L1 and L3 ≤ 7 nt. For large loops, we use the following fitted formula:
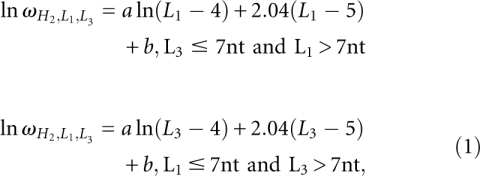 |
where 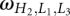 is the number of conformations for given lengths of H2, L1, and L3, and a and b are the coefficient listed in Table 2. The coefficients a and b are functions of the stem length H2 and loop length (L1 or L3). Due to the symmetric spatial arrangement of loops L1 and L3 in the structure, ln
is the number of conformations for given lengths of H2, L1, and L3, and a and b are the coefficient listed in Table 2. The coefficients a and b are functions of the stem length H2 and loop length (L1 or L3). Due to the symmetric spatial arrangement of loops L1 and L3 in the structure, ln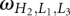 (L3 ≤ 7 nt and L1 > 7 nt) and ln
(L3 ≤ 7 nt and L1 > 7 nt) and ln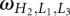 (L1 ≤ 7 nt and L3 > 7 nt) have the similar coefficients (a and b).
(L1 ≤ 7 nt and L3 > 7 nt) have the similar coefficients (a and b).
TABLE 2.
For the longer loops (l > 7 nt), we fit the entropy by 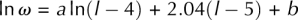

For L1 > 7 nt and L3 > 7 nt, we use the following fitted formula:
where 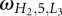 can be calculated from Equation 1.
can be calculated from Equation 1.
The conformational entropy of a coil state can be fitted as 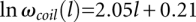 , where l is the chain length of loop L1 or L3, and
, where l is the chain length of loop L1 or L3, and 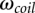 is the number of conformations of the coil state.
is the number of conformations of the coil state.
The entropy change for the formation of the kissing-loop complex is given by 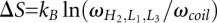 , where
, where 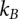 is the Boltzmann constant.
is the Boltzmann constant. 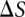 is dependent on the length of stem H2 and the lengths of loops L1 and L3.
is dependent on the length of stem H2 and the lengths of loops L1 and L3.
In summary, based on the Vfold model, we calculate the entropy parameters for the formation of the kissing complex. We note that compared with the Gaussian chain approximation-based entropy calculation (Isambert and Siggia 2000), the present Vfold model has the advantage of explicitly accounting for the excluded volume between helix and loop and between loops. In the following sections, based on the entropy parameters for the kissing-loop complex, we develop a recursive algorithm to compute the partition function and the energy landscape of RNA/RNA kissing complex.
Partition function
At the center of the statistical thermodynamics is the partition function. In a previous study (Cao and Chen 2006a), we developed a method to transform the double-stranded complex into an equivalent single-stranded chain by introducing a 3-nt phantom linker. With the phantom linker, the partition function for the two-strand complex can be evaluated from the effective single-stranded chain through the use of the following two types of structures that are closed by a base pair (a, b):
type-1 if the phantom linker resides inside a closed region a to b (e.g., Fig. 3C,D)
type-0 otherwise (e.g., Supplemental Fig. S1a)
FIGURE 3.

(A) The kissing interaction between two hairpin loops. The curved links in the polymer graph (the right panel) denote base pairs. The straight lines represent RNA backbone chains from 5′ to 3′. The dashed line denotes the phantom link, which is used to connect two RNAs into a single RNA strand (Cao and Chen 2006a). (B) The kissing interaction between a loop and a tail. (C) A type-1 closed kissing conformation 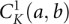 , where nucleotides a and b form base pairings with other nucleotides. We include two type kissing interactions (A) and (B) in the present model. (D) The type-1 open conformation, in which a and b are unpaired (lone) nucleotides. The filled region denotes a helix. We allow other secondary or kissing structures (data not shown in the figure) to be formed in the region (b1, an).
, where nucleotides a and b form base pairings with other nucleotides. We include two type kissing interactions (A) and (B) in the present model. (D) The type-1 open conformation, in which a and b are unpaired (lone) nucleotides. The filled region denotes a helix. We allow other secondary or kissing structures (data not shown in the figure) to be formed in the region (b1, an).
Here a closed region is formed either by a pseudoknotted structure or by a structure whose ends are closed by a base pair, such as the structures for the chain segments from nucleotide ai to nucleotide bi (i = 1, 2,...., n) in Supplemental Figure S1a. In the present study, we extend the previous algorithm, which can only treat RNA secondary structures (Cao and Chen 2006a), to predict the folding thermodynamics and the structure for RNA–RNA complexes with kissing interactions. In particular, we consider two types of kissing interactions (see Fig. 3A,B): kissing contact between hairpin loops (Fig. 3A) and between a hairpin loop and a dangling tail (Fig. 3B). For structures shown in Figure 3, the phantom linker (filled circles) resides inside the region from a to b and thus is a type-1 structure.
A difference between the current study and a previous model (Cao and Chen 2006a) is that we now allow the formation of kissing-loop complexes (Fig. 3C) for the type-1 open conformations 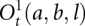 . Here t = L, R, M, and LR represent the different conformational types illustrated below), and l is the number of unpaired nucleotides outside the closed structures (
. Here t = L, R, M, and LR represent the different conformational types illustrated below), and l is the number of unpaired nucleotides outside the closed structures (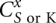 in Fig. 3) plus the number of the closed structures. The four types are defined according to the (a, b) positions relative to the (a1, bn), where a1 is the first nucleotide being paired, and bn is the last nucleotides being paired in 5′ to 3′ direction (see Supplemental Fig. S1b; Chen and Dill 1998):
in Fig. 3) plus the number of the closed structures. The four types are defined according to the (a, b) positions relative to the (a1, bn), where a1 is the first nucleotide being paired, and bn is the last nucleotides being paired in 5′ to 3′ direction (see Supplemental Fig. S1b; Chen and Dill 1998):
type-LR if a1 is adjacent to a (i.e., a1 = a + 1) and bn is adjacent to b (i.e., bn = b − 1)
type-L if only a1 is adjacent to a
type-R if only bn is adjacent to b
type-M if neither a1 nor bn is adjacent to a or b
The purpose of defining four different types of structures is to account for the base pairing at the junctions and hence the viability of the connections between the different structural subunits (Chen and Dill 1995; Zhang and Chen 2001; Cao and Chen 2006a; Kopeikin and Chen 2006; Chen 2008; Liu and Chen 2010).
A key step here is the partition function calculation for the four open structures 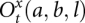 (x = 0, 1; t = M, L, R, LR) for different as and bs. We calculate the partition function for a longer chain from shorter chain segments using the following recursive relationships: Supplemental Figure S2 shows the recursive relationships for the four types of open structures. Though only secondary structures (
(x = 0, 1; t = M, L, R, LR) for different as and bs. We calculate the partition function for a longer chain from shorter chain segments using the following recursive relationships: Supplemental Figure S2 shows the recursive relationships for the four types of open structures. Though only secondary structures (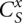 ) are shown in Supplemental Figure S2 (for illustrative purpose), in the actual partition function calculation, kissing structures (
) are shown in Supplemental Figure S2 (for illustrative purpose), in the actual partition function calculation, kissing structures (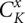 ) are included in the recursive relationships. For the kissing structures, we restrict x = 1 since the phantom linker is always inside the kissing structure (see Fig. 3A,B).
) are included in the recursive relationships. For the kissing structures, we restrict x = 1 since the phantom linker is always inside the kissing structure (see Fig. 3A,B).
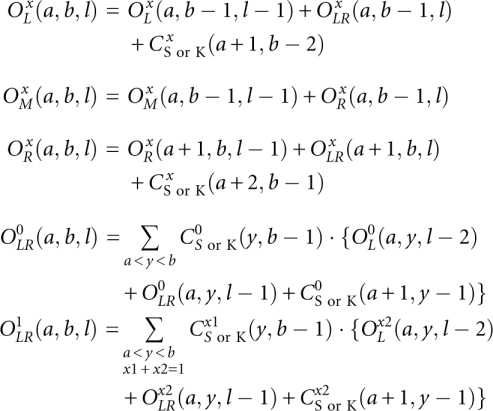 |
The total partition function Qtot(a, b) for a chain from a to b is given by the sum of the partition functions for all the different types of conformations:
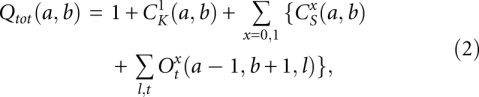 |
where 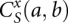 represents the partition function of type-x closed conformation without the kissing structure. From the total partition function, we can obtain the partition function for the complex Z12 from the following equation:
represents the partition function of type-x closed conformation without the kissing structure. From the total partition function, we can obtain the partition function for the complex Z12 from the following equation:
where Z1 and Z2 are the partition functions of strands S1 and S2, respectively.
We define α to quantify the concentration dependence for the formation of the complex as the following:
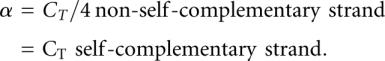 |
Partition function Z, which includes the single strands Z1 and Z2 and the complex Z12, can be calculated from the following formula:
where the value of  is adopted from the reference (Xia et al. 1998):
is adopted from the reference (Xia et al. 1998): 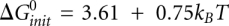 (kcal/mol). T is the temperature. The physical origin of an additional
(kcal/mol). T is the temperature. The physical origin of an additional 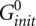 is due to the entropy loss associated with the conversion from two single-stranded RNAs to a single RNA complex, which is independent on the strand concentrations. We define
is due to the entropy loss associated with the conversion from two single-stranded RNAs to a single RNA complex, which is independent on the strand concentrations. We define 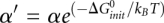 to simplify the expression.
to simplify the expression.
The free energy change 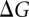 upon the formation of the complex can be derived from the partition function Z(T):
upon the formation of the complex can be derived from the partition function Z(T):
To derive the structure from the free energy, we compute the base-pairing probability 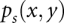 for each base pair between the xth nucleotide and the yth nucleotide for both the double-stranded complex (s = 12) and the single-stranded free molecules (s = 1 or 2):
for each base pair between the xth nucleotide and the yth nucleotide for both the double-stranded complex (s = 12) and the single-stranded free molecules (s = 1 or 2): 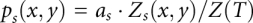 , where
, where 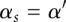 for s = 12 and 1 otherwise. From the base-pairing probability, we can find the probable structures by maximizing the expected pair accuracy S (Do et al. 2006; Lu et al. 2009):
for s = 12 and 1 otherwise. From the base-pairing probability, we can find the probable structures by maximizing the expected pair accuracy S (Do et al. 2006; Lu et al. 2009):
where 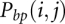 is the probability for nucleotides i and j to form a base pair, and
is the probability for nucleotides i and j to form a base pair, and 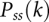 is the probability for nucleotide k to be single-stranded. Depending on the RNA sequence, we may find alternative coexisting structures, corresponding to multiple minima on the free energy landscape.
is the probability for nucleotide k to be single-stranded. Depending on the RNA sequence, we may find alternative coexisting structures, corresponding to multiple minima on the free energy landscape.
Compared to the model developed by Huang et al. (2009), our model is focused on accurately evaluating the entropy parameters for the kissing interactions between two hairpin loops and between the tail and the hairpin loop (see Fig. 3A,B), which have been lacking in the literature. In the current partition function model, we add the two types of kissing motifs to the secondary structural ensemble (Cao and Chen 2006a). The model does not treat the complicated complexes with two or more kissing sites as shown in the reference by Huang et al. (2009). For example, the fhlA/OxyS complex contains two kissing sites and cannot be treated by our model.
RESULTS AND DISCUSSION
Test of energetic parameters
From the temperature-dependence of the partition function Z(T), we can compute the heating capacity melting curve C(T) for a given sequence: 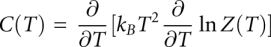 . In the calculation, we use the individual nearest-neighbor hydrogen bonding (INN-HB) model for the stacking energies (Xia et al. 1998). The INN-HB model has been shown to give more accurate base pair predictions than the prior models (Freier et al. 1986). We calculate the melting curves for four RNA duplexes (Fig. 4A,B; Weixlbaumer et al. 2004). To compare with the experimental results, we use the same solution condition as the experimental condition (1 M NaCl solution condition and 9 × 10−6 M for RNA strand concentration) (Weixlbaumer et al. 2004). The predicted melting temperatures, 40°C, 47°C, and 50°C, agree with the experimental results, 40°C, 43.3°C, and 48.4°C for the duplexes D2, D3 and D4, respectively. For D1, we predicted that the melting temperature is 8°C, which cannot be detected in the experiment in which the monitored temperature is higher than the melting temperature. Thus, the INN-HB model provides a good approximation for the stacking energies.
. In the calculation, we use the individual nearest-neighbor hydrogen bonding (INN-HB) model for the stacking energies (Xia et al. 1998). The INN-HB model has been shown to give more accurate base pair predictions than the prior models (Freier et al. 1986). We calculate the melting curves for four RNA duplexes (Fig. 4A,B; Weixlbaumer et al. 2004). To compare with the experimental results, we use the same solution condition as the experimental condition (1 M NaCl solution condition and 9 × 10−6 M for RNA strand concentration) (Weixlbaumer et al. 2004). The predicted melting temperatures, 40°C, 47°C, and 50°C, agree with the experimental results, 40°C, 43.3°C, and 48.4°C for the duplexes D2, D3 and D4, respectively. For D1, we predicted that the melting temperature is 8°C, which cannot be detected in the experiment in which the monitored temperature is higher than the melting temperature. Thus, the INN-HB model provides a good approximation for the stacking energies.
FIGURE 4.

(A) The eight sequences used to calculate the melting curves for experimental test. The calculated melting curves for four duplexes (B) and four kissing complexes (C). In the calculation, the ion condition is 1 M NaCl. The RNA strand concentrations are 9 μM and 10 μM for the duplex and the kissing complexes, respectively. The predicted melting temperatures for the duplexes D2, D3, and D4 are 40°C, 47°C, and 50°C, which agree with the experimental values: 40°C, 43.3°C, and 48.4°C (Weixlbaumer et al. 2004). For sequence D1, we predicted a melting temperature of 8°C. The temperatures for melting the kissing complexes K1, K2, K3, and K4 are 32°C, 55°C, 62°C, and 65°C, which are close to the experimental values: 32°C, 57°C, 64.7°C, and 67.3°C (Weixlbaumer et al. 2004).
To test our theory for the formation of kissing loop complexes, we use the calculated entropy parameters for the kissing loops (see Tables 1, 2) to predict the melting curves of a series of experimentally studied kissing complexes (K1, K2, K3, and K4 in Fig. 4A). In order to make direct comparisons with the experimental data, we again use the same ion concentration 1 M NaCl and RNA strand concentration 10−5 M as used in the experiment. The NMR structures for the kissing complexes show coaxial stacking between stems H1 and H2 and between H2 and H3. Thus, we add a sequence-dependent energy parameters for each coaxial stacking (Walter and Turner 1994). The melting curves for the kissing complexes show two peaks. Our structural calculation for the different temperatures indicate that the low-temperature peak corresponds to the unzipping of the intermolecular base pairs in the kissing complex, and the high-temperature peak corresponds to the unfolding of two single-stranded hairpins. The predicted melting temperatures, 32°C, 55°C, 62°C, and 65°C for K1, K2, K3, and K4, respectively, are in close agreement with the experimental results 32°C, 57°C, 64.7°C, and 67.3°C (see Fig. 4C). The theory-experiment test suggests the validity of our entropy model for the kissing complex. In the following section, we apply the model to investigate folding thermodynamics and the energy landscapes for a series of kissing complexes, including the HIV-1 DIS complex.
Figure 5A shows the predicted native structure for K4 complex at 37°C, which is a kissing complex. By using the entropy of the kissing complex in Table 1, we can estimate the free energy of the K4 complex [ΔG(kissing)]; see Equation 4.
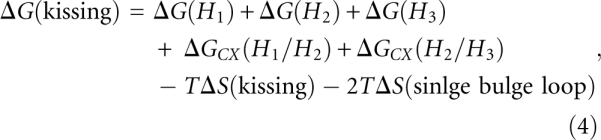 |
where ΔG(H1), ΔG(H2), and ΔG(H3) are the free energies of stems H1, H2, and H3, respectively. ΔGCX(H1/H2) is the coaxial stacking energy between stem H1 and H2, and ΔGCX(H2/H3) is the coaxial stacking energy between stem H2 and H3. ΔS(kissing) is the entropy change associated with the formation of the kissing loop. ΔS(single bulge loop) is the entropy of the single bulge loop A, which connects H1 and H2.
FIGURE 5.

(A–C) The density plot for the base-pairing probabilities and the predicted stable structure for the RNA/RNA complex at the different temperatures. The kissing complex is partially unfolded at 65°C, which corresponds to the first peak in the melting curve. (D–F) The density plot for the base-pairing probabilities and the predicted stable structure for a single stranded RNA at the different temperatures. At 75°C, the population of the kissing complex completely converts to a hairpin structure. The hairpin structure is completely unfolded at 110°C.
Based on the INN-HB model (Xia et al. 1998), we can obtain that ΔG(H1), ΔG(H2), and ΔG(H3) are equal to −15.5, −14.1, and −15.5 kcal/mol, respectively. The coaxial stacking energies 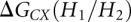 and
and 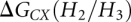 are equal to −4.0 and −3.9 kcal/mol (Walter and Turner 1994), respectively. Equation 5 gives the calculation of the entropy change associated with the formation of the kissing complex:
are equal to −4.0 and −3.9 kcal/mol (Walter and Turner 1994), respectively. Equation 5 gives the calculation of the entropy change associated with the formation of the kissing complex:
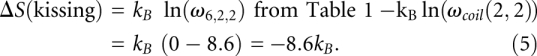 |
The free energy of the kissing complex ΔG(kissing) is equal to:
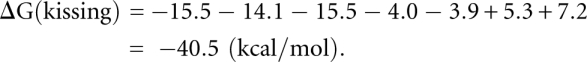 |
In addition, we further test the model's accuracy on predicting the structures of the trans-activating responsive (TAR)–RNA kissing complexes. The RNA aptamer shows a high affinity to bind TAR RNA element by forming the loop–loop kissing interactions. Figure 6 shows the predicted structures of TAR-TAR*(GA) and TAR-R06 complexes at room temperature. In the predicted structures, both TAR-TAR*(GA) and TAR-R06 contain a 6-bp intermolecular kissing interactions. The predicted structures are the same as that of the experimental measured structures (Lebars et al. 2008).
FIGURE 6.

The density plot for the base-pairing probabilities and the predicted stable structure for TAR/TAR*(GA) (A) and TAR/R06 (B) complexes at room temperature. In the calculation, the ion concentration is 0.1 M Na+ and the RNA strand concentration is 1 mM, which are adopted from the experiment (Lebars et al. 2008).
Folding thermodynamics
All the four kissing complexes show two-transition pathways in the equilibrium thermal unfolding (Fig. 4C). To predict the unfolding pathways, we compute the base-pairing probabilities at three different representative temperatures (Fig. 5A–C), corresponding to the temperatures below the lower melting temperature, between the lower and higher melting temperatures, and above the higher melting temperature. In the calculation, the RNA strand concentration is 10−5 M, which is the same as the above melting curve calculation. At low temperature (37°C), the stable structure is the kissing complex. At T = 65°C, the kissing complex is partially unzipped and the single-strand RNA hairpin is partially formed (Fig. 5E). This confirms that the first peak corresponds to the unzipping of the kissing complex. At T = 75°C, the kissing complex is completely converted to the single-strand hairpin structure. The single-strand hairpin structure is much more stable and is disrupted at a high temperature (T = 110°C).
Experimental studies indicate that thermal heating can induce the conformational switch from the kissing complex to the extended-duplex dimer (Muriaux et al. 1996a). Our model for the formation of RNA–RNA kissing complex allows us to quantitatively analyze the transition. For the HIV-1 (Mal) DIS complex, our results show that the kissing complex has a population of 16% at room temperature (Fig. 7). The RNA strand concentration that we used is 150 μM, which is adopted from the experiment (Ennifar et al. 2001). As the temperature is increased, the kissing complex is destabilized. The population of the kissing-loop complex decreases and the population of the extended-duplex dimer increases, which is consistent with the experimental observation (Muriaux et al. 1996a).
FIGURE 7.

The density plot for the base-pairing probabilities and the predicted stable structure for HIV-1 Mal dimer. At room temperature, the kissing-loop dimer and extended-duplex dimer coexist. The extended-duplex dimmer is more stable than the kissing-loop dimer. The kissing-complex dimer converts to the extended-duplex dimer as temperature increases.
Energy landscape of HIV-1 DIS complex and implications on the two-step dimerization process
The dimerization process is essential for the HIV-1 replication. From the structural and functional studies, a two-step dimerization process has been proposed (Muriaux et al. 1996a,b). First, the kissing-loop complex is formed. Due to temperature increase or protein binding, the kissing-loop dimer undergoes a conversion to form the extended-duplex dimer. Due to the lack of the thermodynamic parameters for the kissing-loop dimer, it has been difficult to determine the relative population of each dimer at the different temperatures. Both the kissing-loop dimer and the extended-duplex dimer have been found in the structural measurement by the same research group (Ennifar et al. 1999, 2001). It would be intriguing to know if the kissing-loop dimer is a kinetic intermediate or a thermodynamic stable state at room temperature. Our present model provides a useful tool to quantitatively predict the thermodynamic stabilities for the different dimers by computing the free energy landscape of the two-stranded system.
In the free energy landscape calculation, we use 1 M NaCl concentration and room temperature for the solution condition and 150 μM for the RNA strand concentration (Ennifar et al. 2001). We note that a recent thermodynamic study (Lorenz et al. 2006) suggests that the 1 M NaCl may be equivalent to the physiological ionic concentration. Therefore, the energy landscape in 1 M NaCl might provide useful information for HIV-1 DIS in vivo.
The predicted free energy landscape shows similar shapes for HIV-1 Mal and type-f (Fig. 8). The landscapes show two free energy minima, indicating two coexisting structures (I and II) at room temperature. The energy landscape shows that one sequence encodes two alternative dimeric structures. The result echoes an earlier similar finding for the HDV ribozyme (Schultes and Bartel 2000). Our structural (base-pairing probability) calculations show that the free energy minima correspond to the kissing-complex dimer and extended-duplex dimer, respectively. The free energy of (I, II) is (−29.0 kcal/mol, −28.1 kcal/mol) and (−28.0 kcal/mol, −28.1 kcal/mol) for Mal and type-f, respectively. The extended-duplex dimer in Mal is slightly more stable than that of type-f since the A.G mismatch is more stable than A.A mismatch. The results suggest that the kissing-complex dimer has a comparable stability as the extended-duplex dimer for the two types of HIV-1 DIS that we studied, and the kissing-complex dimer can be formed as a thermodynamically (meta)stable state at room temperature.
FIGURE 8.

The free energy landscape for the HIV-1 dimer at room temperature. Two stable structures (I, II) coexist in the HIV-1 dimer. Structure I corresponds to the extended-duplex dimer, and II corresponds to the kissing-loop dimer. Two different types of species (Mal and Type-f) (A and B, respectively) have the similar energy landscape profile. In the energy landscape, N and NN are the numbers of the native and non-native base pairs, respectively.
Moreover, based on the NMR structure and the computational study, we find that the kissing-complex dimer is stabilized by the coaxial stacking. Binding of protein NCp7 to the kissing-loop complex could disrupt the coaxial stacking and thus destabilize the kissing-loop complex, resulting the transition from the kissing-loop dimer to the extended-duplex dimer. We note that ligand or protein-binding can induce the conformational change and regulate gene expression (Tucker and Breaker 2005; Wickiser et al. 2005; Laederach 2007; Greenleaf et al. 2008; Montange and Batey 2008), and a similar mechanism for protein binding-induced structural change has been proposed for the activation of a conformational switch for yeast U2/U6 spliceosomal RNA complex during the mRNA splicing (Cao and Chen 2006a). Our proposed mechanism is consistent with our predicted unfolding pathways, which show the population of the extended-duplex dimer becomes more dominant as the temperature increases.
3D structures of the dimers
Recently, several models have been developed for the prediction for RNA structures (Michel and Westhof 1990; Tan et al. 2006; Das and Baker 2007; Shapiro et al. 2007; Ding et al. 2008; Parisien and Major 2008; Rother et al. 2011; Westhof et al. 2011). These models are good at predicting some structures at high-accuracy resolution. For example, the de novo prediction models (Das and Baker 2007; Ding et al. 2008; Parisien and Major 2008) can accurately predict the simple and short hairpin structures. However, the models cannot predict the kissing complex. The ability of the Vfold model (Cao and Chen 2011) makes the prediction of kissing complexes possible. In addition, the free energy landscape allows us to go beyond the native state by predicting all the free energy minima.
The virtual bond conformations account only for the coordinates of the P, C4, and N1 or N9 atoms. To predict the all-atom structure, we use a multiscale strategy. First, we use the virtual-bond model to calculate the free energy landscape based on conformations described by base pairs. Our entropy model allows for a rigorous sampling of the conformational space. Second, for each free energy minimum, we construct the 3D structure as illustrated below.
By using the Vfold model for the entropy/free energy calculation, we first predict the energy landscape for HIV-1 dimer (see Fig. 8) The free energy landscape shows two local minima (I and II) at a low temperature. Structure I is an extended duplex, and structure II is a kissing-complex structure with stems (H1, H2, H3) and loops (L1, L2, L3, L4) of lengths (7, 6, 7) bp and (2, 1, 2, 1) nt, respectively. Based on the predicted base pairs (helices), we build the virtual structures for the kissing-complex (Fig. 9A). By using the virtual bond structure as a low-resolution scaffold, we compute the all-atom coordinates using all-atom minimization.
FIGURE 9.

(A) The virtual bond representation of the kissing-loop dimer. (B) The all-atom structure built from the virtual bond structure. (C) The predicted structure for HIV-1 (Mal) kissing-loop dimer after energy minimization. (D–F) The predicted 3D structure (purple-blue) for the kissing-loop dimer and extended-duplex dimer. The all-atom RMSDs are 3.1, 3.3, and 2.9 Å for the three structures. The predicted structures are superimposed on its corresponding experimental structures (color sand). The PDB ids of the experimental structures are 1xpe, 1yxp, and 462d.
Specifically, we extract the all-atom coordinates for the A, U, G, and C nucleotides from an A-form helix. By using these coordinates as the template for base configurations, we add the bases to the virtual backbone structure (Fig. 9B). Because the virtual bond conformations for the loops/junctions are generated in a diamond lattice while here the helices are built according to the atomistic A-form helix structure, the crude atomistic structure at this step may show some artifact. For instance, loops/junctions may not connect to the helices exactly (see Fig. 9B). To remove these artifacts and to relax the structure to an energy minimum based on more realistic force field, we run the Amber minimization.
We first perform 1000 steps minimization with 500.0 kcal/mol restraints on all the residues in the target RNA molecule. Following the 1000 steps minimization, we run another 2000 steps minimization without restraints. We use a 12 Å layer of TIP3PBOX water molecules to explicitly consider the solvent. In the energy refinement, the negative charge in phosphate is neutralized by Na+. We use the command “addions” in AMBER 9 to add Na+ until the total charge of the whole system is zero (Case et al. 2006). The nonbonded interactions are cut at 12 Å. The energy minimization is performed with the sander of AMBER 9 (Pearlman et al. 1995; Case et al. 2005, 2006). In the calculation, we use the AMBER force field version ff99 for RNA (Cornell et al. 1995; Wang et al. 2000). We use the standard input parameters to run the minimization with and without restraints (see the Supplemental Tables 1, 2). In the input, we set ntb = 1 to turn the Particle Mesh Ewald (PME) method on.
We note that the minimization does not cause significant changes in the structure. The purpose of using AMBER minimization is to remove the clashes in the Vfold-predicted coarse-grained structural model (see Fig. 9B). The resultant refined structure (Fig. 9B) has an all-atom root-mean-square deviation (RMSD) of 3.1 Å when we optimally superimposed on the relative NMR structure (Protein Data Bank [PBD] identification, 1xpe) (see Fig. 9D). In addition, we use the same template of Figure 9C to predict the 3D structure of HIV-1 type-f with an all-atom RMSD of 3.3Å (PDB structure, 1yxp). For the extended-duplex dimer (structure I on the energy landscape), using the same method, we can build the 3D structure with an RMSD of 2.9 Å (PDB structure, 462d) (see Fig. 9F; Ennifar et al. 1999). As a future development, either molecular dynamics simulation (Cheatham and Case 2006; Réblová et al. 2007; Sarzyńska et al. 2008) or elastic network modeling (Tirion 1996; Wang et al. 2004; Lu and Ma 2005; Yang et al. 2009) can be used to investigate the fluctuation dynamics of the predicted 3D structures. The dynamic information of the structures would be useful for us to understand the potential relationship between the RMSD ∼ 3 Å and the structural flexibility.
CONCLUSIONS
The reduced (virtual bond) conformational model for RNA allows us to compute the entropy parameters for RNA–RNA kissing complexes. Based on the entropy parameters for the loops/junctions and the nearest neighbor free energy model for the helices, we developed a statistical mechanical model to predict the free energy landscapes and structures from the nucleotide sequence. Tests with the experimental data show good theory-experiment agreements for the thermal stability (such as the melting temperatures).
Application of the theory to the free energy landscape and folding thermodynamics of HIV-1 DIS complex reveals two stable structures at room temperature, corresponding to the kissing-loop dimer and the extended-duplex dimer. In addition, our free energy landscape calculation supports the two-step dimerization process. Binding of protein (such as NCp7) and thermal heating can induce the conformational switch from the kissing-loop dimer to the extended-duplex dimer. Furthermore, using a multiscale approach, we can build the 3D structures for the kissing-loop dimer and extended-duplex dimer. Comparisons with the experimental structural data show a good RMSD of ∼3.0 Å.
Though the theory can treat kissing interactions for RNA–RNA complexes, it is limited by the inability to treat more complex tertiary interactions. For instance, OxyS is a small RNA, which can regulate the gene expression of fhlA. The repression of fhlA is mediated by a complex tertiary interaction between OxyS and fhlA (Argaman and Altuvia 2000). However, the current theory cannot treat for the tertiary interaction in OxyS/fhlA complex. Further development of the current model should include a theory to treat more complex RNA and RNA interactions, such as the ones found in OxyS-fhlA complex.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This research was supported by NIH grant GM063732 and NSF grants MCB0920067 and MCB0920411. Most of the numerical calculations involved in this research were performed on the HPC resources at the University of Missouri Bioinformatics Consortium (UMBC).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.026658.111.
REFERENCES
- Andronescu M, Zhang Z, Condon A 2005. Secondary structure prediction of interacting RNA molecules. J Mol Biol 345: 987–1001 [DOI] [PubMed] [Google Scholar]
- Andronescu MS, Pop C, Condon A 2010. Improved free energy parameters for RNA pseudoknotted secondary structure prediction. RNA 16: 26–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaman L, Altuvia S 2000. fhlA repression by OxyS RNA: kissing complex formation at two sites results in a stable antisense-target RNA complex. J Mol Biol 300: 1101–1112 [DOI] [PubMed] [Google Scholar]
- Arnott S, Hukins DWL 1972. Optimised parameters for RNA double-helices. Biochem Biophys Res Commun 48: 1392–1399 [DOI] [PubMed] [Google Scholar]
- Bartel DP 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297 [DOI] [PubMed] [Google Scholar]
- Bernhart SH, Tafer H, Muckstein U, Flamm C, Stadler PF, Hofacker IL 2006. Partition function and base pairing probabilities of RNA heterodimers. Algorithms Mol Biol 1: 3 doi: 10.1186/1748-7188-1-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunel C, Marquet R, Romby P, Ehresmann C 2002. RNA loop–loop interactions as dynamic functional motifs. Biochimie 84: 925–944 [DOI] [PubMed] [Google Scholar]
- Cao S, Chen S-J 2005. Predicting RNA folding thermodynamics with a reduced chain representation model. RNA 11: 1884–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen S-J 2006a. Free energy landscapes of RNA/RNA complexes: with applications to snRNA complexes in spliceosomes. J Mol Biol 357: 292–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen S-J 2006b. Predicting RNA pseudoknot folding thermodynamics. Nucleic Acids Res 34: 2634–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen S-J 2009. Predicting structures and stabilities for H-type pseudoknots with interhelix loops. RNA 15: 696–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen S-J 2011. Physics-based de novo prediction of RNA 3D structures. J Phys Chem B 115: 4216–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Cheatham TE III, Darden T, Gohlke H, Luo R, Merz KM Jr, Onufriev A, Simmerling C, Wang B, Woods RJ 2005. The Amber biomolecular simulation programs. J Comput Chem 26: 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE III, Simmerling J, Wang RE, Duke R, Luo KM, Merz KM, Pearlman DA, Crowley M, et al. 2006. AMBER 9, University of California, San Francisco [Google Scholar]
- Cheatham TE III, Case DA 2006. Using Amber to simulate DNA and RNA. In Computational studies of DNA and RNA (ed. Sponer J, Lankas F), pp. 45–72 Springer, Dordrecht [Google Scholar]
- Chen S-J 2008. RNA folding: Conformational statistics, folding kinetics, and ion electrostatics. Annu Rev Biophys 37: 197–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S-J, Dill KA 1995. Statistical thermodynamics of double-stranded polymer molecules. J Chem Phys 103: 5802–5813 [Google Scholar]
- Chen S-J, Dill KA 1998. Theory for the conformational changes of double-stranded chain molecules. J Chem Phys 109: 4602–4616 [Google Scholar]
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA 1995. A second generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J Am Chem Soc 117: 5179–5197 [Google Scholar]
- Das R, Baker D 2007. Automated de novo prediction of native-like RNA tertiary structures. Proc Natl Acad Sci 104: 14664–14669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didiano D, Hobert O 2006. Perfect seed pairing is not a generally reliable predictor for miRNA-target interactions. Nat Struct Mol Biol 13: 849–851 [DOI] [PubMed] [Google Scholar]
- Dill KA 1990. Dominant forces in protein folding. Biochemistry 29: 7133–7155 [DOI] [PubMed] [Google Scholar]
- Dimitrov RA, Zuker M 2004. Prediction of hybridization and melting for double-stranded nucleic acids. Biophys J 87: 215–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, Sharma S, Chalasani P, Demidov VV, Broude N, Dokholyan NV 2008. Ab initio RNA folding by discrete molecular dynamics: From structure prediction to folding mechanisms. RNA 14: 1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks RM, Bois JS, Schaeffer JM, Winfree E, Pierce NA 2007. Thermodynamic analysis of interacting nucleic acid strands. SIAM Rev 49: 65–88 [Google Scholar]
- Do CB, Woods DA, Batzoglou S 2006. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics 22: e90–e98 [DOI] [PubMed] [Google Scholar]
- Duarte CM, Pyle AM 1998. Stepping through an RNA structure: a novel approach to conformational analysis. J Mol Biol 284: 1465–1478 [DOI] [PubMed] [Google Scholar]
- Ennifar E, Yusupov M, Walter P, Marquet R, Ehresmann B, Ehresmann C, Dumas P 1999. The crystal structure of the dimerization initiation site of genomic HIV-1 RNA reveals an extended duplex with two adenine bulges. Structure 7: 1439–1449 [DOI] [PubMed] [Google Scholar]
- Ennifar E, Walter P, Ehresmann B, Ehresmann C, Dumas P 2001. Crystal structures of coaxially stacked kissing complexes of the HIV-1 RNA dimerization initiation site. Nat Struct Biol 8: 1064–1068 [DOI] [PubMed] [Google Scholar]
- Ferro DR, Hermans J 1971. A different best rigid-body molecular fit routine. Acta Crystallogr A 33: 345–347 [Google Scholar]
- Freier SM, Kierzek R, Jaeger JA, Sugimoto N, Caruthers MH, Neilson T, Turner DH 1986. Improved free-energy parameters for predictions of RNA duplex stability. Proc Natl Acad Sci 83: 9373–9377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenleaf WJ, Frieda KL, Foster DAN, Woodside MT, Block SM 2008. Direct observation of hierarchical folding in single riboswitch aptamers. Science 319: 630–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FWD, Qin J, Reidys CM, Stadler PF 2009. Partition function and base pairing probabilities for RNA-RNA interaction prediction. Bioinformatics 25: 2646–2654 [DOI] [PubMed] [Google Scholar]
- Isambert H, Siggia ED 2000. Modeling RNA folding paths with pseudoknots: application to hepatitis delta virus ribozyme. Proc Natl Acad Sci 97: 6515–6520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossinet F, Paillart J-C, Westhof E, Hermann T, Skripkin E, Lodmell JS, Ehresmann C, Ehresmann B, Marquet R 1999. Dimerization of HIV-1 genomic RNA of subtypes A and B: RNA loop structure and magnesium binding. RNA 9: 1222–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb FA, Malmgren C, Westhof E, Ehresmann C, Ehresmann B, Wagner EG, Romby P 2000a. An unusual structure formed by antisense-target RNA binding involves an extended kissing complex with a four-way junction and a side-by-side helical alignment. RNA 6: 311–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb FA, Engdahl HM, Slagter-Jäger J, Ehresmann B, Ehresmann C, Westhof E, Wagner EG, Romby P 2000b. Progression of a loop–loop complex to a four-way junction is crucial for the activity of a regulatory antisense RNA. EMBO J 19: 5905–5915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb FA, Westhof E, Ehresmann B, Ehresmann C, Wagner EG, Romby P 2001a. Four-way junctions in antisense RNA-mRNA complexes involved in plasmid replication control: a common theme? J Mol Biol 309: 605–614 [DOI] [PubMed] [Google Scholar]
- Kolb FA, Westhof E, Ehresmann C, Ehresmann B, Wagner EG, Romby P 2001b. Bulged residues promote the progression of a loop–loop interaction to a stable and inhibitory antisense-target RNA complex. Nucleic Acids Res 29: 3145–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopeikin Z, Chen S-J 2006. Folding thermodynamics of pseudoknotted chain conformations. J Chem Phys 124: 154903 doi: 10.1063/1.2188940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC 2003. microRNAs: Runts of the genome assert themselves. Curr Biol 13: R925–R936 [DOI] [PubMed] [Google Scholar]
- Laederach A 2007. Informatics challenges in structured RNA. Brief Bioinform 8: 294–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughrea M, Jetté L 1994. A 19-nucleotide sequence upstream of the 5′ major splice donor is part of the dimerization domain of human immunodeficiency virus 1 genomeric RNA. Biochemistry 33: 13464–13474 [DOI] [PubMed] [Google Scholar]
- Lebars I, Legrand P, Aimé A, Pinaud N, Fribourg S, Di Primo C 2008. Exploring TAR-RNA aptamer loop–loop interaction by X-ray crystallography, UV spectroscopy and surface plasmon resonance. Nucleic Acids Res 36: 7146–7156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB 2003. Prediction of mammalian microRNA targets. Cell 115: 787–798 [DOI] [PubMed] [Google Scholar]
- Li PTX, Bustamante C, Tinoco I Jr 2006. Unusual mechanical stability of a minimal RNA kissing complex. Proc Natl Sci Acad 43: 15847–15852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li PTX, Vieregg J, Tinoco I Jr 2008. How RNA unfolds and refolds. Annu Rev Biochem 77: 77–100 [DOI] [PubMed] [Google Scholar]
- Liu L, Chen S-J 2010. Computing the conformational entropy for RNA folds. J Chem Phys 132: 235104 doi: 10.1063/1.3447385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long D, Lee R, Williams P, Chan CY, Ambros V, Ding Y 2007. Potent effect of target structure on microRNA function. Nat Struct Mol Biol 14: 287–294 [DOI] [PubMed] [Google Scholar]
- Lorenz C, Piganeau N, Schroeder R 2006. Stabilities of HIV-1 DIS type RNA loop–loop interactions in vitro and in vivo. Nucleic Acids Res 34: 334–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Ma J 2005. The role of shape in determining molecular motions. Biophys J 89: 2395–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZJ, Gloor JW, Mathews DH 2009. Improved RNA secondary structure prediction by maximizing expected pair accuracy. RNA 15: 1805–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C 1992. A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell 71: 803–817 [DOI] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C 1994. Dynamic RNA-RNA interactions in the spliceosome. Annu Rev Genet 28: 1–26 [DOI] [PubMed] [Google Scholar]
- Mathews DH, Burkard ME, Freier SM, Wyatt JR, Turner DH 1999. Predicting oligonucleotide affinity to nucleic acid targets. RNA 5: 1458–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F, Westhof E 1990. Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J Mol Biol 216: 585–610 [DOI] [PubMed] [Google Scholar]
- Mitrovich QM, Guthrie C 2007. Evolution of small nuclear RNAs in S. cerevisiae, C. albicans, and other hemiascomycetous yeasts. RNA 13: 2066–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montange RK, Batey RT 2008. Riboswitches: emerging themes in RNA structure and function. Annu Rev Biophys 37: 117–133 [DOI] [PubMed] [Google Scholar]
- Mujeeb A, Clever JL, Billeci TM, James TL, Parslow TG 1998. Structure of the dimer initiation complex of HIV-1 genomic RNA. Nat Struct Biol 5: 432–436 [DOI] [PubMed] [Google Scholar]
- Mujeeb A, Parslow TG, Zarrinpar A, Das C, James TL 1999. NMR structure of the mature dimer initiation complex of HIV-1 genomic RNA. FEBS Lett 458: 387–392 [DOI] [PubMed] [Google Scholar]
- Muriaux D, De Rocquigny H, Roques BP, Paoletti J 1996a. NCp7 activates HIV-1Lai RNA dimerization by converting a transient loop–loop complex into a stable dimer. J Biol Chem 271: 33686–33692 [DOI] [PubMed] [Google Scholar]
- Muriaux D, Fossé P, Paoletti J 1996b. A kissing complex together with a stable dimer is involved in the HIV-1(Lai) RNA dimerization process in vitro. Biochemistry 35: 5075–5082 [DOI] [PubMed] [Google Scholar]
- Nagel JHA, Pleij CWA 2002. Self-induced structural switches in RNA. Bichimie 84: 913–923 [DOI] [PubMed] [Google Scholar]
- Olson WK 1980. Configurational statistics of polynucleotide chains: an updated virtual bond model to treat effects of base stacking. Macromolecules 13: 721–728 [DOI] [PubMed] [Google Scholar]
- Paillart J-C, Skripkin E, Ehresmann B, Ehresmann C, Marquet R 1996. A loop–loop “kissing” complex is the essential part of the dimer linkage of genomic HIV-1 RNA. Proc Natl Acad Sci 93: 5572–5577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillart JC, Shehu-Xhilaga M, Marquet R, Mak J 2004. Dimerization of retroviral RNA genomes: An inseparable pair. Nat Rev Microbiol 2: 461–472 [DOI] [PubMed] [Google Scholar]
- Parisien M, Major F 2008. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 452: 51–55 [DOI] [PubMed] [Google Scholar]
- Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Debolt S, Ferguson D, Seibel G, Kollman P 1995. AMBER: a package of computer-programs for applying molecular mechanics, normal-mode analysis, molecular-dynamics and free-energy calculations to stimulate the structural and energetic properties of molecules. Comput Phys Commun 91: 1–41 [Google Scholar]
- Réblová K, Fadrná E, Sarzynska J, Kulinski T, Kulhánek P, Ennifar E, Koča J, Šponer J 2007. Conformations of flanking bases in HIV-1 RNA DIS kissing complexes studied by molecular dynamics. Biophys J 93: 3932–3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmsmeier M, Steffen P, Höchsmann M, Giegerich R 2004. Fast and effective prediction of microRNA/target duplexes. RNA 10: 1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rother M, Rother K, Puton T, Bujnicki JM 2011. ModeRNA: a tool for comparative modeling of RNA 3D structure. Nucleic Acid Res 39: 4007–4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RS, Liang C, Wainberg MA 2004. Is HIV-1 RNA dimerization a prerequisite for packaging? Yes, no, probably? Retrovirology 1: 23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SantaLucia J Jr, Hicks D 2004. The thermodynamics of DNA structural motifs. Annu Rev Biophys Biomol Struct 33: 415–440 [DOI] [PubMed] [Google Scholar]
- Sarzyńska J, Réblová K, Šponer J 2008. Conformational transitions of flanking purines in HIV-1 RNA dimerization initiation site kissing complexes studied by CHARMM explicit solvent molecular dynamics. Biopolymer 89: 732–746 [DOI] [PubMed] [Google Scholar]
- Sashital DG, Cornilescu G, Butcher SE 2004. U2-U6 RNA folding reveals a group II intron-like domain and a four-helix junction. Nat Struct Mol Biol 11: 1237–1242 [DOI] [PubMed] [Google Scholar]
- Sashital DG, Venditti V, Angers CG, Cornilescu G, Butcher SE 2007. Structure and thermodynamics of a conserved U2 snRNA domain from yeast and human. RNA 13: 328–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultes EA, Bartel DP 2000. One sequence, two ribozymes: Implications for the emergence of new ribozyme folds. Science 289: 448–452 [DOI] [PubMed] [Google Scholar]
- Shapiro BA, Yingling YG, Kasprzak W, Bindewald E 2007. Bridging the gap in RNA structure prediction. Curr Opin Struct Biol 17: 157–165 [DOI] [PubMed] [Google Scholar]
- Skripkin E, Paillart J-C, Marquest R, Ehresmann B, Ehresmann C 1994. Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc Natl Acad Sci 91: 4945–4949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperschneider J, Datta A 2010. DotKnot: pseudoknot prediction using the probability dot plot under a refined energy model. Nucleic Acids Res 38: e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperschneider J, Datta A, Wise MJ 2011. Heuristic RNA pseudoknot prediction including intramolecular kissing hairpins. RNA 17: 27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi KI, Baba S, Chattopadhyay P, Koyanagi Y, Yamamoto N, Takaku H, Kawai G 2000. Structural requirement for the two-step dimerization of human immunodeficiency virus type 1 genome. RNA 6: 96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Baba S, Hayashi Y, Koyanagi Y, Yamamoto N, Takaku H, Kawai G 2005. NMR analysis of intra- and inter-molecular stems in the dimerization initiation site of the HIV-1 genome. J Biochem 138: 583–592 [DOI] [PubMed] [Google Scholar]
- Tan RKZ, Petrov AS, Harvey SC 2006. YUP: A molecular simulation program for coarse-grained and multiscaled models. J Chem Theory Comput 2: 529–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirion M 1996. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys Rev Lett 77: 1905–1908 [DOI] [PubMed] [Google Scholar]
- Tucker BJ, Breaker RR 2005. Riboswitches as versatile gene control elements. Curr Opin Struct Biol 15: 342–348 [DOI] [PubMed] [Google Scholar]
- Ulyanov NB, Mujeeb A, Du Z, Tonelli M, Parslow TG, James TL 2006. NMR structure of the full-length linear dimer of stem-loop-1 RNA in the HIV-1 dimer initiation site. J Biol Chem 281: 16168–16177 [DOI] [PubMed] [Google Scholar]
- Valadlkhan S 2007. The spliceosome: a ribozyme at heart? Biol Chem 388: 693–697 [DOI] [PubMed] [Google Scholar]
- Walter AE, Turner DH 1994. Sequence dependence of stability for coaxial stacking of RNA helixes with Watson-Crick base paired interfaces. Biochemistry 33: 12715–12719 [DOI] [PubMed] [Google Scholar]
- Wang JM, Cieplak P, Kollman PA 2000. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem 21: 1049–1074 [Google Scholar]
- Wang Y, Rader AJ, Bahar I, Jernigan RL 2004. Global ribosome motions revealed with elastic network model. J Struct Biol 147: 302–314 [DOI] [PubMed] [Google Scholar]
- Weixlbaumer A, Werner A, Flamm C, Westhof E, Schroeder R 2004. Determination of thermodynamic parameters for HIV DIS type loop–loop kissing complexes. Nucleic Acids Res 32: 5126–5133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhof E, Masquida B, Jossinet F 2011. Predicting and modeling RNA architecture. Cold Spring Harb Perspect Biol. 3: a003632 doi: 10.1101/cshperspect.a003632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickiser JK, Cheah MT, Breaker RR, Crothers DM 2005. The kinetics of ligand binding by an adenine-sensing riboswitch. Biochemistry 44: 13404–13414 [DOI] [PubMed] [Google Scholar]
- Xia TB, SantaLucia J Jr, Burkard ME, Kierzek R, Schroeder SJ, Jiao XQ, Cox C, Turner DH 1998. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry 37: 14719–14735 [DOI] [PubMed] [Google Scholar]
- Yang L, Song G, Jernigan RL 2009. Protein elastic network models and the ranges of cooperativity. Proc Natl Acad Sci 106: 12347–12352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WB, Chen S-J 2001. Predicting free energy landscapes for complexes of double stranded chain molecules. J Chem Phys 114: 4253–4266 [Google Scholar]