

1.0 INTRODUCTION
Autosomal recessive polycystic kidney disease (ARPKD) is characterized by progressive cystic degeneration of the kidneys resulting in kidney failure and congenital hepatic fibrosis (CHF) often complicated with portal hypertension1–3. Renal cysts in ARPKD originate from dilated collecting ducts. Most ARPKD patients present perinatally or in childhood; a minority present in adulthood.
ARPKD is caused by mutations in the PKHD1 gene that encodes fibrocystin/polyductin, a protein that functions on primary non-motile cilia4,5. ARPKD is the most common childhood-onset ciliopathy with an estimated frequency of 1 in 20, 000. The carrier frequency for pathogenic PKHD1 mutations is estimated to be approximately 1 in 706. Given the recessive pattern of inheritance, parents of ARPKD patients are not expected to develop clinical features. However, this has not been systematically studied.
We performed ultrasound (USG) evaluations on 110 parents and a maternal grandmother from 64 independent ARPKD families. We identified various abnormalities, including multiple liver cysts and increased renal medullary echogenicity. Here we present, the imaging, clinical and molecular findings in this group of ARPKD family members.
2.0 METHODS
2.1 Patients
All patients and their parents were enrolled in the protocol, “Clinical Investigations into the Kidney and Liver Disease in Autosomal Recessive Polycystic Kidney Disease/Congenital Hepatic Fibrosis and other Ciliopathies” (www.clinicaltrials.gov, trial NCT00068224), approved by the NHGRI Institutional Review Board and gave written informed consent. Patients who carried a clinical diagnosis of ARPKD made by a nephrologist were qualified to enroll. Evaluations of the index cases at the NIH Clinical Center included complete abdominal standard probe and high-resolution ultrasound (HR-USG) and magnetic resonance imaging (MRI), comprehensive biochemical testing of blood and urine and sequencing of the PKHD1 gene. The clinical diagnosis of ARPKD for each index case at NIH was based upon established criteria2,7 including typical kidney and liver involvement on imaging and/or biopsy and autosomal recessive inheritance. All parents were evaluated by standard and HR-USG; abdominal MRI and magnetic resonance cholangiopancreatography (MRCP) were performed on parents with abnormalities on standard probe USG.
2.2 Sequencing and Analysis
Complete sequencing of PKHD1 was performed in all patients. When DNA samples were available, mutation analysis in parents was performed to determine the parental origin of the mutations identified in the proband. Complete sequencing of the PKHD1 gene was performed in selected parents with imaging abnormalities.
For the longest open reading frame of PKHD1, coding exons (2–67) and their intronic boundaries were sequenced in two directions using a Beckman CEQ 8000 system (Beckman Coulter, Inc., Fullerton, CA) and a contract with Agencourt (Beverly, MA). Sequencher Software (GeneCodes, Ann Arbor, MI) was used to analyze DNA variants. The pathogenicity of missense variants was evaluated as described8 using segregation analysis, general population frequencies, 3 computational prediction tools (Align GVGD [http://agvgd.iarc.fr/agvgd_input.php]; PolyPhen [http://coot.embl.de/PolyPhen/]; and SNAP [http://cubic.bioc.columbia.edu/services/SNAP/]) and the splice variant interpretation software NetGene2 Server (http://www.cbs.dtu.dk/services/NetGene2/).
2.3 Imaging Studies
Complete abdominal USG evaluations were performed using standard (4 MHz) and high resolution (7 MHz) ultrasonographic probes by a single technologist (K.T.D.) (AVI Sequoia Inc, Mountain View, CA). Magnetic resonance imaging (MRI) was performed on a 1.5 or 3 Tesla machine (Philips Medical Systems, NA, Bothell, Washington; General Electric Healthcare, Waukesha, WI, USA) without intravenous contrast media. USG, MRI and MRCP images were evaluated by a team of 2 radiologists (B.I.T and P.C).
3.0 RESULTS
3.1 Patient Characteristics
In 93 patients the clinical diagnosis of ARPKD was confirmed after NIH evaluations. One family contributed 4 affected siblings, 7 families contributed 2 siblings, and one family contributed an aunt and niece pair leaving 82 independent families. In 73 patients from 63 families the diagnosis was confirmed molecularly; clinical and mutational data from these 73 patients were previously published8,9.
A total of 110 parents (62 mothers and 48 fathers) and a maternal grandmother from 63 independent ARPKD families were available for evaluation. Eighty five parents were from molecularly confirmed families and 25 were from ARPKD families in which the diagnosis was made based on clinical criteria. Mothers’ ages ranged from 27 to 66 years (39 ± 7.2), fathers’ ages ranged from 29 to 58 years (40 ± 6.3) and ages of the total group ranged from 27 to 66 years (39.5 ± 6.8).
3.2 Imaging Findings
Kidney USG showed increased medullary echogenicity in 6 mothers (5.5 %) whose ages ranged from 30 to 62 years (38.8 ± 11.8) (Table 1, Figure 1A–G). Multiple small liver cysts were identified in 10 parents (9 %) including 5 mothers and 5 fathers (Figure 2A–L). The ages of the parents with multiple liver cysts ranged from 36 to 49 years (44.2 ± 4.1). These liver cysts were mostly small and peripherally located. The findings of increased medullary echogenicity and liver cysts in all patients were detected by both standard and high resolution ultrasound probes. MRCP images were inconclusive in terms of the connectivity of the cysts to the biliary system (Figure 2A–F).
Table 1.
Kidney and Liver imaging findings in parents of children with ARPKD
| Parent No | Diagnosis in Offspring | PKHD1 mutation | Sex | Age | Affected status |
|---|---|---|---|---|---|
| 1 | ARPKD-mutation confirmed | p_Arg1624Trp | F | 62 | Bilateral nephrocalcinosis |
| 2 | ARPKD-clinical diagnosis | - | F | 33 | Bilateral nephrocalcinosis |
| 3 | ARPKD-mutation confirmed | p.Thr3035fs or p.Ala293Val** | F | 36 | Bilateral nephrocalcinosis |
| 4 | ARPKD-mutation confirmed | p.Thr36Met or p.lle222Val** | F | 39 | Bilateral nephrocalcinosis |
| 5 | ARPKD-mutation confirmed | p.Thr36Met | F | 30 | Bilateral nephrocalcinosis |
| 6 | ARPKD-clinical diagnosis | - | F | 33 | Bilateral nephrocalcinosis |
| 7 | ARPKD-mutation confirmed | p Thr36Met | F | 45 | Multiple liver cysts |
| 8 | ARPKD-mutation confirmed | p Thr36Met | M | 49 | Multiple liver cysts |
| 9 | ARPKD-clinical diagnosis | - | M | 49 | Multiple liver cysts |
| 10 | ARPKD-mutation confirmed* | - | F | 42 | Multiple liver cysts |
| 11 | ARPKD-mutation confirmed | p.Thr36Met | F | 41 | Multiple liver cysts, 2 kidney cysts |
| 12 | ARPKD-mutation confirmed | p.Phe2374fs | M | 36 | Multiple liver cysts, moderately hyperechoic liver |
| 13 | ARPKD-mutation confirmed | p.Thr36Met | M | 42 | Multiple liver cysts, gall bladder polyp |
| 14 | ARPKD-mutation confirmed | p.Val1741Met | F | 48 | Multiple liver cysts, moderately hyperechoic liver, splenomegaly |
| 15 | ARPKD-mutation confirmed | p.lle246Thr | F | 46 | Multiple liver cysts, splenomegaly, 2 kidney cysts |
| 16 | ARPKD-mutation confirmed | p.Trp2690Arg or IVS55+1G>A** | M | 44 | Multiple liver cysts, unilateral multiple kidney cysts |
| 17 | ARPKD-mutation confirmed | p.Arg2573Cys or unknown** | F | 40 | Unilateral multiple kidney cysts |
| 18 | ARPKD-mutation confirmed | p.Tyr486X | M | 46 | Moderately hyperechoic liver, single liver cyst, single kidney cyst |
| 19 | ARPKD-mutation confirmed | p.Thr3035fs or p.Ala293Val** | M | 36 | Moderately hyperechoic liver, splenomegaly |
| 20 | ARPKD-mutation confirmed | p_Ala1254fs | M | 41 | Gall bladder polyp |
| 21 | ARPKD-mutation confirmed | p.Thr36Met | M | 44 | Gall bladder polyps |
| 22 | ARPKD-clinical diagnosis | - | M | 30 | Splenomegaly |
F: female, M: male,
In this patient only one PKHD1 mutation that was inherited from the father was identified,
In these families parental orgin of the mutations could not be determined since parental DNA was not available.
Figure 1.

Kidney USG images of parents of patients with ARPKD. Standard probe (A) and high resolution (B) renal USG images of 62 years old mother (Table 1, parent no 1), showing increased medullary echogenicity consistent with nephrocalcinosis. Standard probe (C) and high resolution (D) renal USG images of 33 years old mother (Table 1, parent no 6) showing medullary nephrocalcinosis occupying the outer rim of the medulla creating a donut like appearance involving the medullary pyramids. E. Renal USG of 33 year old mother (Table 1, parent no 2) demonstrating echogenic medulla consistent with nephrocalcinosis. F. Multiple echogenic foci within renal medulla of 39 year old mother (Table 1, parent no 4). G. Increased medullary echogenicity of 30 year old mother (Table 1, parent no 5). H. Multiple parapelvic cysts of left kidney of 44 year old father (Table 1, parent no 16), who also had innumerable small liver cysts (see Figure 2A).
Figure 2.
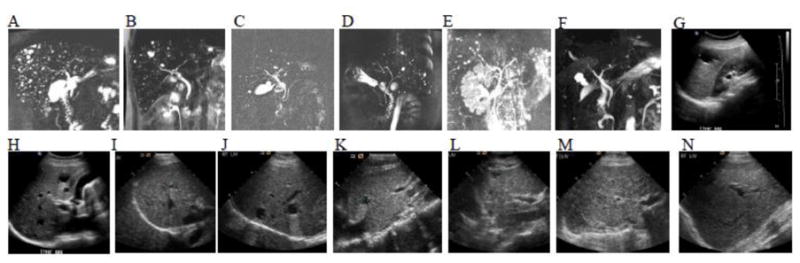
MRCP and USG images of parents of patients with ARPKD showing multiple liver cysts. A. Innumerable small liver cysts ranging from a few mm to 1 cm are visible throughout the liver of 44 year old father (Table 1, parent no 16), who also had multiple parapelvic cysts of the left kidney (see Figure 1H); his right kidney appeared normal. B. MRCP of 41 year old mother (Table 1, parent no 11) showing innumerable small liver cysts; the majority are less than a half cm, a few are about a cm, the largest is about 2 cm. C. Multiple liver cysts, the largest about 1.3 cm adjacent to the gallbladder, others less than a centimeter in a 42 year old father (Table 1, parent no: 13). D. Innumerable small cysts present in both lobes of the liver of a 36 years old father (Table 1, parent no: 12). E. Multiple subcentimeter cysts throughout the liver of 46 years old mother (Table 1. Parent no: 15). MRCP (F) and USG images (G and H) of a 42 years old mother (Table 1, parent no: 10) demonstrating multiple liver cysts, the largest measuring 3 cm. I. Multiple liver cysts on USG of 49 years old father (Table 1. Parent no: 8). J. Fourty nine year old father (Table 1, parent no: 9) with multiple liver cysts. K. Forty five years old mother (Table 1, parent no: 7) with multiple liver cysts on USG. L. USG of a 48 years old mother (Table 1, parent no: 14) demonstrating moderately echogenic liver with multiple cysts. M. Liver USG of the 76 year old grandmother showing heterogeneous appearance of the liver parenchyma with increased echogenicity in comparison to normal (N).
A 44 years old father with multiple liver cysts (Table 1, parent no: 16, Figure 2A) also had unilateral multiple parapelvic kidney cysts (Figure 1H). Multiple unilateral kidney cysts were detected in another parent (#17). A moderately increased liver echo pattern associated with coarse texture suggestive of CHF, was detected in 4 parents (table 1, parents 12, 14, 18 and 19, Figure 2L) including 2 with multiple liver cysts (parents 12 and 14) and two with splenomegaly (parents 14 and 19)10. A total of 9 parents had mild splenomegaly (spleen lengths 15–16 cm); in 5, this was in the absence of fatty changes of the liver. Incidentally, fatty changes in the liver suggested by increased echogenicity in association with smooth echo texture were detected in 20 parents10.
Two other parents with multiple liver cysts (Table 1, parent no 11 and 15) at ages 41 and 46, also had 2 kidney cysts each. Five other fathers ages 38 to 46 had single liver cysts. Two mothers at ages 41 and 46 and a father at age 36 had 2 renal cysts and single renal cysts were detected in 7 parents ages 36 to 49.
Other findings, which may be purely coincidental included gall bladder polyps in 3 parents, liver hemangioma in 3 mothers and a father, left duplicated collecting system in one mother and gall stones in one mother. The remainder of the abdominal USG examinations, including pancreas, were normal in all parents.
3.3 Case of Maternal Grandmother
A 76 year-old maternal grandmother was evaluated because she was documented to have CHF on liver biopsy performed at age 75 prompted by the incidental CT finding of abnormal liver parenchyma. USG imaging at NIH (Figure 2M) showed markedly increased liver echogenicity with moderately coarse echotexture; spleen volume was normal at 100 ml and kidneys were unremarkable.
3.4 Biochemical Findings
Liver and kidney-related serum and urine chemistries including serum cystatin C, PTH, and measurements of calcium and creatinine in 24 hour urines were performed in 6 patients including 3 mothers with increased medullary echogenicity (Table 1, parent no: 1, 3 and 5), 2 mothers with multiple liver cysts (Table 1, parents #10 and 11), and the maternal grandmother. All were within normal limits except for elevated alanine aminotransferase (44 U/L, normal 6–41), aspartate aminotransferase (43 U/L, normal 9–34) and gamma glutamyl transferase (151 U/L, normal 7–38) in the grandmother. Specifically, there was no evidence for hypercalciuria, hyperparathyroidism, or renal tubular acidosis.
3.5 PKHD1 Sequencing Findings
Complete sequencing of the coding exons of PKHD1 was performed on 16 parents with abnormal imaging findings and the maternal grandmother in an affort to identify additional mutations. These sequences confirmed the mutation transmitted to the proband but did not reveal any other pathogenic variants. The maternal grandmother was heterozygous for the p.Thr36Met mutation in PKHD1.
4.0 DISCUSSION
Our findings on 110 ARPKD parents and a maternal grandmother suggest that carrier status for PKHD1 mutations creates a predisposition to multiple liver cysts and renal involvement associated with increased medullary echogenicity on USG.
The ultrasound finding of increased medullary echogenicity is commonly reported as “nephrocalcinosis”, a term that describes the deposition of radiologically-demonstrable calcium salts in renal parenchyma11,12. The differential diagnosis of “nephrocalcinosis” diagnosed radiologically includes true histopathological nephrocalcinosis and medullary sponge kidney (MSK), which itself can be associated with calcium depositions (Table 2)11,13. Most cases of MSK are sporadic; the presence of rare dominantly inherited forms of MSK, and its association with different malformation syndromes, suggest that MSK is an inherited developmental disorder14–16. The molecular basis of MSK remains largely unknown, two sequence variants of the GDNF gene were recently reported in a few MSK patients14. While intravenous pyelography is the gold standard for the diagnosis of MSK, computerized tomography has become the preferred diagnostic modality. MSK is clinically symptomatic in only a subset of patients and, therefore, is probably under-diagnosed15. The prevalence of MSK is estimated at 5 in 10 000 – 100 00014,15. Based upon our findings, we speculate that carrier (heterozygous) status for PKHD1 mutations may be one underlying molecular cause of MSK. Supporting this hypothesis, the microscopic pathology of MSK resembles that of ARPKD; both are characterized by dilated collecting ducts, although MSK involves only the precalyceal collecting ducts14,15. Furthermore, MSK has been reported in patients with autosomal dominant polycystic kidney disease17 and congenital hepatic fibrosis18, most commonly caused by homozygous mutations in PKHD1. Given that the estimated carrier frequency for PKHD1 mutations is 1 in 70 and 5 % of ARPKD parents have increased medullary echogenicity, our data suggest that the frequency of PKHD1-related MSK would be approximately 7 in 10 000.
Table 2.
Conditions associated with the radiological diagnosis of nephrocalcinosis
| Primary hyperparathyroidism (OMIM #145000 and #145001) |
| Distal (type 1) renal tubular acidosis (OMIM #179800) |
| Medullary sponge kidney |
| Neonatal furosemide therapy |
| X-linked hypophosphatemic rickets (Phosphate and vitamin D) (OMIM #307800) |
| Cystinosis (OMIM #219800) |
| Hyperphosphatasia |
| Hypomagnesemia |
| Idiopathic hypercalciuria |
| Vitamin D intoxication |
| Hyperoxaluria (OMIM #259900) |
| Hypothyroidism |
| Cushing’s syndrome |
| Prolonged immobilization |
| Bartter’s syndrome (OMIM #607364) |
| Glycogen storage disease Type 1 (OMIM #232200) |
| Lowe’s oculocerebrorenal syndrome (OMIM #309000) |
| Dent’s Disease (OMIM #300009) |
| Acetazolamide |
| Tuberculosis |
| Sarcoidosis |
| Carrier status for autosomal recessive polycystic kidney disease |
Polycystic liver disease (PLD) occurs most commonly as a part of ADPKD. PLD without renal cysts, often referred to as isolated PLD, is genetically distinct from ADPKD19,20. Clinical diagnostic criteria for PLD are available only for at-risk individuals; 21 in patients younger than 40 years, the presence of any liver cysts is considered to be diagnostic of PLD, while in those older than 40, 4 or more cysts are required to differentiate PLD from simple liver cysts20. These criteria, however, need to be further vetted in a larger cohort of PCLD patients. Isolated PLD is genetically heterogeneous19; approximately 20 to 30% of the isolated cases are due to mutations in either PRKCSH22 or SEC63; the molecular cause of the remainder of isolated PLD is unknown. PRKCSH encodes the beta-subunit of glucosidase II, an ER glucosidase that is involved in quality control of newly synthesized glycoproteins. SEC63 encodes Sec63, an integral ER membrane protein that is a part of the multi-protein translocon, the translocation machinery for integral membrane and secreted proteins. It was recently shown that glucosidase IIβ and Sec63p are required in mice for adequate expression of a functional complex of the polycystic kidney disease gene products, polycystin-1, polycystin-2 and fibrocystin/polyductin encoded by Pkhd123, 24. This is supported by the fact that the histopathological features of isolated PLD are very similar to those of the PLD in ADPKD; in both disorders cysts originate from biliary microhamartomas, also termed von Meyenburg’s complexes25,26. However, the reason why defects in glucosidase IIβ and Sec63 do not cause renal cystic disease is unknown.
The majority of PLD patients are clinically asymptomatic, with normal liver enzymes and intact synthetic function21,27. The incidence of isolated PLD in autopsy series26,28,29 (2–3 in 10 000) is higher than its estimated clinical incidence (< 1 in 10 000), suggesting that most cases remain undetected. Our data suggest that heterozygous carriers of PKHD1 mutations are predisposed to PLD. In all ARPKD parents with multiple liver cysts, these cysts were initially detected by standard probe ultrasound; in every case the number of cysts on initial evaluation exceeded 4. Based on the estimated carrier frequency for PKHD1 mutations of 1 in 70, and our finding that 9 % of ARPKD parents have PLD, the frequency of PKHD1-related PLD would be approximately 1 in 1000. Given the small size of the liver cysts and normal liver-related blood chemistries in ARPKD parents, this condition is more likely to remain undiagnosed, explaining the discrepancy between disease frequencies.
In addition to CHF, ARPKD patients might have liver cysts that are in continuity with the biliary tree, differing form the isolated cysts in PLD. MRCP images of the ARPKD parents were not conclusive in terms of the connectivity of the cysts to biliary ducts. However, the small size and peripheral location of these cysts suggest that they are isolated from the biliary tree, similar to the PLD cysts. This difference in the characteristics of liver cysts between children with ARPKD and their parents is surprising. The increased liver echogenicity associated with coarse echotexture identified in some parents and the biopsy proven CHF in the maternal grandmother presented suggest some PKHD1 carriers are also prone to CHF.
To exclude the possibility that these parents had two PKHD1 mutations, we performed complete sequencing of the gene. However, we found no additional PKHD1 mutations in these individuals. Other genes, including those encoding ciliary proteins, may contribute to cyst formation through synergistic heterozygosity30,31; this could explain why only a subset of PKHD1 mutation carriers develop these liver and or kidney findings.
In summary, our data suggest that carrier status for ARPKD is a predisposition to renal involvement associated with increased medullary echogenicity on USG (possibly MSK), and liver involvement in the form of asymptomatic PLD and CHF in some cases. All these PKHD1 mutation carriers with abnormal kidney and/or liver imaging were clinically asymptomatic. It remains to be determined if some of these individuals might become symptomatic as they age.
Acknowledgments
We thank the ARPKD Alliance for their extensive support and the patients and their families who generously participated in this investigation. Supported by the Intramural Research Programs of the National Human Genome Research Institute, National Cancer Institute and the NIH Clinical Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6.0 References
- 1.Zerres K, Rudnik-Schoneborn S, Deget F, et al. Autosomal recessive polycystic kidney disease in 115 children: clinical presentation, course and influence of gender. Arbeitsgemeinschaft fur Padiatrische, Nephrologie. Acta Paediatr. 1996 Apr;85(4):437–445. doi: 10.1111/j.1651-2227.1996.tb14056.x. [DOI] [PubMed] [Google Scholar]
- 2.Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003 May;111(5 Pt 1):1072–1080. doi: 10.1542/peds.111.5.1072. [DOI] [PubMed] [Google Scholar]
- 3.Gunay-Aygun M, Avner ED, Bacallao RL, et al. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis: summary statement of a first National Institutes of Health/Office of Rare Diseases conference. J Pediatr. 2006 Aug;149(2):159–164. doi: 10.1016/j.jpeds.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Onuchic LF, Furu L, Nagasawa Y, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002 May;70(5):1305–1317. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002 Mar;30(3):259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 6.Zerres K, Rudnik-Schoneborn S, Steinkamm C, Becker J, Mucher G. Autosomal recessive polycystic kidney disease. J Mol Med. 1998 Apr;76(5):303–309. doi: 10.1007/s001090050221. [DOI] [PubMed] [Google Scholar]
- 7.Zerres K, Volpel MC, Weiss H. Cystic kidneys. Genetics, pathologic anatomy, clinical picture, and prenatal diagnosis. Hum Genet. 1984;68(2):104–135. doi: 10.1007/BF00279301. [DOI] [PubMed] [Google Scholar]
- 8.Gunay-Aygun M, Tuchman M, Font-Montgomery E, Lukose L, Edwards H, Garcia A, Ausawarat S, Ziegler S, Piwnica-Worms K, Bryant J, Bernardini I, Fischer R, Huizing M, Guay-Woodford L, Gahl W. PKHD1 Sequence Variations in 78 Children and Adults with Autosomal Recessive Polycystic Kidney Disease and Congenital Hepatic Fibrosis. Molecular Genetics and Metabolism. 2010 doi: 10.1016/j.ymgme.2009.10.010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunay-Aygun M, Font-Montgomery E, Lukose L, et al. Correlation of kidney function, volume and imaging findings, and PKHD1 mutations in 73 patients with autosomal recessive polycystic kidney disease. Clin J Am Soc Nephrol. 2010 Jun;5(6):972–984. doi: 10.2215/CJN.07141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tchelepi H, Ralls PW, Radin R, Grant E. Sonography of diffuse liver disease. J Ultrasound Med. 2002 Sep;21(9):1023–1032. doi: 10.7863/jum.2002.21.9.1023. quiz 1033–1024. [DOI] [PubMed] [Google Scholar]
- 11.Manz F, Jaschke W, van Kaick G, Waldherr R, Willich E. Nephrocalcinosis in radiographs, computed tomography, sonography and histology. Pediatr Radiol. 1980;9(1):19–26. doi: 10.1007/BF00973964. [DOI] [PubMed] [Google Scholar]
- 12.Stout HA, Akin RH, Morton E. Nephrocalcinosis in routine necropsies; its relationship to stone formation. J Urol. 1955 Jul;74(1):8–22. doi: 10.1016/S0022-5347(17)67238-0. [DOI] [PubMed] [Google Scholar]
- 13.Hoppe B, Kemper MJ. Diagnostic examination of the child with urolithiasis or nephrocalcinosis. Pediatr Nephrol. 2010 Mar;25(3):403–413. doi: 10.1007/s00467-008-1073-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torregrossa R, Anglani F, Fabris A, et al. Identification of GDNF gene sequence variations in patients with medullary sponge kidney disease. Clin J Am Soc Nephrol. 2010 Jul;5(7):1205–1210. doi: 10.2215/CJN.07551009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gambaro G, Feltrin GP, Lupo A, Bonfante L, D’Angelo A, Antonello A. Medullary sponge kidney (Lenarduzzi-Cacchi-Ricci disease): a Padua Medical School discovery in the 1930s. Kidney Int. 2006 Feb;69(4):663–670. doi: 10.1038/sj.ki.5000035. [DOI] [PubMed] [Google Scholar]
- 16.Kuiper JJ. Medullary sponge kidney in three generations. N Y State J Med. 1971 Nov;71(22):2665–2669. [PubMed] [Google Scholar]
- 17.Torres VE, Erickson SB, Smith LH, Wilson DM, Hattery RR, Segura JW. The association of nephrolithiasis and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1988 Apr;11(4):318–325. doi: 10.1016/s0272-6386(88)80137-9. [DOI] [PubMed] [Google Scholar]
- 18.Kerr DN, Warrick CK, Hart-Mercer J. A lesion resembling medullary sponge kidney in patients with congenital hepatic fibrosis. Clin Radiol. 1962 Apr;13:85–91. doi: 10.1016/s0009-9260(62)80024-5. [DOI] [PubMed] [Google Scholar]
- 19.Tahvanainen P, Tahvanainen E, Reijonen H, Halme L, Kaariainen H, Hockerstedt K. Polycystic liver disease is genetically heterogeneous: clinical and linkage studies in eight Finnish families. J Hepatol. 2003 Jan;38(1):39–43. doi: 10.1016/s0168-8278(02)00348-3. [DOI] [PubMed] [Google Scholar]
- 20.Qian Q. Isolated polycystic liver disease. Adv Chronic Kidney Dis. 2010 Mar;17(2):181–189. doi: 10.1053/j.ackd.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qian Q, Li A, King BF, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003 Jan;37(1):164–171. doi: 10.1053/jhep.2003.50006. [DOI] [PubMed] [Google Scholar]
- 22.Li A, Davila S, Furu L, et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003 Mar;72(3):691–703. doi: 10.1086/368295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fedeles SV, Tian X, Gallagher AR, et al. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet. 2011;43(7):639–647. doi: 10.1038/ng.860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drenth JP, Martina JA, van de Kerkhof R, Bonifacino JS, Jansen JB. Polycystic liver disease is a disorder of cotranslational protein processing. Trends Mol Med. 2005 Jan;11(1):37–42. doi: 10.1016/j.molmed.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Desmet VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme “ductal plate malformation”. Hepatology. 1992 Oct;16(4):1069–1083. doi: 10.1002/hep.1840160434. [DOI] [PubMed] [Google Scholar]
- 26.Karhunen PJ, Tenhu M. Adult polycystic liver and kidney diseases are separate entities. Clin Genet. 1986 Jul;30(1):29–37. doi: 10.1111/j.1399-0004.1986.tb00565.x. [DOI] [PubMed] [Google Scholar]
- 27.Everson GT, Scherzinger A, Berger-Leff N, et al. Polycystic liver disease: quantitation of parenchymal and cyst volumes from computed tomography images and clinical correlates of hepatic cysts. Hepatology. 1988 Nov-Dec;8(6):1627–1634. doi: 10.1002/hep.1840080626. [DOI] [PubMed] [Google Scholar]
- 28.Melnick PJ. Polycystic liver; analysis of seventy cases. AMA Arch Pathol. 1955 Feb;59(2):162–172. [PubMed] [Google Scholar]
- 29.Feldman M. Polycystic disease of the liver. Am J Gastroenterol. 1958 Jan;29(1):83–86. [PubMed] [Google Scholar]
- 30.Dipple KM, McCabe ER. Phenotypes of patients with “simple” Mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet. 2000 Jun;66(6):1729–1735. doi: 10.1086/302938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vockley J, Rinaldo P, Bennett MJ, Matern D, Vladutiu GD. Synergistic heterozygosity: disease resulting from multiple partial defects in one or more metabolic pathways. Mol Genet Metab. 2000 Sep-Oct;71(1–2):10–18. doi: 10.1006/mgme.2000.3066. [DOI] [PubMed] [Google Scholar]