

Abstract
Quercetin dioxygenase (QDO) catalyzes the oxidation of the flavonol quercetin with dioxygen, cleaving the central heterocyclic ring and releasing CO. The QDO from Bacillus subtilis is unusual in that it has been shown to be active with several divalent metal cofactors such as Fe, Mn, and Co. Previous comparison of the catalytic activities suggest that Mn(II) is the preferred cofactor for this enzyme. We herein report the unprecedented substitution of nitrosyl hydride (HNO) for dioxygen in the activity of Mn-QDO, resulting in the incorporation of both N and O atoms into the product. Turnover is demonstrated by consumption of quercetin and other related substrates under anaerobic conditions in the presence of HNO-releasing compounds and the enzyme. As with dioxygenase activity, a nonenzymatic base-catalyzed reaction of quercetin with HNO is observed above pH 7, but no enhancement of this basal reactivity is found upon addition of divalent metal salts. Unique and regioselective N-containing products (14N/15N) have been characterized by MS analysis for both the enzymatic and nonenzymatic reactions. Of the several metallo-QDO enzymes examined for nitroxygenase activity under anaerobic condition, only the Mn(II) is active; the Fe(II) and Co(II) substituted enzymes show little or no activity. This result represents an enzymatic catalysis which we denote nitroxygenase activity; the unique reactivity of the Mn-QDO suggests a metal-mediated electron transfer mechanism rather than metal activation of the substrate’s inherent base-catalyzed reactivity.
Keywords: nitroxyl, quercetinase
Dioxygenases are enzymes that incorporate both atoms of dioxygen into a targeted substrate, e.g., the enzyme catechol 1,2-dioxygenase cleaves the C–C bond between catechol’s two hydroxyl groups forming the dicarboxylate cis,cis-muconic acid (1). Although highly divergent, there are two broadly defined classes of nonheme dioxygenases: those that directly bind dioxygen at the metal center or those in which dioxygen binds to the substrate that has been activated by interaction with the metal center (2). Such questions also apply to quercetin dioxygenase (QDO), which catalyzes the oxidation of the flavonol quercetin (1) with dioxygen (Fig. 1). During turnover, QDO cleaves the central heterocyclic ring of quercetin, releasing CO, and yielding the corresponding depside. The QDO from Bacillus subtilis is unusual in that it has been shown to be active with several divalent metal cofactors such as Cu, Fe, Mn, and Co. Comparison of the catalytic activities suggest that Mn(II) is the preferred cofactor for this enzyme (3).
Fig. 1.

Schematic of QDO dioxygenase activity.
The simple molecule nitrosyl hydride (HNO) is the singly reduced and protonated form of nitric oxide, NO. HNO has garnered much interest because it demonstrates a physiological reactivity distinctly different from its congener nitric oxide (4, 5). In a series of papers, we have reported the preparation and characterization of the HNO adduct of myoglobin (HNO-Mb) by reduction of NO-Mb (6, 7) and by trapping of free HNO by deoxymyoglobin (Mb-FeII) (8) or the reaction of metmyoglobin with nitrite and borohydride (9). Like the oxyadduct of myoglobin, HNO-Mb is diamagnetic and is uniquely characterized by a 1H NMR signal due to the N-bound hydride found at ca. 15 ppm, well away from other protein resonances, which allowed structural characterization of the heme pocket by 2D NOE and COSY experiments (10). Other ferrous globins also rapidly trap free HNO to directly yield stable HNO adducts, identifiable by changes in electronic absorbances as well as unique 1H and 15N NMR resonances due to the nitrosyl hydride (11).
The anionic form of nitroxyl is isoelectronic with dioxygen and exists as a triplet,  , in aqueous solutions above pH 12; at neutral pH, the protonated singlet 1HNO dominates (12). HNO does not accumulate in aqueous solutions due its rapid dimerization producing N2O between pH 2 to 11 (Eq. 1) (13). Thus HNO cannot be stored as reactant nor can it be identified by direct methods; it is typically generated in situ via precursor compounds that decompose to release HNO (14). By far the most widely used precursor is Angeli’s salt (sodium trioxodinitrate/Na2N2O3), AS, which decomposes to give HNO at pH 4–8 (Eq. 2). Other widely used precursors are the various alkylsulfohydroxamic acids, e.g., benzylsulfohydroxamic acid or Piloty’s acid, which generate HNO upon deprotonation at a high pH (Eq. 3) (15).
, in aqueous solutions above pH 12; at neutral pH, the protonated singlet 1HNO dominates (12). HNO does not accumulate in aqueous solutions due its rapid dimerization producing N2O between pH 2 to 11 (Eq. 1) (13). Thus HNO cannot be stored as reactant nor can it be identified by direct methods; it is typically generated in situ via precursor compounds that decompose to release HNO (14). By far the most widely used precursor is Angeli’s salt (sodium trioxodinitrate/Na2N2O3), AS, which decomposes to give HNO at pH 4–8 (Eq. 2). Other widely used precursors are the various alkylsulfohydroxamic acids, e.g., benzylsulfohydroxamic acid or Piloty’s acid, which generate HNO upon deprotonation at a high pH (Eq. 3) (15).
| [1] |
| [2] |
| [3] |
Much recent interest in HNO-releasing compounds results from their potential pharmacological use for cardiovascular disorders (16–18), cancer (19), and alcoholism (20, 21). But the significance of “free” HNO in nature is as yet uncertain (22). Although several biological pathways for HNO formation have been suggested, e.g., via nitrosothiol decomposition (23, 24) or enzymatic malfunction (25–27), no true physiological role for endogenous HNO has been accepted.
Following our previous characterization of the trapping of free HNO by various O2-binding globins (11), we hypothesized that HNO might similarly interact with O2-dependent oxygenases; we herein report the unprecedented substitution of HNO for O2 in the activity of Mn-QDO, resulting in the incorporation of the nitroxyl-derived N atom into the product, which we denote as nitroxygenase activity.
Results
In QDO oxygenase activity assays, ca. 50 μL of 1.1 mM substrate quercetin in DMSO is mixed with 950 μL of aerated 50 mM Tris buffer at pH 7.5; the assay is initiated by the addition of ca. 10 µM of enzyme (SI Text, Enzyme Expression and Purification), and the loss of substrate concentration is followed at its absorbance at 380 nm. In analogous anaerobic mixtures containing quercetin and QDO, there is no observable change in the substrate concentration. But addition of the HNO-donor AS to such solution leads to a sequential loss of the substrate absorbance due to nitroxygenase activity at a rate close to that of HNO release by AS (SI Text, Nitroxygenase Assay and Control Experiments). Analogous experiments using other nitrogenous reagents such as nitrite and hydroxylamine show no comparable reactivity.
In a typical nitroxygenase assay, solutions of substrate and enzyme are mixed in deaerated pH 7 inorganic phosphate (IP) buffer to give desired concentrations (e.g., 13.7 µM quercetin and 8.5 µM Mn-QDO); the reaction is initiated by the addition of a concentrated stock solution of HNO precursor (137 µM AS) and the loss of substrate concentration is followed by the loss of its absorbance at 380 nm. The quercetin is consumed within an hour, and the resulting reaction mixture was analyzed by liquid chromatography (LC)-MS to identify products (SI Text, Liquid Chromatography-Electrospray Ionization MS Analysis of Assay Products). Negative ion LC-MS analysis of the resulting product mixtures shows complete conversion of quercetin (301 m/z) to a new dominant product ion (134 m/z) that matches a C7H4O2N- anion; LC-MS retention time and fragmentation pattern is consistent with 3,4-dihydroxybenzonitrile, as confirmed with an authentic sample of the product. The use of 15N-labeled AS (specifically 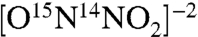 , SI Text, Synthesis and characterization of 15N-labeled Angeli’s Salt) (28) in analogous anaerobic assays results in 15N-labeled product with 135 m/z peak observable in the LC-MS (Fig. 2). Generation of CO during these reactions was confirmed by the conversion of a solution of deoxymyoglobin to its ferrous CO adduct upon exposure to the head gas above the assay mixture (SI Text, Confirmation of CO Formation by Trapping with Deoxy-Myoglobin).
, SI Text, Synthesis and characterization of 15N-labeled Angeli’s Salt) (28) in analogous anaerobic assays results in 15N-labeled product with 135 m/z peak observable in the LC-MS (Fig. 2). Generation of CO during these reactions was confirmed by the conversion of a solution of deoxymyoglobin to its ferrous CO adduct upon exposure to the head gas above the assay mixture (SI Text, Confirmation of CO Formation by Trapping with Deoxy-Myoglobin).
Fig. 2.

Total ion chromatogram (Left) and mass spectra (Right) corresponding to the initial LC peak obtained in analysis of product mixtures from the reactions of (A) labeled 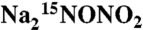 and (B) natural abundance Na2N2O3 with quercetin (1). (C) Chromatogram and mass spectrum obtained using authentic 3,4 dihydroxy benzonitrile (6).
and (B) natural abundance Na2N2O3 with quercetin (1). (C) Chromatogram and mass spectrum obtained using authentic 3,4 dihydroxy benzonitrile (6).
Further confirmation of the nitroxygenase activity was obtained by assays utilizing the analogous flavonol substrates quercetin-5-methoxy and quercetin-5-sulfonic acid, compounds 2 and 3 in Fig. 3. Analysis of the resulting product mixtures by LC-MS showed similar major ion peaks consistent with 3,4-dihydroxy-5-methoxy benzonitrile and 3,4-dihydroxy-5-sulfonic benzonitrile products (SI Text, LC-MS of Products of Nitroxygenase Mn-QDO Assay with Modified Quercetin Derivatives Quercetin-5-Sulfonic Acid and Quercetin-5-Methoxy), which were not seen in control experiments of anaerobic reactivity of these 5-substituted quercetin derivatives with AS in the absence of Mn-QDO at pH 7.
Fig. 3.

Schematic of Mn-QDO nitroxygenase activity substrates and products.
Initial kinetic analysis was performed and analyzed using the sequential reactions depicted in Eqs. 4–7. Rate analysis of the nitroxygenase reactivity is complicated by the slow decomposition rate of the HNO-donor AS and the competitive dimerization of free HNO (29).* The slow release of HNO from AS would be predicted as the rate-limiting step, but at relatively low substrate concentrations, the initial rates of reaction show dependence on both [QDO] and [AS], implying the catalytic turnover and dimerization are competitive in this regime (SI Text, Rates of Kinetic Runs of Nitroxygenase Activity Under Varying Conditions). Previous mechanistic studies of Mn-QDO have suggested that formation of a quercetin/QDO complex, KM = 4 μM, precedes reaction with dioxygen (3). If quantitative quercetin/QDO complex formation is assumed at 100-fold excess substrate to enzyme, modeling of the rate of reaction under these conditions gives a rate constant of 3.3 × 104 M-1 s-1 for this pseudobimolecular reaction (Eq. 7) (SI Text, Kinetic Modeling of Nitroxygenase Reaction Sequence).
| [4] |
| [5] |
| [6] |
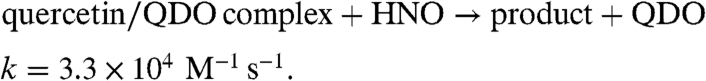 |
[7] |
The dioxygenase activity of Mn-QDO has been shown to have distinctive pH dependence between pH 5.5 and 8, with a maximum rate of ca. pH 7.0 (3). The pH dependence of the nitroxygenase activity was investigated from pH 5 to 8, shown as the plot in Fig. 4. For comparison, the decomposition rate of AS under the experimental conditions is also plotted, essentially constant from pH 4 to 8 (30). Above pH 7, a nonenzymatic consumption of quercetin is observed; correcting for this background reactivity demonstrates that the pH dependence of the Mn-QDO nitroxygenase catalysis mimics that of its oxygenase activity.
Fig. 4.

A comparative plot of effect of pH on rate of decomposition of Angeli’s salt (○); loss of quercetin absorbance at 380 nm during reaction with Angeli’s salt in the presence of Mn-QDO (▪) at 1∶10∶100, Mn-QDO/AS/quercetin; and same after subtraction of nonenzymatic rate (□).
A nonenzymatic, basal reactivity of AS with quercetin initiates above pH 7 (SI Text, Nonenzymatic Reaction of HNO With Quercetin). LC-MS analysis of the product mixtures obtained a large product peak consistent with 3,4 dihydroxybenzonitrile (compound 6 in Fig. 3), therefore retaining the regioselectivity of the enzymatic reaction. The observed rates of the nonenzymatic reaction become competitive to the enzymatic rates above pH 7.5.
The analogous nonenzymatic reaction of quercetin with dioxygen has long been known to proceed at similar pH range (31). The pH and oxygen dependence of the rate of reaction supports a direct reaction of the monoanionic form of quercetin (pKa 7.03) (32) with dioxygen, originally postulated as a simple nucleophilic attack of a carbon-centered anion on dioxygen (33). Subsequent reports have suggested an outersphere single electron transfer (SET) generates a quercetin-based C radical which may then react rapidly with dioxygen or superoxide. Both the base-catalyzed (34) and SET (35) mechanisms have been suggested in aqueous/organic mixtures. The observed nonenzymatic nitroxygenase reactivity is also consistent with a base-catalyzed mechanism; HNO is well known to undergo facile and rapid nucleophilic attack by anions such as thiolates and amides (36).
A unique feature of the Mn-QDO nitroxygenase reactivity is the regioselectivity of N-atom incorporation into the product (Fig. 5). The presumed initial product, 2-((3,4-dihydroxyphenyl)(imino)methoxy)-4,6-dihydroxybenzoate, undergoes a 1,3 proton transfer to generate the observed nitrile and phenolic products (37); this is analogous to the hydration of the initial phenolic ester seen at basic pH, resulting in carboxylic and phenolic products (33). For the observed nitroxygenase products, the N-atom addition occurs selectively at the site corresponding to that suggested for initial dioxygen reaction in previously proposed dioxygenase mechanisms—i.e., of initial nucleophilic attack by a substrate anion or coupling of the substrate and diatom radicals. The role of the metal cofactor in enhancing this reactivity may be attributed to either substrate activation (i.e., promotion of the reactive tautomeric anion of the flavonol) or to a promotion of electron transfer between the substrate and diatom, generating and orienting the radicals needed.
Fig. 5.

Comparison of regioselectivity of QDO oxygenase and nitroxygenase reactivity.
As previously noted, flavonol dioxygenases have been identified that act with various metal-ion cofactors (38, 39). For instance, several species of Aspergillus have flavonol dioxygenases with Cu cofactors (40–42); a QDO from Streptomyces sp. designated strain FLA (flavonol utilization) was characterized with divalent Fe, Ni, and Co cofactors (43). The QDO from B. subtilis, has been characterized with divalent Fe, Mn, and Co cofactors (44) and tested for activity with divalent Mg, Ni, and Cd (45). It has been suggested that exchange of the active-site metal ion may allow biological modulation of enzyme activity (45).
To test the metal-ion dependence of nitroxygenase activity, samples of Fe-QDO were expressed in Escherichia coli cells grown in LB medium, and Co-QDO was expressed in M9 minimal medium in the presence of added Co(II) salts, analogous to the expression of Mn-QDO (44). Metal-ion incorporation in each batch was corroborated by inductively coupled plasma (ICP)-MS and atomic absorption spectrometric analysis of the reconstituted enzymes (SI Text, Analysis of Metal Content in Metallo-QDOs by Inductively Coupled Plasma and Atomic Absorption Spectrometry), which showed only minor odd-metal contamination of the Mn and Fe-QDO samples, with the Co-QDO sample containing ca. 10% Mn (Table 1).
Table 1.
ICP-MS and atomic absorption spectrometric metal analysis of metallo-QDOs
| QDO | % metal content | ||
| Mn | Fe | Co | |
| Mn | 95.34 | 4.66 | ND |
| Fe | 0.83 | 99.17 | ND |
| Co | 10.40 | 2.32 | 87.28 |
ND, not detected.
The dioxygenase activities of the metal-substituted QDO samples were examined (Fig. 6Upper), which confirms the comparative values previously determined (i.e., Mn > Fe > Co) (3). In contrast, only the Mn-QDO demonstrates nitroxygenase activity under standard assay conditions at pH 5.5 (Fig. 6Lower) (the minor activity seen for the Co-QDO sample may be due to the ca. 10% Mn-QDO content).
Fig. 6.

Normalized time course absorbance plots of catalytic reaction of metal-substituted QDOs, as followed by loss of quercetin absorbance at 380 nm in IP buffer pH 6.5. (Upper) Dioxygenase assays in aerobic solutions (130 μM O2): Co-QDO (2.85 μM, [Q]: 14.25 μM, dashed), Fe-QDO (2.48 μM, [Q]: 12.42 μM, dotted), and Mn-QDO (2.31 μM, [Q]: 11.55 μM, solid). (Lower) Nitroxygenase assays in anaerobic solutions: Co-QDO (2.98 μM, [Q]: 14.90 μM, [AS]: 148 μM, dashed), Fe-QDO (2.85 μM, [Q]: 14.2 μM, [AS]: 140 μM, dotted), and Mn-QDO (3.73 μM, [Q]: 18.63 μM, [AS]: 184 μM, solid).
Discussion
A continuing question regarding the native dioxygenase activity of QDO is whether dioxygen binds to the metal cofactor prior to insertion into the substrate. The unique reactivity of the Mn-QDO in the nitroxygenase activity argues against simple substrate activation by metal coordination, as the Lewis acidity of divalent Mn, Fe, and Co may be assumed to be roughly comparable (46). Under the SET hypothesis, the metal ion acts as a conduit for an internal electron transfer between the metal-bound flavonol and HNO, also orienting the resultant organic radicals to facilitate coupling.
Similar tertiary complexes have recently been crystallographically characterized for site-specific mutants of homoprotocatechuate 2,3-dioxygenase (2,3-HPCD): The substrate 4-nitrocatechol and dioxygen were found bound to the active-site Fe ion in several distinct intermediate states (47). EPR and Mossbauer spectroscopy characterization of both the Fe- and Mn-substituted 2,3-HPCD provide evidence of MIII-radical intermediates, which suggests sequential oxidation and reduction of the metal cofactor may take place during such dioxygenase activity (48, 49).
Fig. 7 display possible reaction pathways for both the dioxygenase and nitroxygenase activities of Mn-QDO. By analogy to the 2,3-HPCD mechanistic studies (47–49), the Mn-QDO dioxygenase reaction is proposed to go through an initial tertiary complex, A, in which an initial SET has yielded an MnIII-superoxide species. A second SET from substrate to metal allows subsequent radical coupling between the metal-bound superoxide and substrate. Nucleophilic attack on the ketone by the resulting alkyl-peroxide, C, generates a bridging dioxalane, D, which then decomposes with the release of CO to depside product, E.
Fig. 7.

Proposed reaction pathway for Mn-QDO catalyzed dioxygenase (I) and nitroxygenase (II) reactivities, as described in the text.
The corresponding sequence for nitroxygenase activity is given below, with an initial MnIII-aminoxyl radical adduct, A′, and subsequent intracomplex SET to generate the substrate radical, B′. Radical coupling would then form an alkylhydroxamate intermediate, C′, and nucleophilic attack would generate an isoxazolidine bridge, D′, which then decomposes to the parent carboximidic ester, E′.
The unique reactivity of the Mn-QDO suggests a metal-mediated electron transfer mechanism rather than metal-activation of the substrate’s inherent base-catalyzed reactivity. There has been much discussion over the electronic nature of bonding in transition metal HNO complexes (50), for instance in the putative catalytic intermediate in nitric oxide reductase P450nor (51). An HNO-MnII species with a d5 electronic configuration is unprecedented, as all isolable HNO-metal complexes are diamagnetic with low-spin d6 metal ions. But there exists precedence for reduction of a metal-bound HNO in the catalytic cycles of various nitrite reductases (52–54). The singly reduced form of HNO, the aminoxyl radical anion (HNO-), is generated by photolysis of the stable HNO adduct of deoxymyoglobin (55); and published theoretical calculations suggest that a proton-coupled oxidation HNO/FeII to H2NO/FeIII is enthalpically favorable (56, 57).
A second related issue is the radical coupling step. Previously proposed dioxygenase mechanisms contend that the pendant beta-oxygen atom of the metal-bound superoxide (47), B in Fig. 7, couples to the organic radical. But the observed regioselectivity of the nitroxygenase reactivity demands that an N radical, the alpha-atom attached to the metal in B′, couples to the substrate radical. The HNO adduct is depicted as N bound, as found in characterized end-on HNO-metal complexes (58). There is much recent evidence for O-bound linkage isomers of metal-nitrosyls, termed isonitrosyls, but these are typically only observed after photolysis at low temperature within a solid or crystalline material (59–62). The analogous O-bound HNO, isonitroxyl, has never been characterized to our knowledge; theorectical calculations have predicted the O-bound isonitroxyl adduct of Fe porphyrinates would be over 50 kJ/mol higher in energy than the corresponding N-bound nitroxyl (63).
Materials and Methods
AS was purchased from Cayman chemicals and used as received. Quercetin and 3,4-dihydroxy benzonitrile were purchased from Tokyo Chemical Industry America; quercetin-5-sulfonic acid and quercetin-5-methoxy were purchased from Indofine Chemical Company. Metal-substituted QDOs were prepared by growing E. coli BL21 (DE3) carrying plasmid pQuer4, as previously described (3). Specific data on the metal analysis of metal-substituted QDOs by ICP and atomic absorption spectrometry is available in SI Text, Analysis of Metal Content in Metallo-QDOs by Inductively Coupled Plasma and Atomic Absorption Spectrometry. LC-electrospray ionization MS analysis of assay products were analyzed on an Accela liquid chromatograph coupled to a Linear Trap Quadrupole Orbitrap Discovery mass spectrometer, procedures given in SI Text, Liquid Chromatography-Electrospray Ionization MS Analysis of Assay Products. Unless otherwise stated, all reaction solutions were prepared inside an anaerobic glove box.
Assays of Mn-QDO Nitroxygenase Activity.
Assay reactions with varying amounts of quercetin, Mn-QDO, and the HNO-precursor AS were carried out in a screw-capped UV cuvette and monitored by following the decrease of the substrate absorbance at 380 nm. In a typical assay, 13.7 µM quercetin is mixed with 8.5 µM of enzyme in deareated IP buffer at pH 7; the reaction is initiated by the addition of a stock AS solution to give final concentration of 137 µM. The reaction was initiated by gently shaking the cuvette before placing it in the spectrometer, followed by observing the loss of absorbance at 380 nm.
The Nonenzymatic Reaction of HNO with Quercetin.
HNO-precursor AS (1.3 µM) was added to IP buffer at appropriate pH containing quercetin (0.13 µM). The reaction was followed by observing the loss of absorbance at 380 nm.
Dioxygenase Assays Using Metal-Substituted QDOs.
In a typical experiment, quercetin and enzyme was added to the IP buffer at pH 6.5. The reaction was initiated by adding oxygen-saturated solution (130 μM). The reaction was followed by loss of quercetin absorbance at 380 nm.
Nitroxygenase Assays Using Metal-Substituted QDOs.
The nitroxygenase assays utilized of AS with the enzyme Co- and Fe-QDOs and quercetin were carried out in IP buffer at a range of pH. No significant losses of quercetin absorbance were observed under conditions studied.
Supplementary Material
Acknowledgments.
This material is based upon work supported by the National Science Foundation (CHE-1057942 to P.J.F., and MCB-0745236 to W.A.F.), the National Institutes of Health (1R21ES016441-01 to P.J.F.), and startup funds from Baylor University.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1111488108/-/DCSupplemental.
*A reviewer has pointed out that reliance on the decomposition rates of AS as a measure of HNO production is a major assumption in our kinetic analysis, as certain reports suggest that HNO production may not necessarily be constant in the pH 4–8 range.
References
- 1.Costas M, Mehn MP, Jensen MP, Que L., Jr Dioxygen activation at mononuclear nonheme iron active sites: Enzymes, models, and intermediates. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 2.Kovaleva EG, Lipscomb JD. Versatility of biological non-heme Fe(II) centers in oxygen activation reactions. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaab MR, Barney BM, Francisco WA. Kinetic and spectroscopic studies on the quercetin dioxygenase from Bacillus subtilis. Biochemistry. 2006;45:1009–1016. doi: 10.1021/bi051571c. [DOI] [PubMed] [Google Scholar]
- 4.Fukuto JM, et al. The physiological chemistry and biological activity of nitroxyl (HNO): The neglected, misunderstood, and enigmatic nitrogen oxide. Chem Res Toxicol. 2005;18:790–801. doi: 10.1021/tx0496800. [DOI] [PubMed] [Google Scholar]
- 5.Fukuto JM, Switzer CH, Miranda KM, Wink DA. Nitroxyl (HNO): Chemistry, biochemistry, and pharmacology. Annu Rev Pharmacol Toxicol. 2005;45:335–355. doi: 10.1146/annurev.pharmtox.45.120403.095959. [DOI] [PubMed] [Google Scholar]
- 6.Lin R, Farmer PJ. The HNO adduct of myoglobin: Synthesis and characterization. J Am Chem Soc. 2000;122:2393–2394. [Google Scholar]
- 7.Pervitsky D. 2008. Generation and reactivity of HNO adducts of horse skeletal myoglobin and human hemoglobin. PhD Dissertation (Univ of California, Ivine, CA) [Google Scholar]
- 8.Sulc F, Immoos CE, Pervitsky D, Farmer PJ. Efficient trapping of HNO by deoxymyoglobin. J Am Chem Soc. 2004;126:1096–1101. doi: 10.1021/ja0376184. [DOI] [PubMed] [Google Scholar]
- 9.Kumar MR, Fukuto JM, Miranda KM, Farmer PJ. Reactions of HNO with heme proteins: New routes to HNO-heme complexes and insight into physiological effects. Inorg Chem. 2010;49:6283–6292. doi: 10.1021/ic902319d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sulc F, Fleischer E, Farmer PJ, Ma D, La Mar GN. 1H NMR structure of the heme pocket of HNO-myoglobin. J Biol Inorg Chem. 2003;8:348–352. doi: 10.1007/s00775-002-0422-7. [DOI] [PubMed] [Google Scholar]
- 11.Kumar MR, et al. Nitrosyl hydride (HNO) as an O2 analogue: Long-lived HNO-adducts of ferrous globins. Biochemistry. 2009;48:5018–5025. doi: 10.1021/bi900122r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shafirovich V, Lymar SV. Spin-forbidden deprotonation of aqueous nitroxyl (HNO) J Am Chem Soc. 2003;125:6547–6552. doi: 10.1021/ja034378j. [DOI] [PubMed] [Google Scholar]
- 13.Lymar SV, Shafirovich V, Poskrebyshev GA. One-electron reduction of aqueous nitric oxide: A mechanistic revision. Inorg Chem. 2005;44:5212–5221. doi: 10.1021/ic0501317. [DOI] [PubMed] [Google Scholar]
- 14.Hughes MN, Cammack R. Synthesis, chemistry, and applications of nitroxyl ion releasers sodium trioxodinitrate or Angeli’s salt and Piloty’s acid. Methods Enzymol. 1999;301:279–287. doi: 10.1016/s0076-6879(99)01092-7. [DOI] [PubMed] [Google Scholar]
- 15.King SB, Nagasawa T. Chemical approaches toward generation of nitroxyl. Methods Enzymol. 1999;301:211–220. doi: 10.1016/s0076-6879(99)01084-8. [DOI] [PubMed] [Google Scholar]
- 16.Paolocci N, et al. Positive inotropic and lusitropic effects of HNO/NO- in failing hearts: Independence from β-adrenergic signaling. Proc Natl Acad Sci USA. 2003;100:5537–5542. doi: 10.1073/pnas.0937302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tocchetti CG, et al. Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ Res. 2007;100:96–104. doi: 10.1161/01.RES.0000253904.53601.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai T, et al. Nitroxyl increases force development in rat cardiac muscle. J Physiol. 2007;580:951–960. doi: 10.1113/jphysiol.2007.129254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norris AJ, et al. Nitroxyl inhibits breast tumor growth and angiogenesis. Int J Cancer. 2007;122:1905–1910. doi: 10.1002/ijc.23305. [DOI] [PubMed] [Google Scholar]
- 20.Demaster EG, Redfern B, Nagasawa HT. Mechanisms of inhibition of aldehyde dehydrogenase by nitroxyl, the active metabolite of the alcohol deterrent agent cyanamide. Biochem Pharmacol. 1998;55:2007–2015. doi: 10.1016/s0006-2952(98)00080-x. [DOI] [PubMed] [Google Scholar]
- 21.Nagasawa HT, Shirota FN, DeMaster EG. Alcoholism: Aldehyde dehydrogenase inhibitors as alcohol deterrent agents. In: Torrence PF, editor. Biomedical Chemistry: Applying Chemical Principles to the Understanding and Treatment of Disease. New York: Wiley; 2000. pp. 73–97. [Google Scholar]
- 22.Fukuto JM, Carrington SJ. HNO signaling mechanisms. Antioxid Redox Signal. 2011;14:1649–1657. doi: 10.1089/ars.2010.3855. [DOI] [PubMed] [Google Scholar]
- 23.Kirsch M, Buscher A-M, Aker S, Schulz R, de Groot H. New insights into the S-nitrosothiol-ascorbate reaction: The formation of nitroxyl. Org Biomol Chem. 2009;7:1954–1962. doi: 10.1039/b901046g. [DOI] [PubMed] [Google Scholar]
- 24.Spencer NY, Patel NK, Keszler A, Hogg N. Oxidation and nitrosylation of oxyhemoglobin by S-nitrosoglutathione via nitroxyl anion. Free Radic Biol Med. 2003;35:1515–1526. doi: 10.1016/j.freeradbiomed.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 25.Rusche KM, Spiering MM, Marletta MA. Reactions catalyzed by tetrahydrobiopterin-free nitric oxide synthase. Biochemistry. 1998;37:15503–15512. doi: 10.1021/bi9813936. [DOI] [PubMed] [Google Scholar]
- 26.Xia Y, Zweier JL. Direct measurement of nitric oxide generation from nitric oxide synthase. Proc Natl Acad Sci USA. 1997;94:12705–12710. doi: 10.1073/pnas.94.23.12705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donzelli S, et al. Generation of nitroxyl by heme protein-mediated peroxidation of hydroxylamine but not N-hydroxy-L-arginine. Free Radic Biol Med. 2008;45:578–584. doi: 10.1016/j.freeradbiomed.2008.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith PAS, Hein GE. The alleged role of nitroxyl in certain reactions of aldehydes and alkyl halides. J Am Chem Soc. 1960;82:5731–5740. [Google Scholar]
- 29.Yi J, Namjou K, McCaan PJ, Richter-Addo GB. Simultaneous gas-phase detection of nitric oxide (NO) and nitrous oxide (N2O) from the decomposition of Angeli’s salt (Na2N2O3) at different pHs using tunable-diode laser absorption spectroscopy. Am J Biomed Sci. 2009;1:38–46. [Google Scholar]
- 30.Hughes MN, Wimbledon PE. The chemistry of trioxodinitrates. Part I. Decompostion of sodium trioxodinitrate (Angeli’s salt) in aqueous solution. J Chem Soc Dalton Trans. 1976;8:703–707. [Google Scholar]
- 31.Nishinaga A, Tojo T, Tomita H, Matsuura T. Base-catalyzed oxygenolysis of 3-hydroxyflavones. J Chem Soc Perkin Trans. 1979;10:2511–2525. [Google Scholar]
- 32.Lema’nska K, et al. The influence of pH on antioxidant properties and the mechanism of antioxidant action of hydroxyflavones. Free Radic Biol Med. 2001;31:869–881. doi: 10.1016/s0891-5849(01)00638-4. [DOI] [PubMed] [Google Scholar]
- 33.Brown SB, Rajananda V, Holroyd JA, Evans E, Glyn V. A study of the mechanism of quercetin oxygenation by oxygen-18 labeling. A comparison of the mechanisms with that of heme degradation. Biochem J. 1982;205:239–244. doi: 10.1042/bj2050239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balogh-Hergovich E, Speier G. Kinetics and mechanism of the base-catalyzed oxygenation of flavonol in DMSO-H2O solution. J Org Chem. 2001;66:7974–7978. doi: 10.1021/jo015517n. [DOI] [PubMed] [Google Scholar]
- 35.Kaizer J, Speier G. Radical-initiated oxygenation of flavonols by dioxygen. J Mol Catal A Chem. 2001;171:33–36. [Google Scholar]
- 36.Bartberger MD, Fukuto JM, Houk KN. On the acidity and reactivity of HNO in aqueous solution and biological systems. Proc Natl Acad Sci USA. 2001;98:2194–2198. doi: 10.1073/pnas.041481598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larcheveque M, Debal A. A convenient synthesis of γ-hydroxy and γ-keto nitriles. Synth Commun. 1980;10:49–57. [Google Scholar]
- 38.Hund HK, et al. Flavonol 2,4-dioxygenase from Aspergillus niger DSM 821, a type 2 Cu(II)-containing glycoprotein. Eur J Biochem. 1999;263:871–878. doi: 10.1046/j.1432-1327.1999.00574.x. [DOI] [PubMed] [Google Scholar]
- 39.Barney BM, Schaab MR, LoBrutto R, Francisco WA. Evidence for a new metal in a known active site: Purification and characterization of an iron-containing quercetin dioxygenase from Bacillus subtilis. Protein Expr Purif. 2004;35:131–141. doi: 10.1016/j.pep.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 40.Oka T, Simpson FJ. Quercetinase, a dioxygenase containing copper. Biochem Biophys Res Commun. 1971;43:1–5. doi: 10.1016/s0006-291x(71)80076-1. [DOI] [PubMed] [Google Scholar]
- 41.Reddi K, et al. Surface-associated material from the bacterium Acinobacillus actinomycetemcomitans contains a peptide which, in contrast to lipopolysaccharide, directly stimulates fibroblast interleukin-6 gene transcription. Eur J Biochem. 1996;236:871–876. doi: 10.1111/j.1432-1033.1996.00871.x. [DOI] [PubMed] [Google Scholar]
- 42.Fusetti F, et al. Crystal structure of the copper-containing quercetin dioxygenase from Aspergillus japonicas. Structure. 2002;10:259–268. doi: 10.1016/s0969-2126(02)00704-9. [DOI] [PubMed] [Google Scholar]
- 43.Merkens H, Kappl R, Jakob RP, Schmid FX, Fetzner S. Quercetinase QueD of Streptomyces sp. FLA, a monocupin dioxygenase with a preference for Nickel and Cobalt. Biochemistry. 2008;47:12185–12196. doi: 10.1021/bi801398x. [DOI] [PubMed] [Google Scholar]
- 44.Bowater L, Fairhurst SA, Just VJ, Bornemann S. Bacillus subtilis YxaG is a novel Fe-containing quercetin dioxygenase. FEBS Lett. 2004;557:45–48. doi: 10.1016/s0014-5793(03)01439-x. [DOI] [PubMed] [Google Scholar]
- 45.Gopal B, Madan LL, Betz SF, Kossiakoff A. The crystal structure of a quercetin dioxygenase from Bacillus subtilis suggests modulation of enzyme activity by a change in the metal ion at the active site(s) Biochemistry. 2005;44:193–201. doi: 10.1021/bi0484421. [DOI] [PubMed] [Google Scholar]
- 46.Ho T-L. The hard soft acids bases (HSAB) principle and organic chemistry. Chem Rev. 1975;75:1–20. [Google Scholar]
- 47.Kovaleva EG, Lipscomb JD. Crystal structures of Fe2+ dioxygenase superoxo, alkylperoxo, and bound product intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gunderson WA, et al. Electron paramagnetic resonance detection of intermediates in the enzymatic cycle of an extradiol dioxygenase. J Am Chem Soc. 2008;130:14465–14467. doi: 10.1021/ja8052255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mbughuni MM, et al. Trapping and spectroscopic characterization of an FeIII-superoxo intermediate from a nonheme mononuclear iron-containing enzyme. Proc Natl Acad Sci USA. 2010;107:16788–16793. doi: 10.1073/pnas.1010015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farmer PJ, Sulc F. Coordination chemistry of the HNO ligand with hemes and synthetic coordination complexes. J Inorg Biochem. 2005;99:166–184. doi: 10.1016/j.jinorgbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 51.Daiber A, et al. Isotope effects and intermediates in the reduction of NO by P450(NOR) J Inorg Biochem. 2002;88:343–352. doi: 10.1016/s0162-0134(01)00386-5. [DOI] [PubMed] [Google Scholar]
- 52.Einsle O, Messerschmidt A, Huber R, Kroneck PMH, Neese F. Mechanism of the six-electron reduction of nitrite to ammonia by cytochrome c nitrite reductase. J Am Chem Soc. 2002;124:11737–11745. doi: 10.1021/ja0206487. [DOI] [PubMed] [Google Scholar]
- 53.Averill BA. Dissimilatory nitrite and nitric oxide reductases. Chem Rev. 1996;96:2951–2954. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]
- 54.Blair E, Sulc F, Farmer PJ. Biomimetic NOx reductions by heme models and proteins. In: Zagal JH, Bedioui F, Dodelet JP, editors. N4 Macrocyclic Metal Complexes. New York: Springer; 2006. pp. 149–190. [Google Scholar]
- 55.Pervitsky D, Immoos C, Van der Veer W, Farmer PJ. Photolysis of the HNO adduct of myoglobin: Transient generation of the aminoxyl radical. J Am Chem Soc. 2007;129:9590–9591. doi: 10.1021/ja073420y. [DOI] [PubMed] [Google Scholar]
- 56.Dutton AS, Fukuto JM, Houk KN. Theoretical reduction potentials for nitrogen oxides from CBS-QB3 energetics and (C)PCM solvation calculations. Inorg Chem. 2005;44:4024–4028. doi: 10.1021/ic048734q. [DOI] [PubMed] [Google Scholar]
- 57.Lind J, Gábor MG. Kinetic and thermodynamic properties of the aminoxyl (NH2O•) radical. J Phys Chem A. 2006;110:192–197. doi: 10.1021/jp054747t. [DOI] [PubMed] [Google Scholar]
- 58.Doctorovich F, et al. Nitroxy (azanone) trapping by metalloporphyrins. Coord Chem Rev. 2011;255:2764–2784. [Google Scholar]
- 59.Carducci MD, Pressprich MR, Coppens P. Diffraction studies of photoexcited crystals: Metastable nitrosyl-linkage isomers of sodium nitroprusside. J Am Chem Soc. 1997;119:2669–2678. [Google Scholar]
- 60.Wondimagegn T, Ghosh A. A quantum chemical survey of metalloporphyrin-nitrosyl linkage isomers: Insights into the observation of multiple FeNO conformations in a recent crystallographic determination of nitrophorin. J Am Chem Soc. 2001;123:5680–5683. doi: 10.1021/ja004314y. [DOI] [PubMed] [Google Scholar]
- 61.Bitterwolf TE. Photochemical nitrosyl linkage isomerism/metastable states. Coord Chem Rev. 2006;250:1196–1207. [Google Scholar]
- 62.Lee Dong-Heon, Mondal Biplab, Karlin KD. Nitrogen monoxide and nitrous oxide binding and reduction. In: Tolman WB, editor. Activation of Small Molecules. Berlin: Wiley; 2006. pp. 43–79. [Google Scholar]
- 63.Linder DP, and Rodgers KR. Structural, electronic, and vibrational characterization of Fe-HNO porphyrinates by density functional theory. Inorg Chem. 2005;44:8259–8264. doi: 10.1021/ic0504745. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.