

Abstract
The renin-angiotensin-aldosterone system is inappropriately activated in obesity. In individuals at risk for diabetes, inhibition of the renin-angiotensin-aldosterone system protects against kidney and heart disease, and also reduces the incidence of diabetes in large clinical trials. At a cellular level, angiotensin II and aldosterone induce insulin resistance by increasing oxidative stress and altering insulin signaling, leading to decreased glucose transport. Angiotensin II also contributes to oxidative stress, inflammation, and apoptosis in pancreatic β-cells. Aldosterone diminishes glucose-stimulated insulin secretion in vivo and in vitro from isolated pancreatic islets and cultured β-cells through a mineralocorticoid receptor-independent mechanism. We review these findings in the context of pharmacological strategies to interrupt the renin-angiotensin-aldosterone system to highlight the potential application of these strategies to the prevention of diabetes progression.
The renin-angiotensin-aldosterone system and diabetes
Approximately 246 million adults worldwide have diabetes.1 This number is expected to grow to 380 million by the year 2025.1,2 Diabetes is associated with a dramatically increased risk of heart attack, stroke, and renal failure, as well as disability.3 The cost of diabetes-related health care was estimated to be $232 billion in 2007. With an expected 7 million incident cases of type 2 diabetes mellitus (T2DM) per year worldwide,2 there is a pressing need to develop strategies to prevent the development of T2DM in high risk individuals, such as those with the metabolic syndrome. Because recent clinical studies demonstrate a protective effect of renin-angiotensin-aldosterone system (RAAS) blockade on T2DM incidence, we review the mechanisms by which these drugs may confer protection.
The RAAS is essential to increasing sodium and fluid reabsorption when the body is challenged by sodium restriction or fluid loss (Figure 1). In the kidney, renin secretion by the juxtaglomerular apparatus is triggered by reduced sodium chloride delivery to macula densa cells in the distal nephron. Renin converts angiotensinogen into angiotensin I (Ang I), which is largely inactive. Ang I is cleaved by Ang I converting enzyme (ACE) to angiotensin II (Ang II). Ang II activates the Ang II type 1 (AT1), Ang II type 2 (AT2), and other receptors. AT1 activation produces intense vasoconstriction in the systemic vasculature and raises blood pressure, whereas AT2 produces vasodilatation. In the adrenal gland, Ang II stimulates aldosterone secretion from glomerulosa cells via AT1. Aldosterone increases renal sodium chloride and water reabsorption by activation of the mineralocorticoid receptor (MR) in the principal cells in the distal nephron. An expanded discussion of the RAAS is provided elsewhere.4
Figure 1. The renin-angiotensin-aldosterone system.

The cascade is initiated by renin secretion, and subsequent conversion of angiotensinogen to angiotensin I (Ang I). Angiotensin II (Ang II) produces vasoconstriction and stimulates adrenal aldosterone secretion via AT1 receptors. Pharmacologic agents are available to block this pathway at nearly every step in the pathway (highlighted in red). Aliskiren is the only currently available renin inhibitor. Angiotensin I converting enzyme (ACE) inhibitors prevent conversion of Ang I to Ang II. Mineralocorticoid receptor (MR) antagonists (e.g. spironolactone, eplerenone) block the effects of aldosterone in the kidney. Downstream targets of aldosterone include the epithelial sodium channel (ENaC) which is upregulated by the MR and mediates sodium reabsorption in the distal kidney.
The RAAS is pathologically activated in certain conditions such as essential hypertension, renal artery stenosis, and congestive heart failure. Hyperaldosteronism can result from inappropriate secretion by the adrenal and produces resistant hypertension with hypokalemia and metabolic alkalosis. Drugs which block the RAAS at various steps in the pathway are widely available (Figure 1), and include direct renin inhibitors, ACE inhibitors (e.g., ramipril, lisinopril, captopril, etc.), AT1 receptor blockers (ARBs; e.g., valsartan, losartan, etc.), and MR antagonists (spironolactone and eplerenone). Under conditions of adequate sodium intake, the RAAS is suppressed, and pharmacologic blockade has little effect on blood pressure. RAAS blockade effectively reduces blood pressure in essential hypertension, however, and this effect is accentuated by diuretic administration.
ACE inhibitors and ARBs have long been used to prevent cardiovascular and renal damage in diabetic patients.5-7 Recent studies suggest that drugs that decrease the formation or actions of Ang II may also reduce the incidence of diabetes.8,9 We review here the evidence that the RAAS is activated in obesity and diabetes, including the mechanisms by which Ang II induces insulin resistance and affects pancreatic inflammation and apoptosis. We also examine recent data implicating aldosterone as a major effector in the development of insulin resistance and decreased pancreatic β-cell function. Finally, we consider the implications of these studies for the development of pharmacologic strategies to reduce incident diabetes.
The renin-angiotensin-aldosterone system is activated in obesity
Adipocytes express angiotensinogen (a precursor to Ang II), renin, and the Ang II type 1 (AT1) receptor, with some species variability.10 The local formation of Ang II appears to be increased in obesity and leads to activation of nuclear factor kappa B (NFκB) and the formation of inflammatory cytokines.10 Ang II also inhibits preadipocyte recruitment, leading to the storage of lipids outside of adipose tissue, lipotoxicity and insulin resistance, effects than can be blocked by AT1 receptor inhibition.11 In animal models of obesity, aldosterone is inappropriately elevated during high fat feeding, demonstrated by an increase in circulating aldosterone despite net positive sodium balance.12 In humans, plasma aldosterone concentrations correlate with body mass index or indices of insulin resistance.13 Cell culture studies demonstrate that human adipocytes produce aldosterone-stimulating substances, such as oxidized derivates of linoleic acid.14,15 Conversely, weight loss in humans results in decreased circulating angiotensinogen, plasma renin activity, and aldosterone concentrations, as well as lipid angiotensinogen.16-18 These data suggest that adipocyte-derived factors inappropriately activate the RAAS, and provide a rationale for targeting this system in obese patients to prevent cardiovascular and metabolic complications.
Angiotensin converting-enzyme inhibitors and angiotensin receptor blockers decrease the incidence of diabetes in high-risk populations
ACE inhibitors and ARBs decrease the incidence of new onset diabetes in large randomized clinical trials in patients with heart failure or at risk for coronary artery disease.8 In the first prospective randomized clinical trial designed to test the hypothesis that interrupting the RAAS would improve glucose homeostasis, the Diabetes Reduction Assessment with Ramipril and Rosiglitazone Medication (DREAM) study, treatment with the ACE inhibitor ramipril did not significantly reduce the incidence of new-onset diabetes in patients with impaired fasting glucose (IFG) (18.1% versus 19.5%, P=0.15).19 A greater proportion of subjects randomized to ramipril achieved fasting normoglycemia (42.5 vs. 38.2%; P<0.001 vs. control), however, and the median 2-hour OGTT glucose was lower in the ramipril group (135.1 vs. 140.5 mg/dl; P=0.01).
The NAVIGATOR study compared the effect of the ARB valsartan versus placebo on the development of diabetes in patients with impaired glucose tolerance and cardiovascular risk factors or disease.9 The incidence of diabetes was modestly but significantly lower in the valsartan-treated group compared to the placebo group (31% versus 33%; hazard ratio in the valsartan group, 0.86 with a 95% confidence interval [CI] of 0.80 to 0.92; P<0.001).
Compared to traditional anti-diabetic agents, ACE inhibition and ARBs have modest effects on glycemia. Treatment with the biguanide metformin reduced the incidence of T2DM in the Diabetes Prevention Program study, as did troglitazone.20,21 In the DREAM study, rosiglitazone significantly decreased the incidence of new onset T2DM.22 Thiazolidinediones are also associated with increased risk of cardiovascular events, however.23,24 Therefore, the need to develop pharmacological strategies to reduce the incidence of T2DM without increasing the risk of cardiovascular events remains. Randomized trials have established the cardiovascular benefits of ACE inhibitors, ARBs, and renin inhibition in high-risk populations.25-27
Angiotensin II and aldosterone decrease insulin sensitivity
The mechanism(s) whereby preventing the formation or action of Ang II could decrease the incidence of diabetes probably involves many pathways, including improved blood flow and decreased sympathetic activity. Activation of the RAAS also causes insulin resistance directly (Figure 2).
Figure 2. Angiotensin II and aldosterone induce insulin resistance.

Angiotensin II (Ang II) and aldosterone activate nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase to generate reactive oxygen species (O2-). Activation of redox-sensitive serine kinases such as c-jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK)-1 leads to the phosphorylation of serines in insulin receptor substrate-1 (IRS-1) and decreased interaction with phosphatidylinositol 3-kinase (PI3K). This in turn leads to decreased activation of protein kinase B (Akt) and PKC, decreased translocation of GLUT4 to the membrane and decreased glucose transport. Blockade of the Ang II subtype 1 receptor (AT1R) or the mineralocorticoid receptor prevents these effects of Ang II and aldosterone, respectively. Direct renin inhibitors and angiotensin-converting enzyme (ACE) inhibitors improve insulin resistance by decreasing the formation of Ang II and aldosterone. ACE inhibitors also increase glucose transport via a nitric oxide-dependent mechanism.
In muscle, Ang II inhibits phosphorylation of insulin receptor substrate (IRS)-1, preventing increases in phosphatidylinositol 3 (PI3)-kinase and subsequent translocation of glucose transporter (GLUT) 4 to the cell membrane.28-30 The mechanism through which Ang II decreases insulin-dependent GLUT-4 translocation involves activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and the formation of reactive oxygen species.31,32 Conversely, ACE inhibitors and ARBs increase GLUT 4 translocation to the membrane and improve skeletal muscle glucose uptake in animal models.33,34 ARBs can also increase GLUT4 translocation and improve insulin sensitivity in cultured adipocytes and rodent models.35-37 Renin inhibition also improves insulin sensitivity and muscle glucose uptake in transgenic rats which harbor and express murine renin in extra-renal tissues (TG(mRen2)27 rats (Ren2)).38 The direct renin inhibitor aliskiren improves insulin resistance, as determined by an insulin tolerance test, in the db/db mouse model.39
Studies provide conflicting data as to the role of nitric oxide (NO) in the pathogenesis of insulin resistance during activation of the RAAS. On the one hand, increased NO and cyclic guanosine monophosphate (cGMP) have been reported to contribute the favorable effects of ACE inhibition and Ang II receptor blockade on muscle glucose uptake.34,40 For example, treatment of diabetic mice with an ACE inhibitor decreased circulating glucose and insulin concentrations and increased skeletal muscle glucose uptake through a bradykinin B2receptor- and NO synthase (NOS)-dependent pathway.34,41 Likewise, NOS inhibition prevented the favorable effects of the ARB valsartan on muscle glucose uptake.40 On the other hand, Csibi et al. have reported that Ang II induces of tyrosine nitration of Akt, resulting in inhibition of Akt phosphorylation and decreased translocation of GLUT 4, an effect that could be blocked by NOS inhibition.42
Recent data suggest that Ang II may cause insulin resistance in part through aldosterone, or at least mineralocorticoid receptor (MR)-dependent, effects. For example, the aldosterone inhibitor spironolactone decreased oxidative stress and increased insulin-induced muscle glucose uptake in the Ren2 rat.43 Likewise, aldosterone induces oxidative stress, inflammation and insulin resistance in rodent adipocytes, effects that are blocked by eplerenone, an MR inhibitor.44 Aldosterone induces oxidative stress, degradation of insulin receptor substrates 1 and 2, and insulin resistance in cultured 3T3-L1 cells; in this case, the effect is blocked by a glucocorticoid receptor (GR) antagonist, but not by an MR antagonist.45 Aldosterone inhibits uncoupling protein-1 and induces insulin resistance in brown adipocytes as well.46 In a recent study in humans, however, Urbanet et al. reported no effect of physiological or supraphysiological concentrations of aldosterone on insulin sensitivity in visceral adipose tissue ex vivo, whereas pharmacological concentration of aldosterone decreased insulin sensitivity through a GR-dependent but MR-independent mechanism.47 Thus, pharmacological strategies to decrease aldosterone formation may be expected to improve insulin sensitivity.
The renin-angiotensin-aldosterone system and β-cell function
Many studies of the role of the RAAS in modulating glucose homeostasis have focused on insulin sensitivity, but the importance of adequate insulin secretion by the pancreas has recently gained attention. Although obesity and the metabolic syndrome are characterized by insulin resistance, the maintenance of glucose homeostasis in this setting requires increased secretion and/or decreased insulin clearance to maintain effective insulin concentrations.48 Studies in insulin-resistant populations indicate that the progression from normal to impaired glucose tolerance, and from impaired glucose tolerance to T2DM is characterized by declining β-cell function (or, inappropriate insulin secretion).49 In this setting, insulin concentrations are increased compared to insulin-sensitive subjects, but are inappropriate in the setting of insulin resistance because they fail to meet the demands of insulin resistant tissues and cannot maintain normoglycemia. Results from two independent genome-wide association studies support the hypothesis that insulin secretory capacity is important in T2DM pathogenesis.50,51 These studies demonstrated that genes involved in pancreatic islet development and function are associated with increased T2DM risk. Although genetic and dietary components determine insulin secretory capacity, many other factors remain unexplored, and an expanded understanding should lead to additional targeted interventions.48
Evidence suggests that the RAAS affects insulin secretion, and pharmacologic inhibition could reduce diabetes risk by preserving beta cell function. Ang II may impair insulin secretion indirectly by producing vasoconstriction and reducing islet blood flow. This effect has been demonstrated during pancreatic perfusion studies in rats.52 Similarly, Ang II infusion in humans decreases the amplitude of basal insulin oscillation and impairs glucose tolerance and insulin response during an oral glucose tolerance test.53
Just as there is a vascular RAAS, many components of the RAAS are expressed in the endocrine pancreas, further supporting a role in regulation of insulin secretion (Figure 3).52 Renin and ACE have been localized to the vasculature and microvasculature of the pancreatic islet. In the islet, β-cells express angiotensinogen, and AT1 and AT2 receptors. Islet α-cells also express angiotensinogen and the δ cell expresses the AT2 receptor. The MR is expressed in both mouse and human islets and appears to localize primarily in δ-cells and PP-cells of the pancreas.54
Figure 3. Angiotensin II and aldosterone decrease insulin secretion.
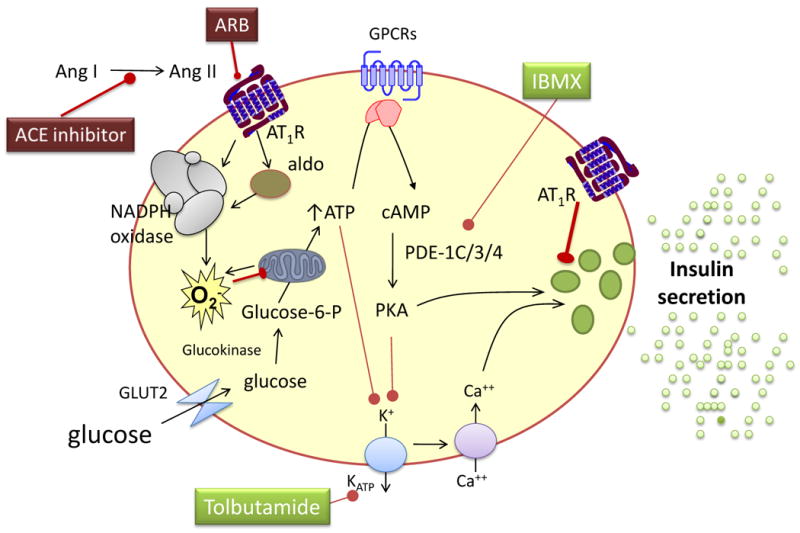
Chronic hyperglycemia and free fatty acids increase the expression of the angiotensin II subtype 1 receptor (AT1R) and uncoupling protein 2, as well as the activity of protein kinase C and nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase, leading to inflammation, the formation of reactive oxygen species (O2-), β-cell apoptosis and decreased insulin formation and secretion. These effects are reversed by AT1 receptor blockade (ARB). Aldosterone decreases insulin secretion without affecting the insulin content of β-cells, through a mechanism that also involves reactive oxygen species. This effect is not mediated via the mineralocorticoid receptor. ATP indicates adenosine triphosphate, cAMP cyclic adenosine monophosphate, GLU2 glucose transporter 2, IBMX 3-isobutyl-1-methylxanthine, PDE phosphodiesterase, PKA protein kinase A, GPCR G-protein coupled receptors.
Chronic exposure to high glucose concentrations or to high fat increases AT1 receptor expression as well as oxidative stress, inflammation and apoptosis in β-cells, in isolated islets, and in the intact pancreas.54-56 These changes are accompanied by decreased insulin content. Exposure to an ACE inhibitor or ARB reverses these effects.55,56 Direct renin inhibition increased pancreatic β-cell area, insulin secretion, and glucose tolerance in the genetically obese, diabetic KK-A(y) mice.57 These data suggest that RAAS blockade could improve glucose homeostasis by improving the insulin secretory response.
Although the effects of RAAS blockade on insulin secretion in humans have not been studied extensively, recent studies are encouraging. In subjects with impaired fasting glucose or impaired glucose tolerance, treatment with valsartan for 26 weeks modestly increased glucose-stimulated insulin secretion, as measured via hyperglycemic clamp, and improved insulin sensitivity, measured by euglycemic clamp.58 There was no effect of the ARB on glucose-independent insulin secretion, assessed by L-arginine stimulation.58 Similarly, treatment with valsartan for 16 weeks reduced glucose excursion and increased insulin response during glucose tolerance tests in obese, hypertensive subjects. The benefits of RAAS blockade may be due to effects on both peripheral insulin sensitivity and insulin secretion, and further studies are needed to better define these responses and durability of the effect.
Aldosterone decreases insulin secretion in vitro and in vivo
A recent study using hyperglycemic clamps in aldosterone synthase-deficient mice suggests that endogenous aldosterone suppresses glucose-stimulated insulin secretion in vivo.54 Aldosterone also decreases glucose- and IMBX-stimulated insulin secretion from cultured or perifused islets and from MIN6 cells, a β-cell line.54 Several lines of evidence suggest that aldosterone decreases glucose-stimulated insulin secretion through an MR-independent mechanism. First, aldosterone decreased insulin secretion from the MIN6 β-cell line, which minimally express MR.54 Second, the effect of aldosterone was not blocked by spironolactone, eplerenone, or the water soluble MR antagonist RU-28318; nor was the effect blocked by GR antagonism.54
During treatment with an MR antagonist, plasma renin activity, Ang II, and aldosterone concentrations all increased, due to loss of feedback inhibition and induction of aldosterone synthesizing enzymes.59 If aldosterone decreases insulin secretion through an MR-independent mechanism, then increased aldosterone concentrations during MR antagonism could result in impaired β-cell function and hyperglycemia in patients with the metabolic syndrome. Consistent with an MR-independent effect of aldosterone on insulin secretion, in patients with an aldosterone-producing adenoma, surgical excision of the adenoma reduced circulating glucose concentrations, whereas pharmacological treatment with spironolactone did not.60 In one randomized, placebo-controlled, crossover study in 50 patients with T2DM and resistant hypertension, spironolactone improved blood pressure but increased circulating Ang II concentrations and significantly worsened glycemic control.61 Among patients with heart failure, treatment with spironolactone resulted in increased hemoglobin A1C and cortisol concentrations and decreased adiponectin, whereas these findings were not seen in the eplerenone-treated group.62 In a recent observational study, the incidence of diabetes was increased in spironolactone-treated patients compared to patients treated with the ARB losartan.63 Additional prospective clinical studies are needed to elucidate the effect of MR antagonism on glucose metabolism and insulin secretion.
Concluding remarks
Ang II and aldosterone promote oxidative stress and interfere with insulin signaling, to decrease glucose uptake in insulin-sensitive cells via AT1 receptor- and mineralocorticoid receptor-dependent mechanism, respectively. Pharmacologic strategies to reduce the production of Ang II and aldosterone or to block these two receptors improve insulin sensitivity both in vitro and in animal models. Increasing evidence suggests that Ang II and aldosterone also decrease pancreatic β-cell function. In particular, Ang II contributes to glucose and lipid induced oxidative stress, inflammation, and apoptosis via the AT1 receptor. Aldosterone decreases insulin secretion without decreasing insulin content of β-cells, through a mineralocorticoid receptor-independent mechanism that also involves oxidative stress. AT1 receptor blockade improves insulin secretion in animal models and in humans. Strategies to reduce the production of Ang II should have similar effects. By contrast MR antagonism may be expected to decrease insulin secretion because aldosterone concentrations are increased due to lack of feedback inhibition. Thus, the net effect of MR antagonism may depend on the balance between improved insulin sensitivity and worsened insulin secretion.
The extent to which endogenous aldosterone contributes to the observed effects of Ang II on insulin sensitivity and insulin secretion remains an important question. During chronic AT1 receptor antagonism or ACE inhibition, aldosterone concentrations “escape” back to normal concentrations.64 If aldosterone contributes significantly to the effects of Ang II on insulin sensitivity or secretion, this could lead to inconsistent effects of ARBs and ACE inhibitors on the incidence of diabetes. Further strategies to prevent “aldosterone escape” could therefore be beneficial in individuals at high risk for diabetes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.International Diabetes Federation. Diabetes Atlas. 3rd Edition edn. International Diabetes Federation; 2006. [Google Scholar]
- 2.Hossain P, et al. Obesity and diabetes in the developing world--a growing challenge. N Engl J Med. 2007;356:213–215. doi: 10.1056/NEJMp068177. [DOI] [PubMed] [Google Scholar]
- 3.Haffner SM, et al. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. see comments. [DOI] [PubMed] [Google Scholar]
- 4.Briet M, Schiffrin EL. Aldosterone: effects on the kidney and cardiovascular system. Nat Rev Nephrol. 2010;6:261–273. doi: 10.1038/nrneph.2010.30. [DOI] [PubMed] [Google Scholar]
- 5.Lewis EJ, et al. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329:1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 6.Husten L. Calcium antagonist stopped in ABCD study. Appropriate Blood Pressure Control in Diabetes. Lancet. 1998;351:731. doi: 10.1016/s0140-6736(05)78505-6. [DOI] [PubMed] [Google Scholar]
- 7.Brenner BM, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 8.Scheen AJ. Prevention of type 2 diabetes mellitus through inhibition of the Renin-Angiotensin system. Drugs. 2004;64:2537–2565. doi: 10.2165/00003495-200464220-00004. [DOI] [PubMed] [Google Scholar]
- 9.McMurray JJ, et al. Effect of valsartan on the incidence of diabetes and cardiovascular events. N Engl J Med. 2010;362:1477–1490. doi: 10.1056/NEJMoa1001121. [DOI] [PubMed] [Google Scholar]
- 10.Engeli S, et al. The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome? Int J Biochem Cell Biol. 2003;35:807–825. doi: 10.1016/s1357-2725(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 11.Sharma AM, et al. Angiotensin blockade prevents type 2 diabetes by formation of fat cells. Hypertension. 2002;40:609–611. doi: 10.1161/01.hyp.0000036448.44066.53. [DOI] [PubMed] [Google Scholar]
- 12.Rocchini AP, et al. Pathogenesis of weight-related changes in blood pressure in dogs. Hypertension. 1989;13:922–928. doi: 10.1161/01.hyp.13.6.922. [DOI] [PubMed] [Google Scholar]
- 13.Goodfriend TL, et al. Relationships among plasma aldosterone, high-density lipoprotein cholesterol, and insulin in humans. Hypertension. 1995;25:30–36. doi: 10.1161/01.hyp.25.1.30. [DOI] [PubMed] [Google Scholar]
- 14.Ehrhart-Bornstein M, et al. Human adipocytes secrete mineralocorticoid-releasing factors. Proc Natl Acad Sci U S A. 2003;100:14211–14216. doi: 10.1073/pnas.2336140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodfriend TL, et al. Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension. 2004;43:358–363. doi: 10.1161/01.HYP.0000113294.06704.64. [DOI] [PubMed] [Google Scholar]
- 16.Engeli S, et al. Weight Loss and the Renin-Angiotensin-Aldosterone System. Hypertension. 2005 doi: 10.1161/01.HYP.0000154361.47683.d3. [DOI] [PubMed] [Google Scholar]
- 17.Dall'Asta C, et al. Effect of weight loss through laparoscopic gastric banding on blood pressure, plasma renin activity and aldosterone levels in morbid obesity. Nutr Metab Cardiovasc Dis. 2009;19:110–114. doi: 10.1016/j.numecd.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Tuck ML, et al. The effect of weight reduction on blood pressure, plasma renin activity, and plasma aldosterone levels in obese patients. N Engl J Med. 1981;304:930–933. doi: 10.1056/NEJM198104163041602. [DOI] [PubMed] [Google Scholar]
- 19.The DREAM Trial Investigators et al. Effect of ramipril on the incidence of diabetes. N Engl J Med. 2006;355:1551–1562. doi: 10.1056/NEJMoa065061. [DOI] [PubMed] [Google Scholar]
- 20.Knowler WC, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knowler WC, et al. Prevention of type 2 diabetes with troglitazone in the Diabetes Prevention Program. Diabetes. 2005;54:1150–1156. doi: 10.2337/diabetes.54.4.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DREAM (Diabetes REduction Assessment with ramipril and rosiglitazone et al. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet. 2006;368:1096–1105. doi: 10.1016/S0140-6736(06)69420-8. [DOI] [PubMed] [Google Scholar]
- 23.Home PD, et al. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet. 2009;373:2125–2135. doi: 10.1016/S0140-6736(09)60953-3. [DOI] [PubMed] [Google Scholar]
- 24.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 25.Parving HH, et al. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008;358:2433–2446. doi: 10.1056/NEJMoa0708379. [DOI] [PubMed] [Google Scholar]
- 26.Yusuf S, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–153. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- 27.Pfeffer MA, et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med. 2003;349:1893–1906. doi: 10.1056/NEJMoa032292. [DOI] [PubMed] [Google Scholar]
- 28.Velloso LA, et al. Cross-talk between the insulin and angiotensin signaling systems. Proc Natl Acad Sci U S A. 1996;93:12490–12495. doi: 10.1073/pnas.93.22.12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Folli F, et al. Angiotensin II inhibits insulin signaling in aortic smooth muscle cells at multiple levels. A potential role for serine phosphorylation in insulin/angiotensin II crosstalk. J Clin Invest. 1997;100:2158–2169. doi: 10.1172/JCI119752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andreozzi F, et al. Angiotensin II impairs the insulin signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate-1 on Ser312 and Ser616 in human umbilical vein endothelial cells. Circ Res. 2004;94:1211–1218. doi: 10.1161/01.RES.0000126501.34994.96. [DOI] [PubMed] [Google Scholar]
- 31.Diamond-Stanic MK, Henriksen EJ. Direct inhibition by angiotensin II of insulin-dependent glucose transport activity in mammalian skeletal muscle involves a ROS-dependent mechanism. Arch Physiol Biochem. 2010;116:88–95. doi: 10.3109/13813451003758703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei Y, et al. Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells. J Biol Chem. 2006;281:35137–35146. doi: 10.1074/jbc.M601320200. [DOI] [PubMed] [Google Scholar]
- 33.Henriksen EJ, et al. Selective angiotensin II receptor receptor antagonism reduces insulin resistance in obese Zucker rats. Hypertension. 2001;38:884–890. doi: 10.1161/hy1101.092970. [DOI] [PubMed] [Google Scholar]
- 34.Shiuchi T, et al. ACE inhibitor improves insulin resistance in diabetic mouse via bradykinin and NO. Hypertension. 2002;40:329–334. doi: 10.1161/01.hyp.0000028979.98877.0c. [DOI] [PubMed] [Google Scholar]
- 35.Fujimoto M, et al. An angiotensin II AT1 receptor antagonist, telmisartan augments glucose uptake and GLUT4 protein expression in 3T3-L1 adipocytes. FEBS Lett. 2004;576:492–497. doi: 10.1016/j.febslet.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 36.Juan CC, et al. Angiotensin II enhances insulin sensitivity in vitro and in vivo. Endocrinology. 2005;146:2246–2254. doi: 10.1210/en.2004-1136. [DOI] [PubMed] [Google Scholar]
- 37.Munoz MC, et al. Long-term treatment with an angiotensin II receptor blocker decreases adipocyte size and improves insulin signaling in obese Zucker rats. J Hypertens. 2009;27:2409–2420. doi: 10.1097/HJH.0b013e3283310e1b. [DOI] [PubMed] [Google Scholar]
- 38.Lastra G, et al. Direct renin inhibition improves systemic insulin resistance and skeletal muscle glucose transport in a transgenic rodent model of tissue renin overexpression. Endocrinology. 2009;150:2561–2568. doi: 10.1210/en.2008-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang YS, et al. Aliskiren improves insulin resistance and ameliorates diabetic vascular complications in db/db mice. Nephrol Dial Transplant. 2011;26:1194–1204. doi: 10.1093/ndt/gfq579. [DOI] [PubMed] [Google Scholar]
- 40.Shiuchi T, et al. Angiotensin II type-1 receptor blocker valsartan enhances insulin sensitivity in skeletal muscles of diabetic mice. Hypertension. 2004;43:1003–1010. doi: 10.1161/01.HYP.0000125142.41703.64. [DOI] [PubMed] [Google Scholar]
- 41.Carvalho CR, et al. Effect of captopril, losartan, and bradykinin on early steps of insulin action. Diabetes. 1997;46:1950–1957. doi: 10.2337/diab.46.12.1950. [DOI] [PubMed] [Google Scholar]
- 42.Csibi A, et al. Angiotensin II inhibits insulin-stimulated GLUT4 translocation and Akt activation through tyrosine nitration-dependent mechanisms. PLoS One. 2010;5:e10070. doi: 10.1371/journal.pone.0010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lastra G, et al. Low-dose spironolactone reduces reactive oxygen species generation and improves insulin-stimulated glucose transport in skeletal muscle in the TG(mRen2)27 rat. Am J Physiol Endocrinol Metab. 2008;295:E110–E116. doi: 10.1152/ajpendo.00258.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirata A, et al. Blockade of mineralocorticoid receptor reverses adipocyte dysfunction and insulin resistance in obese mice. Cardiovasc Res. 2009;84:164–172. doi: 10.1093/cvr/cvp191. [DOI] [PubMed] [Google Scholar]
- 45.Wada T, et al. Aldosterone inhibits insulin-induced glucose uptake by degradation of insulin receptor substrate (IRS) 1 and IRS2 via a reactive oxygen species-mediated pathway in 3T3-L1 adipocytes. Endocrinology. 2009;150:1662–1669. doi: 10.1210/en.2008-1018. [DOI] [PubMed] [Google Scholar]
- 46.Kraus D, et al. Aldosterone inhibits uncoupling protein-1, induces insulin resistance, and stimulates proinflammatory adipokines in adipocytes. Horm Metab Res. 2005;37:455–459. doi: 10.1055/s-2005-870240. [DOI] [PubMed] [Google Scholar]
- 47.Urbanet R, et al. Analysis of insulin sensitivity in adipose tissue of patients with primary aldosteronism. J Clin Endocrinol Metab. 2010;95:4037–4042. doi: 10.1210/jc.2010-0097. [DOI] [PubMed] [Google Scholar]
- 48.Kahn SE, et al. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 49.Weyer C, et al. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saxena R. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 51.Sladek R, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 52.Leung PS. Pancreatic RAS. Adv Exp Med Biol. 2010;690:89–105. doi: 10.1007/978-90-481-9060-7_6. [DOI] [PubMed] [Google Scholar]
- 53.Fliser D, et al. Angiotensin II affects basal, pulsatile, and glucose-stimulated insulin secretion in humans. Hypertension. 1997;30:1156–1161. doi: 10.1161/01.hyp.30.5.1156. [DOI] [PubMed] [Google Scholar]
- 54.Luther JM, et al. Aldosterone decreases glucose-stimulated insulin secretion in vivo in mice and in murine islets. Diabetologia. 2011;54:2152–2163. doi: 10.1007/s00125-011-2158-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saitoh Y, et al. Candesartan attenuates fatty acid-induced oxidative stress and NAD(P)H oxidase activity in pancreatic beta-cells. Diabetes Res Clin Pract. 2010;90:54–59. doi: 10.1016/j.diabres.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 56.Yuan L, et al. Effects of renin-angiotensin system blockade on islet function in diabetic rats. J Endocrinol Invest. 2010;33:13–19. doi: 10.1007/BF03346544. [DOI] [PubMed] [Google Scholar]
- 57.Iwai M, et al. Direct renin inhibition improved insulin resistance and adipose tissue dysfunction in type 2 diabetic KK-A(y) mice. J Hypertens. 2010;28:1471–1481. doi: 10.1097/HJH.0b013e32833bc420. [DOI] [PubMed] [Google Scholar]
- 58.van der Zijl NJ, et al. Valsartan improves {beta}-cell function and insulin sensitivity in subjects with impaired glucose metabolism: a randomized controlled trial. Diabetes Care. 2011;34:845–851. doi: 10.2337/dc10-2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weinberger MH, et al. Effects of eplerenone versus losartan in patients with low-renin hypertension. Am Heart J. 2005;150:426–433. doi: 10.1016/j.ahj.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 60.Sindelka G, et al. Insulin action in primary hyperaldosteronism before and after surgical or pharmacological treatment. Exp Clin Endocrinol Diabetes. 2000;108:21–25. doi: 10.1055/s-0032-1329211. [DOI] [PubMed] [Google Scholar]
- 61.Swaminathan K, et al. Spironolactone for poorly controlled hypertension in type 2 diabetes: conflicting effects on blood pressure, endothelial function, glycaemic control and hormonal profiles. Diabetologia. 2008;51:762–768. doi: 10.1007/s00125-008-0972-5. [DOI] [PubMed] [Google Scholar]
- 62.Yamaji M, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A(c) levels in patients with chronic heart failure. Am Heart J. 2010;160:915–921. doi: 10.1016/j.ahj.2010.04.024. [DOI] [PubMed] [Google Scholar]
- 63.Arase Y, et al. Losartan reduces the onset of type 2 diabetes in hypertensive Japanese patients with chronic hepatitis C. J Med Virol. 2009;81:1584–1590. doi: 10.1002/jmv.21572. [DOI] [PubMed] [Google Scholar]
- 64.McKelvie RS, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation. 1999;100:1056–1064. doi: 10.1161/01.cir.100.10.1056. [DOI] [PubMed] [Google Scholar]