

Abstract
The c-fes proto-oncogene encodes a unique non-receptor protein-tyrosine kinase (c-Fes) that contributes to the differentiation of myeloid hematopoietic, vascular endothelial, and some neuronal cell types. Although originally identified as the normal cellular homolog of the oncoproteins encoded by avian and feline transforming retroviruses, c-Fes has recently been implicated as a tumor suppressor in breast and colonic epithelial cells. Structurally, c-Fes consists of a unique N-terminal region harboring an FCH domain, two coiled-coil motifs, a central SH2 domain, and a C-terminal kinase domain. In living cells, c-Fes kinase activity is tightly regulated by a mechanism that remains unclear. Previous studies have established that c-Fes forms high molecular weight oligomers in vitro, suggesting that the dual coiled-coil motifs may regulate the interconversion of inactive monomeric and active oligomeric states. Here we show for the first time that c-Fes forms oligomers in live cells independently of its activation status using a YFP bimolecular fluorescence complementation assay. We also demonstrate that both N-terminal coiled-coil regions are essential for c-Fes oligomerization in transfected COS-7 cells as well as HCT 116 colorectal cancer and K-562 myeloid leukemia cell lines. Together, these data provide the first evidence that c-Fes, unlike c-Src, c-Abl and other non-receptor tyrosine kinases, is constitutively oligomeric in both its repressed and active states. This finding suggests that conformational changes, rather than oligomerization, may govern its kinase activity in vivo.
The human c-fes proto-oncogene encodes a structurally unique, 93 kDa non-receptor protein-tyrosine kinase (c-Fes) expressed in myeloid hematopoietic, vascular endothelial, and some neuronal cells where it has been linked to signaling pathways controlling differentiation (1,2). Early work showed that restoring wild-type c-Fes expression in the chronic myelogenous leukemia cell line K-562 suppresses cell proliferation and primes the cells for differentiation to macrophages by phorbol esters (3,4). Similarly, active c-Fes mutants induced GM-CSF-independent proliferation in addition to cell attachment and spreading in the cytokine-dependent myeloid leukemia cell line TF-1, consistent with differentiation along the monocyte-macrophage pathway (5). In the monocytic precursor cell line U-937, an active c-Fes mutant also induced cell adherence, macrophage morphology, and differentiation marker expression (6). Over-expression of wild-type c-Fes accelerated NGF-induced neurite extension in PC12 cells, while active c-Fes mutants induced spontaneous neurite formation in this cell line, suggestive of a role in neuronal differentiation (7,8). Finally, over-expression of wild-type c-Fes induced FGF-2-independent tube formation by cultured brain capillary endothelial cells, implicating c-Fes in angiogenesis (9).
More recently, c-Fes expression has been detected in epithelial cells, where it may serve a tumor suppressor function. Greer and colleagues determined that tumor onset in a breast cancer model occurred more rapidly in mice targeted with either null or kinase-inactivating c-fes mutations, and that a c-fes transgene restored the latency of tumor formation (10). Our group found that c-Fes protein is strongly expressed in normal human colonic epithelial cells, while expression was reduced or absent in 67% of colon tumor sections from the same individuals (11). In addition, we have established that the expression of functional c-fes transcripts is downregulated in colorectal cancer cell lines via promoter methylation (12). Furthermore, re-expression of wild-type or active c-Fes suppressed anchorage-independent growth of two colorectal cancer cell lines, HCT 116 and HT-29, both of which are negative for c-Fes protein expression (11). Kinase-inactivating mutations have also been reported for c-Fes in colorectal cancer cell lines (10,11,13), providing further support for a tumor suppressor function for c-Fes in some tissue types.
Structurally, c-Fes consists of a long unique N-terminal region, with a tubulin-binding Fes/CIP4 homology (FCH) domain and two coiled-coil homology motifs, followed by a central Src-homology 2 (SH2) domain and a C-terminal kinase domain (1,2) (Figure 1). The c-Fes FCH and first coiled-coil domain together belong to the family of F-BAR homology domains, which have been linked to regulation of membrane curvature by other proteins (14). Unlike other non-receptor protein-tyrosine kinases, c-Fes lacks negative regulatory features such as an SH3 domain or the negative regulatory tail associated with Src-family kinases (15). Despite this, c-Fes kinase activity remains strictly regulated in mammalian cells. When wild-type c-Fes is ectopically expressed in rodent fibroblasts, little or no transforming activity is apparent (5,16,17). However, mutation or deletion of the first coiled-coil domain results in strong upregulation of kinase activity and release of transforming potential in fibroblasts (3,5). Similarly, en bloc substitution of the c-Fes SH2 domain with that of v-Src also causes loss of negative regulation in vitro (18), implicating the SH2 domain as well as the N-terminal coiled-coil motif in the regulation of kinase activity.
Figure 1.
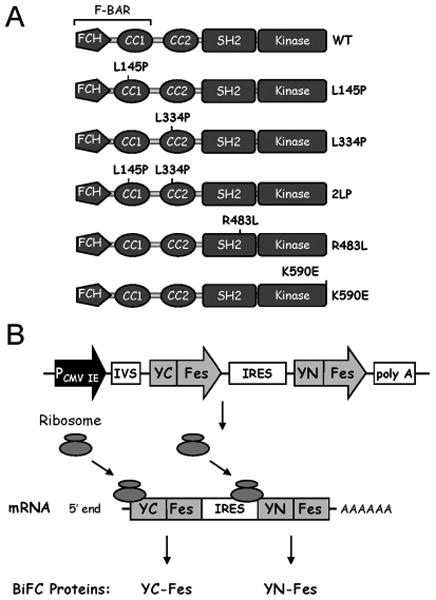
c-Fes constructs and BiFC experimental design. A) The structure of the wild-type (WT) c-Fes protein is shown at the top, which includes a unique N-terminal region harboring a Fes/CIP4 homology (FCH) domain, two coiled-coil homology motifs (CC1 and CC2), a central SH2 domain, and a C-terminal kinase domain. The FCH and CC1 domains together constitute an F-BAR domain. Coiled-coil domain mutants include leucine to proline substitutions in CC1 (L145P) and CC2 (L334P), as well as the corresponding double mutant (2LP). c-Fes proteins with inactivating mutations in the SH2 domain (R483L) as well as the kinase domain (K590E) are also shown. B) pIRES YFP BiFC system. The coding sequences for c-Fes were fused in frame with the nucleotide sequences of the N-terminal portion of YFP (encoding amino acids 1-154) to create the YN-Fes fusion protein and to the sequences of the C-terminal part of YFP (encoding amino acids 155-238) to create YC-Fes. The resulting c-Fes fusion proteins were subcloned into the pIRES vector as shown. Use of the IRES construct ensures simultaneous expression of both YN-Fes and YC-Fes BiFC partners from the same transcript as shown.
Gel-filtration and cross-linking studies have established that c-Fes forms higher order oligomers in vitro (up to a pentamer), and that the two N-terminal coiled-coil domains are responsible for oligomerization (5,19). In particular, gel-filtration analyses involving a small fragment of the N-terminal region showed that point mutations disrupting both coiled-coil motifs prevent oligomerization (5). This result led to a hypothetical model for c-Fes activation in which the coiled-coil domains mediate interconversion between inactive monomeric and active oligomeric states (5). In this model, the more N-terminal coiled-coil was proposed to interact intramolecularly with the second to hold c-Fes in an inactive monomeric conformation. Supporting this model is the observation that negative regulation imparted by the first coiled-coil domain is dramatically perturbed by mutation (19,20). However, no evidence for an inactive monomer has been generated either in vitro or in vivo, and it is unclear how oligomerization modulates c-Fes activity in living cells.
In this study, we investigated c-Fes oligomerization in live cells using a bimolecular fluorescence complementation (BiFC) assay based on yellow fluorescent protein (YFP) as originally developed by Kerppola and colleagues for the study of transcription factors (21,22). BiFC provides a useful technique to examine protein-protein interaction in a normal cellular environment, as it is sensitive enough to detect interactions between proteins expressed at physiological levels (21,22). In this approach, oligomerization partners (c-Fes in this case) are fused to non-fluorescent N- and C-terminal portions of the YFP coding sequence and co-expressed in the same cell. Oligomerization brings the two YFP fragments into close proximity, resulting in structural complementation and fluorescence. Using the BiFC technique, we made the surprising discovery that c-Fes exists as a constitutive oligomer in cells, regardless of its autophosphorylation state. We further demonstrated that both of the c-Fes coiled-coil regions are essential for oligomerization in transfected COS-7 cells as well as cell lines where c-Fes is known to exert biological effects (HCT 116 colorectal cancer cells and K-562 CML cells). Together, these data demonstrate that c-Fes oligomerization is independent of activation and suggest that conformational changes, rather than oligomerization, govern c-Fes kinase activation and downstream signaling in vivo. The discovery that c-Fes exists as a pre-formed oligomer in vivo makes it unique among non-receptor protein-tyrosine kinases, and suggests new strategies for the design of small molecules that may modulate its activity in vivo. Such compounds may be of utility for the differentiation therapy of certain types of tumors.
Experimental Procedures
Construction of plasmid vectors
Fes point mutants L145P, L334P, L145P-L334P (2LP), R483L, and K590E have been described elsewhere (5,7,20) (Figure 1A). To create the vectors required for BiFC analysis, sequences encoding the non-fluorescent N- and C-terminal portions of YFP (YN: amino acids 1-154, YC: amino acids 155-238) were amplified by PCR from pEYFP-C1 (Clontech) and subcloned into separate pcDNA3.1(+) vectors (Invitrogen). Full-length YFP was cloned in a similar manner for creation of YFP-Fes fusion plasmids. Each of the c-Fes cDNAs was fused to the C-terminal end of either YN or YC, creating YN-Fes and YC-Fes BiFC fusion pairs. Each BiFC fusion pair was subcloned from pcDNA3.1+ into the mammalian expression vector pIRES (Clontech). In this construct, YC-Fes was subcloned directly downstream of the CMV promoter with YN-Fes downstream of the IRES sequence (Figure 1B). This cloning strategy was also applied to the pIRES-Fes BiFC control plasmids, which either express YC-Fes or YN-Fes alone. All c-Fes constructs used in this study encode a C-terminal FLAG epitope tag.
Cell culture and transfection
Cell lines were maintained at 37 °C in a 5% CO2 humidified incubator. COS-7, HCT 116, and K-562 cells were obtained through the ATCC and cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen), McCoy's Modified 5A medium (Invitrogen), or RPMI 1640 (Invitrogen), respectively. All culture media were supplemented with 10% fetal bovine serum (FBS; Atlanta Biological) and Antibiotic/Antimycotic (Invitrogen). Transient transfection was performed as follows: COS-7 cells (2.25×105) were plated in 60 mm dishes and transfected one day later with 2 μg of total plasmid DNA using Fugene 6 (Roche). HCT 116 cells (6×105) were seeded in 6-well plates and transfected one day later with 4 μg of total plasmid DNA using Lipofectamine 2000. K-562 cells (6×105) were seeded in 12-well plates and transfected immediately with 2 μg of total plasmid DNA using Lipofectamine 2000 (Invitrogen). All transfections were performed using serum-free Opti-MEM (Invitrogen) as diluent, and transfected cells were grown in Antibiotic/Antimycotic-free medium. Following incubation at 37 °C for 20 h, transfected cells were switched to room temperature for 2 h to promote fluorophore maturation prior to fluorescence microscopy.
Immunofluorescence microscopy and fluorescence imaging
Transfected COS-7 or HCT 116 cells were fixed with 4% paraformaldehyde in PBS for 10 min followed by 2 washes with PBS and permeabilized with 0.2% Triton X-100 in PBS for 15 min. Cells were then blocked in PBS containing 2% BSA for 30 min and incubated for 60 min with either anti-Fes (1:250 dilution; Fes C19, Santa Cruz Biotechnology) or anti-Fes phosphospecific primary antibodies [pFes; 1:1,000 dilution; recognizes phosphotyrosine 713 in the activation loop (23)]. Immunostained cells were visualized with secondary antibodies conjugated to Alexa Fluor 594 (Invitrogen) or Texas red (Southern Biotech). Fluorescent images were recorded at 400× magnification using a Nikon TE300 inverted microscope with epifluorescence capability and a SPOT CCD high-resolution digital camera and software (Diagnostic Instruments). Images were recorded at 600× using either a Nikon TE2000 inverted microscope with epifluorescence capability (Figure 2) or an Olympus IX81 and with Fluoview software (Figure 4). Following acquisition of black & white images, appropriate color palettes were applied (green for YFP or BiFC, red for c-Fes protein, pTyr-713, or RFP), and minimal histogram, brightness, and contrast adjustments were performed to improve image clarity. Identical manipulations were applied to all images in a given experiment.
Figure 2.
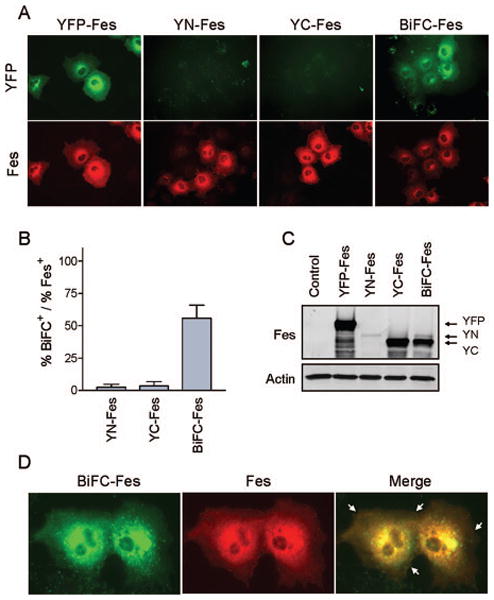
Wild-type c-Fes forms oligomers in living cells. COS-7 cells were transfected with plasmids encoding YFP-Fes, YN-Fes alone, YC-Fes alone, and YN-Fes + YC-Fes (BiFC-Fes). Twenty hours after transfection, cells were incubated at room-temperature for 2 hours and either fixed and immunostained with a c-Fes-specific antibody or lysed for immunoblot analysis. A) Fluorescent images of representative cells expressing YFP-Fes, YN-Fes, YC-Fes, and BiFC-Fes. YFP-Fes, YN-Fes, YC-Fes, and BiFC-Fes proteins were examined for YFP fluorescence (top row). Secondary antibodies conjugated to Alexa Fluor 594 were used to visualize transfected cells immunoreactive for the c-Fes antibody (bottom row). YFP/BiFC and Alexa Fluor 594 are represented as green and red, respectively. B) BiFC fluorescence intensity was normalized to the total c-Fes fluorescence intensity as determined by antibody staining using Metamorph (version 7.5.6). Each experiment was repeated in triplicate and the results are presented as the mean ratios ± S.D. C) Lysates from the transfected cell populations shown in part A were analyzed by immunoblotting with antibodies to c-Fes (top) and to actin as a loading control (bottom). The arrows indicate the positions of the YFP-Fes (YFP), YN-Fes (YN), and YC-Fes (YC) fusion proteins. D) High magnification images (600×) reveal subcellular localization of BiFC-Fes. BiFC-Fes proteins were examined for YFP fluorescence (left panel) and immunoreactivity to a c-Fes antibody (center panel); a merged image is shown as well (right panel).
Figure 4.
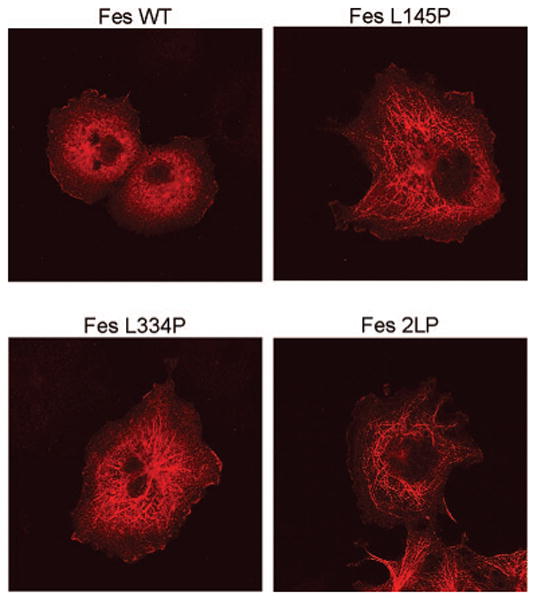
Coiled-coil domain mutations do not alter plasma membrane localization of c-Fes. Wild-type c-Fes, as well as the L145P, L334P, and 2LP coiled-coil domain mutants were transiently expressed in COS-7 cells as YFP fusion proteins. Twenty hours later, cells were fixed and stained with a c-Fes-specific antibody. Secondary antibodies conjugated to Alexa Fluor 594 were used to visualize c-Fes-positive cells. Representative images are shown.
Immunoblotting and antibodies
Transiently transfected COS-7, K-562, and HCT 116 cells were washed with PBS, resuspended in Fes lysis buffer [50 mM Tris/HCl (pH 7.4), 1 mM EDTA, 50 mM NaCl, 1 mM MgCl2, 0.1% Triton X-100, 2 mM PMSF, 2 mM sodium orthovanadate, 25 mM sodium fluoride and Protease Inhibitor Cocktail Set III (Calbiochem)], and sonicated for 10 s at 4 °C. The cell lysates were clarified by centrifugation, diluted with 2× SDS-PAGE sample buffer (125 mM Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 172 mM 2-mercaptoethanol, 0.05% bromophenol blue), and heated to 95 °C for 10 min. Lysates were subjected to immunoblot analysis with antibodies to c-Fes (C-19; 1 μg/ml), pFes (1:1000 dilution), phosphotyrosine (PY99; 1 μg/ml, Santa Cruz Biotechnology), and Hck (N30; 1 μg/ml, Santa Cruz Biotechnology) as required. Lysates were also blotted with anti-actin antibodies (Chemicon MAB1501; 1:10000 dilution) as a loading control. Immunoreactive bands were detected using an alkaline phosphatase-conjugated secondary antibody followed by colorimetric detection with NBT/BCIP.
Results
Visualization of c-Fes oligomers in live cells
Previous data from our laboratory suggested that c-Fes kinase activity may be regulated by interconversion of inactive monomeric and active oligomeric configurations (see Introduction) (3,5,19). These studies implied that wild-type c-Fes, whose kinase activity is strongly repressed in cells, naturally adopts a monomeric conformation, as is the case for the downregulated forms of several other non-receptor tyrosine kinases such as c-Abl (24,25), Hck (26,27), and c-Src (28,29). To determine the oligomerization status of wild-type c-Fes in live cells, we applied a BiFC approach in which complementary fragments of YFP (YN: amino acids 1-154, YC: amino acids 155-238) were fused to the amino-terminus of full-length wild-type c-Fes and subcloned into a single pIRES expression vector (Figure 1B). In parallel, we created a plasmid where full-length YFP was fused to c-Fes for use as a positive control for subcellular localization. COS-7 cells were transfected with plasmids encoding YFP-Fes, YN-Fes alone, YC-Fes alone, as well as both YN-Fes plus YC-Fes and monitored by fluorescence microscopy (Figure 2A). Transfected cultures were also immunostained with a Fes-specific antibody to identify the c-Fes-positive cells. The BiFC fluorescence intensity was then normalized to the total c-Fes fluorescence intensity in each culture (Figure 2B). Cells expressing YN-Fes or YC-Fes alone failed to exhibit detectable fluorescent signals. In contrast, more than 50% of the cells expressing both YN-Fes and YC-Fes fusion proteins exhibited strong cytoplasmic and perinuclear fluorescent signals, indicative of c-Fes oligomerization in vivo. Immunoblots shown in Figure 2C confirm expression of full-length YFP-Fes, YN-Fes, and YC-Fes. Note that YN-Fes is expressed at lower levels than YC-Fes, most likely because its translation is controlled by the IRES in the bicistronic transcript (Figure 1B). This observation shows that c-Fes oligomerization in vivo is not an artifact of over-expression, as the c-Fes BiFC signal is readily observed despite the limiting amount of YN-Fes protein present. These results demonstrate for the first time that wild-type c-Fes forms oligomers in living cells.
The diffuse cytoplasmic and perinuclear distribution of the c-Fes BiFC signal is similar to that observed with YFP-Fes, and is consistent with previous data for cells transfected with wild-type downregulated c-Fes (7,18,30,31). This observation suggests that the YN and YC fusions do not alter c-Fes subcellular localization. Imaging of the Fes BiFC-positive cells at higher magnification revealed that wild-type c-Fes oligomers also localized to the plasma membrane (Figure 2D), consistent with recent work demonstrating that the c-Fes F-BAR domain localizes to the cell periphery in transfected COS-7 cells (32).
The coiled-coil homology domains are essential for c-Fes oligomerization in vivo
Located within the unique N-terminal region of c-Fes are two coiled-coil motifs that mediate c-Fes oligomerization in vitro and play a key role in the regulation of c-Fes kinase activity in vivo (3,5,19). To assess the role of the coiled-coil motifs in c-Fes oligomerization in cells, BiFC assays were performed using coiled-coil leucine to proline point mutants previously shown to disrupt the function of these domains (5) (Figure 1A). Expression vectors that co-express both YN-Fes and YC-Fes coiled-coil mutants from the same transcript were generated as described above, and tested for oligomerization via BiFC in COS-7 cells. As shown in Figure 3, coiled-coil domain mutations (L145P, L334P, or 2LP) dramatically reduced the Fes-BiFC fluorescence intensity, indicating a requirement for both coiled-coil domains in maintaining c-Fes oligomerization in vivo. These results also provide an essential control for the wild-type c-Fes BiFC result, as they show that mutations in the coiled-coil domains of c-Fes previously established to contribute to oligomerization in vitro are required for fluorescence complementation in cells. Thus homotypic c-Fes interactions, and not the YFP fragments themselves, are the driving force behind the BiFC signal. A final interesting feature of the coiled-coil mutants is that while they fail to form oligomers in vivo (negative BiFC signal), they all show a staining pattern consistent with localization to the microtubule network. This result suggests that association with microtubules does not require oligomerization, and is consistent with our previous data showing that c-Fes movement to microtubules is dependent upon both Fes-mediated tubulin phosphorylation and subsequent binding via the c-Fes SH2 domain (7). Indeed, immunoblots show that the YN- and YC-Fes coiled-coil domain mutants are strongly autophosphorylated, consistent with this idea (Figure 3C and Supplemental Figure S1).
Figure 3.
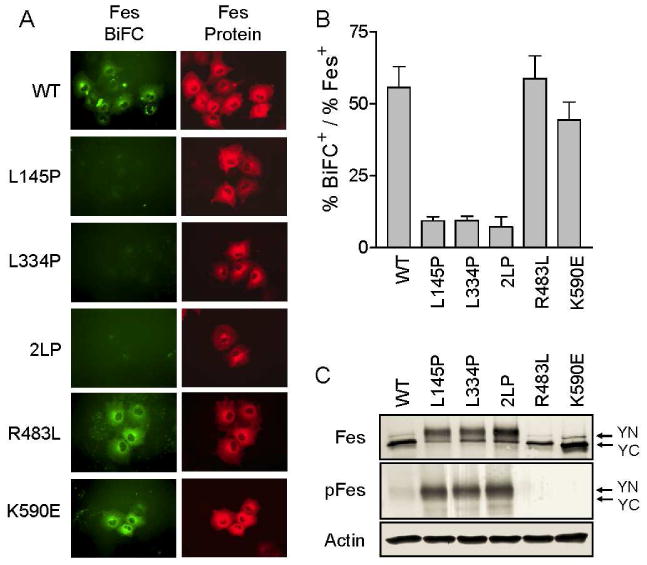
Coiled-coil homology domains mediate c-Fes oligomerization in vivo. Plasmids encoding wild-type, L145P, L334P, 2LP, R483L, and K590E c-Fes BiFC partners (YN and YC fusions; see Figure 1) were transiently expressed in COS-7 cells. Twenty-hours after transfection, cells were incubated at room-temperature for 2 hours and either fixed and stained with a c-Fes-specific antibody or lysed for immunoblot analysis. A) Representative fluorescent images of cells expressing the c-Fes BiFC partners for each of the point mutants shown as well as the wild-type (WT) control (top). Secondary antibodies conjugated to Alexa Fluor 594 were used to visualize c-Fes-positive immunostained cells. The BiFC signal and Alexa Fluor 594 fluorescence are represented as green and red, respectively. Images were recorded at 400× magnification. B) BiFC fluorescence intensity was normalized to the total c-Fes fluorescence intensity using Metamorph (version 7.5.6). Each experiment was repeated three times, and the results are presented as the mean ratios ± S.D. C) Lysates from the transfected cell populations shown in part A were analyzed by immunoblotting for c-Fes protein (top), c-Fes autophosphorylation (pFes; middle) and actin as a loading control (bottom). The arrows indicate the positions of the YN-Fes (YN), and YC-Fes (YC) fusion proteins.
In addition to the coiled-coil domains, we also assessed the contribution of the c-Fes SH2 and kinase domains to oligomerization using mutant proteins. Unlike the coiled-coil mutants, inactivating mutations in the SH2 domain (R483L) or kinase domain (K590E) did not alter the capacity of c-Fes to form oligomers, as both mutants produced BiFC signals with intensities similar to wild-type c-Fes. These mutants exhibited a diffuse cytoplasmic distribution and failed to track to microtubules, consistent with lack of kinase activity in vivo. Immunoblots shown in Figure 3C are consistent with this conclusion, as neither of these mutants reacted with the c-Fes phosphospecific antibody. Taken together, these results strongly suggest that both c-Fes coiled-coil domains, but neither the SH2 nor the kinase domain, are required for c-Fes oligomerization in living cells.
Coiled-coil mutations do not alter membrane localization of c-Fes
Data presented in Figure 3 directly implicate the coiled-coil domains as mediators of c-Fes oligomerization in vivo. Because the coiled-coil mutations are located near amino acid residues critical for phosphoinositide binding within the F-BAR domain, we investigated whether these mutations affected c-Fes membrane localization (32). COS-7 cells expressing wild-type Fes as well as the L145P, L334P, and 2LP coiled-coil domain mutants were immunostained with a c-Fes-specific antibody and imaged by confocal microscopy. As shown in Figure 4, wild-type Fes as well as each of the coiled-coil mutants localized to the cell periphery. This observation suggests that disruption of the coiled-coil domains did not alter the integrity of the F-BAR domain in terms of its membrane-targeting function. In addition, the images clearly show that mutation of the coiled-coil domains resulted in localization of c-Fes to the microtubule network, similar to what was observed with the BiFC constructs of the Fes coiled-coil domain mutants (Figure 2). One possible caveat is that membrane-targeting mechanisms may be distinct between wild-type and mutant Fes proteins. Based on this analysis alone, we cannot rule out that F-BAR/phosphoinositide binding may be affected by coiled-coil mutations and that these changes may be complemented by a targeting mechanism unique to these monomeric PTKs.
Activation does not influence wild-type c-Fes oligomerization
Results presented in Figure 2 strongly suggest that c-Fes naturally adopts an oligomeric conformation in vivo. To evaluate the effect of kinase activation on c-Fes oligomerization in living cells, the YFP-Fes and BiFC-Fes plasmids used in Figure 2 were co-transfected with a plasmid encoding an active form of Hck (Hck-YF) (33). Hck is a member of the Src kinase family expressed in myeloid cells that has been previously demonstrated to activate wild-type c-Fes in vivo (7,9). To identify cells expressing active c-Fes, the transfected cultures were also immunostained with a phosphospecific antibody (pFes) that recognizes autophosphorylation of Tyr-713 in the c-Fes activation loop (23). As shown in Figure 5A, YFP-Fes alone exhibited a diffuse cytoplasmic localization and only a trace amount of staining with the phosphospecific antibody. On the other hand, co-expression with Hck-YF induced strong YFP-Fes activation as judged by pFes antibody staining. In addition, active YFP-Fes relocalized to the prominent microtubule network present in COS cells, consistent with our previous observations (7). Paralleling results for YFP-Fes, Figure 5B shows that cells co-expressing YN-Fes plus YC-Fes exhibited a strong BiFC signal but only a trace of autophosphorylation indicating that wild-type c-Fes oligomers are catalytically inactive. Co-expression with Hck-YF induced strong phosphorylation of the c-Fes oligomers at Tyr-713, as judged by phosphospecific antibody staining. In addition, activation of c-Fes by Hck-YF induced localization to microtubules, demonstrating that active c-Fes remains oligomeric in living cells and moves to microtubules as an oligomer. Immunoblot analysis confirmed that wild-type YFP-Fes, YN-Fes and YC-Fes are weakly autophosphorylated when expressed alone, but are robustly phosphorylated in the presence of Hck-YF (Figure 5C). These results illustrate that activation does not alter wild-type c-Fes oligomerization in living cells, as c-Fes remains oligomeric irrespective of kinase domain autophosphorylation.
Figure 5.
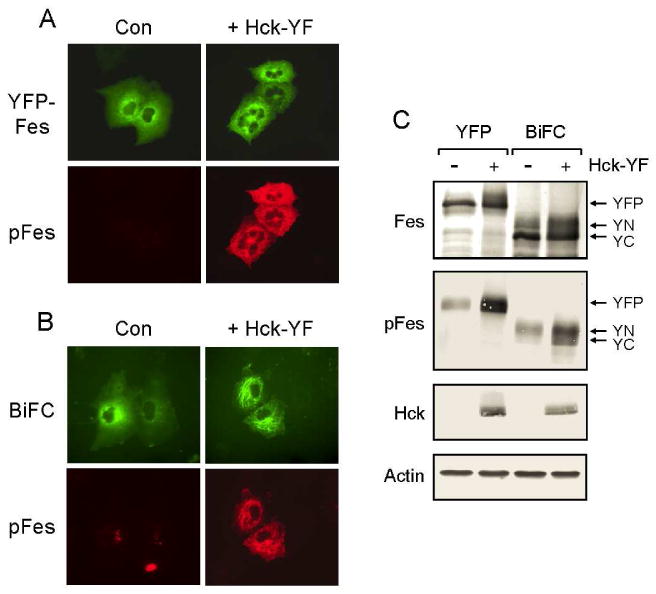
Active wild-type c-Fes remains oligomeric in vivo. Wild-type YFP-Fes and c-Fes BiFC fusion vectors were transiently expressed in COS-7 cells. Cells were also co-transfected with an active form of the Src family kinase Hck as indicated. The active form of Hck in these experiments has a phenylalanine substitution for the negative regulatory tyrosine in the C-terminal tail (Hck-YF). Twenty hours later, cells were incubated at room-temperature for 2 hours and either fixed and stained with c-Fes phosphospecific antibodies or lysed for immunoblot analysis. Note that cells expressing active Hck alone do not cross-react with the c-Fes phosphospecific antibody (data not shown). A) Representative fluorescent images of COS-7 cells expressing YFP-Fes plus and minus (Con) active Hck. Secondary antibodies conjugated to Texas Red were used to visualize pFes staining. YFP-Fes and Texas red (pFes) fluorescence are represented as green and red, respectively. Images were recorded at 400× magnification. B) Representative fluorescent images of COS-7 cells expressing BiFC-Fes (YN-Fes & YC-Fes) plus and minus (Con) Hck. The BiFC-Fes fluorescent signal (BiFC) and Texas red (pFes) fluorescence are represented as green and red, respectively. Images were recorded at 400× magnification. C) Immunoblot analyses of cell lysates from part A for c-Fes protein levels (Fes) and c-Fes autophosphorylation with the phosphospecific antibody (pFes); arrows indicate the position of YFP-Fes (YFP), YN-Fes (YN), and YC-Fes (YC). Blots were also performed for Hck protein expression and actin as a loading control.
c-Fes forms coiled-coil-mediated oligomers in human colorectal cancer and chronic myelogenous leukemia cell lines
Previous work has demonstrated that c-Fes is expressed in normal colonic epithelium as well as myeloid hematopoietic cells, and that loss of c-Fes expression correlates with tumor progression in both colorectal cancer and CML (4,11,34). Restoring c-Fes expression to HCT 116 colorectal cancer and K-562 CML cells results in a growth-suppressive effect (3,4,11). To determine whether c-Fes forms oligomeric complexes in these biologically relevant cellular contexts, BiFC analysis was performed using wild-type c-Fes and the double coiled-coil c-Fes mutant (2LP) as a negative control. Transient expression of the YN- and YC-Fes fusion proteins as well as the YFP-Fes control was achieved in HCT 116 cells using the pIRES vector system. Figure 6A shows that wild-type c-Fes exhibited strong BiFC-dependent fluorescence similar to the YFP-Fes control in HCT 116 cells. The diffuse cytoplasmic BiFC signal was observed in nearly 50% of the cells (Figure 6B), consistent with the results in COS7 cells. On the other hand, the 2LP coiled-coil mutant reduced the BiFC fluorescence intensity to less than 10% in HCT 116 cells (Figure 6B). Together, these results indicate that c-Fes forms oligomers in colorectal carcinoma cells in a coiled-coil-dependent fashion.
Figure 6.
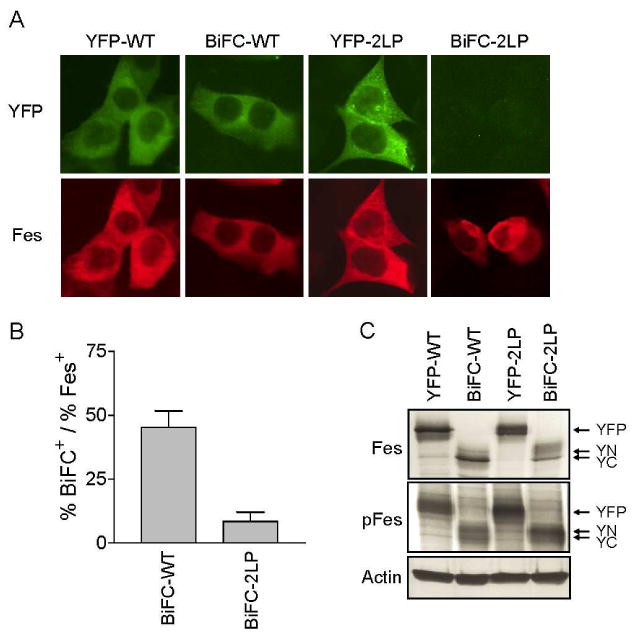
Coiled-coil-dependent oligomerization of c-Fes in HCT 116 colorectal carcinoma cells. HCT 116 cells were transfected with plasmids encoding wild-type c-Fes or the double coiled-coil domain mutant (2LP) as YFP fusions or as BiFC partners using the pIRES vector shown in Figure 1. Twenty hours after transfection, cells were incubated at room temperature for 2 hours and either fixed and stained with a c-Fes-specific antibody or lysed for immunoblot analysis. A) Representative fluorescent images of cells expressing wild-type YFP-Fes (YFP-WT) or the corresponding BiFC partners (BiFC-WT) as well as the YFP fusion of the coiled-coil domain double mutant (YFP-2LP) or the BiFC partners of this mutant (BiFC-2LP). Secondary antibodies conjugated to Alexa Fluor 594 were used to visualize Fes-positive immunostained cells. YFP/ BiFC and Alexa Fluor 594 fluorescence are represented as green and red, respectively. Images were recorded at 40× magnification. B) BiFC fluorescence intensity was normalized to the total c-Fes fluorescence intensity using Metamorph (version 7.5.6). Each experiment was repeated twice, and the results are presented as the mean ratios ± S.D. C) Lysates from the transfected cell populations shown in part A were analyzed by immunoblotting for c-Fes protein (top), c-Fes autophosphorylation (pFes; middle) and actin as a loading control (bottom). The arrows indicate the positions of the YFP-Fes (YFP), YN-Fes (YN), and YC-Fes (YC) fusion proteins.
Immunoblots were performed on HCT 116 cell lysates to verify the expression and activation status of the c-Fes fusion proteins. As shown in Figure 5C, the YFP, YN, and YC fusion proteins of both the wild-type and 2LP c-Fes proteins are clearly present. Interestingly, immunoblots of the same cell extracts with the c-Fes phosphospecific antibody (pFes) show that wild-type c-Fes is strongly phosphorylated on activation loop tyrosine residue Tyr-713. This is in contrast to transfected COS-7 cells, where wild-type c-Fes autophosphorylation is downregulated in comparison to the coiled-coil domains mutants [Figures 4 and 5; see also Ref. (7)]. Activation of wild-type c-Fes in transfected colon carcinoma cells may be due to direct phosphorylation by Src-family kinases, which are often constitutively active in colorectal cancer cells (35).
In K-562 CML cells, we were unable to detect translation of the YN-Fes protein from the pIRES plasmid (data not shown). To analyze c-Fes oligomerization in this cell line, therefore, separate expression vectors encoding the YN-Fes and YC-Fes fusion proteins were co-transfected together with an RFP expression plasmid to monitor transfection efficiency. As in previous experiments, wild-type and 2LP mutant YFP-Fes fusion proteins were included as controls. As shown in Figure 7A, K-562 cells co-expressing the wild-type c-Fes BiFC partners exhibited strong cytoplasmic fluorescence, similar to that observed with the YFP-Fes control. The BiFC signal intensity was observed in greater than 60% of the transfected cells (Figure 7B). In contrast, BiFC analysis of the Fes-2LP mutant reduced the fluorescence complementation intensity to nearly 10%, indicating that c-Fes oligomerization is coiled-coil-dependent in cells of myeloid lineage. Transfected cells were readily identified in the BiFC-2LP cell population, as indicated by strong fluorescence from the RFP control. Immunoblot analysis of cell lysates verified expression of the YN-, YC- and YFP-Fes fusion proteins in each of the transfected cell populations (Figure 7C). Immunoblots with the pFes phosphospecific antibody showed that wild-type YFP-Fes autophosphorylation was repressed in K-562 cells, but strongly activated by the double coiled-coil (2LP) mutation. In K-562 cells expressing the c-Fes BiFC partners, a low level of autophosphorylation was observed with wild-type c-Fes which was enhanced with the YN- and YC-Fes 2LP mutants, consistent with the results in COS-7 cells.
Figure 7.
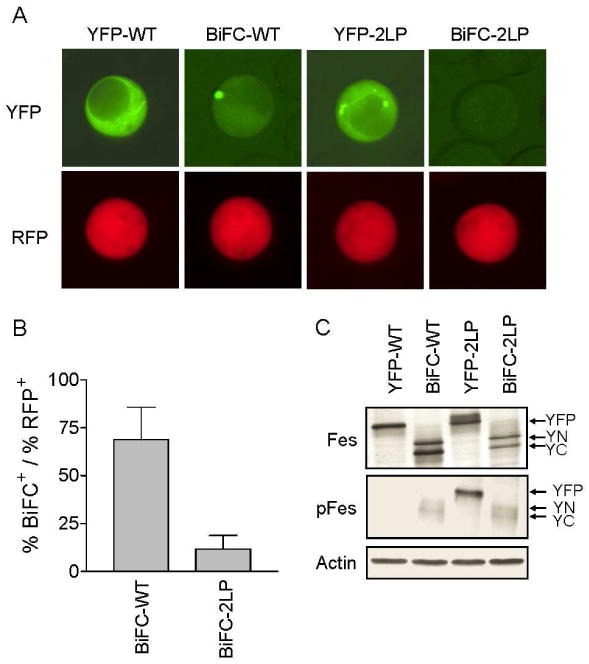
Coiled-coil-dependent oligomerization of c-Fes in K562 chronic myelogenous leukemia cells. K-562 cells were transfected with individual plasmids encoding wild-type c-Fes or the double coiled-coil domain mutant (2LP) as YFP fusions or as BiFC partners (YN-Fes + YC-Fes fusions). Cells were co-transfected with RFP as a marker for transfection efficiency. A) Representative fluorescent images of cells expressing wild-type YFP-Fes (YFP-WT) or the corresponding BiFC partners (BiFC-WT) as well as the YFP fusion of the coiled-coil domain double mutant (YFP-2LP) or the BiFC partners of this mutant (BiFC-2LP). YFP/BiFC and RFP fluorescence are represented as green and red, respectively. Images were recorded at 40× magnification. B) BiFC intensity was normalized to RFP intensity (a marker of transfection efficiency) using Metamorph version 7.5.6. Each experiment was repeated twice, and the results are presented as the mean ratios ± S.D. C) Lysates from the transfected cell populations shown in part A were analyzed by immunoblotting for c-Fes protein (left), c-Fes autophosphorylation (pFes; right top) and actin as a loading control (right bottom). The arrows indicate the positions of the YFP-Fes (YFP), YN-Fes (YN), and YC-Fes (YC) fusion proteins.
Oligomerization-defective Fes mutants transphosphorylate kinase-dead Fes
One additional mechanistic issue involves whether or not loss of oligomerization function, while resulting in kinase domain upregulation, affects intermolecular autophosphorylation. To test this, COS-7 cells were co-transfected with wild-type Fes or each of the three coiled-coil domain mutants together with a kinase-defective Fes mutant (Fes-KE) as a potential phosphoacceptor. N-terminal YFP fusions of the wild-type and coiled-coil domain Fes proteins were used to allow separation from Fes-KE by SDS-PAGE. Immunoblot analyses were performed with antibodies to the Fes protein to verify expression as well as the phosphospecific Fes antibody to detect phosphoryation of the kinase domain activation loop. As shown in Figure 8, all three of the active point mutants, despite lacking functional coiled-coil domains, were able to transphosphorylate Fes-KE. These data suggest that active Fes does not require functional coiled-coil domains to induce activation of other Fes molecules via trans-phosphorylation of the activation loop. This result is consistent with our observation that Src family kinases, which lack coiled-coil domains, can also activate Fes by direct transphosphorylation [e.g. Figure 5 and Refs. (7,9)] However, these data are still consistent with an important role for the coiled-coil domains in the negative regulation of Fes activity (see Discussion).
Figure 8.
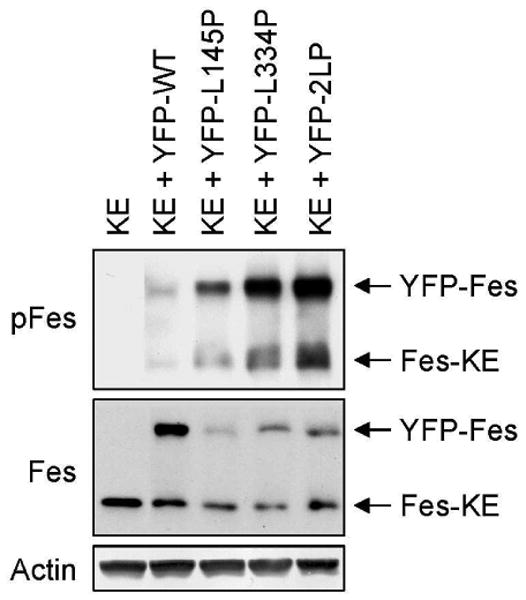
Oligomerization-defective Fes mutants transphosphorylate kinase-dead Fes. COS-7 cells were transfected with a kinase-defective Fes mutant in which the conserved arginine in the ATP-binding site was replaced with glutamate (KE) either alone or together with N-terminal YFP fusions of wild-type Fes (YFP-WT), the L145P mutant (YFP-L145P), the L334P mutant (YFP-L334P) or the double coiled-coil domain mutant (YFP-2LP). Lysates from the transfected cells were analyzed by immunoblotting for c-Fes activation loop tyrosine phosphorylation (pFes; top), Fes protein (Fes; middle) and actin as a loading control (bottom). The arrows indicate the positions of the YFP-Fes fusion proteins as well as Fes-KE. This experiment was repeated three times with comparable results.
Discussion
The non-receptor protein-tyrosine kinases encoded by the three classic tyrosine kinase proto-oncogenes, c-Src, c-Abl, and c-Fes, are strictly regulated with respect to kinase activity in vivo. In the case of the c-Src and c-Abl, extensive X-ray crystallographic studies have provided tremendous insight as to the structural mechanisms responsible for downregulation of kinase activity in vivo (15,36). In both cases, intramolecular interactions cause these kinases to adopt a monomeric, downregulated conformation. In contrast, no structural information is available for the full-length c-Fes kinase, and the mechanism responsible for suppression of its kinase activity in vivo remains unclear. Previous work from our laboratory has implicated the coiled-coil domains as key regulators of c-Fes kinase activity in vivo, and initially led us to a model in which c-Fes self-regulates its kinase activity through coiled-coil-mediated monomer (inactive) to oligomer (active) transition (3,5,23). Prior to our study, however, the oligomeric nature of c-Fes had not been directly examined in living cells, and no evidence for the putative inactive monomer existed.
In this study, we explored c-Fes oligomerization in live cells by developing a YFP bimolecular fluorescence complementation (BiFC) assay, based on previous work of Kerppola and colleagues for the analysis of transcription factors that also contain coiled-coil oligomerization domains (21,22). Here we establish for the first time that c-Fes, unlike c-Src and c-Abl, is a constitutive oligomer in living cells. Using COS-7 cells as a model system, we used BiFC to demonstrate that wild-type c-Fes adopts an oligomeric conformation irrespective of kinase domain autophosphorylation. Fluorescence resonance energy transfer (FRET) analyses, which require interacting partners to be separated by 10-100 Å (37), were also performed, but did not provide clear results (data not shown). Thus, for proteins such as c-Fes, where the structural basis for interactions is not known, BiFC provides an ideal alternative because protein-protein interactions can be detected when partners are separated by distances greater than 100 Å (21).
Using previously described point mutations that disrupt the function of each major c-Fes domain (5,7,20), we determined that both coiled-coil domains are required for oligomerization in living cells. Mutation of either coiled-coil domain alone or together (2LP) substantially reduced the percentage of oligomeric c-Fes molecules (Figure 3). This result agrees with previous gel-filtration experiments using the same mutations in the context of a shorter c-Fes N-terminal protein construct encompassing only the coiled-coils and the intervening protein sequence (5). In these prior studies, mutation of both coiled-coils was required for elution of the recombinant protein as a monomer. In contrast to the coiled-coil domains, disruption of either SH2 function (R483L) or kinase activity (K590E) was without effect on oligomerization, as both of these mutants formed oligomers with BiFC efficiencies similar to wild-type c-Fes in vivo (Figure 3). Previous chemical cross-linking studies have also suggested that the SH2 and kinase domains are not involved in the oligomerization of c-Fes (19).
Upon establishing the coiled-coil domains as mediators of c-Fes oligomerization in living cells, we next assessed whether c-Fes also form oligomers in cell lines where it has been shown to produce a biological effect. Previous studies have demonstrated that re-expression of c-Fes suppresses anchorage-independent growth of HCT 116 colorectal cancer cells and causes growth arrest and terminal differentiation in K-562 CML cells (3,4,11). In these cell lines, we determined that wild-type c-Fes forms coiled-coil mediated oligomers (Figures 6 and 7), paralleling our results in COS-7 cells and strongly suggesting that oligomerization is essential to c-Fes function. Unexpectedly, wild-type c-Fes was also found to be constitutively autophosphorylated upon expression in HCT 116 cells (Figure 6C). This observation may be due to the presence of active Src-family kinases in colorectal cancer cell lines (35), which can directly activate c-Fes by phosphorylating its activation loop tyrosine residue (Figure 5) (7).
With the surprising revelation that c-Fes intrinsically adopts an oligomeric conformation regardless of the activation status of its kinase domain, the question remains as to what mechanism governs the tight regulation of c-Fes catalytic activity in vivo. c-Fes may self-regulate its catalytic activity through conformational changes involving interplay of the unique N-terminal region and SH2-kinase unit, an idea originally suggested by Greer (1). When c-Fes is inactive, the coiled-coil domains may lock c-Fes in a conformation that hinders the association of the SH2 domain with the kinase domain. In response to upstream stimuli, such as proteins that bind to the coiled-coils in trans (38,39), the conformational restraints imparted by the coiled-coil domains may be disrupted, enabling the SH2 domain to interact with and prime the c-Fes kinase domain for trans-autophosphorylation and cellular signaling. This revised model suggests that small molecules designed to specifically bind to the coiled-coil domains may be potent c-Fes agonists. Such molecules may have utility in the differentiation therapy of tumor cells where the c-Fes protein is expressed but its catalytic activity is repressed. The proposed role of the SH2 domain in this model of c-Fes regulation strikes a contrast to that of Src-family kinases, where SH2 is essential for negative regulation of kinase activity (15).
Support for the activation mechanism described above comes from several previous studies as well as new data presented here. First, deletion of the c-Fes SH2 domain has been shown to diminish autophosphorylation and substrate phosphorylation both in vitro and in cell-based assays (23,40). Furthermore, an intact SH2 domain is required for c-Fes biological activity (fibroblast transformation), as an activated c-Fes variant (myristoyl-Fes) can be rendered biologically inert by SH2 domain deletion or substitution with a heterologous SH2 domain (18). Second, c-Fes has been shown to bind directly to its own SH2 domain (40), and this interaction is mediated at least in part by the kinase domain (T. Smithgall, unpublished results). In addition, early studies involving a viral counterpart of c-Fes (v-Fps) suggest that the SH2 domain interacts in cis with the kinase domain to form the active kinase conformation (2). Mild proteolysis of v-Fps released a stable globular fragment containing the SH2 and kinase domains, consistent with the idea that SH2-kinase domain interaction is essential for full kinase activity (41,42).
Very recently, the X-ray crystal structure of the c-Fes SH2-kinase region has been solved (43). This structure reveals that the SH2 domain indeed makes multiple contacts with the N-terminal lobe of the kinase domain, including the critical αC helix involved in conformational regulation of the active site. Mutagenesis of key residues at the SH2:kinase interface dramatically reduced kinase activity, firmly establishing that c-Fes kinase domain function is dependent upon interaction with the SH2 domain. When considered in the context of the full-length structure, it is reasonable to postulate that N-terminal sequences may interfere with the formation of this positive regulatory interface, thus repressing kinase activity. Along these lines, data presented in Figure 4 show that mutational disruption of the N-terminal coiled-coil domains causes a complete loss of oligomerization (no BiFC signal), yet result in very strong kinase domain autophosphorylation as well as c-Fes movement to the microtubule network. Note that our previous work has established that microtubule association of c-Fes correlates strongly with kinase activation in COS cells (7). Thus the coiled-coil domains contribute not only to oligomerization, but also to repression of kinase activity, possibly by affecting the SH2:kinase domain interaction described above. In addition, the findings that oligomerization-defective, active mutants of Fes as well as oligomerization-competent wild-type Fes activated by Hck all localize to microtubules show that microtubule localization, while activity-dependent, does not require oligomerization.
Alternatively, catalytic activation of c-Fes may involve an equilibrium shift from lower-order to higher-order oligomers. Cross-linking studies suggest that c-Fes forms trimers, while gel-filtration experiments support the existence of oligomers as large as pentamers (19). Note that the BiFC analysis used here cannot distinguish between individual oligomerization states, and cannot rule out the presence of Fes monomers in cells. Rather, BiFC only reports in a binary fashion as to whether or not c-Fes has interacted with itself in vivo. While further clarification of the contribution of oligomerization to c-Fes catalytic activity and biological function is required, it is tempting to speculate that c-Fes is primed for autophosphorylation and signaling through the constitutive oligomeric nature imparted upon its overall structure by its unique N-terminal coiled-coil oligomerization domains.
Supplementary Material
Acknowledgments
The authors would like to thank Kira Lathrop of the University of Pittsburgh Eye and Ear Institute for her assistance with the confocal microscopy.
Abbreviations
- FCH
Fes/CIP4 homology
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- NGF
nerve growth factor
- FGF
fibroblast growth factor
- SH
Src homology
- BiFC
bimolecular fluorescence complementation
- YFP
yellow-shifted variant of green fluorescent protein
- CML
chronic myelogenous leukemia
- CC
coiled-coil
Footnotes
This work was supported by National Institutes of Health Grant CA123756.
Supporting Information Available: A supplemental figure (Figure S1) showing immunoblots resulting from the individual and combined expression of the YN-Fes and YC-Fes constructs in COS-7 cells is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Greer PA. Closing in on the biological functions of Fps/Fes and Fer. Nature Rev Mol Cell Biol. 2002;3:278–289. doi: 10.1038/nrm783. [DOI] [PubMed] [Google Scholar]
- 2.Smithgall TE, Rogers JA, Peters KL, Li J, Briggs SD, Lionberger JM, Cheng H, Shibata A, Scholtz B, Schreiner S, Dunham NA. The c-Fes Family of Protein-Tyrosine Kinases. Critical Rev Oncogenesis. 1998;9:43–62. doi: 10.1615/critrevoncog.v9.i1.40. [DOI] [PubMed] [Google Scholar]
- 3.Cheng HY, Rogers JA, Dunham NA, Smithgall TE. Regulation of c-Fes tyrosine kinase and biological activities by N-terminal coiled-coil oligomerization domains. Mol Cell Biol. 1999;19:8335–8343. doi: 10.1128/mcb.19.12.8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu G, Smithgall TE, Glazer RI. K562 leukemia cells transfected with the human c-fes gene acquire the ability to undergo myeloid differentiation. J Biol Chem. 1989;264:10276–10281. [PubMed] [Google Scholar]
- 5.Cheng HY, Schiavone AP, Smithgall TE. A point mutation in the N-terminal coiled-coil domain releases c-Fes tyrosine kinase activity and survival signaling in myeloid leukemia cells. Mol Cell Biol. 2001;21:6170–6180. doi: 10.1128/MCB.21.18.6170-6180.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim J, Feldman RA. Activated Fes protein tyrosine kinase induces terminal macrophage differentiation of myeloid progenitors (U937 cells) and activation of the transcription factor PU.1. Mol Cell Biol. 2002;22:1903–1918. doi: 10.1128/MCB.22.6.1903-1918.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laurent CE, Delfino FJ, Cheng HY, Smithgall TE. The human c-Fes tyrosine kinase binds tubulin and microtubules through separate domains and promotes microtubule assembly. Mol Cell Biol. 2004;24:9351–9358. doi: 10.1128/MCB.24.21.9351-9358.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shibata A, Laurent CE, Smithgall TE. The c-Fes protein-tyrosine kinase accelerates NGF-induced differentiation of PC12 cells through a PI3K-dependent mechanism. Cell Signal. 2003;15:279–288. doi: 10.1016/s0898-6568(02)00089-x. [DOI] [PubMed] [Google Scholar]
- 9.Kanda S, Lerner EC, Tsuda S, Shono T, Kanetake H, Smithgall TE. The non-receptor protein-tyrosine kinase c-Fes is involved in FGF-2-induced chemotaxis of murine brain capillary endothelial cells. J Biol Chem. 2000;275:10105–10111. doi: 10.1074/jbc.275.14.10105. [DOI] [PubMed] [Google Scholar]
- 10.Sangrar W, Zirgnibl RA, Gao Y, Muller WJ, Jia Z, Greer PA. An identity crisis for fps/fes: oncogene or tumor suppressor? Cancer Res. 2005;65:3518–3522. doi: 10.1158/0008-5472.CAN-04-3468. [DOI] [PubMed] [Google Scholar]
- 11.Delfino FJ, Stevenson HM, Smithgall TE. A growth-suppressive function for the c-Fes protein-tyrosine kinase in colorectal cancer. J Biol Chem. 2006;281:8829–8835. doi: 10.1074/jbc.M507331200. [DOI] [PubMed] [Google Scholar]
- 12.Shaffer JM, Smithgall TE. Promoter methylation blocks FES protein-tyrosine kinase gene expression in colorectal cancer. Genes Chromosomes Cancer. 2009;48:272–284. doi: 10.1002/gcc.20638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bardelli A, Parsons DW, Silliman N, Ptak J, Szabo S, Saha S, Markowitz S, Willson JK, Parmigiani G, Kinzler KW, Vogelstein B, Velculescu VE. Mutational analysis of the tyrosine kinome in colorectal cancers. Science. 2003;300:949. doi: 10.1126/science.1082596. [DOI] [PubMed] [Google Scholar]
- 14.Itoh T, De CP. BAR, F-BAR (EFC) and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature. Biochim Biophys Acta. 2006;1761:897–912. doi: 10.1016/j.bbalip.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 15.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 16.Feldman RA, Lowy DR, Vass WC, Velu TJ. A highly efficient retroviral vector allows detection of the transforming activity of the human c-fps/fes protooncogene. J Virol. 1989;63:5469–5474. doi: 10.1128/jvi.63.12.5469-5474.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greer PA, Meckling-Hansen K, Pawson T. The human c-fps/fes gene product expressed ectopically in rat fibroblasts is nontransforming and has restrained protein-tyrosine kinase activity. Mol Cell Biol. 1988;8:578–587. doi: 10.1128/mcb.8.2.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogers JA, Cheng HY, Smithgall TE. Src homology 2 domain substitution modulates the kinase and transforming activities of the Fes protein-tyrosine kinase. Cell Growth Differ. 2000;11:581–592. [PubMed] [Google Scholar]
- 19.Read RD, Lionberger JM, Smithgall TE. Oligomerization of the Fes tyrosine kinase: Evidence for a coiled-coil domain in the unique N-terminal region. J Biol Chem. 1997;272:18498–18503. doi: 10.1074/jbc.272.29.18498. [DOI] [PubMed] [Google Scholar]
- 20.Rogers JA, Read RD, Li J, Peters KL, Smithgall TE. Autophosphorylation of the Fes tyrosine kinase: Evidence for an intermolecular mechanism involving two kinase domain tyrosine residues. J Biol Chem. 1996;271:17519–17525. doi: 10.1074/jbc.271.29.17519. [DOI] [PubMed] [Google Scholar]
- 21.Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 22.Hu CD, Kerppola TK. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol. 2003;21:539–545. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takashima Y, Delfino FJ, Engen JR, Superti-Furga G, Smithgall TE. Regulation of c-Fes tyrosine kinase activity by coiled-coil and SH2 domains: analysis with Saccharomyces cerevisiae. Biochemistry. 2003;42:3567–3574. doi: 10.1021/bi0272499. [DOI] [PubMed] [Google Scholar]
- 24.Nagar B, Hantschel O, Seeliger M, Davies JM, Weis WI, Superti-Furga G, Kuriyan J. Organization of the SH3-SH2 unit in active and inactive forms of the c-Abl tyrosine kinase. Mol Cell. 2006;21:787–798. doi: 10.1016/j.molcel.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 25.Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti-Furga G, Kuriyan J. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 2003;112:859–871. doi: 10.1016/s0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 26.Schindler T, Sicheri F, Pico A, Gazit A, Levitzki A, Kuriyan J. Crystal structure of Hck in complex with a Src family-selective tyrosine kinase inhibitor. Mol Cell. 1999;3:639–648. doi: 10.1016/s1097-2765(00)80357-3. [DOI] [PubMed] [Google Scholar]
- 27.Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- 28.Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3:629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- 29.Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- 30.Haigh J, McVeigh J, Greer P. The Fps/Fes tyrosine kinase is expressed in myeloid, vascular endothelial, epithelial and neuronal cells and is localized to the trans-golgi network. Cell Growth and Differentiation. 1996;7:931–944. [PubMed] [Google Scholar]
- 31.Takahashi S, Inatome R, Hotta A, Qin Q, Hackenmiller R, Simon MC, Yamamura H, Yanagi S. Role for Fes/Fps tyrosine kinase in microtubule nucleation through is Fes/CIP4 homology domain. J Biol Chem. 2003;278:49129–49133. doi: 10.1074/jbc.C300289200. [DOI] [PubMed] [Google Scholar]
- 32.McPherson VA, Everingham S, Karisch R, Smith JA, Udell CM, Zheng J, Jia Z, Craig AW. Contributions of F-BAR and SH2 domains of Fes protein tyrosine kinase for coupling to the FcepsilonRI pathway in mast cells. Mol Cell Biol. 2009;29:389–401. doi: 10.1128/MCB.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Briggs SD, Smithgall TE. SH2-kinase linker mutations release Hck tyrosine kinase and transforming activities in rat-2 fibroblasts. J Biol Chem. 1999;274:26579–26583. doi: 10.1074/jbc.274.37.26579. [DOI] [PubMed] [Google Scholar]
- 34.Smithgall TE, Yu G, Glazer RI. Identification of the differentiation-associated p93 tyrosine protein kinase of HL-60 leukemia cells as the product of the human c-fes locus and its expression in myelomonocytic cells. J Biol Chem. 1988;263:15050–15055. [PubMed] [Google Scholar]
- 35.Johnson FM, Gallick GE. SRC family nonreceptor tyrosine kinases as molecular targets for cancer therapy. Anticancer Agents Med Chem. 2007;7:651–659. doi: 10.2174/187152007784111278. [DOI] [PubMed] [Google Scholar]
- 36.Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- 37.Sekar RB, Periasamy A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol. 2003;160:629–633. doi: 10.1083/jcb.200210140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Smithgall TE. Co-expression with Bcr induces activation of the Fes tyrosine kinase and phosphorylation of specific N-terminal Bcr tyrosine residues. J Biol Chem. 1996;271:32930–32936. doi: 10.1074/jbc.271.51.32930. [DOI] [PubMed] [Google Scholar]
- 39.Delfino FJ, Shaffer JM, Smithgall TE. The KRAB-associated co-repressor KAP-1 is a coiled-coil binding partner, substrate, and activator of the c-Fes protein-tyrosine kinase. Biochem J. 2006;399:141–150. doi: 10.1042/BJ20060194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hjermstad SJ, Peters KL, Briggs SD, Glazer RI, Smithgall TE. Regulation of the human c-fes protein-tyrosine kinase (p93c-fes) by its src homology 2 domain and major autophosphorylation site (tyr 713) Oncogene. 1993;8:2283–2292. [PubMed] [Google Scholar]
- 41.Koch CA, Moran M, Sadowski I, Pawson T. The common src homology region 2 domain of cytoplasmic signaling proteins is a positive effector of v-fps tyrosine kinase function. Mol Cell Biol. 1989;9:4131–4140. doi: 10.1128/mcb.9.10.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weinmaster G, Hinze E, Pawson T. Mapping of multiple phosphorylation sites within the structural and catalytic domains of the Fujinami avian sarcoma virus transforming protein. J Virol. 1983;46:29–41. doi: 10.1128/jvi.46.1.29-41.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Filippakopoulos P, Kofler M, Hantschel O, Gish G, Grebien F, Salah E, Neudecker P, Kay LE, Turk BE, Superti-Furga G, Pawson T, Knapp S. Structural coupling of Fes and Abl SH2-tyrosine kinase domains links substrate recognition and kinase activation. Cell. 2008;134:793–803. doi: 10.1016/j.cell.2008.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.