

SUMMARY
Disrupted-in Schizophrenia 1 (DISC1), a susceptibility gene for major mental disorders, encodes a scaffold protein that has a multifaceted impact on neuronal development. How DISC1 regulates different aspects of neuronal development is not well understood. Here we show that Fasciculation and Elongation Protein Zeta-1 (FEZ1) interacts with DISC1 to synergistically regulate dendritic growth of newborn neurons in the adult mouse hippocampus, and that this pathway complements a parallel DISC1-NDEL1 interaction that regulates cell positioning and morphogenesis of newborn neurons. Furthermore, genetic association analysis of two independent cohorts of schizophrenia patients and healthy controls reveals an epistatic interaction between FEZ1 and DISC1, but not between FEZ1 and NDEL1, for risk of schizophrenia. Our findings support a model in which DISC1 regulates distinct aspects of neuronal development through its interaction with different intracellular partners and such epistasis may contribute to increased risk for schizophrenia.
INTRODUCTION
An abiding principle of brain organization holds that the precise synaptic connectivity of neuronal networks determines brain functions. Conversely, pathological disturbances of this neuronal and synaptic patterning may contribute to the symptomatology of many neurological and psychiatric illnesses. Therefore, understanding molecular mechanisms that regulate neuronal development and connectivity can generate insight into the processes that govern the functional integrity of the developing and adult brain. In the hippocampus of the adult mammalian brain, new neurons are continually generated from neural stem cells throughout the lifespan of the organism (Lledo et al., 2006; Ming and Song, 2005; Zhao et al., 2008). Adult neurogenesis recapitulates the complete process of embryonic neuronal development, including proliferation and fate specification of neural progenitors, morphogenesis, axon and dendritic growth, migration, and synapse formation of neuronal progeny (Duan et al., 2008; Ming and Song, 2011). Many signaling pathways play conserved roles during embryonic and adult neurogenesis and disruption of many of these same pathways have also been implicated in the etiology of psychiatric disorders (Harrison and Weinberger, 2005; Kempermann et al., 2008).
There is a growing body of evidence demonstrating a convergent effect of genetic mutations that both confer susceptibility to psychiatric diseases and result in dysregulation of neuronal development, supporting a neurodevelopmental origin of these diseases. One prominent example of this genetic convergence is disrupted in schizophrenia-1 (DISC1), a gene initially identified at the breakpoint of a balanced (1;11) (q42;q14) chromosome translocation in a large Scottish family that segregates with schizophrenia and other major mental disorders (Blackwood et al., 2001; Millar et al., 2000). Additional linkage studies with DISC1 mutations further support its role in influencing risk for psychosis and autistic spectrum disorders (Chubb et al., 2008). Functional studies in animal models suggest that DISC1 plays a multifaceted role in both embryonic and postnatal neurogenesis in vivo. Exogenous manipulation of DISC1 results in a spectrum of neuronal abnormalities, depending on the timing and anatomical locus of perturbation. During embryonic cortical development, knockdown of DISC1 in E13 embryos accelerates cell cycle exit and neuronal differentiation (Mao et al., 2009), whereas knockdown at E14.5 leads to inhibition of neuronal migration and disorganized dendritic arbors (Kamiya et al., 2005). During adult hippocampal neurogenesis, suppression of DISC1 also leads to decreased proliferation of neural progenitors (Mao et al., 2009) and an array of neurodevelopmental defects in newborn dentate granule cells, including soma hypertrophy, mis-positioning, impaired axonal targeting, and accelerated dendritic growth and synaptogenesis (Duan et al., 2007; Faulkner et al., 2008; Kim et al., 2009).
The signaling mechanisms by which DISC1 regulates neurogenesis in vivo have just begun to be explored. For example, DISC1 regulates proliferation of neural progenitors through interaction with GSK3β (Mao et al., 2009), whereas it regulates development of newborn dentate granule cells through direct interaction with KIAA1212/Girdin in the hippocampus (Enomoto et al., 2009; Kim et al., 2009). NDEL1 (nuclear distribution gene E-like homolog 1) also directly interacts with DISC1(Morris et al., 2003; Ozeki et al., 2003). Knockdown of NDEL1 in newborn neurons in the adult hippocampus leads to primary defects in neuronal positioning and appearance of ectopic dendrites, representing some, but not all, of phenotypes observed with DISC1 suppression (Duan et al., 2007). This result suggests the existence of additional mechanisms by which DISC1 regulates other aspects of neuronal development. Indeed, early biochemical and yeast two-hybrid screens have identified a large number of DISC1 binding partners, many of which are known to be involved in neurodevelopmental processes (Camargo et al., 2007). While these studies established DISC1 as a scaffold protein, the functional role of the majority of these potential interactions in neuronal development remains to be demonstrated in vivo. Understanding mechanisms by which DISC1 differentially regulates distinct neurodevelopmental processes through its binding partners may reveal how dysfunction of DISC1 contributes to a wide spectrum of psychiatric and mental disorders.
Fasciculation and Elongation Protein Zeta-1 (FEZ1) is one of the first identified binding partners of DISC1 (Miyoshi et al., 2003). FEZ1 is a mammalian ortholog of the C. elegans UNC-76 protein, thought to be involved in nerve growth and fasciculation (Bloom and Horvitz, 1997; Kuroda et al., 1999). FEZ1 expression is developmentally regulated and appears to be abundant in the adult mouse dentate gyrus (Miyoshi et al., 2003; Sakae et al., 2008). In vitro, FEZ1 co-localizes with DISC1 at neuronal growth cones and regulates neurite outgrowth of PC12 cells (Miyoshi et al., 2003). The role of FEZ1 in mammalian neuronal development in vivo is not well understood. Fez1 null mice exhibit hyperactivity and enhanced responsiveness to psychostimulants (Sakae et al., 2008), supporting a potential contribution of FEZ1 dysfunction to schizophrenia. Single nucleotide polymorphism (SNP) and haplotype association analyses of the FEZ1 locus with schizophrenia have demonstrated a positive association in one cohort of patients (Yamada et al., 2004), but not in others (Hodgkinson et al., 2007; Koga et al., 2007; Nicodemus et al., 2010; Rastogi et al., 2009). Interestingly, there is a significant reduction of FEZ1 mRNA in both hippocampus and dorsolateral prefrontal cortex of schizophrenia patients and an association of the DISC1 genotype and FEZ1 mRNA levels (Lipska et al., 2006). These findings raise the possibility that FEZ1 and DISC1 may cooperate to regulate both neuronal development and risk for schizophrenia.
In the present study, we used adult mouse hippocampal neurogenesis as an in vivo cellular model to dissect signaling mechanisms by which DISC1 regulates different aspects of neuronal development. We showed that interaction between FEZ1 and DISC1 regulates dendritic development of newborn dentate granule cells in the adult brain. This functional association complements the parallel DISC1-NDEL1 interaction, which regulates positioning and morphogenesis of newborn neurons. Biochemically, endogenous DISC1 interacts with both FEZ1 and NDEL1, whereas FEZ1 and NDEL1 do not appear to interact without DISC1. Furthermore, genetic association analyses in two clinical cohorts reveal an epistatic interaction between FEZ1 and DISC1, but not between FEZ1 and NDEL1, for an increased risk for schizophrenia. Together, our findings support a model in which DISC1 interacts with different partners to regulate distinct aspects of neuronal development and epistatic interactions between DISC1 and these genes may exacerbate neurodevelopmental deficits and confer an increased risk for schizophrenia.
RESULTS
Regulation of new neuron development by FEZ1 in the adult brain
To explore signaling pathways underlying DISC1-dependent regulation of neuronal development, we generated retroviral vectors co-expressing GFP and specific short-hairpin RNAs (shRNAs) against mouse fez1 (See Experimental Procedures). We first examined their efficacy in knocking down the expression of endogenous FEZ1 in cultured adult mouse neural progenitors, which expressed FEZ1 during the proliferation state and after induced neuronal differentiation (Figure S1A). Two shRNAs against mouse fez1 (shRNA-F1 and shRNA-F2), but not a control shRNA (shRNA-C1) (Ma et al., 2008), were very effective in knocking down the expression of endogenous FEZ, but not DISC1 or NDEL1, at the protein level (Figures 1A and S1B).
Figure 1. Role of FEZ1 in regulating development of newborn neurons in the adult mouse dentate gyrus.

(A) Validation of the efficacy of shRNAs against mouse fez1. Retroviruses expressing shRNAs were used to infect mouse adult neural progenitors in culture and equal amount of samples from cell lysates were subject to Western Blot for FEZ1 and GAPDH. Shown are sample Western Blot analysis and quantification after expression of shRNA-control (C1), shRNA-FEZ1#1 (F1) or shRNA-FEZ1#2 (F2). Values represent mean ± SEM (n = 3; *: P < 0.01; ANOVA).
(B) Morphological development of newborn neurons in the adult brain. Shown are sample confocal single section images of GFP and DAPI at 14 days post injection (dpi) of retroviruses co-expressing GFP and shRNA in the adult dentate gyrus. The domains of cell body localization are indicated along the sample images (Area 1: inner granule cell layer; Area 2: middle granule cell layer; Area 3: outer granule cell layer; Area 4: molecular layer). Scale bar: 10 μm. Also shown is a summary of the soma size of GFP+ cells under different conditions. Numbers associated with each bar graph refer to the total number of GFP+ neurons analyzed from at least four animals under each condition. Values represent mean ± SEM (*: P < 0.01; ANOVA).
(C-E) Dendritic development of newborn neurons in the adult dentate gyrus. Shown in (C) are sample confocal projection images of GFP+ neurons at 14 dpi. Scale bar: 10 μm. Also shown are summary of the total dendritic length (D) and Sholl analysis of dendritic complexity (E) of GFP+ newborn neuron expressing different shRNAs. The same sets of GFP+ cells as in (B) were analyzed. Values represent mean ± SEM (*: P < 0.01; ANOVA).
To assess the potential function of FEZ1 in regulating development of newborn neurons in the adult brain, we stereotaxically injected retroviruses co-expressing shRNA and GFP into the dentate gyrus of the adult mice brain. GFP+ newborn neurons were examined with confocal microscopy at 14 days post injection (dpi). When compared to GFP+ neurons expressing shRNA-C1, there was a significant increase in the soma size of GFP+ neurons expressing either shRNA-F1 or shRNA-F2 (Figure 1B). Furthermore, GFP+ neurons expressing either shRNA-F1 or shRNA-F2 exhibited accelerated dendritic development with significant increases in both total dendritic length and complexity as shown by the Sholl analysis (Figures 1C to 1E). Interestingly, increased dendritic growth and soma hypertrophy have also been observed with DISC1 knockdown in these newborn dentate granule cells in the adult hippocampus (Duan et al., 2007). On the other hand, GFP+ neurons with FEZ1 knockdown did not exhibit ectopic primary dendrites, aberrant neuronal positioning (Figure S1C), or mossy fiber axonal mis-targeting (Figure S1D), other characteristic defects that result from DISC1 knockdown (Duan et al., 2007; Faulkner et al., 2008; Kim et al., 2009). Thus, FEZ1 knockdown leads to a specific subset of, but not all, developmental defects observed in newborn neurons with DISC1 knockdown during adult neurogenesis.
Rescue of development defects from FEZ1 knockdown by wild-type FEZ1 expression
The similarity of phenotypes from two shRNAs against different regions of the fez1 gene suggests a specific role of FEZ1 in the development of newborn neurons in the adult brain. To further confirm the specificity of the shRNA manipulation, in vivo rescue experiments were performed. We engineered two sets of retroviruses: the first co-expressing GFP and wild-type (WT) mouse fez1 cDNA without the 3′ untranslated region (3′ UTR; pCUXIE-mFEZ1), or GFP alone (pCUXIE); the second co-expressing mCherry and shRNA-F1 (Figure S2A). The shRNA-F1 targets the 3′UTR of the mouse fez1 gene, thus it does not affect mFEZ1 expression from the rescue vector (pCUXIE-mFEZ1). The two types of engineered retroviruses were co-injected into the adult dentate gyrus (Figure 2A). Expression of shRNA-F1 and mCherry resulted in significant increases in the total dendritic length and soma size in comparison to those expressing shRNA-C1, whereas overexpression of mFEZ1 itself did not lead to any obvious effects (Figures 2B and 2C), except for a modest change in the dendritic complexity, but not the total dendritic length (Figure S2B). Importantly, co-expression of mFEZ1, but not vector control, largely normalized increased dendritic growth and soma hypertrophy by shRNA-F1 (Figures 2B and 2C). Under all conditions, no significant effects on the number of primary dendrites and neuronal positioning were detected (Figures S2D and S2E). Taken together, these rescue experiments further support specific roles of FEZ1 in regulating distinct aspects of new neuron development in the adult brain.
Figure 2. Rescue of developmental defects of newborn neurons from FEZ1 knockdown by overexpression of wild-type FEZ1.

(A) Sample confocal images of double-labeled newborn neurons in the adult dentate gyrus. Retroviruses co-expressing mCherry and shRNA-FEZ1#1 (F1) and those co-expressing GFP and mouse FEZ1 (mFEZ1) or GFP alone (pCUXIE) were co-injected into the adult dentate gyrus and animals were analyzed at 14 dpi after immunostaining for GFP. Shown are sample confocal projection image of GFP staining (left) and images of GFP staining (green), mCherry (red), DAPI (blue) and merged at lower magnification. Scale bar: 10 μm.
(B and C) Summary of total dendritic length (B) and soma size (C) of newborn neuron in the adult brain expressing shRNA-control (C1), mFEZ1, shRNA-FEZ1#1 (F1), or co-expressing F1 and mFEZ1 (F1 + mFEZ1) or control vector (F1 + pCUXIE). Numbers associated with each bar graph refer to the total number of neurons analyzed from at least two animals under each condition. Values represent mean ± SEM (*: P < 0.01; ANOVA).
Synergistic interaction between FEZ1 and DISC1 in regulating development of newborn neurons in the adult brain
The similar effect of DISC1 and FEZ1 knockdown on dendritic growth and soma size of newborn neurons in the adult brain and reported direct interaction between these two proteins (Miyoshi et al., 2003) suggest that they may functionally interact in regulating neuronal development. We previously generated a collection of shRNAs against mouse disc1 that exhibit different knockdown efficacy (Duan et al., 2007). Expression of the strong shRNA against disc1 (shRNA-D1) in newborn neurons led to the full spectrum of phenotypes at 14 dpi, whereas expression of the weak shRNA against disc1 (shRNA-D3) by itself led to a modest phenotype that manifests at 28 dpi, but not at 14 dpi (Figures 3A, 3C to 3E, S3B and S3C) (Duan et al., 2007). To examine a potential interaction between FEZ1 and DISC1, we employed an in vivo double knockdown approach (Figure S3A). Interestingly, co-expression of shRNA-F1 and shRNA-D3 exacerbated the dendritic growth phenotype compared to expression of shRNA-F1 alone, as shown in both total dendritic length and complexity (Figures 3B, 3C and 3D). On the other hand, no apparent synergistic effect was observed for soma size (Figure 3E), number of ectopic dendrites, or positioning of newborn neurons (Figures S3B and S3C). These results suggest that FEZ1 and DISC1 functionally interact to regulate dendritic development of newborn neurons in the adult brain.
Figure 3. Functional interaction between FEZ1 and DISC1 in regulating development of newborn neurons in the adult brain.

(A-E) Effect of double knockdown of DISC1 and FEZ1 on development of newborn neurons in the adult dentate gyrus. Shown in (A) are sample confocal projection images of GFP+ newborn neurons expressing shRNA-control (C1), shRNA-FEZ1#1 (F1), a weak shRNA against mouse disc1 (shRNA-DISC1#3; D3), or a strong shRNA against mouse disc1 (shRNA-DISC1#1; D1) at 14 dpi. Shown in (B) are sample confocal images of double-labeled newborn neurons co-expressing shRNA-DISC1#3 (D3)/GFP and shRNA-FEZ1#1 (F1)/mCherry at 14 dpi. Scale bars: 20 μm. Also shown are summary of the total dendritic length (C), Sholl analysis of dendritic complexity (D) and soma size (E) of newborn neuron in the adult brain expressing shRNA-control (C1), shRNA-FEZ1#1 (F1), shRNA-DISC1#3 (D3), or shRNA-DISC1#1 (D1), or co-expressing F1 and D3 (F1 + D3), at 14 dpi. The same group of cells for C1 and F1 as in Figure 1 were plotted for comparison. Values represent mean ± SEM (*: P < 0.01; ANOVA).
(F-H) Effect of expression of a DISC1 blocking peptide on development of newborn neurons in the adult dentate gyrus. Retroviruses co-expressing GFP and a peptide encoding a DISC1 domain that interacts with FEZ1 (a.a. 446-633; Figure S3D) were stereotaxically injected into adult dentate gyrus to infect proliferating progenitors. Shown are summaries of the total dendritic length (F), Sholl analysis (G) and soma size (H) of GFP+ neurons at 14 dpi, similarly as in (C-E).
To further assess the interaction between endogenous DISC1 and FEZ1 in regulating neuronal development, we explored a blocking peptide using the DISC1 domain that interacts with FEZ1 (a.a. 446-633 of mouse DISC1; Figure S3D) (Miyoshi et al., 2003). In the coimmunoprecipitation (co-IP) analysis, expression of this peptide in HEK293 cells attenuated interaction between exogenous DISC1 and FEZ1, but not between DISC1 and endogenous KIAA1212/Girdin (Kim et al., 2009) or Kendrin (Shimizu et al., 2008), two other proteins interacting with DISC1 at sites that overlap with this region (Figure S3D). Furthermore, over-expression of this peptide significantly reduced the interaction between endogenous DISC1 and FEZ1 in adult neural progenitors, but had no effect on the interaction between endogenous DISC1 and NDEL1 (Figure S3E), supporting the specificity of the blocking peptide on the DISC1 and FEZ1 interaction. Retrovirus-mediated co-expression of the DISC1 peptide and GFP in newborn neurons in the adult dentate gyrus led to increased total dendritic length and complexity as well as soma hypertrophy of GFP+ neurons at 14 dpi (Figures 3F to 3H), but no effect on the number of primary dendrites or neuronal positioning (Figures S3F and S3G), fully recapitulating the FEZ1 knockdown phenotype. Taken together, these results support that FEZ1 regulates specific aspects of new neuron development in the adult brain through functional interaction with DISC1.
Lack of synergistic interaction between FEZ1 and NDEL1 in regulating development of newborn neurons in the adult brain
NDEL1 is another DISC1 interacting protein that regulates neuronal development in vivo (Duan et al., 2007; Sasaki et al., 2005; Shu et al., 2004). Consistent with our previous findings (Duan et al., 2007), expression of a specific shRNA against mouse ndel1 (shRNA-N1) led to developmental defects of newborn dentate granule cells, mostly in the appearance of ectopic dendrites and aberrant positioning (Figure 4). Thus, FEZ1 and NDEL1 appear to mediate DISC1 signaling in a complementary set of neuronal developmental processes. To determine whether FEZ1 and NDEL1 also functionally interact to regulate development of newborn neurons, we performed double knockdown experiments in vivo. The effect of co-expressing shRNA-F1 and shRNA-N1 on dendritic growth and soma size of newborn neurons was very similar to those expressing shRNA-F1 alone (Figures 4A to 4C), whereas the effect on ectopic dendrites and neuronal positioning was similar to those expressing shRNA-N1 alone (Figures 4D and 4E). Thus, concomitant suppression of NDEL1 and FEZ1 only leads to additive effects of individual knockdown, instead of a synergistic action. These results further support the notion that FEZ1 and NDEL1 differentially regulate distinct aspects of new neuron development in the adult brain.
Figure 4. Lack of functional interaction between FEZ1 and NDEL1 in regulating development of newborn neurons in the adult brain.

Summary of total dendritic length (A), Sholl analysis of dendritic complexity (B), soma size (C), number of primary dendrites (D), and distribution of cell body position within different domains (E, as defined in Figure 1B).of newborn neuron in the adult brain expressing shRNA-control (C1), shRNA-FEZ1#1 (F1), shRNA-NDEL1 (N1), or co-expressing F1 and N1 (F1 + N1), at 14 dpi. The same group of cells for C1 and F1 as in Figure 1 were plotted for comparison. Values represent mean ± SEM (*: P < 0.01; #: P > 0.05; ANOVA).
KIAA1212/Girdin is also a DISC1 binding partner that regulates development of newborn dentate granule cells in the hippocampus (Enomoto et al., 2009; Kim et al., 2009). We next examined whether KIAA1212 interacts with FEZ1 or NDEL1 in regulating neuronal development. Consistent with previous findings, DISC1 was co-IPed with each of the three proteins, NDEL1, FEZ1 or KIAA1212, when each pair was co-expressed in the heterologous system (Figure S4A). Furthermore, these four proteins could be co-IPed together with DISC1 when all were co-expressed (Figure S4A). Also consistent with the previous finding (Kim et al., 2009), overexpression of KIAA1212 led to increased total dendritic length, number of primary dendrites, and soma size in newborn neurons in the adult dentate gyrus (Figures S4B to S4D). Compared to KIAA1212 overexpression or FEZ1 knockdown alone, co-manipulation exacerbated phenotypes of increased dendritic length and soma size, but not the number of primary dendrites and positioning of newborn neurons (Figures S4B to S4E). On the other hand, simultaneous KIAA1212 overexpression and NDEL1 knockdown exhibited phenotypes very similar to those of NDEL1 knockdown alone (Figures S4B to S4E). Taken together, these results support a model that DISC1 interacts with FEZ1 and KIAA1212 mainly to regulate dendritic growth and soma size of newborn neurons during adult neurogenesis, whereas DISC1 interacts with NDEL1 mainly to regulate positioning of newborn neurons (Table 1).
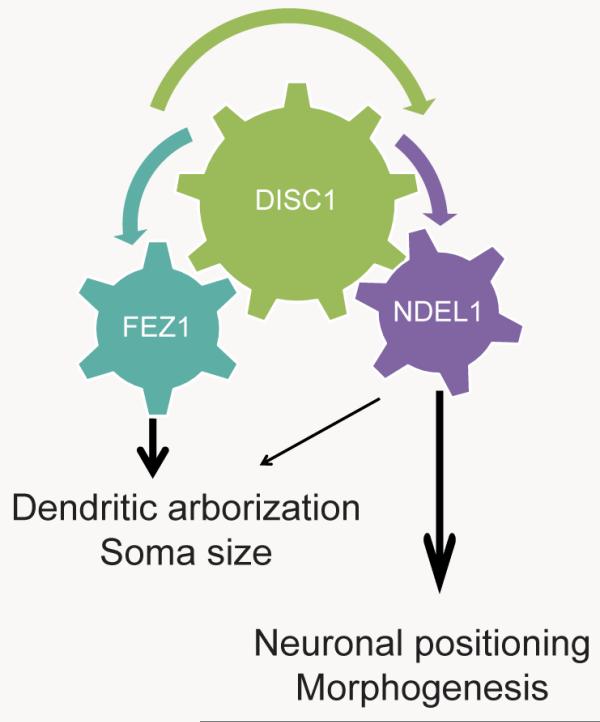
Table 1. A model of interaction between DISC1 with FEZ1 or NDEL1 in regulating distinct aspects of newborn neuron development in the adult mouse hippocampus.
Shown on the left is a schematic diagram of the model on signaling mechanisms of DISC1 in regulating new neuron development during adult hippocampal neurogenesis. Shown on the right is a summary table of effects of different genetic manipulations that lead to defects in neuronal positioning, soma hypertrophy, ectopic dendrites, and enhanced dendritic outgrowth of newborn neurons in the adult dentate gyrus at 14 dpi (“o” represents normal and the severity of phenotypes is represented by the numbers of “+”).
| shRNA | Neuronal positioning |
Soma hypertrophy |
Ectopic dendrites |
Enhanced dendritic outgrowth |
|
|---|---|---|---|---|---|
|
Control | O | O | O | O |
| DISC1#1 | +++ | +++ | +++ | +++ | |
| FEZ1 | O | +++ | O | ++ | |
| NDEL1 | ++ | + | ++ | + | |
| DISC1#3 | O | O | O | O | |
| FEZ1+DISC1#3 | O | +++ | O | ++++ | |
| FEZ1+NDEL1 | ++ | +++ | ++ | +++ | |
| DISC1#3+NDEL1 | ++ | +++ | +++ | +++ |
Biochemical interaction among FEZ1, DISC1 and NDEL1
To examine the biochemical basis of cooperation among FEZ1, DISC1 and NDEL1 in regulating neuronal development, we examined interactions of endogenous proteins using co-IP analysis of protein lysates from adult mouse neural progenitors in culture. Consistent with previous findings from studies of exogenous proteins (Kamiya et al., 2005; Miyoshi et al., 2003), endogenous DISC1 was co-IPed with FEZ1 and NDEL1, and vice versa (Figures 5A and S1A). Furthermore, endogenous NDEL1 was co-IPed with FEZ1, and vice versa (Figure 5A). We obtained similar results with protein lysates from adult mouse hippocampal tissue and with two different anti-DISC1 antibodies (Figures S5A to S5C). These results suggest that FEZ1, DISC1 and NDEL1 comprise a protein complex or complexes in vivo.
Figure 5. Biochemical interaction among FEZ1, DISC1 and NDEL1.

(A) Association of endogenous DISC1, FEZ1 and NDEL1. Cultured adult mouse neural progenitors were subjected to co-IP analysis using antibodies against DISC1, NDEL1 or FEZ1, respectively. A summary of quantification of co-IP efficacy is also shown. Values represent mean ± SEM (n = 3; P > 0.05; ANOVA).
(B) Lack of interaction between FEZ1 and NDEL1 in adult neural progenitors with DISC1 knockdown. Adult neural progenitors were infected with retroviruses to express shRNA-DISC1 (D1) or shRNA-control (C1). After 48 hours, cell lysates were subjected to Western Blot analysis for expression of DISC1, FEZ1 or NDEL1, or subjected to co-IP analysis using antibodies against FEZ1 or NDEL1, respectively. A summary of quantification of co-IP efficacy is also shown. Values represent mean ± SEM (n = 3; *: P < 0.01; ANOVA).
(C) Independent interaction between DISC1 and FEZ1 and between DISC1 and NDEL1. Adult neural progenitors were infected with retroviruses to express shRNA-NDEL1 (N1), shRNA-FEZ1 (F1), or shRNA-control (C1). After 48 hours, cell lysates were subjected to co-IP analysis using antibodies against DISC1. A summary of quantification of co-IP efficacy is also shown. Values represent mean ± SEM (n = 3; P > 0.05; ANOVA).
To determine whether FEZ1 and NDEL1 interact through the common binding partner DISC1 or independently of DISC1, we performed co-IP experiments using adult mouse neural progenitors expressing shRNA-D1 (Figure 5B). DISC1 knockdown did not affect the endogenous protein expression level of either NDEL1 or FEZ1 in adult neural progenitors (Figure 5B). Interestingly, DISC1 knockdown led to a significant decrease in the co-IP efficacy between FEZ1 and NDEL1 (Figure 5B). In contrast, NDEL1 knockdown did not affect the co-IP efficacy of FEZ1 and FEZ1 knockdown did not affect the co-IP efficacy of NDEL1 using anti-DISC1 antibodies (Figure 5C). Furthermore, FEZ1 overexpression in HEK293 cells did not appear to hinder the interaction between DISC1 and NDEL1, and vice versa, suggesting a lack of apparent competition between FEZ1 and NDEL1 for binding to DISC1 (Figure S5D).
Taken together, these results suggest that DISC1 interacts with both NDEL1 and FEZ1, whereas NDEL1 and FEZ1 appear to form a complex through DISC1, but not directly in vivo. These findings are consistent with our findings of a synergistic interaction between DISC1 and FEZ1 (Figure 3), and between DISC1 and NDEL1 (Duan et al., 2007), but not between FEZ1 and NDEL1 (Figure 4), in regulating distinct aspects of new neuron development in the adult brain (Table 1).
Epistasis between FEZ1 and DISC1 for risk of schizophrenia
In parallel to examining the FEZ1 role in neuronal development in an animal model, we conducted a genetic association study of FEZ1 in schizophrenia with a cohort of 279 Caucasian patients with schizophrenia and schizoaffective disorder and 249 Caucasian healthy controls (ZHH cohort) (Burdick et al., 2008). We assessed 4 SNPs within the FEZ1 gene, spanning B36 positions 124834271 to 124858699 (rs12224788; rs10893385; rs618900; rs2849222; Figure 6A). The linkage disequilibrium (LD) among the 4 SNPs comprising the haplotypes was high with D-prime values of 0.93 or greater. All SNPs were in Hardy-Weinberg equilibrium (HWE; data not shown). However, χ2 analyses revealed that none of the four genotyped SNPs were associated with a significant risk for schizophrenia (Supplementary Table 1A). In addition, there were no significant haplotype associations with schizophrenia susceptibility (Supplementary Table 1A).
Figure 6. Epistatic interaction between FEZ1 and DISC1 for the risk of schizophrenia.

(A) Linkage disequilibrium (χ2) structure of FEZ1 haplotype block using Haploview 3.32. The 4 SNPs formed a single haplotype block within the FEZ1 gene.
(B) A summary plot of χ2 analyses with Zucker Hillside Hospital (ZHH) samples (left) and the Genetic Association Information Network (GAIN) samples (right) to depict the DISC1-FEZ1 genotype interaction graphically. Subjects are grouped by genotype at both FEZ1 rs12224788 and DISC1 Ser704Cys. The X and Y axis represent FEZ1 genotype group and odds ratio respectively. The likelihood ratio test from the regression analysis indicated a significant FEZ1 x DISC1 interaction (P = 0.029 for ZHH sample and P = 0.05 for the GAIN sample).
(C) Summary of the likelihood ratio test in a backward stepwise regression with ZHH and GAIN samples (See Table S1B for sample size and composition). Genotypes are dichotomized for regression analyses. The same sets of samples as in (B) were analyzed. (*: P < 0.05)
Several converging lines of evidence have suggested that DISC1 Ser704Cys is of particular importance in increasing risk for schizophrenia through modifying DISC1 protein interactions (Burdick et al., 2008; Callicott et al., 2005; DeRosse et al., 2007; Lipska et al., 2006). We thus examined potential epistatic interactions between each of the 4 FEZ1 SNPs and the DISC1 Ser704Cys locus. Owing to the low frequency of the Cys allele, DISC1 Ser704Cys genotypes were grouped to compare subjects carrying at least one copy of the Cys allele (Cys) with subjects homozygous for the Ser allele (SerSer). For all FEZ1 SNPs, we grouped minor allele carriers and major allele homozygotes to optimize power based on genotype frequencies (Supplementary Table 1B). We first investigated the possible influence that an interaction between FEZ1 and DISC1 might have on risk for schizophrenia by carrying out 4 separate χ2 analyses with one for each FEZ1 SNP, while conditioning the sample on DISC1 Ser704Cys status. These analyses revealed that the C allele at FEZ1 rs12224788 increased risk for schizophrenia in the context of a DISC1 SerSer background (χ2 = 4.75; df = 1; P = 0.029; OR = 2.55; 95% CI: 1.1-6.0; Fisher’s exact P value = 0.046), but was not significant in patients carrying 1 or 2 copies of the Cys allele (χ2 = 0.18; df = 1; P = 0.67; OR = 0.82; 95% CI: 0.3-2.1; Fisher’s exact P value = 0.815; Figure 6B). Likewise, FEZ1 rs10893385 χ2 results indicated a potential interaction with DISC1 Ser704Cys such that T allele carriers were at significantly increased risk for schizophrenia in the background of DISC1 SerSer (χ2 = 3.84; df = 1; P = 0.050; OR = 0.49; 95% CI: 0.2-1.0), but not in DISC1 Cys Carriers (χ2 = 0.14; df = 1; P = 0.711; OR = 1.16; 95% CI: 0.5-2.5; Figure S6A). Neither FEZ1 rs618900 nor rs2849222 showed any evidence of interaction at the χ2 level (Figures S6B and S6C). Therefore, to test for statistical evidence of a true epistatic interaction, we used the likelihood ratio test in a backward stepwise regression and included DISC1 Ser704Cys genotype, FEZ1 rs12224788 genotype, FEZ1 rs10893385, and an interaction term for each FEZ1 SNP (DISC1 x FEZ1). While none of the main effects for genotype were significant [DISC1 Ser704Cys (Beta = −0.27, P = 0.69); FEZ1 rs12224788 (Beta = −0.18, P = 0.701); FEZ1 rs10893385 (Beta = −0.08, P = 0.888)], the FEZ1 rs12224788 x DISC1 Ser704Cys interaction term was significant (Beta = 0.54, P = 0.028), indicating evidence of epistasis (Figure 6C). Despite the χ2 results, the interaction term for FEZ1 rs10893385 x DISC1 Ser704Cys was not significant (Beta = −0.42, P = 0.211), suggesting that this is not an epistatic relationship. The final model classified subject type with 59% accuracy with a χ2 = 4.88 (P = 0.027).
Subsequent analyses were conducted in the much larger Genetic Association Information Network (GAIN) sample set to test for a replication of our ZHH results. The GAIN sample consisted of 1351 schizophrenia cases (29.9% female; mean age 43.3 + 11.4 years) and 1378 healthy controls (54.0% female; mean age 51.1 + 17.0 years) for which genotype data at the four FEZ1 SNPs were available (Supplementary Table 1A). No significant results were detected on susceptibility to schizophrenia for each of the four FEZ1 SNPs, which is consistent with the result from the ZHH cohort (Supplementary Table 1A). The platform used to genotype GAIN (Affymetrix 6.0 chip) did not include the DISC1 Ser704Cys marker but has a perfect proxy for this SNP (rs1754605; r2 = 1.0). We then performed a backward stepwise regression to test for an interaction between the proxy SNP for DISC1 Ser704Cys and FEZ1 rs12224788 (Figure 6C). As this approach was to serve as a replication of the findings in the ZHH dataset, we included only the FEZ1 SNP with statistical evidence of epistasis (FEZ1 rs12224788) in the GAIN sample regression model (Supplementary Table 1B). Variables retained in the best fit model included FEZ1 genotype (Beta = 0.72; P = 0.041), DISC1 genotype (Beta = 0.53; P = 0.044), and the interaction term FEZ1 x DISC1 (Beta = −0.45; P = 0.039). While the significant interaction is consistent with results from the ZHH dataset, the Beta term is negative, suggesting a somewhat different pattern of interaction in the GAIN sample as compared with the ZHH sample. Specifically, χ2 analyses revealed only a trend-level association for the C allele at FEZ1 rs12224788 in DISC1 Ser homozygotes (χ2 = 1.53; df = 1; P = 0.12; OR = 1.2) and a significant association for the FEZ1 GG genotype in the context of a DISC1 Cys background (χ2 = 2.83; df = 1; P = 0.05; OR = 0.77; Figure 6B). While not identical, this pattern of risk association related to the interaction is consistent with that found in the ZHH sample.
We also performed a series of χ2 tests in the same way to test for a potential interaction between our NDEL1 risk SNP (rs1391768) (Burdick et al., 2008) and each of the four FEZ1 SNPs using the ZHH sample. We carried out 4 separate χ2 analyses with one for each FEZ1 SNP, while conditioning the sample on NDEL1 rs1391768 status. The results from these analyses provided no significant evidence of interaction among these four FEZ1 SNPs and NDEL1 rs1391768 (all P > 0.10; Figure S6D and data not shown).
Taken together, these genetic interaction results from clinical cohorts mirror the biochemical and cell biological findings of a synergistic interaction between FEZ1 and DISC1, but not between FEZ1 and NDEL1, in regulating neuronal development in the animal model.
DISCUSSION
Cumulative evidence supports a significant neurodevelopmental contribution to the pathophysiology of schizophrenia and other major mental disorders (Lewis and Levitt, 2002; Rapoport et al., 2005; Weinberger, 1987), yet underlying molecular mechanisms are far from clear. DISC1 has emerged as a general risk factor for schizophrenia, schizoaffective disorder, bipolar disorder, major depression, autism and Asperger Syndrome (Chubb et al., 2008; Muir et al., 2008). In support of a role of DISC1 in mental disorders, dysfunction of DISC1 in rodent models leads to several schizophrenia and/or depression-related behavioral phenotypes (Johnstone et al., 2011). At the cellular level, expression of DISC1 is developmentally regulated within the nervous system (Miyoshi et al., 2003) and DISC1 in turn regulates multiple processes of both embryonic and adult neurogenesis (Christian et al., 2010). At the molecular level, a large number of potential DISC1 binding partners have been identified from a yeast two-hybrid screen (Chubb et al., 2008), many of which are also involved in neurodevelopmental processes implicated in the pathophysiology of psychiatric diseases. Regarded as an “edge piece” of psychiatric genetics, DISC1 may thus provide an entry point to understand molecular mechanisms and etiology underlying complex psychiatric disorders. Using a combinatorial approach to analyze the effect of genetic manipulations on individual neurons in the animal model, biochemical interactions of endogenous proteins in a homogenous cell population, and genetic associations in clinical cohorts, we demonstrate two parallel pathways for FEZ1 and NDEL1 that independently cooperate with DISC1 to regulate different aspects of neuronal development and risk for schizophrenia.
In the dentate gyrus of the hippocampus, a region implicated in schizophrenia pathophysiology (Harrison, 2004), neurogenesis continues throughout life in all mammals and contributes to specific brain functions (Zhao et al., 2008). Adult hippocampal neurogenesis provides a unique model system for dissecting signaling mechanisms that regulate neurodevelopment and offers several distinct advantages for molecular analysis, including a prolonged developmental time-course for more precise temporal resolution, a single neuronal subtype, and amenability to birth-dating, lineage tracing and genetic manipulations (Christian et al., 2010). Using this in vivo model system, we have identified novel functions of FEZ1 in regulating dendritic growth and soma size of newborn dentate granule cells in the adult hippocampus (Figure 1). Furthermore, results from concomitant suppression of DISC1 and FEZ1 support a synergistic interaction between these two proteins in regulating dendritic growth in vivo (Figure 3). In parallel, the NDEL1-DISC1 interaction regulates a complementary subset of developmental processes, namely, neuronal positioning and development of primary dendrites (Duan et al., 2007). Interestingly, there is no apparent synergistic interaction between FEZ1 and NDEL1 in regulating neuronal development (Figure 4) and no protein-protein interaction in the absence of DISC1 (Figure 5). These results illustrate two discrete pathways associated with the DISC1 interactome that, in conjunction, account for most of the DISC1-mediated effects in orchestrating development of newborn neurons during adult hippocampal neurogenesis (Table 1). Despite a large number of potential DISC1 interactors and multiple functions of DISC1 at different developmental stages and in different brain regions, our study demonstrates the feasibility of dissecting complex signaling pathways to identify molecules that mediate each of these specific aspects of DISC1 function in vivo.
Schizophrenia is a heterogeneous disease with complex genetic contributions. There are at least two non-mutually exclusive models to explain how genetic variations contribute to the risk for schizophrenia. In the “common disease - common alleles” model, an increased risk of schizophrenia stems from combined effects of multiple common polymorphisms that incrementally impact the overall susceptibility (Chakravarti, 1999). In the “common disease - rare alleles” model, schizophrenia is a common disease precipitated by the presence of rare alleles that individually confer significant risk with high penetrance (McClellan et al., 2007). In the case of DISC1, the chromosome translocation that disrupted DISC1 in the original Scottish family increased the risk of developing schizophrenia and other major mental disorders by about 50 fold compared to the general population (Blackwood et al., 2001), supporting the model of “common disease - rare alleles”. So far, genome-wide association studies (GWAS) of schizophrenia, including a recent large meta-analysis (Mathieson et al., 2011), have not yet shown a significant association with the DISC1 locus. Association of DISC1 haplotypes with schizophrenia and other mental illness has been found in some populations, but not others (Chubb et al., 2008). For example, one DISC1 SNP on exon 11 (rs821616, Ser704) has been identified as a risk allele (Callicott et al., 2005) and associated with positive symptoms in schizophrenia only in some populations (DeRosse et al., 2007). A number of studies identified other genes, including FEZ1, which indicate susceptibility in some populations, but cannot be confirmed in others. The failures to replicate risk association of specific genes might reflect small marginal effects, while the possibility of interaction is often overlooked due to computational and statistical limitations in the absence of preexisting hypotheses of specific gene pairs. In fact, epistatic interactions have been suggested as a major component of the “missing heritability” witnessed by GWAS (Eichler et al., 2010). Our analysis of a cohort of 279 patients with schizophrenia and 249 healthy controls suggests a lack of significant direct association of variation within the FEZ1 gene and risk for schizophrenia. Instead, we found an epistatic interaction between FEZ1 rs12224788 and DISC1 Ser704Cys, which significantly influences schizophrenia susceptibility. Specifically, an approximate 2.5-fold increased risk for schizophrenia is seen in individuals carrying the C allele at FEZ1 rs12224788, but only in the context of a DISC1 Ser704Ser background with no significant effect in DISC1 Cys carriers. Importantly, our analysis of a second cohort from the GAIN date set with independent and larger number of samples supports a similar conclusion (Figure 6). Whether our statistical evidence of epistasis reflects disruption of molecular interactions between DISC1 and FEZ1 involving coding variants in linkage disequilibrium with rs12224788, or whether rs12224788 tags a regulatory variant, remains unclear and is an interesting lead for future studies. Interestingly, there is also an epistatic interaction between NDEL1 rs1391768 and DISC1 Ser704Cys only in the context of a DISC1 Ser704Ser background (Burdick et al., 2008). On the other hand, we did not find any significant epistatic interaction between the four FEZ1 SNPs and four NDEL1 SNPs (Figure S6 and data not shown), although we cannot rule out the possibility of an epistatic interaction of these two genes at other SNPs. In a recent study of epistasis based on machine learning algorithms and functional magnetic resonance imaging (fMRI) analysis, significant interaction was found between DISC1, CIT and NDEL1 SNPs (Nicodemus et al., 2010). Taken together, these findings put DISC1 at the center of a signaling complex in which its interaction with different partners confers increased risk for schizophrenia as well as regulating different aspects of neuronal development.
Our results may begin to reconcile the contrasting views on genetically determined disease susceptibility. DISC1 is a multivariate modulator of risk conference with high penetrability and represents an essential component of divergent pathways that regulate disease and development. Due to this partial functional overlap between DISC1 and its binding partners, DISC1 emerges as a key player in disease susceptibility, whereas genes regulating a subset of DISC1 functions may only incrementally increase overall risk. Our results thus demonstrate how the two prevailing views of genetically conferred disease susceptibility are compatible in mechanistic terms. We provide evidence in support of large effects from the disruption of a single gene (DISC1) and how polymorphisms in DISC1 and associated genes can work synergistically, through epistatic mechanisms, resulting in increased risk for schizophrenia. Importantly, this synergistic interaction also reveals how genetic context is critical in determining the extent of susceptibility to disease pathogenesis. As shown in our association results, an individual SNP confers differential risk effects depending on the genetic background of the patient. Because of the prohibitively large number of genes that have been identified as potential risk factors for schizophrenia and related disorders, an efficient method to determine the relevant genetic interactions is through biochemical and cellular assays based on functional analysis. We provide an example of how a targeted investigation of molecular pathways associated with DISC1 functions can generate testable hypotheses of genetic interactions in the patient population. By continuing to investigate functional interactions of key genes associated with disease susceptibility at the cellular level, we can begin to unravel the complex genetic contributions to neuropsychiatric disorders.
EXPERIMENTAL PROCEDURES
Constructs, Cell Culture and Biochemistry
Engineered self-inactivating murine oncoretroviruses were used to co-express shRNAs under the U6 promoter and GFP or mCherry under the EF1α promoter (pUEG/pUEM vector), or to co-express mouse fez1 cDNA (without the 3′ UTR) under the Ubiquitin C promoter and GFP following the IRES sequence (pCUXIE vector), specifically in proliferating cells and their progeny in vivo (Duan et al., 2007). shRNAs against mouse disc1 (shRNA-D1, shRNA-D3) and ndel1 (shRNA-N1) have been previously characterized (Duan et al., 2007; Faulkner et al., 2008). Two fez1 shRNAs were designed to target the 3′ UTR of mouse fez1 gene with following sequences: shRNA-FEZ1#1 (F1): 5′-CTTATACTCTTAAGACTAA-3′; shRNA-FEZ1#2 (F2): 5′-GCGTGTATTTAAACGTGTA-3′. The control shRNA vector (shRNA-C1; C1) contains a scrambled sequence without homology to any known mammalian mRNA: 5′-TTCTCCGAACGTGTCACGT-3′ (Qiagen).
Neural progenitors were isolated from adult mice hippocampi (C57BL/6) and cultured as a monolayer as previously described (Kim et al., 2009; Ma et al., 2008). At 48 hrs after retroviral infection, cell lysates were prepared in the lysis buffer containing 10% glycerol, 1% nonylphenoxypolyethoxy ethanol (Nonidet P-40), 50 mM Tris-Cl (pH 7.5), 200 mM NaCl, 2 mM MgCl2, 0.2 mM Na3VO4, and 1 μg/ml protease inhibitor cocktail (Roche). Protein lysates were subjected to Western Blot analysis for FEZ1 (goat, 1:1000; Novus), DISC1 (goat, 1:1000; Santa Cruz), NDEL1 (rat, 1:1000; gift of A. Sawa) (Kamiya et al., 2005), and GAPDH (mouse, 1:1000; Abcam). For co-IP analysis, both adult mouse neural progenitors at 48 hrs after retroviral infection and dissected hippocampal tissues from adult mice were used as previously described (Kim et al., 2009). Samples were immunoprecipitated with antibodies against DISC1 (goat, 1:100; Santa Cruz, or rabbit, 1:100; Zymed), FEZ1, or NDEL1, and then subjected to Western Blot analysis. Blots were stripped and reblotted with the same antibodies used for their immunoprecipitation to ensure equal loading. For quantification, the densitometry measurement of each band (Image J) was first normalized to that of GAPDH and then averaged from at least three independent experiments.
In vivo Genetic Manipulation of Neural Progenitors with Engineered Retroviruses
High titers of engineered retroviruses were produced as previously described (Duan et al., 2007). Adult female C57BL/6 mice (7-8 weeks old; Charles River) housed under standard conditions were anaesthetized. Concentrated retroviruses were stereotaxically injected into the dentate gyrus at 4 sites (0.5 μl per site at 0.25 μl/min) with the following coordinates (in mm; posterior = 2 from Bregma, lateral = + 1.6, ventral = 2.5; posterior = 3 from Bregma, lateral = + 2.6, ventral = 3.2) as previously described (Duan et al., 2007). All animal procedures used in this study were performed in accordance with the protocols approved by the Institutional Animal Care and Use Committee.
Immunohistology, Confocal Imaging and Analysis
Coronal brain sections (40 μm thick) were prepared from injected mice at 14 dpi and processed for immunostaining as previously described (Duan et al., 2007; Ge et al., 2006). For rescue experiments (Figure 2), anti-GFP antibodies (rabbit, 1:1000, Abcam) were used to enhance the signaling for all conditions in parallel. The sections were incubated for 30 min in 4′,6′-diaminodino-2-phenylindole (DAPI, 1:5000) before washing and mounting. Images were acquired on a META multiphoton confocal system (Zeiss LSM 510) using a multi-track configuration. At least two animals for each condition were analyzed. Statistical comparison of data was performed by ANOVA.
For analysis of cell morphology, Z-series stacks of confocal images were taken and a single confocal image slice with the largest soma area for individual GFP+ neurons was used for quantification using NIH ImageJ program. For analysis of neuronal positioning, single section confocal images of GFP+ neurons with DAPI staining were used to determine the cell localization within four areas as defined in Figure 1B. For analysis of dendritic development, three dimensional (3D) reconstruction of entire dendritic processes of each GFP+ neuron was made from Z-series stacks of confocal images. The 2D projection images were traced with NIH ImageJ. All GFP+ dentate granule cells with largely intact dendritic trees were analyzed for total dendritic length as previously described (Duan et al., 2007; Ge et al., 2006). The measurements did not include corrections for inclinations of dendritic process and therefore represented projected lengths. Sholl analysis for dendritic complexity was carried out by counting the number of dendrites that crossed a series of concentric circles at 10 μm intervals from the cell soma as previously described (Duan et al., 2007). Analysis of axonal development was carried out as previously described (Faulkner et al., 2008). Briefly, cross-sections (50 μm thick) taken perpendicular to the long axis (septal-temporal) were prepared from dissected hippocampi of fixed mouse brain at 14 dpi and processed for immunostaining using anti-GFP antibodies (goat, 1:500, Rockland) and DAPI (1:5000). 3D reconstructions of entire images were obtained using stitch and maximum intensity projection functions in the Zen software (Zeiss).
Analysis of Cohorts of Schizophrenia Patients and Healthy Controls
For the ZHH sample, the same study group as previously analyzed for DISC1 and NDEL1 interaction were used (Burdick et al., 2008). Briefly, the cohort included 279 Caucasian patients with schizophrenia or schizoaffective disorder (SZ; 35.5% female) with a mean age of 38.6 + 10.8 years and an estimated IQ of 96.6 + 8.5 (based on WRAT-3 Reading). All subjects provided written informed consent to an Institutional Review Board of the North Shore-Long Island Jewish Health System (NSLIJHS)-approved protocol. Clinical characteristics that were collected included duration of illness (17.0 + 10.8 years) and global assessment of function (GAF; 38.7 + 15.6). Caucasian healthy control subjects (HC; n = 249) were recruited from the general population. Subjects were excluded if they had an Axis I diagnosis, active or recent substance abuse, or if they had a first degree relative with a known or suspected Axis I disorder based on family history questionnaire. Controls were 52.2% female, had a mean age of 48.4 + 18.6 years and an estimated IQ of 105.0 + 12.4. All subjects were Caucasian by self-report and drawn from a single geographic location (Glen Oaks, NY area). Although population stratification is a potential confound in any case-control study, we have previously demonstrated that undetected substructure is not present in our geographically homogeneous population. In a genome-wide association study of a case-control cohort collected by the same methods described above (Lencz et al., 2007), we tested for stratification using 210 ancestry informative markers selected for maximal informativeness and observed no differences between patients and controls beyond chance levels. Moreover, none of the subjects in the cohort deviated from a single population as assessed by the STRUCTURE program (Pritchard et al., 2000). Patient diagnosis was established through structured interview (Structured Clinical Interview-DSM-IV; SCID-IV) (First et al., 1998) and confirmed by diagnostic consensus conference, which utilizes expert clinical opinion alongside SCID-IV data and corroborating medical record information. Healthy controls for the project were assessed using the Structured Clinical Interview for DSM-IV, Non-Patient edition, specifically designed for healthy subjects to rule out Axis I diagnoses. In addition to the structured diagnostic interview, potential subjects were screened to rule out any history of CNS trauma, neurological disorder, or diagnosed learning disability.
FEZ1 genotyping procedures were carried out using the Affymatrix 500K platform as previously described (Lencz et al., 2007). Briefly, genomic DNA was extracted from whole blood and hybridized to two chips containing ~262,000 and ~238,000 SNPs based on manufacturer′s specifications. Patients and controls were proportionally distributed on each 96-well plate. Genotype calls were made using Bayesian Robust Linear Model with Mahalanobis distance classifier algorithm threshold at 0.5 applied to batches of 100 samples. Mean call rates < 90% on both chips, or < 85% on one chip, were rejected, resulting in a mean call rate for the retained sample of 97%. Allele frequencies, Hardy-Weinberg equilibrium (HWE), and linkage disequilibrium structure were examined using Haploview 3.32 (Barrett et al., 2005). The four SNPs formed a single haplotype block as shown in Figure 6A.
For FEZ1 association with illness, none of the 4 SNPs in the haplotype deviated from HWE (P > 0.05). χ2 test statistics were used to test for allelic and genotypic associations to schizophrenia for single SNPs, as well as for group differences on haplotype frequencies. For FEZ1 and DISC1 epistasis, we tested for a genotype interaction between each of the 4 genotyped FEZ1 SNPs and DISC1 Ser704Cys by utilizing a likelihood ratio test in an unconditional logistic regression model, with subject type (SZ versus HC) as the dependent measure and entering FEZ1 rs12224788 genotype, DISC1 Ser704Cys genotype, and the interaction term FEZ1 x DISC1 in a backward stepwise model. In addition, χ2 analyses were conducted to determine odds ratios for the significant interaction, by conditioning the sample on DISC1 Ser704Cys. Identical statistical methods were utilized when investigating the potential interactions between FEZ1 and NDEL1, focusing on our previously identified NDEL1 risk SNP (rs1391768) (Burdick et al., 2008) and testing each of the four FEZ1 SNPS for interaction.
The Molecular Genetics of Schizophrenia (MGS) sample from the Genetic Association Information Network (GAIN) included 1351 Caucasian schizophrenia cases and 1378 healthy controls with available genotype data at the four FEZ1 SNPs. The platform used to genotype the GAIN samples was the Affymetrix 6.0 array (Shi et al., 2009). The schizophrenia sample was 29.9% female (mean age: 43.3 + 11.4 years). The GAIN controls were 54.0% female (mean age: 51.1 + 17.0 years). Analyses were carried out using the identical methodology as those used for the ZHH sample. First, χ2 analyses were conducted to test for association of the four FEZ1 SNPs with risk for schizophrenia. Next, we carried out a backward stepwise regression to test for an interaction between the proxy SNP for DISC1 Ser704Cys and FEZ1 rs12224788. Only the FEZ1 SNP with statistical evidence of epistasis (FEZ1 rs12224788) in the ZHH analyses was included in the GAIN sample regression model, as this was meant to serve as a replication cohort.
Supplementary Material
Highlights.
FEZ1 regulates specific aspects of adult hippocampal neurogenesis
FEZ1 interacts with DISC1 in regulating neuronal development
FEZ1 and NDEL1 differentially mediate DISC1-dependent neuronal development
FEZ1 and DISC1 interact epistatically to affect the risk for schizophrenia
ACKNOWLEDGEMENT
We thank D. Weinberg, D. Valle, and members of Ming and Song Laboratories for critical comments, L. Liu, Y. Cai and H. Qasim for technical support, A. Sawa and A. Kamiya for anti-NDEL1 antibodies. This work was supported by NIH (NS048271, HD069184), NARSAD, and MSCRF to G-l.M., by NIH (NS047344, AG024984, MH084018, MH087874), IMHRO and Johns Hopkins BSI to H.S., by MH79800, MH080173 and the Donald and Barbara Zucker Foundation to A.K.M., and by MH077807 to K.E.B. J.Y.K. and K.C. were partially supported by postdoctoral fellowships from MSCRF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Blackwood DH, Fordyce A, Walker MT, St Clair DM, Porteous DJ, Muir WJ. Schizophrenia and affective disorders--cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet. 2001;69:428–433. doi: 10.1086/321969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom L, Horvitz HR. The Caenorhabditis elegans gene unc-76 and its human homologs define a new gene family involved in axonal outgrowth and fasciculation. Proc Natl Acad Sci U S A. 1997;94:3414–3419. doi: 10.1073/pnas.94.7.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick KE, Kamiya A, Hodgkinson CA, Lencz T, DeRosse P, Ishizuka K, Elashvili S, Arai H, Goldman D, Sawa A, Malhotra AK. Elucidating the relationship between DISC1, NDEL1 and NDE1 and the risk for schizophrenia: evidence of epistasis and competitive binding. Hum Mol Genet. 2008;17:2462–2473. doi: 10.1093/hmg/ddn146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callicott JH, Straub RE, Pezawas L, Egan MF, Mattay VS, Hariri AR, Verchinski BA, Meyer-Lindenberg A, Balkissoon R, Kolachana B, et al. Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc Natl Acad Sci U S A. 2005;102:8627–8632. doi: 10.1073/pnas.0500515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, Bonnert TP, Whiting PJ, Brandon NJ. Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry. 2007;12:74–86. doi: 10.1038/sj.mp.4001880. [DOI] [PubMed] [Google Scholar]
- Chakravarti A. Population genetics--making sense out of sequence. Nat Genet. 1999;21:56–60. doi: 10.1038/4482. [DOI] [PubMed] [Google Scholar]
- Christian K, Song H, Ming GL. Adult neurogenesis as a cellular model to study schizophrenia. Cell Cycle. 2010;9:636–637. doi: 10.4161/cc.9.4.10932. [DOI] [PubMed] [Google Scholar]
- Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ, Millar JK. The DISC locus in psychiatric illness. Mol Psychiatry. 2008;13:36–64. doi: 10.1038/sj.mp.4002106. [DOI] [PubMed] [Google Scholar]
- DeRosse P, Hodgkinson CA, Lencz T, Burdick KE, Kane JM, Goldman D, Malhotra AK. Disrupted in schizophrenia 1 genotype and positive symptoms in schizophrenia. Biol Psychiatry. 2007;61:1208–1210. doi: 10.1016/j.biopsych.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Duan X, Chang JH, Ge S, Faulkner RL, Kim JY, Kitabatake Y, Liu XB, Yang CH, Jordan JD, Ma DK, et al. Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell. 2007;130:1146–1158. doi: 10.1016/j.cell.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Kang E, Liu CY, Ming GL, Song H. Development of neural stem cell in the adult brain. Curr Opin Neurobiol. 2008;18:108–115. doi: 10.1016/j.conb.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto A, Asai N, Namba T, Wang Y, Kato T, Tanaka M, Tatsumi H, Taya S, Tsuboi D, Kuroda K, et al. Roles of disrupted-in-schizophrenia 1-interacting protein girdin in postnatal development of the dentate gyrus. Neuron. 2009;63:774–787. doi: 10.1016/j.neuron.2009.08.015. [DOI] [PubMed] [Google Scholar]
- Faulkner RL, Jang MH, Liu XB, Duan X, Sailor KA, Kim JY, Ge S, Jones EG, Ming GL, Song H, Cheng HJ. Development of hippocampal mossy fiber synaptic outputs by new neurons in the adult brain. Proc Natl Acad Sci U S A. 2008;105:14157–14162. doi: 10.1073/pnas.0806658105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- First MB, Spitzer R, Williams JBW, Gibbon M. Structured clinical interview for Axis I DSM IV disorders (scid/p, vERSION 20) Biometric Research Department; New York: 1998. [Google Scholar]
- Ge S, Goh EL, Sailor KA, Kitabatake Y, Ming GL, Song H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. 2006;439:589–593. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ. The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology (Berl) 2004;174:151–162. doi: 10.1007/s00213-003-1761-y. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- Hodgkinson CA, Goldman D, Ducci F, DeRosse P, Caycedo DA, Newman ER, Kane JM, Roy A, Malhotra AK. The FEZ1 gene shows no association to schizophrenia in Caucasian or African American populations. Neuropsychopharmacology. 2007;32:190–196. doi: 10.1038/sj.npp.1301177. [DOI] [PubMed] [Google Scholar]
- Johnstone M, Thomson PA, Hall J, McIntosh AM, Lawrie SM, Porteous DJ. DISC1 in schizophrenia: genetic mouse models and human genomic imaging. Schizophr Bull. 2011;37:14–20. doi: 10.1093/schbul/sbq135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y, Sawamura N, Park U, Kudo C, Okawa M, et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol. 2005;7:1167–1178. doi: 10.1038/ncb1328. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Krebs J, Fabel K. The contribution of failing adult hippocampal neurogenesis to psychiatric disorders. Curr Opin Psychiatry. 2008;21:290–295. doi: 10.1097/YCO.0b013e3282fad375. [DOI] [PubMed] [Google Scholar]
- Kim JY, Duan X, Liu CY, Jang MH, Guo JU, Pow-anpongkul N, Kang E, Song H, Ming GL. DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron. 2009;63:761–773. doi: 10.1016/j.neuron.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga M, Ishiguro H, Horiuchi Y, Albalushi T, Inada T, Iwata N, Ozaki N, Ujike H, Muratake T, Someya T, Arinami T. Failure to confirm the association between the FEZ1 gene and schizophrenia in a Japanese population. Neurosci Lett. 2007;417:326–329. doi: 10.1016/j.neulet.2007.02.055. [DOI] [PubMed] [Google Scholar]
- Kuroda S, Nakagawa N, Tokunaga C, Tatematsu K, Tanizawa K. Mammalian homologue of the Caenorhabditis elegans UNC-76 protein involved in axonal outgrowth is a protein kinase C zeta-interacting protein. J Cell Biol. 1999;144:403–411. doi: 10.1083/jcb.144.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencz T, Morgan TV, Athanasiou M, Dain B, Reed CR, Kane JM, Kucherlapati R, Malhotra AK. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry. 2007;12:572–580. doi: 10.1038/sj.mp.4001983. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Peters T, Hyde TM, Halim N, Horowitz C, Mitkus S, Weickert CS, Matsumoto M, Sawa A, Straub RE, et al. Expression of DISC1 binding partners is reduced in schizophrenia and associated with DISC1 SNPs. Hum Mol Genet. 2006;15:1245–1258. doi: 10.1093/hmg/ddl040. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Alonso M, Grubb MS. Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci. 2006;7:179–193. doi: 10.1038/nrn1867. [DOI] [PubMed] [Google Scholar]
- Ma DK, Chiang CH, Ponnusamy K, Ming GL, Song H. G9a and Jhdm2a regulate embryonic stem cell fusion-induced reprogramming of adult neural stem cells. Stem Cells. 2008;26:2131–2141. doi: 10.1634/stemcells.2008-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136:1017–1031. doi: 10.1016/j.cell.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson I, Munafo MR, Flint J. Meta-analysis indicates that common variants at the DISC1 locus are not associated with schizophrenia. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan JM, Susser E, King MC. Schizophrenia: a common disease caused by multiple rare alleles. Br J Psychiatry. 2007;190:194–199. doi: 10.1192/bjp.bp.106.025585. [DOI] [PubMed] [Google Scholar]
- Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, Clair DM, Muir WJ, Blackwood DH, Porteous DJ. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–250. doi: 10.1146/annurev.neuro.28.051804.101459. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi K, Honda A, Baba K, Taniguchi M, Oono K, Fujita T, Kuroda S, Katayama T, Tohyama M. Disrupted-In-Schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol Psychiatry. 2003;8:685–694. doi: 10.1038/sj.mp.4001352. [DOI] [PubMed] [Google Scholar]
- Morris JA, Kandpal G, Ma L, Austin CP. DISC1 (Disrupted-In-Schizophrenia 1) is a centrosome-associated protein that interacts with MAP1A, MIPT3, ATF4/5 and NUDEL: regulation and loss of interaction with mutation. Hum Mol Genet. 2003;12:1591–1608. doi: 10.1093/hmg/ddg162. [DOI] [PubMed] [Google Scholar]
- Muir WJ, Pickard BS, Blackwood DH. Disrupted-in-Schizophrenia-1. Curr Psychiatry Rep. 2008;10:140–147. doi: 10.1007/s11920-008-0025-2. [DOI] [PubMed] [Google Scholar]
- Nicodemus KK, Callicott JH, Higier RG, Luna A, Nixon DC, Lipska BK, Vakkalanka R, Giegling I, Rujescu D, St Clair D, et al. Evidence of statistical epistasis between DISC1, CIT and NDEL1 impacting risk for schizophrenia: biological validation with functional neuroimaging. Hum Genet. 2010;127:441–452. doi: 10.1007/s00439-009-0782-y. [DOI] [PubMed] [Google Scholar]
- Ozeki Y, Tomoda T, Kleiderlein J, Kamiya A, Bord L, Fujii K, Okawa M, Yamada N, Hatten ME, Snyder SH, et al. Disrupted-in-Schizophrenia-1 (DISC-1): mutant truncation prevents binding to NudE-like (NUDEL) and inhibits neurite outgrowth. Proc Natl Acad Sci U S A. 2003;100:289–294. doi: 10.1073/pnas.0136913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport JL, Addington AM, Frangou S, Psych MR. The neurodevelopmental model of schizophrenia: update 2005. Mol Psychiatry. 2005;10:434–449. doi: 10.1038/sj.mp.4001642. [DOI] [PubMed] [Google Scholar]
- Rastogi A, Zai C, Likhodi O, Kennedy JL, Wong AH. Genetic association and post-mortem brain mRNA analysis of DISC1 and related genes in schizophrenia. Schizophr Res. 2009;114:39–49. doi: 10.1016/j.schres.2009.06.019. [DOI] [PubMed] [Google Scholar]
- Sakae N, Yamasaki N, Kitaichi K, Fukuda T, Yamada M, Yoshikawa H, Hiranita T, Tatsumi Y, Kira J, Yamamoto T, et al. Mice lacking the schizophrenia-associated protein FEZ1 manifest hyperactivity and enhanced responsiveness to psychostimulants. Hum Mol Genet. 2008;17:3191–3203. doi: 10.1093/hmg/ddn215. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Mori D, Toyo-oka K, Chen A, Garrett-Beal L, Muramatsu M, Miyagawa S, Hiraiwa N, Yoshiki A, Wynshaw-Boris A, Hirotsune S. Complete loss of Ndel1 results in neuronal migration defects and early embryonic lethality. Mol Cell Biol. 2005;25:7812–7827. doi: 10.1128/MCB.25.17.7812-7827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Matsuzaki S, Hattori T, Kumamoto N, Miyoshi K, Katayama T, Tohyama M. DISC1-kendrin interaction is involved in centrosomal microtubule network formation. Biochem Biophys Res Commun. 2008;377:1051–1056. doi: 10.1016/j.bbrc.2008.10.100. [DOI] [PubMed] [Google Scholar]
- Shu T, Ayala R, Nguyen MD, Xie Z, Gleeson JG, Tsai LH. Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron. 2004;44:263–277. doi: 10.1016/j.neuron.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- Yamada K, Nakamura K, Minabe Y, Iwayama-Shigeno Y, Takao H, Toyota T, Hattori E, Takei N, Sekine Y, Suzuki K, et al. Association analysis of FEZ1 variants with schizophrenia in Japanese cohorts. Biol Psychiatry. 2004;56:683–690. doi: 10.1016/j.biopsych.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.